
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exposing elusive cationic magnesium–chloro aggregates in aluminate complexes through donor control†
Etienne V.
Brouillet
a,
Alan R.
Kennedy
a,
Konrad
Koszinowski
*b,
Ross
McLellan
a,
Robert E.
Mulvey
a and
Stuart D.
Robertson
*a
aWestCHEM, Department of Pure and Applied Chemistry, University of Strathclyde, Glasgow, G1 1XL, UK. E-mail: stuart.d.robertson@strath.ac.uk
bInstitut für Organische und Biomolekulare Chemie, Georg-August-Universität Göttingen, Tammannstr. 2, 37077 Göttingen, Germany. E-mail: konrad.koszinowski@chemie.uni-goettingen.de
First published on 22nd February 2016
Abstract
The cationic magnesium moiety of magnesium organohaloaluminate complexes, relevant to rechargeable Mg battery electrolytes, typically takes the thermodynamically favourable dinuclear [Mg2Cl3]+ form in the solid-state. We now report that judicious choice of Lewis donor allows the deliberate synthesis and isolation of the hitherto only postulated mononuclear [MgCl]+ and trinuclear [Mg3Cl5]+ modifications, forming a comparable series with a common aluminate anion [(Dipp)(Me3Si)NAlCl3]−. By pre-forming the Al–N bond prior to introduction of the Mg source, a consistently reproducible protocol is reported. Usage of the green solvent 2-methyltetrahydrofuran in place of THF in the context of Mg/Al battery electrolyte type complexes is also promoted.
Introduction
A key area of study for post-lithium ion batteries can be found in neighbouring group 2, specifically magnesium,1 due to its considerably greater natural abundance, which as a consequence makes it more economically viable long-term.2 Furthermore, the neutral metal/cation redox couple is a two-electron process, giving magnesium a higher volumetric capacity than lithium (3833 mA h cm−3cf. 2062 mA h cm−3), while its high reduction potential of −2.37 V (vs. SHE) is conducive to high energy density and high voltage batteries, provided other drawbacks, such as development of suitable electrolytes and cathodes, can be adequately surmounted. One main impediment of magnesium based electrolytes is that, unlike lithium, its neutral inorganic metal salts are incapable of reversibly conducting magnesium ions in aprotic solvents sufficiently, forming passivating films on the electrode surface, while the strong reducing nature of Grignard reagents gives them low anodic stability. A possible way to circumvent these problems is to move to a bimetallic ate complex3 such as a magnesium aluminate,4 which typically takes the form [Mg2Cl3]+ [RxAlCl4−x]− and can be generated from reaction of Lewis basic magnesium and Lewis acidic aluminium precursors.5 Moreover, their solvent separated ion pairing enhances their conductivity. Inorganic only haloaluminates (x = 0) have been studied but suffer from poor solubility even in THF,6 although more recent studies on this system have confirmed its enhanced oxidative stability.7 The solubility can be increased by grafting an organo group onto the aluminium (e.g. R = Et,8 Ph![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7a,9) but their nucleophilicity can render them incompatible with the electrophilic sulfur cathodes typically employed. To prevent this problem, a bulky non-nucleophilic amido group (NR2) can be utilized instead,10 which maintains the benefit of increased solubility but without the propensity toward undesirable side reactions. Chloride ligands have been suggested as the likely culprit for corrosion of magnesium electrolytes,11 a problem when using a non-noble metal electrode; although purity of the starting materials has also been implicated as a potential cause.12 Despite this possibility, magnesium organohaloaluminates continue to dominate the landscape in Mg battery research,13 including theoretical calculations on the nuclearity of the active cation.14 Indeed, it was recently suggested that free chloride anions in the electrolyte solution adsorb at the electrode surface, enhancing magnesium electrodeposition.15
7a,9) but their nucleophilicity can render them incompatible with the electrophilic sulfur cathodes typically employed. To prevent this problem, a bulky non-nucleophilic amido group (NR2) can be utilized instead,10 which maintains the benefit of increased solubility but without the propensity toward undesirable side reactions. Chloride ligands have been suggested as the likely culprit for corrosion of magnesium electrolytes,11 a problem when using a non-noble metal electrode; although purity of the starting materials has also been implicated as a potential cause.12 Despite this possibility, magnesium organohaloaluminates continue to dominate the landscape in Mg battery research,13 including theoretical calculations on the nuclearity of the active cation.14 Indeed, it was recently suggested that free chloride anions in the electrolyte solution adsorb at the electrode surface, enhancing magnesium electrodeposition.15
While the dinuclear cation is the most common structurally characterized motif within these systems due to its thermodynamic stability, other aggregated cationic moieties have been implicated as playing an important role evidenced through techniques such as mass spectrometry, although they have never been isolated nor characterized crystallographically in a magnesium organoaluminate species. These include mononuclear [MgCl]+ and trinuclear [Mg3(μ3-Cl)2(μ2-Cl)3]+ (Fig. 1)16 that can all be found in a complicated equilibrium in solution which is difficult to resolve due to, for example, lack of appropriate NMR handles in the [MgxCl2x−1·nTHF] cations. Indeed, Muldoon has contended that this equilibrium can conceivably affect Mg electrochemistry in solution and consequently it should not be assumed that the crystallographically verified dinuclear complex is solely responsible,17 a hypothesis supported by an X-ray absorption near edge structure (XANES) study which suggests that it is a different (unidentified) magnesium species which is electrochemically active.18 The suggestion that these different cationic oligomers were involved was particularly interesting to us given our long standing interest in the bimetallic ‘ate’ chemistry of the main group metals, including magnesium and aluminium.19 We felt our synthetic expertise could be exploited to shed light on these different, important oligomeric cations in the presence of a common anion, the results of such a study we now present herein.
 | ||
| Fig. 1 THF solvated magnesium chloride cations implicated in magnesium aluminate solution chemistry. | ||
Results and discussion
Synthesis and molecular structures
We commenced by formulating a simple synthetic protocol designed to consistently produce the desired crystalline material. Instead of transferring the organo anion from Mg to AlCl3, as many previous studies had done (Scheme 1, eqn (1)–(3)), we decided to follow the recent work of Liu and co-workers (Scheme 1, eqn (4)),7a and Zhao-Karger and co-workers (Scheme 1, eqn (5)),8b who pre-formed the Al–C bond prior to the introduction of the magnesium source. Thus, our first step was to make the Al–N bond via a salt metathesis reaction of equimolar amounts of lithium amide and AlCl320 prior to introducing the magnesium reagent. The bulky aryl/silyl amide [(Dipp)(Me3Si)N]− (Dipp = 2,6-diisopropylphenyl) was chosen as it has a complementary combination of steric and electronic properties which can stabilize low valent or low coordination main group and transition metal species21 and so seems ideally suited for purpose. Our focus was on an aluminium mono secondary amide since a higher Cl:R ratio is understood to lead to a higher oxidative potential.8a Addition of nBuMgCl, followed by slow diffusion of hexane into the resulting THF solution furnished crystals of [(Dipp)(Me3Si)NAlCl3]− [Mg2(μ2-Cl)3·6THF]+, 1 (Scheme 1, eqn (6) and Fig. 2a, b show the molecular structures of the anion and cation, respectively).
 | ||
| Scheme 1 Reactions accessing crystallographically authenticated magnesium aluminates (R, R′ = Dipp, Me3Si). Stoichiometries of reagents and identities of by-products are not shown for brevity. | ||
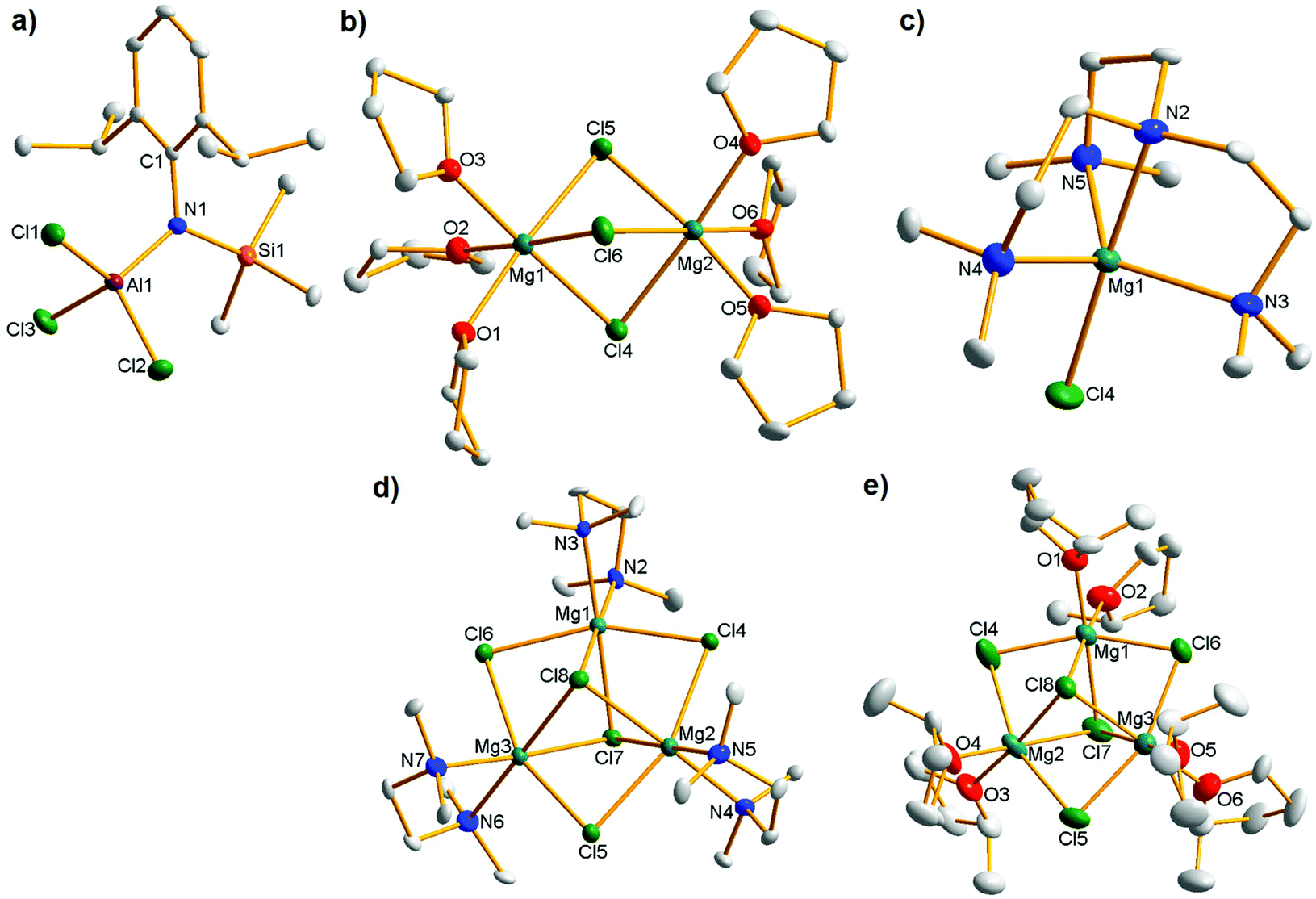 | ||
| Fig. 2 Molecular structures of (a) anionic moiety of 1 [(Dipp)(Me3Si)NAlCl3]−; (b) cationic moiety of 1 [Mg2(μ2-Cl)3·6THF]+; (c) cationic moiety of 2 [MgCl·Me6TREN]+; (d) cationic moiety of 3 [Mg3(μ3-Cl)2(μ2-Cl)3·3TMEDA]+; (e) cationic moiety of 4 [Mg3(μ3-Cl)2(μ2-Cl)3·6MeTHF]+. All ellipsoids are displayed at 50% probability and H atoms and minor disordered components have been omitted for clarity. The anion is consistent across all four complexes so only that of 1 is shown for brevity. For selected bond parameters of the anion in all four cases see Table S1;† for bond lengths of all four cations see Table S2;† for bond angles of all four cations see Tables S3–S6.† Selected average bond parameters (Å/°) of cation 1: Mg–Cl, 2.5027; Mg–O, 2.0808; O–Mg–Cltrans, 176.91; O–Mg–Clcis, 92.98; Cl–Mg–Cl, 84.61; O–Mg–O, 89.36; Mg–Cl–Mg, 77.98; cation 2: Mg–Cl, 2.3229; Mg–Nax, 2.219; Mg–Neq, 2.182; Nax–Mg–Cl, 178.28; Neq–Mg–Cl, 98.98; Nax–Mg–Neq, 81.06; Neq–Mg–Neq, 117.61; cation 3: Mg–Cl(μ2), 2.489; Mg–Cl(μ3), 2.573; Mg–N, 2.190; N–Mg–Cl(μ3)trans, 178.32; N–Mg–Cl(μ3)cis, 95.48; N–Mg–Cl(μ2)cis, 98.38; Cl(μ2)–Mg–Cl(μ3), 81.67; Cl(μ2)–Mg–Cl(μ2), 157.31; Cl(μ3)–Mg–Cl(μ3), 84.92; N–Mg–N, 84.12; Mg–Cl(μ3)–Mg, 79.42; Mg–Cl(μ2)–Mg, 82.65; cation 4: Mg–Cl(μ2), 2.4929; Mg–Cl(μ3), 2.5555; Mg–O, 2.034; O–Mg–Cl(μ3)trans, 176.52; O–Mg–Cl(μ3)cis, 93.95; O–Mg–Cl(μ2)cis, 98.11; Cl(μ2)–Mg–Cl(μ3), 81.58; Cl(μ2)–Mg–Cl(μ2), 157.25; Cl(μ3)–Mg–Cl(μ3), 83.84; O–Mg–O, 88.28; Mg–Cl(μ3)–Mg, 80.24; Mg–Cl(μ2)–Mg, 82.69. | ||
As seen previously in other relevant magnesium aluminates, the cation of 1 has a non-crystallographic C3 axis of symmetry passing through the two magnesium atoms, which are in a virtually octahedral environment consisting of three bridging fac-chloride anions and three terminal THF molecules.
With respect to mononuclear species, the predominant form of the [MgCl]+ cation in the solid state is the penta-THF solvate, although to the best of our knowledge it has never been seen as the cationic moiety in a magnesium organohaloaluminate complex, which appear to prefer to adopt the thermodynamically more stable [Mg2(μ2-Cl)3]+ cationic structure.22 We do note that [MgCl·5THF]+ has been crystallographically characterized with an [AlCl4]− counteranion,23 suggesting that in the presence of THF, cation nuclearity can be dictated by crystal packing effects and is thus difficult to control or predict. Given that THF seems an unsuitable Lewis donor to stabilize [MgCl]+ on demand, we utilized the hemisphere-capping tripodal tetraamine Me6TREN [N(CH2CH2NMe2)3], previously exploited by us to trap sensitive mononuclear organometallic species24 and which Hazari has shown can stabilize [MgBr]+ and [MgMe]+ cations.25 Repeating the synthesis of 1 but with an equivalent of Me6TREN added prior to crystallization yielded [(Dipp)(Me3Si)NAlCl3]− [MgCl·Me6TREN]+, 2 (see Fig. 2c for the cationic moiety: the anion is the same as structure 1 so omitted for clarity). This unequivocally confirmed our view that a mononuclear MgCl cation could be prepared on demand by fully occupying one hemisphere of the metal and represents the first example of [MgCl·Me6TREN]+ to be synthesized and crystallographically characterized. Moreover, this represents potentially a key breakthrough for the solution study of mononuclear magnesium aluminate battery electrolyte complexes (vide infra) since the previously synthesized THF solvate displays poor solubility even in polar THF.9a Its Mg centre lies in a trigonal bipyramidal environment with the central nitrogen and chloride occupying axial positions with the three pendant arm N donor atoms occupying equatorial sites.
The number of crystallographically authenticated [Mg3(halide)5]+ cations is limited and none are THF solvated (we have identified five diethyl ether solvates26 and one TMEDA solvate in the CCDB,27 none of which have an aluminate counteranion). Some relevant THF solvated trinuclear Mg cations, with three μ2-Cl ligands and two μ2-alkoxide/aryloxide ligands were reported recently and shown to be promising in the battery electrolyte context,17,28 making a reproducible synthetic protocol for [Mg3Cl5]+ species particularly timely. We therefore moved away from THF and rather utilized bidentate chelating TMEDA. The resulting product [(Dipp)(Me3Si)NAlCl3]− [Mg3(μ3-Cl)2(μ2-Cl)3·3TMEDA]+, 3 was trinuclear as hoped (Fig. 2d) although its THF solubility was poor making it difficult to adequately characterize in solution (vide infra). We then moved to 2-methyltetrahydrofuran (MeTHF), whose slightly increased bulk vis-a-vis THF might prevent tris-solvation of the metal centre, and that enforced bis-solvation would consequently promote trinuclear cation formation. MeTHF is a greener alternative to THF with some similar properties29 and has previously found use as an electrolyte solvent for rechargeable lithium batteries30 so any progress using this Lewis donor would constitute a welcome development in the magnesium battery sector. Introducing MeTHF as the bulk solvent proved a successful protocol, with the resulting crystalline material shown by XRD to be [(Dipp)(Me3Si)NAlCl3]− [Mg3(μ3-Cl)2(μ2-Cl)3·6MeTHF]+, 4 (Fig. 2e). The cations contain a six atom (MgCl)3 ring capped on each side by another chloride anion. In 3 and 4, the Mg centres are in a distorted octahedral environment made up of two mutually cis Lewis donor atoms (N, 3; O, 4), two trans μ2 chloride anions and two mutually cis μ3 chloride anions. Looking at the synthetic work as a whole, the yields of the isolated O-donor complexes 1 and 4 were excellent (88/93% respectively with respect to magnesium) while those of N-donor complexes 2 (65%) and 3 (31%) were lower. In each case, elemental analysis confirmed the bulk purity of the samples.
Following the success of our reactions with polydentate N-donors TMEDA and Me6TREN, we repeated our protocol using the related ligand N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA), expecting that this tridentate ligand could act as an isodentate surrogate for three molecules of THF, yielding a magnesium aluminate with a dinuclear N-solvated cation. However, the resulting product was shown by single crystal X-ray diffraction to be the neutral magnesium dichloride complex MgCl2·PMDETA (5, Fig. 3). We note that the related tridentate O-donor bis(2-methoxyethyl)ether (diglyme) also results in a neutral magnesium complex when used in this context, specifically dimeric [MgCl2·diglyme]2.10b This may suggest that acyclic tridentate donors do not possess the correct spatial conformation of their donor atoms to adequately protect one end of a [Mg2Cl3]+ fragment as they are aligned for mer rather than fac coordination to an octahedral metal centre, while they do not have the requisite number of donor atoms to protect a mononuclear or trinuclear cation.
 | ||
| Fig. 3 Molecular structure of one of the independent molecules of complex 5 [MgCl2·PMDETA]. All ellipsoids are displayed at 50% probability and H atoms have been omitted for clarity. Selected bond parameters (Å/°): Mg1–Cl1, 2.3913(7) [2.3770(6)]; Mg1–Cl2, 2.3108(7) [2.3371(6)]; Mg1–N1, 2.1932(15) [2.1886(13)]; Mg1–N2, 2.3230(13) [2.2959(13)]; Mg1–N3, 2.2142(15) [2.2226(13)]; Cl1–Mg1–Cl2, 106.26(3) [105.26(3)]; Cl1–Mg1–N1, 93.44(4) [93.19(4)]; Cl2–Mg1–N1, 109.78(4) [106.25(4)]; Cl1–Mg1–N2, 156.89(4) [156.78(4)]; Cl2–Mg1–N2, 96.84(4) [98.09(4)]; Cl1–Mg1–N3, 93.20(4) [92.27(4)]; Cl2–Mg1–N3, 109.25(4) [113.79(4)]; N1–Mg1–N2, 78.91(5) [79.78(5)]; N1–Mg1–N3, 136.63(5) [136.49(5)]; N2–Mg1–N3, 78.42(5) [78.48(5)]. Bond parameters of second independent molecule are in parentheses. | ||
NMR spectroscopy
Although a longstanding cornerstone characterisational tool in organometallic chemistry, NMR spectroscopy does not reveal much regarding the behaviour of magnesium organohaloaluminates in solution, particularly their cation nuclearity. Conventionally studied nuclei such as 1H and 13C are only present in the Lewis donor ligands of the cation and do not present evidence for their aggregation state while 25Mg is a low sensitivity, low abundance, quadrupolar nucleus. Nevertheless, given that we now have a coherent series of magnesium chloride cationic moieties in the presence of a common aluminate anion we felt it important to consider this technique to see if any light could be shed on the solution constitution of this series.1H and 13C NMR spectra, collected in d8-THF, confirmed the solvent separated ion pairing was maintained in solution with the anions giving identical spectra in all four cases (see Table S8† for a summary). The only 1H/13C containing fragments of the cations were the donor ligands making characterization challenging. However, the resonances of the Lewis donors in complexes 2 and 3 were noticeably deshielded with respect to the free ligand in the same solvent (see Fig. S1† for full details) showing that THF had not replaced Me6TREN or TMEDA. For complex 1, the 1H NMR spectroscopic THF resonances were very close to those of free THF, probably as a consequence of OC4H8/OC4D8 exchange while for 4, the MeTHF resonances were identical to those of free MeTHF (see Fig. S1 and S2†) suggesting that MeTHF is replaced by d8-THF, although this does not shed light on the aggregation state of the magnesium containing species in solution. 27Al NMR spectroscopy was uninformative, with well-resolved singlets absent due to the lack of high symmetry at the aluminium centre.
Complexes 1, 2 and 4 were also sufficiently soluble in C6D6 to obtain NMR spectra in the absence of bulk Lewis donor. Surprisingly, despite the anion being identical in all three cases, there were noticeable differences in their 1H NMR spectra (Fig. 4) suggesting strong ion-pairing in less polar solvent.
 | ||
| Fig. 4 Selected sections of 1H NMR spectra of complexes 1, 2 and 4, each containing the same anion but with a different cationic aggregation state, in C6D6 solution at 300 K. The parent amine is also included for comparison. Unlabeled resonances belong to donor ligand solvating Mg, or NMR solvent. | ||
While the amido resonances of the di- and trinuclear complexes are fairly similar, those of the mononuclear complex (2) are clearly different. Specifically, the aryl resonances are well resolved into triplet (para) and doublet (meta), while the iPr and SiMe3 resonances are deshielded with respect to those in the di- and trinuclear complexes (1 and 4 respectively). Furthermore the iPr methyl resonance is resolved into a pair of doublets (1.64/1.53 ppm) suggesting inequivalence. There is a slight separation of these resonances in 1 and 4 but they still overlap at around 1.32 ppm in each. Temperature effects can be ruled out since all spectra were obtained at 300 K, while a variable concentration NMR experiment revealed identical spectra, showing that this change is not a consequence of concentration variation. The reason for these differences is not instantly clear, although one could perhaps speculate that the unique feature of mononuclear complex 2, namely a terminal Mg–Cl bond, is somehow able to interact with the anionic moiety in weakly-donating benzene solvent.
Mass spectrometry
Due to the inherent difficulty thus far in characterizing the cations in solution we turned to Electrospray-ionization (ESI) mass spectrometry. This method has been effective for the characterization of complex inorganic and organometallic ions in solution.31 Negative-ion mode ESI-MS of a THF solution of [(Dipp)(Me3Si)NAlCl3]− [MgCl·Me6TREN]+ (2) led to the exclusive detection of the anionic component of this salt (Fig. S3 and S4†). For all other salts investigated, virtually the same result was obtained (Fig. S5–S9†). These findings strongly suggest that solutions of these salts all contain the crystallographically characterized free [(Dipp)(Me3Si)NAlCl3]− to a significant extent.In positive ion mode, we looked first at the N-chelate containing complexes 2 and 3, in THF solution. For 2, the cations [MgCl(Me6TREN)]+ and [Mg2Cl3(Me6TREN)2]+ were observed (Fig. 5a, S10 and S11†). The former corresponds to the cationic component of the salt, the latter to its dinuclear homologue. It is not clear whether the dinuclear ion was already present in the original sample solution or whether it only formed during the ESI process. The ESI process produces charged nanodroplets, which permanently lose solvent molecules due to evaporation. The increased effective concentration in these nanodroplets can lead to shifts of aggregation equilibria and, thus, to formation of the observed dinuclear ions.32 Both [MgCl(Me6TREN)]+ and [Mg2Cl3(Me6TREN)2]+ exhibit a 1:1 stoichiometry of magnesium and ligand, which reflects the latter's polydentate nature. Likewise, the absence of any THF adducts points to the lack of empty coordination sites at the magnesium centre. In addition, ions containing the protonated ligand were detected (Fig. 5a and S12†). Because of its high Brønsted basicity, the ligand can easily react with traces of protic contaminants remaining in the used glassware or the ESI source. For 3, which was run at a lower concentration due to its poor solubility, vide supra, we observed ions belonging to the homologous series [MgnCl2n−1(TMEDA)n]+, n = 1–3 (Fig. 5b and S13–S15†). Like in the case of the Me6TREN-containing ions, these chelated ions display a 1:1, Mg:ligand stoichiometry and do not bind any THF.
 | ||
| Fig. 5 (a) positive-ion mode ESI mass spectrum of a 20 mM solution of [(Dipp)(Me3Si)NAlCl3]− [MgCl·Me6TREN]+ (2) in THF (L = Me6TREN); (b) positive-ion mode ESI mass spectrum of a saturated solution (c ≤ 10 mM) of [(Dipp)(Me3Si)NAlCl3]− [Mg3(μ3-Cl)2(μ2-Cl)3·3TMEDA]+ (3) in THF; (c) positive-ion mode ESI mass spectrum of a 20 mM solution of [(Dipp)(Me3Si)NAlCl3]− [Mg2(μ2-Cl)3·6THF]+ (1) in THF; (d) concentration dependence of the normalized abundances of mono-, di-, and trinuclear cations observed upon positive-ion mode ESI of solutions of (1) in THF; (e) positive-ion mode ESI mass spectrum of a 20 mM solution of [(Dipp)(Me3Si)NAlCl3]− [Mg3(μ3-Cl)2(μ2-Cl)3·6MeTHF]+ (4) in MeTHF; (f) positive-ion mode ESI mass spectrum of a 20 mM solution of (1) in MeTHF. | ||
Moving to the THF-solvated dinuclear complex [(Dipp)(Me3Si)NAlCl3]− [Mg2(μ2-Cl)3·6THF]+ (1) in THF, small amounts of [MgCl(THF)3]+ as well as [Mg2Cl3(THF)n]+, n = 3–5, and [Mg3Cl5(THF)n]+, n = 5 and 6, were detected (Fig. 5c and S16–S19†). Concentration-dependent measurements (Fig. 5d) showed that the relative abundance of the mononuclear ion decreased as a function of concentration, as expected on the basis of the law of mass action. The fraction of the dinuclear ions slightly increased with higher concentrations, whereas that of their trinuclear counterparts decreased slightly, the reason for this decrease not being obvious.
Analysis of solutions of the MeTHF-solvated complex [(Dipp)(Me3Si)NAlCl3]− [Mg3(μ3-Cl)2(μ2-Cl)3·6MeTHF]+ (4) in THF gave similar mass spectra (Fig. S20†). This finding proves that the MeTHF molecules coordinating to Mg centres are easily exchanged by excess of less bulky THF and corroborates our NMR findings. Repeating this experiment in MeTHF resulted in the detection of the trinuclear ions [Mg3Cl5(MeTHF)n]+, n = 4 and 5 (Fig. 5e and S21†). In comparison with THF-solvated 1 in THF, the nuclearity of the observed complexes was significantly shifted toward higher aggregation states. Accordingly, the behavior of these salts in solution appears to parallel their behavior in the solid state.
Next we performed the reverse control experiment and dissolved the THF-containing salt 1 in MeTHF (Fig. 5f and S22†). In this case, the recorded ESI mass spectrum showed mainly ions coordinated by MeTHF, but a few complexes retaining a single THF molecule as well. This incomplete exchange again indicates that THF binds to the magnesium cations more strongly than MeTHF.
Finally, further information was obtained from the gas-phase fragmentation experiments (Fig. S10–S20†). The Mg complexes binding THF and MeTHF exclusively dissociated by losing one or two solvent molecules (Fig. S28–S33†). For the larger and more fully solvated ions, the loss of one THF or MeTHF molecule occurred so easily that it proceeded even without the application of any extra excitation energy, as also the poorer mass resolution of the isotope patterns for these ions indicated.33 For the smaller and less solvated ions, the loss of one THF or MeTHF molecule occurred less easily and required the concomitant addition of one water molecule to avoid a decrease in the coordination number (the ion trap mass spectrometer inevitably contains a low partial pressure of background water). The TMEDA-containing ions exchanged a ligand for water only to a minor extent, but mainly decomposed by expulsion of a neutral [MgCl2(TMEDA)] fragment (Fig. S26 and S27†). The analogous loss of neutral [MgCl2(Me6TREN)] was also observed for the dinuclear complex [Mg2Cl3(Me6TREN)2]+ (Fig. S25†) whereas such a fragmentation reaction was not feasible for its mononuclear counterpart. This mononuclear ion only underwent partial decomposition of the ligand (Fig. S23†). This deviating behavior of the TMEDA- and Me6TREN-containing complexes reflects the significantly stronger binding energies of these chelating ligands in comparison with monodentate THF and MeTHF.
Conclusion
In summary, mono- and trinuclear chloromagnesium cations (charge-balanced by a common organohaloaluminate counter-anion), implicated previously as key solution species in magnesium aluminate battery electrolytes, have now been rationally and selectively prepared for the first time by controlling the magnesium solvating Lewis donor additive. Paired alongside the thermodynamically favoured dinuclear derivative, these reproducible synthetic protocols represent a significant step forward since access to such a family opens the door to a greater understanding of their solution chemistry, particularly due to their excellent solubility. The present study has already taken the first steps in this direction and demonstrated the particular suitability of ESI mass spectrometry for this purpose. We have also introduced 2-methyl-THF as a coordinating Lewis donor into this chemistry and shown that it promotes and stabilizes formation of a trinuclear, magnesium rich cationic species, both in the solid state and in bulk MeTHF solution. Given its green credentials, we further intend to pursue this solvents applicability in systems such as these and hope that other research groups, inspired by our own findings, may follow suit.Acknowledgements
We gratefully acknowledge the Royal Society of Edinburgh (BP Trust Fellowship to SDR), the Royal Society (Wolfson research merit award to REM), the EPSRC (grant award no. EP/L027313/1 and EP/K001183/1) and the Deutsche Forschungsgemeinschaft (grant no. KO 2875/4-1).References
- (a) H. D. Yoo, I. Shterenberg, Y. Gofer, G. Gershinsky, N. Pour and D. Aurbach, Energy Environ. Sci., 2013, 6, 2265–2279 RSC; (b) R. Van Noorden, Nature, 2014, 507, 26–28 CrossRef CAS PubMed.
- http://www.greentechmedia.com/articles/read/Is-There-Enough-Lithium-to-Maintain-the-Growth-of-the-Lithium-Ion-Battery-M .
- T. D. Gregory, R. J. Hoffman and R. C. Winterton, J. Electrochem. Soc., 1990, 137, 775–780 CrossRef CAS.
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Nature, 2000, 407, 724–727 CrossRef CAS PubMed.
- Magnesium aluminates have many other practical uses: (a) For trapping of CO2 with a magnesium aluminate complex see: C.-C. Chang, M.-C. Liao, T.-H. Chang, S.-M. Peng and G.-H. Lee, Angew. Chem., Int. Ed., 2005, 44, 7418–7420 CrossRef CAS PubMed; (b) For uses in iron-catalyzed cross-coupling see: S. Kawamura, K. Ishizuka, H. Takaya and M. Nakamura, Chem. Commun., 2010, 46, 6054–6056 RSC; (c) S. Kawamura, T. Kawabata, K. Ishizuka and M. Nakamura, Chem. Commun., 2012, 48, 9376–9378 RSC; (d) For uses in aromatic functionalization see: A. Unsinn, S. H. Wunderlich, A. Jana, K. Karaghiosoff and P. Knochel, Chem. – Eur. J., 2013, 19, 14687–14696 CrossRef CAS PubMed.
- Y. Viestfrid, M. D. Levi, Y. Gofer and D. Aurbach, J. Electroanal. Chem., 2005, 576, 183–195 CrossRef CAS.
- (a) T. Liu, Y. Shao, G. Li, M. Gu, J. Hu, S. Xu, Z. Nie, X. Chen, C. Wang and J. Liu, J. Mater. Chem. A, 2014, 2, 3430–3438 RSC; (b) R. E. Doe, R. Han, J. Hwang, A. J. Gmitter, I. Shterenberg, H. D. Yoo, N. Pour and D. Aurbach, Chem. Commun., 2014, 50, 243–245 RSC.
- (a) H. Gizbar, Y. Vestrid, O. Chusid, Y. Gofer, H. E. Gottlieb, V. Marks and D. Aurbach, Organometallics, 2004, 23, 3826–3831 CrossRef CAS; (b) Z. Zhao-Karger, J. E. Mueller, X. Zhao, O. Fuhr, T. Jacob and M. Fichtner, RSC Adv., 2014, 4, 26294–26297 RSC.
- (a) N. Pour, Y. Gofer, D. T. Major and D. Aurbach, J. Am. Chem. Soc., 2011, 133, 6270–6278 CrossRef CAS PubMed; (b) J. Muldoon, C. B. Bucur, A. G. Oliver, T. Sugimoto, M. Matsui, H. S. Kim, G. D. Allred, J. Zajicek and Y. Kotani, Energy Environ. Sci., 2012, 5, 5941–5950 RSC.
- (a) H. S. Kim, T. S. Arthur, G. D. Allred, J. Zajicek, J. G. Newman, A. E. Rodnyansky, A. G. Oliver, W. C. Boggess and J. Muldoon, Nat. Commun., 2011, 2, 427–432 CrossRef PubMed; (b) Z. Zhao-Karger, X. Zhao, O. Fuhr and M. Fichtner, RSC Adv., 2013, 3, 16330–16335 RSC.
- J. Muldoon, C. B. Bucur, A. G. Oliver, J. Zajicek, G. D. Allred and W. C. Boggess, Energy Environ. Sci., 2013, 6, 482–487 CAS.
- C. B. Bucur, T. Gregory, A. G. Oliver and J. Muldoon, J. Phys. Chem. Lett., 2015, 6, 3578–3591 CrossRef CAS PubMed.
- (a) A. Benmayza, M. Ramanathan, T. S. Arthur, M. Matsui, F. Mizuno, J. Guo, P.-A. Glans and J. Prakash, J. Phys. Chem. C, 2013, 117, 26881–26888 CrossRef CAS; (b) C. J. Barile, R. Spatney, K. R. Zavadil and A. A. Gewirth, J. Phys. Chem. C, 2014, 118, 20694–10699 CrossRef; (c) Y. Cheng, R. M. Stolley, K. S. Han, Y. Shao, B. W. Arey, N. M. Washton, K. T. Mueller, M. L. Helm, V. L. Sprenkle, J. Liu and G. Li, Phys. Chem. Chem. Phys., 2015, 17, 13307–13314 RSC.
- P. Canepa, S. Jayaraman, L. Cheng, N. N. Rajput, W. D. Richards, G. S. Gautam, L. A. Curtiss, K. A. Persson and G. Ceder, Energy Environ. Sci., 2015, 8, 3718–3730 CAS.
- (a) K. A. See, K. W. Chapman, L. Zhu, K. M. Wiaderek, O. J. Borkiewicz, C. J. Barile, P. J. Chupas and A. A. Gewirth, J. Am. Chem. Soc., 2016, 138, 328–337 CrossRef CAS PubMed; (b) J. L. Esbenshade, C. J. Barile, T. T. Fister, K. L. Bassett, P. Fenter, R. G. Nuzzo and A. A. Gewirth, J. Phys. Chem. C, 2015, 119, 23366–23372 CrossRef CAS.
- (a) T. Liu, J. T. Cox, D. Hu, X. Deng, J. Hu, M. Y. Hu, J. Xiao, Y. Shao, K. Tang and J. Liu, Chem. Commun., 2015, 51, 2312–2315 RSC; (b) C. J. Barile, E. C. Barile, K. R. Zavadil, R. G. Nuzzo and A. A. Gewirth, J. Phys. Chem. C, 2014, 118, 27623–27630 CrossRef CAS.
- J. Muldoon, C. B. Bucur and T. Gregory, Chem. Rev., 2014, 114, 11683–11720 CrossRef CAS PubMed.
- T. S. Arthur, P.-A. Glans, M. Matsui, R. Zhang, B. Ma and J. Guo, Electrochem. Commun., 2012, 24, 43–46 CrossRef CAS.
- See the following & references cited therein: (a) A. J. Martinez-Martinez, D. R. Armstrong, B. Conway, B. J. Fleming, J. Klett, A. R. Kennedy, R. E. Mulvey, S. D. Robertson and C. T. O′Hara, Chem. Sci., 2014, 5, 771–781 RSC; (b) D. R. Armstrong, E. Crosbie, E. Hevia, R. E. Mulvey, D. L. Ramsay and S. D. Robertson, Chem. Sci., 2014, 5, 3031–3045 RSC.
- This RR′NAlCl2 species has previously been prepared by ligand exchange of RR′NAlMe2 and Me3SnCl: M. Schiefer, H. Hatop, H. W. Roesky, H.-G. Schmidt and M. Noltemeyer, Organometallics, 2002, 21, 1300–1303 CrossRef CAS.
- (a) For an Al(I) species see: M. Schiefer, N. D. Reddy, H. W. Roesky and D. Vidovic, Organometallics, 2003, 22, 3637–3638 CrossRef CAS; (b) For a 2-coordinate Si(II) species see: A. V. Protchenko, K. H. Birjkumar, D. Dange, A. D. Schwarz, D. Vidovic, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2012, 134, 6500–6503 CrossRef CAS PubMed; (c) For a 2-coordinate Ni(II) complex see: M. I. Lipschutz and T. D. Tilley, Chem. Commun., 2012, 48, 7146–7148 RSC; (d) For a 2-coordinate Fe(I) complex see: M. I. Lipschutz, T. Chantarojsiri, Y. Dong and T. D. Tilley, J. Am. Chem. Soc., 2015, 137, 6366–6372 CrossRef CAS PubMed; (e) For a series of 2-coordinate Fe/Co/Ni complexes see: C.-Y. Lin, J.-D. Guo, J. C. Fettinger, S. Nagase, F. Grandjean, G. J. Long, N. F. Chilton and P. P. Power, Inorg. Chem., 2013, 52, 13584–13593 CrossRef CAS PubMed.
- L. F. Wan and D. Prendergast, J. Am. Chem. Soc., 2014, 136, 14456–14464 CrossRef CAS PubMed.
- P. Sobota, T. Pluzinski, J. Utko and T. Lis, Inorg. Chem., 1989, 28, 2217–2219 CrossRef CAS.
- (a) M. G. Davidson, D. Garcia-Vivo, A. R. Kennedy, R. E. Mulvey and S. D. Robertson, Chem. – Eur. J., 2011, 17, 3364–3369 CrossRef CAS PubMed; (b) T. Cadenbach, E. Hevia, A. R. Kennedy, R. E. Mulvey, J.-A. Pickrell and S. D. Robertson, Dalton Trans., 2012, 41, 10141–10144 RSC; (c) D. R. Armstrong, M. G. Davidson, D. Garcia-Vivo, A. R. Kennedy, R. E. Mulvey and S. D. Robertson, Inorg. Chem., 2013, 52, 12023–12032 CrossRef CAS PubMed; (d) A. R. Kennedy, R. E. Mulvey, R. I. Urquhart and S. D. Robertson, Dalton Trans., 2014, 43, 14265–14274 RSC; (e) S. D. Robertson, A. R. Kennedy, J. J. Liggat and R. E. Mulvey, Chem. Commun., 2015, 51, 5452–5455 RSC.
- L. M. Guard and N. Hazari, Organometallics, 2013, 32, 2787–2794 CrossRef CAS.
- (a) B. Werner, T. Kräter and B. Neumüller, Z. Anorg. Allg. Chem., 1995, 621, 346–358 CrossRef CAS; (b) D. Loos, K. Eichkorn, J. Magull, R. Ahlrichs and H. Schnöckel, Z. Anorg. Allg. Chem., 1995, 621, 1582–1588 CrossRef CAS; (c) W. Zhang, J.-P. Hu, X.-F. Ding, Y.-J. Wu and Z.-W. Ye, Inorg. Chem. Commun., 2003, 6, 1185–1187 CrossRef CAS; (d) J. K. Vohs, L. E. Downs, M. E. Barfield, K. Latibeaudiere and G. H. Robinson, J. Organomet. Chem., 2003, 666, 7–13 CrossRef CAS; (e) P. C. Andrews, P. C. Junk, I. Nuzhnaya, L. Spiccia and N. Vanderhoek, J. Organomet. Chem., 2006, 691, 3426–3433 CrossRef CAS.
- R. B. Bedford, P. B. Brenner, E. Carter, P. M. Cogswell, M. F. Haddow, J. N. Harvey, D. M. Murphy, J. Nunn and C. H. Woodall, Angew. Chem., Int. Ed., 2014, 53, 1804–1808 CrossRef CAS PubMed.
- C. Liao, B. Guo, D.-E. Jiang, R. Custelcean, S. M. Mahurin, X.-G. Sun and S. Dai, J. Mater. Chem. A, 2014, 2, 581–584 CAS.
- V. Pace, P. Hoyos, L. Castoldi, P. Dominguez de Maria and A. R. Alcantara, ChemSusChem, 2012, 5, 1369–1379 CrossRef CAS PubMed.
- V. R. Koch and J. H. Young, Science, 1979, 204, 499–501 CAS.
- (a) B. H. Lipshutz, K. L. Stevens, B. James, J. G. Pavlovich and J. P. Snyder, J. Am. Chem. Soc., 1996, 118, 6796–6797 CrossRef CAS; (b) K. Koszinowski, J. Am. Chem. Soc., 2010, 132, 6032–6040 CrossRef CAS PubMed; (c) J. E. Fleckenstein and K. Koszinowski, Organometallics, 2011, 30, 5018–5026 CrossRef CAS; (d) D. Schröder, L. Ducháčková, J. Tarábek, M. Karwowska, K. J. Fijalkowski, M. Ončák and P. Slaviček, J. Am. Chem. Soc., 2011, 133, 2444–2451 CrossRef PubMed; (e) M. A. Henderson, T. K. Trefz, S. Collins, M. Y. Wang and J. S. McIndoe, Organometallics, 2013, 32, 2079–2083 CrossRef CAS; (f) T. K. Trefz, M. A. Henderson, M. Linnolahti, S. Collins and J. S. McIndoe, Chem. – Eur. J., 2015, 21, 2980–2991 CrossRef CAS PubMed.
- (a) V. B. Di Marco and G. G. Bombi, Mass Spectrom. Rev., 2006, 25, 347–379 CrossRef CAS PubMed; (b) N. G. Tsierkezos, J. Roithová, D. Schröder, M. Ončak and P. Slaviček, Inorg. Chem., 2009, 48, 6287–6296 CrossRef CAS PubMed.
- The peaks broadened because of dissociation reactions occuring during the m/z scan.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1431065–1431068 and 1443823. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt00531d |
| This journal is © The Royal Society of Chemistry 2016 |