DOI:
10.1039/C6DT00538A
(Paper)
Dalton Trans., 2016,
45, 6449-6455
Binding of molecular oxygen by an artificial heme analogue: investigation on the formation of an Fe–tetracarbene superoxo complex†
Received
6th February 2016
, Accepted 26th February 2016
First published on 26th February 2016
Abstract
The dioxygen reactivity of a cyclic iron(II) tetra–NHC-complex (NHC: N-heterocyclic carbene) is investigated. Divergent oxidation behavior is observed depending on the choice of the solvent (acetonitrile or acetone). In the first case, exposure to molecular oxygen leads to an oxygen free Fe(III) whereas in the latter case an oxide bridged Fe(III) dimer is formed. In acetone, an Fe(III)-superoxide can be trapped, isolated and characterized as intermediate at low temperatures. An Fe(III)–O–Fe(III) dimer is formed from the Fe(III) superoxide in acetone upon warming and the molecular structure has been revealed by single crystal X-ray diffraction. It is shown that the oxidation of the Fe(II) complex in both solvents is a reversible process. For the regeneration of the initial Fe(II) complex both organic and inorganic reducing agents can be used.
Introduction
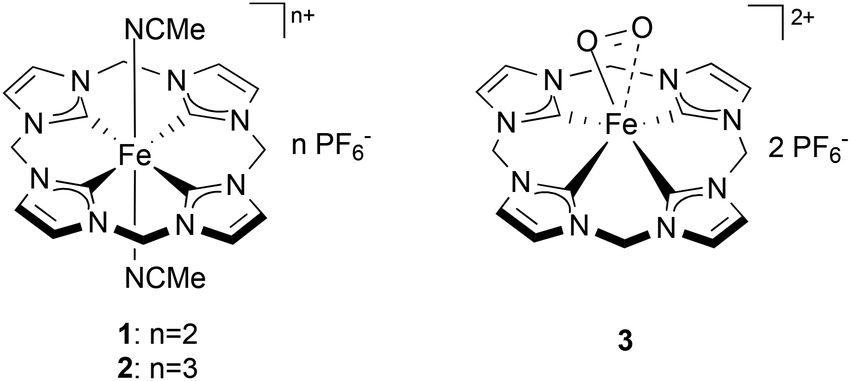
Understanding the nature of the intermediates of dioxygen activation by iron complexes is of high importance given the role that they play in catalysis and in a number of metabolic reactions.1 Amongst them, Fe superoxo species recently have been proposed as key intermediates in the oxidation mechanism of a number of non-heme Fe complexes and enzymes.2 Direct spectroscopic evidence (by Raman, Mössbauer, EPR, UV/Vis) of non-heme Fe superoxo species has been reported for artificial3 and natural4 Fe-based species upon exposure to dioxygen in solution. Structural information on Fe superoxo intermediates has been obtained crystallographically for a few biological Fe-complexes5 and, recently, also for a synthetic, non-heme, species.6 Nevertheless, the direct isolation of non-heme superoxo species remains challenging and no examples of truly organometallic systems (i.e. containing Fe–C bonds) have been reported so far. Recently, a biomimetic non-heme Fe-complex bearing an N-heterocyclic tetracarbene ligand (1, Chart 1) was synthesized, showing remarkable structural and electronic affinity with heme systems.7 Complex 1, and especially its oxidized analogue (2), have proven to be excellent epoxidation catalysts using aqueous hydrogen peroxide as an oxidant.8 Similar Fe(II) NHC compounds were recently reported as active catalysts for olefin epoxidation, aromatic hydroxylation, and the oxidation of unreactive C–H bonds.9 In this work the interaction of 1 with dioxygen is investigated in different solvents and under various experimental conditions. During this study, the formation of a new, isolable complex (3, Chart 1) was observed from the reaction of 1 and O2 at low temperature. The superoxidic nature of 3 has been investigated and it is supported by EPR spin trap experiments, NMR spectroscopy and DFT calculations. The interplay between 1, superoxide 3 and the other oxidation products of 1 is highlighted by reactivity studies showing the reversibility of the oxidative pathways arising from this compound and the involvement of 3 as an intermediate in these reactions.
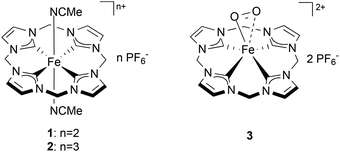 |
| | Chart 1 Fe tetracarbene complexes 1, 2 and the suggested structure of the intermediate 3. | |
Results and discussion
Preliminary observations and UV-Vis spectroscopy studies
The reactivity of Fe complexes with dioxygen has been previously studied. The reaction leads to dimeric Fe(III)–O–Fe(III) species according with the mechanism presented in Scheme 1.10
 |
| | Scheme 1 Stepwise oxidation of an Fe(II) complex to a dimeric Fe(III)–O–Fe(III) species by molecular oxygen. | |
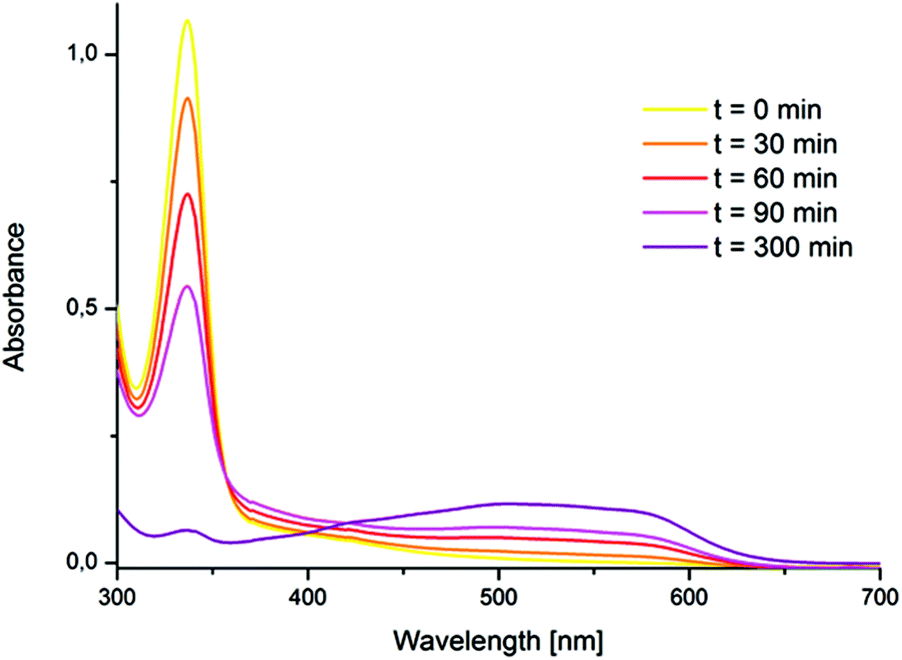
The formation of a tetra NHC ligated FeOFe complex with oxygen in acetonitrile was reported by Meyer et al.11 In spite of very similar structural features between 1 and the “Meyer-system”, the reaction of 1 with oxygen in acetonitrile did not yield the expected FeOFe complex. In this case, oxygen-free species 2 was irreversibly formed and isolated. (Scheme 2, Path A) 2 was reported previously, but its preparation was achieved by oxidation of 1 with stoichiometric amounts of thianthrenyl hexafluorophosphate.8 In spite of the absence of a Fe–O bond in 2, the transformation of 1 to 2 was found to proceed with the intake of nearly 1 equiv. O2, being indicative for a stoichiometric reaction of 1 and O2 (see the ESI,† section 6). Accordingly, the process of formation of 2 from 1 in acetonitrile without observable formation of a FeOFe dimer, is analogous to the oxidation of hemoglobin (FeII) by O2 to afford methemoglobin (FeIII) and a superoxide.12 The reaction of 1 with O2 (1 bar) in acetonitrile to afford compound 2 was accompanied by a shift in the maximum UV/Vis absorption from 337 nm to 505 nm (Fig. 1, see also Fig. S1†). The prominent absorption of 1 at 337 nm disappears in the presence of molecular oxygen. Simultaneously, a broad absorption band forms at 505 nm. This band can be assigned to an Fe(III)–NHC LMCT transition.13 The broad shape of the absorption is very similar to that reported by Meyer, Kühn et al. for the oxidized Fe(II)–tetracarbene,13 although it is slightly blue shifted (λmax = 505 nm for 2versus λmax = 540 nm). At 1 bar O2, the oxidation of 1 by molecular oxygen in acetonitrile is a slow process that could be monitored by UV/Vis spectroscopy (Fig. 1); the complete conversion of 1 to 2 was observed within 5 h at room temperature. The relatively low reaction rate under these conditions could be due to the competing coordination of either acetonitrile or oxygen at the Fe(II) center.
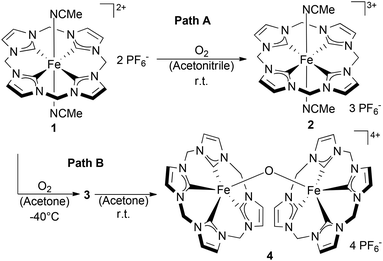 |
| | Scheme 2 Divergent oxidation behavior of 1 in acetone and in acetonitrile leading to complexes 3, 4 and 2 respectively. | |
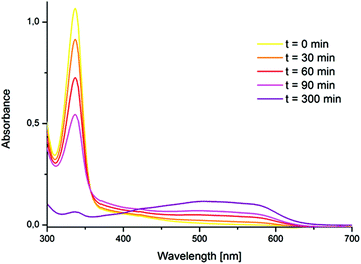 |
| | Fig. 1 UV/Vis kinetics of the formation of 2 from 1 (initial concentration 2 × 10−4 M) and O2 in acetonitrile (r.t., 1 bar). 1 (λmax = 337 nm; ε337 = 5450 l mol−1 cm−1); 2 (λmax = 505 nm; ε505 = 1100 l mol−1 cm−1). | |
This hypothesis is corroborated by the observation that in the presence of DMSO (10% v/v) no formation of 2 or 3 is detected under ambient conditions. Instead, the formation of a stable DMSO-substituted derivative of 1 is observed.7 Moreover, the amount of complex 2 forming within 1 h by the reaction with O2 increases with the partial O2 pressure (see in the ESI,† section 7). Taken together, these findings indicate that the key step in the oxidation of 1 to 2 is an oxygen-axial ligand exchange that is hampered by axial ligand coordination (MeCN, DMSO, etc.). The overall process is assumed to proceed with the initial displacement of one of the acetonitrile ligands in the axial position and with the formation of a superoxo FeIII–O2 adduct complex or of a dimeric FeIII–O–O–FeIII species. The fate of the (presumably) formed superoxide or peroxide anion upon formation of 2 is not clear as no additional products of oxidation could be found in solution. One plausible reaction pathway might involve a non-innocent role of the solvent. It is known that acetonitrile can be oxidized to glycolonitrile by molecular oxygen in the presence of FeIII–O–O–FeIII complexes. The reaction has been proposed to proceed via α-proton abstraction by the superbasic peroxo-complex and formation of a cyanomethide intermediate.14
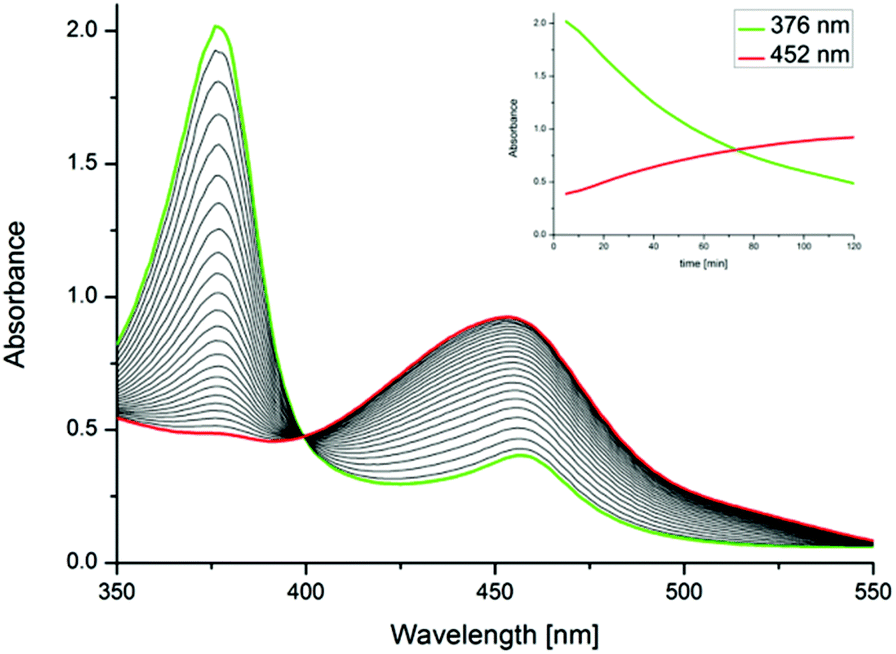
Indeed, when different nitriles such as benzonitrile or pivalonitrile, are used as solvents for the reaction of 1 and O2, the formation of Fe(III)–O–Fe(III) complex dimer 4 (Scheme 2) was observed instead of 2. This indicates that in the absence of an abstractable α-proton at the nitrile moiety the reaction between 1 and dioxygen proceeds along the path displayed in Scheme 1. Furthermore, it is known that the metabolism of acetonitrile by the Fe-containing enzyme sMMO leads to the formation of cyanide via glycolonitrile as an intermediate.15 Therefore, the formation and rapid decomposition of chemically unstable glycolonitrile16 to afford minute amounts of gaseous products might explain the absence of additional oxygenated products detectable in solution upon formation of 2 in acetonitrile. Further observations on the reactivity of isolated superoxide 3 with acetonitrile (vide infra) seem to support this hypothesis. In acetone, a different reactivity pattern (Scheme 2, Path B) was observed leading to dimeric complex 4. This pathway proceeds possibly through the mechanism depicted in Scheme 1. The formation of Fe(III)–O–Fe(III) dimers from Fe(II) and molecular oxygen is well known for both heme and non-heme iron compounds.10 Interestingly, when the reaction of 1 with O2 is carried out at low temperatures in acetone an intermediate compound was observed by UV/Vis spectroscopy (Fig. 2). The formation of the new species from 1 at −20 °C is accompanied by the rise of a single absorption in the UV/Vis spectrum at 452 nm and by the disappearance of the absorption of 1 at 376 nm (Fig. 2). This compound was proposedly identified as the Fe(O2) species 3 (Scheme 2) based on EPR and 1H NMR spectroscopies, reactivity studies and theoretical calculations (vide infra). When the solution of 3 is warmed up, dimeric complex 4 is formed. At 0 °C, the band at 452 nm disappears within 4 h due to decay of 3 to 4, the latter being insoluble in acetone. The formation of 3 in acetone was observed to occur already at low temperatures (approximately −70 °C), albeit at very low rates. Quantitative conversion of 1 to 3 was observed at ca. −40 °C. At this temperature, 3 is stable for several hours in solution and can be isolated by precipitation with cold diethyl ether with the resulting orange solid being stable at −30 °C for some days.
 |
| | Fig. 2 UV/Vis kinetics of the formation of 3 from 1 and O2 in acetone, sampled every 5 min for 2 h. ([1]0 = 4 × 10−4 M at −20 °C, 1 bar O2). 1, (λmax = 376 nm; ε376 = 5400 l mol−1 cm−1); 3, (λmax = 452 nm; ε452 = 2700 l mol−1 cm−1). Insert: time dependent trace of the absorptions at 376 and 452 nm. | |
EPR spectroscopy
EPR studies were carried out in order to investigate the composition of 3. Paramagnetic, oxygen-free compound 2 displays a singlet signal at g = 2.10 in acetone.8 Due to their diamagnetic nature, complex 1 and in situ generated compound 3 do not show any EPR activity in acetone, with the exception of a small EPR signal arising from minor decomposition of 3 to 2.8 To confirm the presence of an iron-bound superoxide, 5,5-dimethyl-1-pyrroline N-oxide (DMPO) was utilized as superoxide trapping reagent. Oxygen centered radicals can react with DMPO to form EPR active oxidation products.17 A blank experiment using pure DMPO did not show any EPR activity (Fig. 3, left). After the reaction of 2 with DMPO under inert gas, a triplet signal with a g = 1.97 (Fig. 3, left) was observed, while the signal at g = 2.10 (relative to pure 2) disappeared. To a minor extent, this signal was also found for 1 in the presence of DMPO, most likely arising from partial oxidation of 1 to 2 by the N-oxide. When 2 was reacted with DMPO in the presence of O2; no difference with the oxygen-free reaction was observed for 2 + DMPO + O2 (g = 1.97), as Fe(III) cannot reduce molecular oxygen. For 1 + DMPO + O2 a triplet signal with g = 1.95 was obtained (Fig. 3, middle).18
 |
| | Fig. 3 X-band EPR spectra (298 K) in acetone of: pure DMPO, 1 + DMPO and 2 + DMPO under an inert atmosphere (left), 1 + DMPO in the presence of oxygen (middle) and of isolated 3 + DMPO (right). | |
Finally, the reaction of isolated 3 with DMPO under an inert atmosphere gives rise to two independent signals with g = 1.97 and g = 1.95 (Fig. 3, right). The signal at g = 1.95 correlates to that found for 1 + DMPO + O2, whereas the signal at g = 1.97 is identical to that observed upon reaction of Fe(III) species complex 2 with DMPO under inert atmosphere (as well as in the presence of O2) and hints at a partial decomposition of 3 to complex 2 under the reaction conditions. These experiments suggest that 3 should contain the same kind of oxygen centered radical formed upon reaction of 1 and O2. As the reaction of 3 + DMPO was performed under an inert gas atmosphere, the oxygen radical should be bound to the Fe center of compound 3. These findings strongly support the proposed superoxidic nature of species 3.
1H-NMR studies
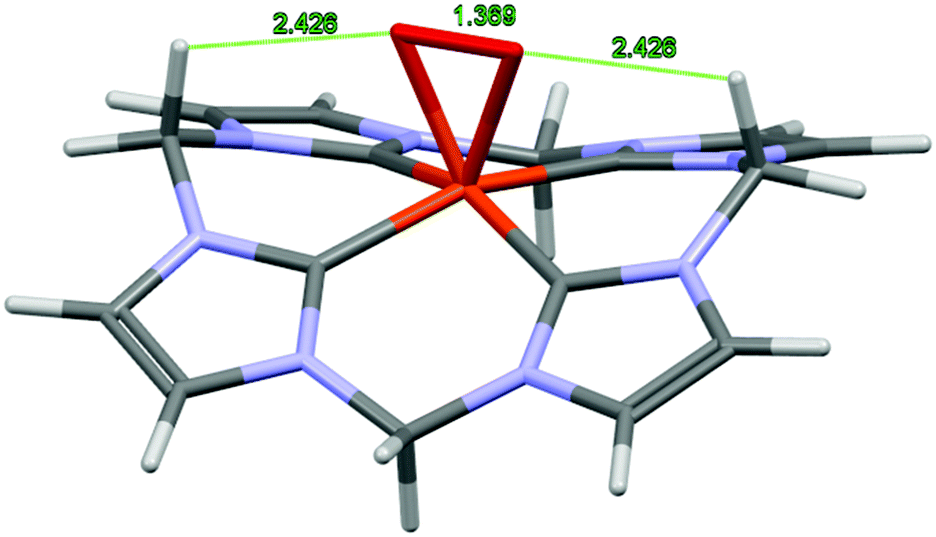
Compound 3 is diamagnetic and therefore suitable for 1H NMR investigations (see Fig. S2, S4 and S5 in the ESI†). The diamagnetic nature of 3 can be explained by the antiferromagnetic coupling of the unpaired electrons on the Fe(III) center and the O2− ligand.3a,19 In agreement with the structure obtained from density functional theory (DFT) studies (Fig. 4) the 1H NMR of 3 does not indicate coordinating acetonitrile ligands. The CH2 bridges of the backbone of 3 display signals at 6.48 ppm and 6.31 ppm. The splitting (2JHH = 13.1 Hz) suggests a square-pyramidal geometry for 3 that is similar to the nitrosyl derivative of 1 that has been previously described by our group.7 A fast fluxional behavior between side-on and end-on coordination of O2− on an NMR scale cannot be excluded. The suggested side-on coordination of the superoxide is based on DFT calculations.
 |
| | Fig. 4 DFT-derived structure of the cationic fragment [Fe(O2)CCCC]2+ of 3. Selected calculated bond lengths (Å) and angles (°): Fe1–C1: 1.966, Fe1–C5: 1.956, Fe1–O1: 1.883, O1–O1*: 1.369, O1–H8: 2.426, C1–Fe1–C5: 87.62 C5–Fe1–C9: 89.73, O1–Fe–O1*: 42.63, C8–H8–O1: 105.79. | |
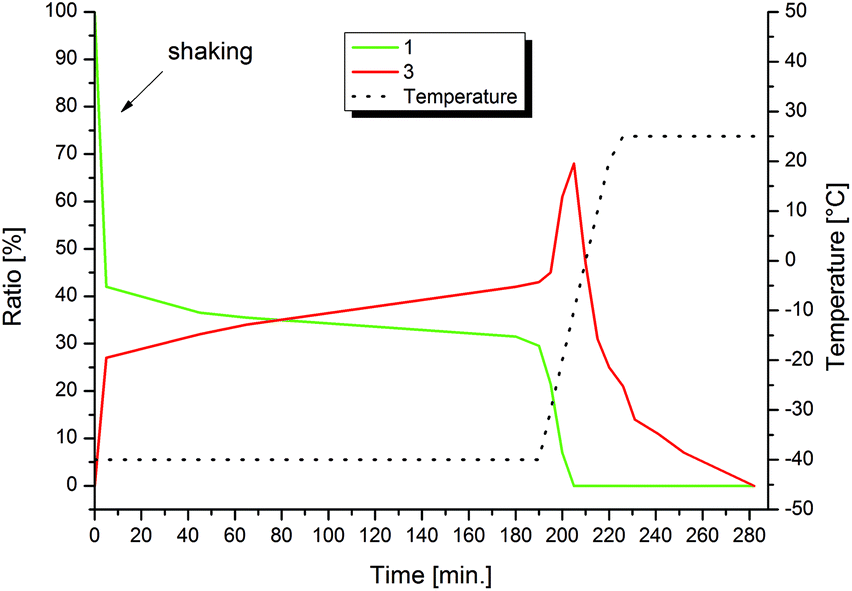
A valid energetic minimum for the geometry optimization was obtained only in case of side-on coordination of the superoxide and the respective square pyramidal geometry with a calculated O–O bond length of 1.369 Å. This value confirms the expected lengthening of the dioxygen bond upon coordination (free O2: 1.21 Å/free O2−: 1.33 Å).20 Interestingly, an interaction between the oxygen atoms and two of the protons of the CH2 bridges is indicated with O–H distances of 2.43 Å, suggesting a stabilizing effect on 3. Comparable observations were made by various groups reporting on molecular structures of Fe(IV) oxo complexes, where O–H interactions with similar distances were revealed.11,211H NMR kinetics of the formation and decay of 3 in acetone are in agreement with the UV/Vis experiments (Fig. 5, see also section 5 of the ESI†). After exposing a solution of 1 in acetone-d6 to O2 at −40 °C, rapid formation of the characteristic signals of 3 (6.48 ppm and 6.31 ppm) is observed, while the signals of 1 (7.86 ppm, 6.63 ppm and of Fe-coordinated MeCN at 1.7 ppm) decrease. After 3 h at −40 °C a mixture of 31% 1 and 43% 3 – relative to the initial amount of 1 – is obtained whereas the remaining 26% precipitate as the blue compound 4. Allowing the reaction to warm up to 25 °C within 40 min leads to complete conversion to 3 and its simultaneous decomposition to 4. Complete conversion of 1 is observed at −10 °C with a maximum yield of 68% of 3 (see Fig. 6). At temperatures above −10 °C, 3 readily decomposes to 4 within less than 1 h. At the end of the experiment a blue solid precipitated. This precipitate could be identified as pure 4 by means of 1H-NMR spectroscopy in CD3CN (see the ESI†).
 |
| | Fig. 5 Formation of 3 from 1 and O2 and subsequent decay of 3 to 4, monitored by 1H NMR in acetone-d6. (1 bar O2, T = −40 °C to 25 °C, [1]0 = 1 × 10−1 M). | |
 |
| | Fig. 6 Formation and decay of 3 (red) from 1 (orange) and O2 in acetone, monitored by 1H NMR based on the integration of the signals at 7.85 (1 + 3), 6.59 (1) and 6.31 (3) ppm (internal standard Et2O). (1 bar O2, T = −40 °C to 25 °C, [1]0 = 1 × 10−1 M). | |
Reactivity studies on 3
As 3 can be generated in acetone from 1 (Scheme 2) it appears reasonable to assume that 3 can also be formed by the reaction of 2 with KO2 (Scheme 3). Indeed, monitoring the reaction of 2 with an excess KO2 at temperatures from −80 °C to 25 °C by 1H NMR confirms the suggested reaction pathway (for details and spectra see Fig. S7 in the ESI†). Upon warming from −80 °C to −40 °C complete conversion of 2 to 3 was observed in the presence of KO2. Once again, a further increase in temperature up to r.t. leads to the disappearance of all 1H-NMR signals and to the precipitation of 4 as a dark blue solid. 4 could be reduced to 1 in acetonitrile by addition of KO2, thus closing the ideal redox circle. Therefore, O2− may serve as a reducing agent for Fe(III) compounds (2 and 4), whereas O2 acts as an oxidant for Fe(II) complex 1. Dimeric Fe(III)–O–O–Fe(III) compounds have been reported to be paramagnetic and to be irreversibly formed from oxygen and Fe(II) precursors.14
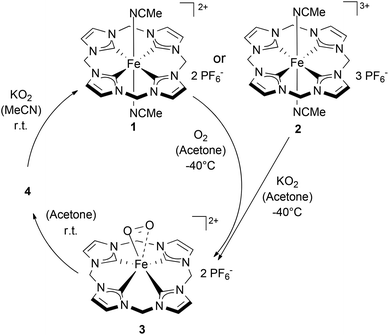 |
| | Scheme 3 Reactivity of 1 with O2 or 2 with KO2 in acetone affording 3 at −40 °C and subsequently 4 after warming. 4 can be converted to 1 by reaction with KO2 thus ideally closing a reversible redox cycle of 1. | |
Compound 3 however, is diamagnetic and it can be reversibly converted to an Fe(II) complex analogous to 1 by addition of more nucleophilic ligands such as PPh3 or DMSO in acetone (Scheme 4). PPh3 and DMSO are not oxidized during this process, behaving exclusively as displacing ligands under formation of 5 or 6 (see Fig. S6 in the ESI†). The addition of acetonitrile (10 equiv.) in acetone leads to the formation of complex 2 (compare with Scheme 3). This is in good agreement with previous reports on superoxo species22 and with the observed reactivity between of 1 and O2 in acetonitrile leading to complex 2.
 |
| | Scheme 4 Reactivity of 3 with PPh3, DMSO, and MeCN in acetone. | |
Preparation and characterization of 4
Complex 4 was prepared in high purity by adding oxygen to a concentrated acetone solution of 1 (>50 mM) at −40 °C followed by slow warming to r.t. Product formation was indicated by the precipitation of a dark blue powder in good yields (60–70%). Instead of acetone also benzonitrile or pivalonitrile as solvents afforded good yields of 4. Additionally 4 could also be prepared by using air instead of dry oxygen at r.t., albeit in significantly lower yields (∼10%). Both Fe(III) centers in 4 formally consist of 15 valence electrons; nevertheless, 4 is diamagnetic and therefore suitable for NMR-spectroscopy. This is attributed to antiferromagnetic coupling of the unpaired electrons over the bridging oxide, which is known for similar systems.11 In 1H NMR a singlet at 7.57 ppm is observed for 4. In addition, two doublets of the bridging CH2 groups with geminal couplings of 13.1 Hz arise at 6.08 and 5.94 ppm. The 13C NMR spectrum of 4 shows two sharp signals at 122.72 and 64.05 ppm, which are assigned to the backbone CH groups and the CH2 bridges, respectively. The signal of the coordinating carbons only appears as a broad peak at 175 ppm. Slow diffusion of oxygen-enriched diethyl ether into an acetone solution of 1 allowed collection of single crystals of 4 suitable for single crystal X-ray diffraction (SC-XRD, Fig. 7). The molecular structure shows a slightly distorted square pyramidal geometry with the tetradentate cyclic ligand coordinated in saddle distorted fashion. The Fe–CNHC distances of 4 with its saddle-distorted ligand conformation average to 1.949 Å, which is slightly shorter than for comparable Fe(III)OFe(III) tetracarbene complexes (∼1.99 Å) reported by Meyer et al.11 The Fe–O distance is 1.732 Å and the FeOFe angle is 162.7°.
 |
| | Fig. 7 Ortep-style drawing of the cationic fragment of O[Fe(III)CCCC]24+ of 4. Hydrogen atoms and PF6− anions are omitted for clarity and thermal ellipsoids are shown at a 50% probability level. Selected bond lengths (Å) and angles (°): Fe1–C1: 1.948(4), Fe1–C5: 1.941(1), Fe1–C9: 1.943(4), Fe1–C13: 1.966(4), Fe1–O1: 1.7322(7), C1–Fe1–C5: 87.28(16) C5–Fe1–C9: 87.84(17), C9–Fe1–C13: 87.20(17), C1–Fe1–N13: 86.92(16), C1–Fe1–O1: 101.55(16), C5–Fe1–O1: 100.80(12), C9–Fe1–O1: 102.30(16), C13–Fe1–O1: 105.47(12), Fe1–O1–Fe1a: 162.7(2). | |
Reactivity of 4
Compound 4 is able to act as an oxidizing agent. For instance, PPh3 is stoichiometrically oxidized to OPPh3 (Scheme 5) under formation of 1 (see the ESI†). Reduction of 4 to 1 is observed in the presence of reducing agents such as Zn powder, Fe-powder, hydroquinone (H2Q), or KO2. At r. t. in CD3CN one third of the initial amount of 4 is reduced to 1 after approximately 5 d. (see Fig. S9 in the ESI†). As reported for the case of the oxidation of 1 to 2 by oxygen in acetonitrile, oxidation products cannot be observed by 1H- or 2D-NMR. In the absence of other reducing agents, very slow oxidation of acetonitrile by 4 is observed at r.t. Upon heating an acetonitrile solution of 4 to reflux, full conversion to 1 takes place within 5 min. Furthermore, 4 can also be deoxygenated to quantitatively yield 2 by the simple addition of stoichiometric amounts of strong Brønsted acids (e.g. HBF4/HPF6) at r.t.
 |
| | Scheme 5 | |
Conclusions
The reactivity of an heme analogue Fe(II) (1) compound with molecular oxygen was investigated and three different Fe(III) compounds, have been identified and characterized. In their structural characteristics, all of them are closely related to heme systems. A strong influence of the solvent on the reactivity is observed, resulting in two different products from the oxidation of 1 with molecular oxygen: the oxide bridged Fe(III) dimer 4 in acetone and the metheme like Fe(III) complex 2 in acetonitrile. The intermediate proposed in both cases, the Fe(III) superoxide (3), as well as the oxide bridged Fe(III)dimer (4) have been studied. Indirect chemical and spectroscopic evidence (EPR spin trap experiments, NMR spectroscopy) of the superoxodic nature of 3 has been presented. 3 is prone to ligand exchange reactions, resulting in reductive dissociation of dioxygen upon addition of nucleophilic ligands such as PPh3 and DMSO. In the latter case the oxide bridged Fe(III) dimer 4 acts as an oxidant while being reduced to 1. Also, 4 can be deoxygenated under the retention of its formal oxidation state to yield 2. With respect to iron-based systems in nature, which are widely used as oxidation catalysts, oxygen activation by artificial systems is of high interest. A deeper understanding of the reactivity of such complexes can help to improve the quality of artificial, bio-inspired iron systems for various applications.
Acknowledgements
M. R. A. and S. H. gratefully acknowledge support by the TUM Graduate School. This project was supported through a collaboration with the King Abdullah University of Saudi-Arabia (Grant No. KSA-C0069/UKC0020).
Notes and references
-
(a) W. Nam, Acc. Chem. Res., 2015, 48, 2415 CrossRef CAS PubMed;
(b) Y. You and W. Nam, Chem. Sci., 2014, 5, 4123 RSC;
(c) P. C. A. Bruijnincx, G. van Koten and R. J. M. Klein Gebbink, Chem. Soc. Rev., 2008, 37, 2716 RSC;
(d) H. Chen, M. Ikeda-Saito and S. Shaik, J. Am. Chem. Soc., 2008, 130, 14778 CrossRef CAS PubMed;
(e) E. G. Kovaleva, M. B. Neibergall, S. Chakrabarty and J. D. Lipscomb, Acc. Chem. Res., 2007, 40, 475 CrossRef CAS PubMed;
(f) S. V. Kryatov, E. V. Rybak-Akimova and S. Schindler, Chem. Rev., 2005, 105, 2175 CrossRef CAS PubMed.
-
(a) K. Ray, F. F. Pfaff, B. Wang and W. Nam, J. Am. Chem. Soc., 2014, 136, 13942 CrossRef CAS PubMed;
(b) S. P. de Visser, J. H. Rohde, Y.-M. Lee, J. Cho and W. Nam, Coord. Chem. Rev., 2013, 257, 381 CrossRef CAS;
(c) D. Mandon, H. Jaafar and A. Thibon, New J. Chem., 2011, 35, 1986 RSC;
(d) W. A. van der Donk, C. Krebs and J. M. Bollinger Jr., Curr. Opin. Struct. Biol., 2010, 20, 673 CrossRef CAS PubMed;
(e) G. Dong, S. Shaik, W. Lai, S. P. de Visser, S. Shaik and W. Nam, Chem. Commun., 2012, 48, 2189 RSC;
(f) R. Latifi, L. Tahsini, W. Nam and S. P. de Visser, Phys. Chem. Chem. Phys., 2012, 14, 2518 RSC;
(g) F. Bonnot, T. Molle, S. Ménage, Y. Moreau, S. Duval, V. Favaudon, C. Houée-Levin and V. Nivière, J. Am. Chem. Soc., 2012, 134, 5120 CrossRef CAS PubMed;
(h) H. Chen, K.-B. Cho, W. Lai, W. Nam and S. Shaik, J. Chem. Theory Comput., 2012, 8, 915 CrossRef CAS PubMed;
(i) L. W. Chung, X. Li, H. Hirao and K. Morokuma, J. Am. Chem. Soc., 2011, 133, 20076 CrossRef CAS PubMed.
-
(a) F. Oddon, Y. Chiba, J. Nakazawa, T. Ohta, T. Ogura and S. Hikichi, Angew. Chem., Int. Ed., 2015, 54, 7336 CrossRef CAS PubMed;
(b) C.-W. Chiang, S. T. Kleespies, H. D. Stout, K. K. Meier, P. Y. Li, E. L. Bominaar, L. Que Jr., E. Münck and W.-Z. Lee, J. Am. Chem. Soc., 2014, 136, 10846 CrossRef CAS PubMed.
-
(a) M. M. Mbughuni, M. Chakrabarti, J. A. Hayden, E. L. Bominaar, M. P. Hendrich, E. Münck and J. D. Lipscomb, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16788 CrossRef CAS PubMed;
(b) C. Costentin, H. Dridi and J.-M. Savéant, J. Am. Chem. Soc., 2015, 137, 13535 CrossRef CAS PubMed;
(c) K. Duerr, O. Troeppner, J. Olah, J. Li, A. Zahl, T. Drewello, N. Jux, J. N. Harvey and I. Ivanović-Burmazović, Dalton Trans., 2012, 41, 546 RSC;
(d) W. Xu, K. Dziedzic-Kocurek, M. Yu, Z. Wu and A. Marcelli, RSC Adv., 2014, 4, 46399 RSC.
-
(a) J.-H. Jeoung, M. Bommer, T.-Y. Lin and H. Dobbek, Proc. Natl.
Acad. Sci. U. S. A., 2013, 110, 12625 CAS;
(b) E. G. Kovaleva and J. D. Lipscomb, Science, 2007, 316, 453 CrossRef CAS PubMed.
- S. Hong, K. D. Sutherlin, J. Park, E. Kwon, M. A. Siegler, E. I. Solomon and W. Nam, Nat. Commun., 2015, 5, 5440 CrossRef PubMed.
- M. R. Anneser, S. Haslinger, A. Pöthig, M. Cokoja, J. M. Basset and F. E. Kühn, Inorg. Chem., 2015, 54, 3797 CrossRef CAS PubMed.
- J. W. Kück, M. R. Anneser, B. Hofmann, A. Pöthig, M. Cokoja and F. E. Kühn, ChemSusChem, 2015, 8, 4056 CrossRef PubMed.
-
(a) J. W. Kück, A. Raba, I. I. E. Markovits, M. Cokoja and F. E. Kühn, ChemCatChem, 2014, 6, 1882 CrossRef;
(b) A. Raba, M. Cokoja, W. A. Herrmann and F. E. Kühn, Chem. Commun., 2014, 50, 11454 RSC;
(c) S. Haslinger, A. Raba, M. Cokoja, A. Pöthig and F. E. Kühn, J. Catal., 2015, 331, 147 CrossRef CAS.
-
(a) I. V. Korendovych, O. P. Kryatova, W. M. Reiff and E. V. Rybak-Akimova, Inorg. Chem., 2007, 46, 4197 CrossRef CAS PubMed;
(b) D. J. Liston and B. O. West, Inorg. Chem., 1985, 24, 1568 CrossRef CAS;
(c) D.-H. Chin, N. Gerd, G. N. La Mar and A. N. Balch, J. Am. Chem. Soc., 1980, 102, 4344 CrossRef CAS.
- S. Meyer, I. Klawitter, S. Demeshko, E. Bill and F. Meyer, Angew. Chem., Int. Ed., 2013, 52, 901 CrossRef CAS PubMed.
- J. Umbreit, Am. J. Hematol., 2007, 82, 134 CrossRef CAS PubMed.
- I. Klawitter, M. R. Anneser, S. Dechert, S. Meyer, S. Demeshko, S. Haslinger, A. Pöthig, F. E. Kühn and F. Meyer, Organometallics, 2015, 34, 2819 CrossRef CAS.
- C. E. Tinberg and S. J. Lippard, Biochemistry, 2010, 49, 7902 CrossRef CAS PubMed.
- D. E. Feierman and A. I. Cederbaum, Chem. Res. Toxicol., 1989, 2, 359 CrossRef CAS PubMed.
-
T. Foo and A. Panova, Process for the Synthesis of Glycolonitrile, US7368492, 2008 Search PubMed.
-
(a) R. Lia, H. Kobayashi, X. Yana and J. Fan, Catal. Today, 2014, 233, 140 CrossRef;
(b) J.-L. Clement, N. Ferré, D. Siri, H. Karoui, A. Rockenbauer and P. Tordo, J. Org. Chem., 2005, 70, 1198 CrossRef CAS PubMed;
(c) G. R. Buettner, Free Radical Res. Commun., 1993, 19, 79 CrossRef.
- Z. Barbierikova, M. Mihalikova and V. Brezova, Photochem. Photobiol., 2012, 88, 1442 CrossRef CAS PubMed.
- J. P. Collman, C. J. Sunderland, K. E. Berg, M. A. Vance and E. I. Solomon, J. Am. Chem. Soc., 2003, 125, 6648 CrossRef CAS PubMed.
-
CRC Handbook of Chemistry & Physics, ed. D. R. Lide, Taylor and Francis, 90th edn, 2009 Search PubMed.
- J. Li, B. C. Noll, A. G. Oliver, C. E. Schulz and W. R. Scheidt, J. Am. Chem. Soc., 2013, 135, 15627 CrossRef CAS PubMed.
- P. L. Holland, Dalton Trans., 2010, 39, 5415 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, UV/Vis spectra of 1–4, kinetic data, spectroscopic data of all synthesized compounds, EPR of 2, and X-ray data for 4 in CIF format. CCDC 1428063. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt00538a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence