
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemistry and catalytic properties of amphiphilic vitamin B12 derivatives in nonaqueous media†
M.
Giedyk
a,
H.
Shimakoshi
*b,
K.
Goliszewska
a,
D.
Gryko
*a and
Y.
Hisaeda
*b
aInstitute of Organic Chemistry Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: dorota.gryko@icho.edu.pl; Web: http://ww2.icho.edu.pl/gryko_group
bDepartment of Chemistry and Biochemistry, Graduate School of Engineering, Kyushu University, Fukuoka, 819-0395, Japan. E-mail: shimakoshi@mail.cstm.kyushu-u.ac.jp; yhisatcm@mail.cstm.kyushu-u.ac.jp
First published on 2nd March 2016
Abstract
The reduction pathway of cobalester (CN)Cble, an amphiphilic vitamin B12 derivative, was investigated in organic solvents under electrochemical conditions and compared with mono- and dicyanocobyrinates. The redox characteristics were determined using cyclic voltammetry and spectroelectrochemical methods. The presence of a nucleotide moiety in B12-derivative impedes the in situ formation of dicyano-species thus facilitating the (CN)Co(III) to Co(I) reduction. The (CN)Cble shows stepwise reduction to Co(I) via (CN)Co(II). The reduction of (CN)Co(II)/Co(I) was found to depend on cyanide-solvent exchange equilibrium with weakly coordinating solvents and bulky peripheral chains promoting intact (CN)Co(II) species existence. The studied complexes were also utilized as catalysts in bulk electrolysis of benzyl bromide affording bibenzyl in very good yield.
Introduction
Vitamin B12 (cyanocobalamin 1) is a highly functionalised organometalic complex comprising a corrin ring with the central cobalt cation and peripheral amide chains as shown in Fig. 1.1,2 As a cofactor for B12-dependent enzymes it plays an important role in the normal functioning of mammalian organisms.3–5 Inspired by nature, scientists successfully employed cyanocobalamin (1) and its derivatives as catalysts in organic synthesis, due to their ability to form and selectively cleave Co–C bonds.6–9 The scope of B12-catalyzed reactions is continuously expanding, now including such processes as dehalogenation, addition to double bonds, ring expansion, C–H functionalization etc.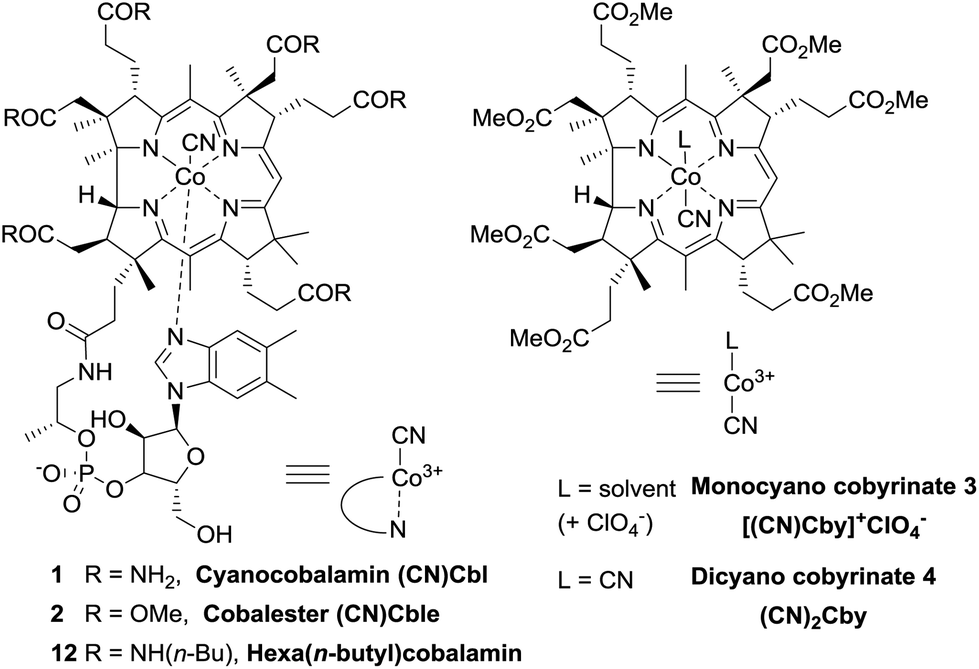 | ||
| Fig. 1 Structures and abbreviations of vitamin B12 derivatives. | ||
The catalytic properties of vitamin B12 derivatives rely on redox chemistry of the central cobalt cation with the reduction of Co(III) to active Co(II) or Co(I) forms being critical.10 Therefore, the understanding of their electrochemistry becomes an essential element for the rational design and successful application of B12-catalyzed reactions.
Even though the natural, nontoxic, and stable cyanocobalamin (1) may appear as an ideal catalyst, it suffers from low solubility in synthetically useful organic solvents such as DCM, MeCN, THF etc. Hence, modification of its structure is commonly performed giving access to more lipophilic derivatives.11 For example, the synthesis and utilization of heptamethyl cobyrinates (e.g. monocyanocobyrinate 3) have been well established.12–17 An alternative approach was recently reported by Gryko et al. who synthesized cobalester ((CN)Cble, 2) – a derivative possessing six ester groups on the periphery of the corrin ring with the nucleotide fragment intact that displays good solubility both in aqueous and in organic media.18 A direct comparison of derivatives with and without the nucleotide loop became thereby possible.
Whereas the redox characteristics of cobyrinate derivatives in organic solvents have been extensively studied by Murakami and Hisaeda,19–21 no suitable data are available for cobalester (2). We wondered whether the nucleotide fragment influences the redox behavior and catalytic activity of cobalamin derivatives in organic solvents, preferred in organic synthesis.
Herein, we present the redox characteristics of cobalester (2) with regard to factors influencing the reduction of Co(III) to catalytically active Co(II) and Co(I) species and demonstrate its application as a catalyst in potential-controlled electrolysis of benzyl bromide.
Results and discussion
Cyclic voltammetry of vitamin B12 derivatives in organic solvents
In order to understand the influence of the nucleotide fragment on the redox behaviour of cobalesters (2) in organic solvents, a series of cyclic voltammograms was recorded. The use of a glassy carbon working electrode proved superior over Pt, allowing for the reproducible observation of spectra in a broad range of potentials with clear detection of reduction and oxidation peaks. The spectrum of cobalester (2) in MeCN displayed apparent one quasi-reversible reduction peak (Ered) at ca. −0.9 V vs. Ag–AgCl which corresponds to two-step reduction of complex 2, leading to Co(I) species via (CN)Co(II) species (Fig. 2A). Such redox behaviour is in agreement with that observed by Lexa and Savéant for cyanocobalamin in water and DMSO,22,23 but is distinctively different from heptamethyl cobyrinates 3 and 4 for which Co(III)/Co(II) and Co(II)/Co(I) redox couples are well separated (Fig. 2C and D). This indicates an efficient formation of Co(I) species of cobalester (2) at less negative potentials compared to cobyrinate derivatives having the cyanide ligand.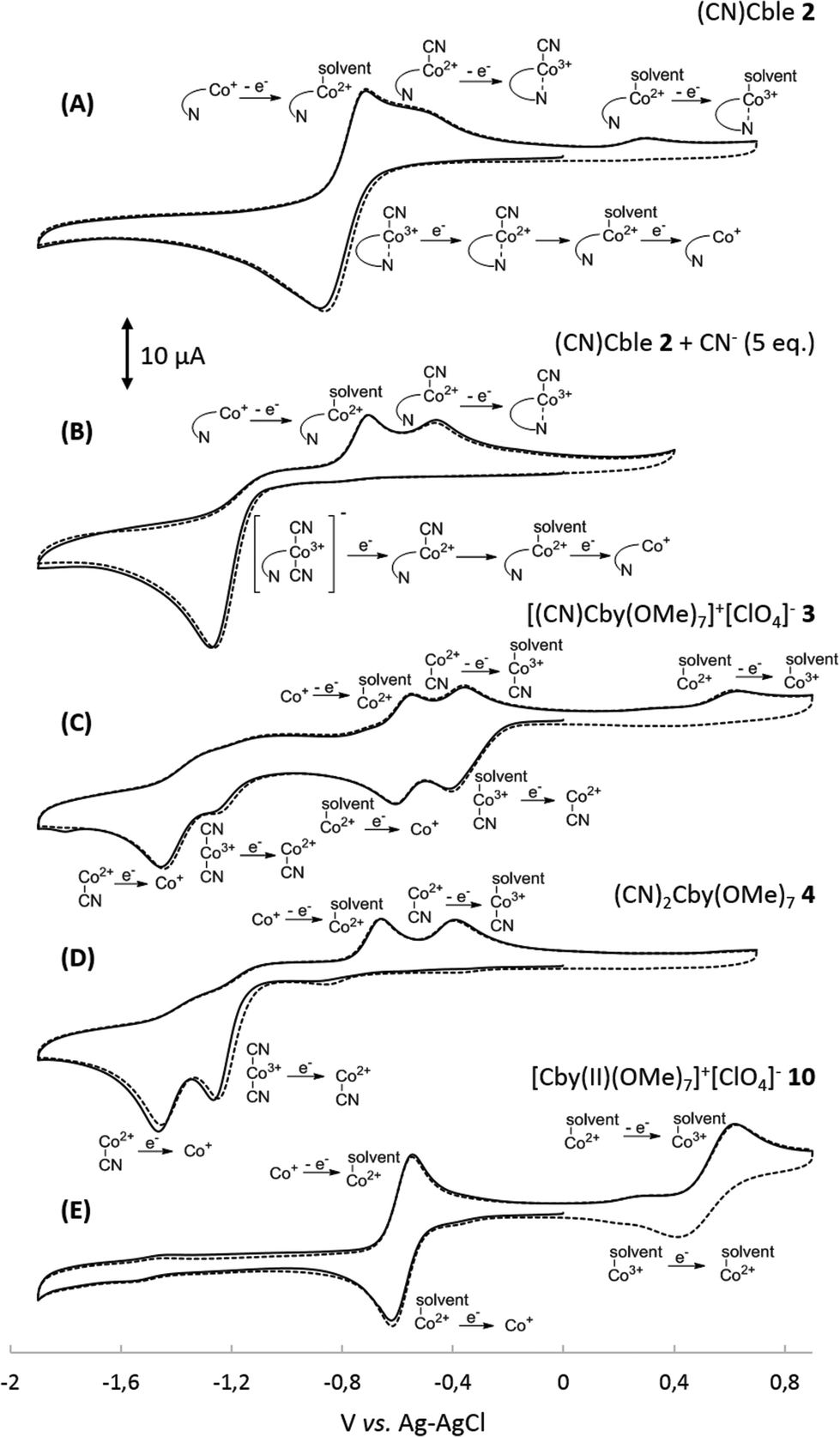 | ||
| Fig. 2 Cyclic voltammograms of 1.0 mM MeCN solutions of: (A) cyanocobalester (2), (B) cyanocobalester (2) with addition of Bu4N+CN− (5 eq.), (C) monocyanocobyrinate (3), (D) dicyanocobyrinate, and (E) cobalt(II) cobyrinate (4). All solutions contained 0.1 M Bu4N+ClO4− and measurements were carried out at room temperature under a nitrogen atmosphere using the GC working electrode and Pt counter electrode; sweep rate: 0.1 V s−1. | ||
Subsequently, we evaluated if and how the presence of an excess of the cyanide anion influences the redox properties of cobalester (2). A series of measurements were performed, proving that the two-electron reduction peak shifts towards the cathodic side (Ered = −1.2 V vs. Ag–AgCl) with the total current remaining the same (Fig. 2B). Supposedly, this was caused by the formation of the base-off (CN)2Co(III) form with an electron rich cobalt centre which is more difficult to reduce. A similar observation has already been described for cyanocobalamin (1).22
Electronic spectra of vitamin B12 derivatives in organic solvents
Subsequently, cyanocobalester (2) was examined using controlled-potential electronic spectroscopy. The spectrum recorded after 15 min of electrolysis at −1.0 V vs. Ag–AgCl revealed the presence of expected Co(I) species with the absorption maximum at 390 nm, which was accompanied by a by-product exhibiting maximum absorption at 370 and 580 nm wavelengths that are characteristic for (CN)2Co(III) species (Fig. 3A).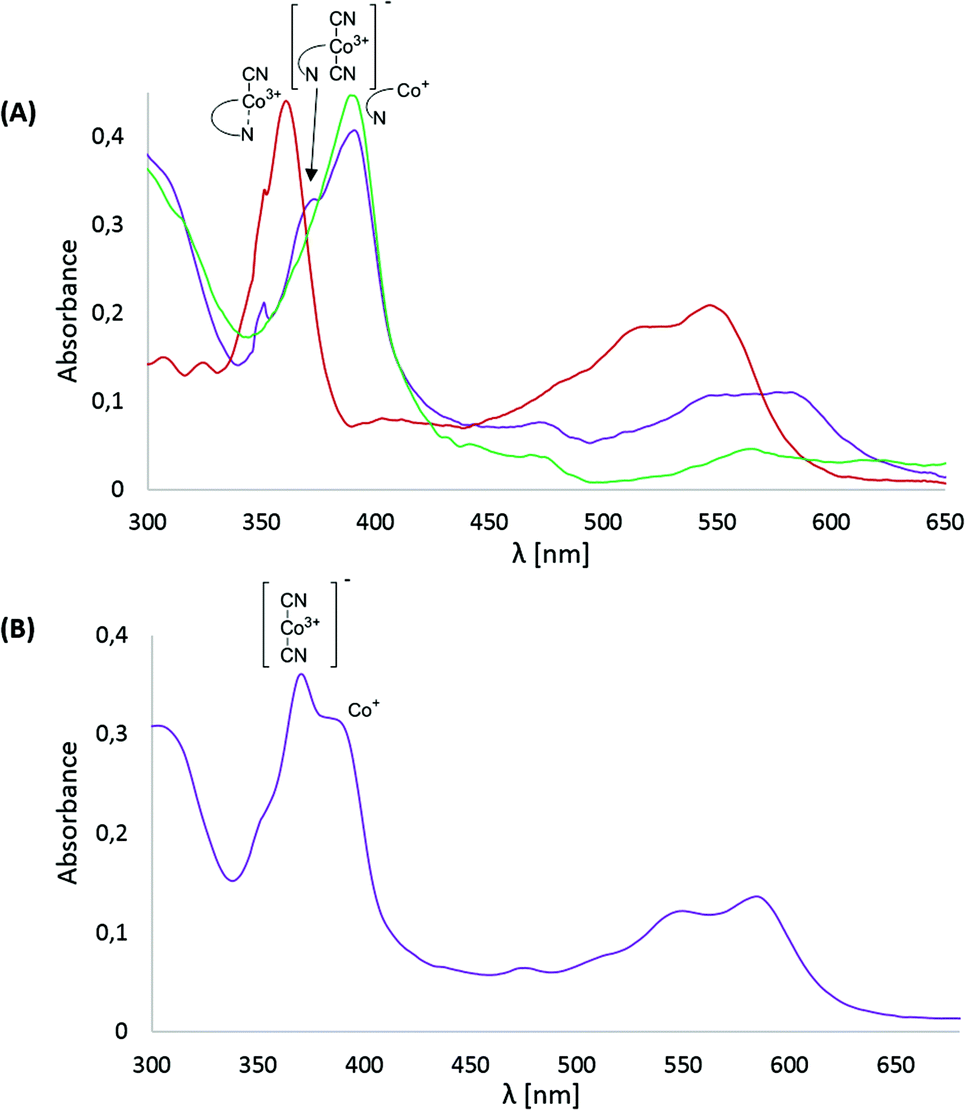 | ||
| Fig. 3 Electronic spectra of 0.33 mM solution in MeCN of: (A) (CN)Cble (2) (red), (CN)Cble (2) subjected to 15 min electrolysis at −1.0 V vs. Ag–AgCl (purple) and fully reduced (CN)Cble (2) (green, 15 min electrolysis at −1.4 V vs. Ag–AgCl); (B) [(CN)Cby]+ClO4− (3) subjected to 15 min reduction at −1.0 V vs. Ag–AgCl. All solutions contained 0.1 M Bu4N+ClO4− and measurements were carried out at room temperature under a nitrogen atmosphere using the carbon grid ITO working electrode and Pt counter electrode. | ||
The absorption bands did not disappear upon prolonging the time of electrolysis, but once the potential was lowered to −1.4 V vs. Ag–AgCl, a mixture quantitatively transformed into a final Co(I) form. Such a behaviour proves the in situ formation of the (CN)2Co(III) complex from cyanocobalester (2) and CN− released during its electrolysis (Fig. 3A). The working hypothesis was confirmed by superimposing the spectrum of the mixture with the one recorded for the solution of [(CN)2Cble]− (prepared by mixing cyanocobalester with 5 eq. of Bu4N+CN−) (see the ESI†). The formation of the cobalt(III) dicyano-complex was also observed when monocyanocobyrinate 3 was subjected to electrolysis at −1.0 V vs. Ag–AgCl (Fig. 3B). In this case the presence of the (CN)2Co(III) complex was even more pronounced as it was dominating over Co(I) species. The difference in the amount of (CN)2Co(III) formation was also detectable by cyclic voltammetry. No formation of dicyanocobalester was observed in 2 (Fig. 2A) within the time scale of the measurement, while the spectrum of cobyrinate 3 having no nucleotide fragment showed a reduction peak at ca. −1.2 V vs. Ag–AgCl (Fig. 2C), revealing the presence of (CN)2Co(III) complex 4.
Mechanistic aspects of the reduction process
The results presented above clearly demonstrate the existence of (CN)Co(III)/(CN)2Co(III) equilibrium during the reduction of compounds 2 and 3 (Scheme 1), with the cyanide binding equilibrium constant for monocyano cobyrinate 3 much higher than the one for cobalester 2 (KIIICble < KIIICby). This stays in agreement with reports by Zelder, who studied cyanide complexation by cobalamin and cobinamide derivatives.24,25 After identification of dicyano-compounds 4 and 8 as important intermediates in the reduction pathways, the attention was focused on the mechanism of the Co(I) species formation. Murakami et al. proposed (CN)Co(II) complex 9 as an intermediate formed in the first step upon monocyano cobyrinate 3 reduction in DMF.20 This in turn supposedly undergoes further reduction to Co(I) species 11 at ca. −1.4 V vs. Ag–AgCl. While this is consistent with our observations for monocyano cobyrinate 3 in MeCN, the spectrum of cobalester (2) did not reveal such a negatively located signal in CV. Therefore, an exchange of the cyanide anion coordinating (CN)Co(II) 5 for a solvent molecule was considered (Scheme 1). In the first step of the reduction (at ca. −0.9 V and −0.4 V vs. Ag–AgCl for compounds 2 and 3 respectively), (CN)Co(II) species 5 and 9 were formed. Additionally to direct reduction at −1.4 V vs. Ag–AgCl, they could also undergo axial ligand exchange to yield (solvent)Co(II) complexes 6 and 10, which are readily reduced at more positive potentials. This stays in contrast to what was proposed for cyanocobalamin (1) in protic solvents, where no (CN)Co(II) intermediate was considered in the absence of purposely added cyanide.23 Furthermore, as it is generally accepted that the benzimidazole moiety in vitamin B12 possesses stronger electron donating abilities than MeCN, the mechanism involving base-on Co(II) form 7 would require more negative potentials. On the other hand, the equilibrium of Co(II)base-on/Co(II)base-off should not favour the base-on form in MeCN as it does in water. This allowed us to exclude the pathway involving the base-on Co(II) complex as less favoured.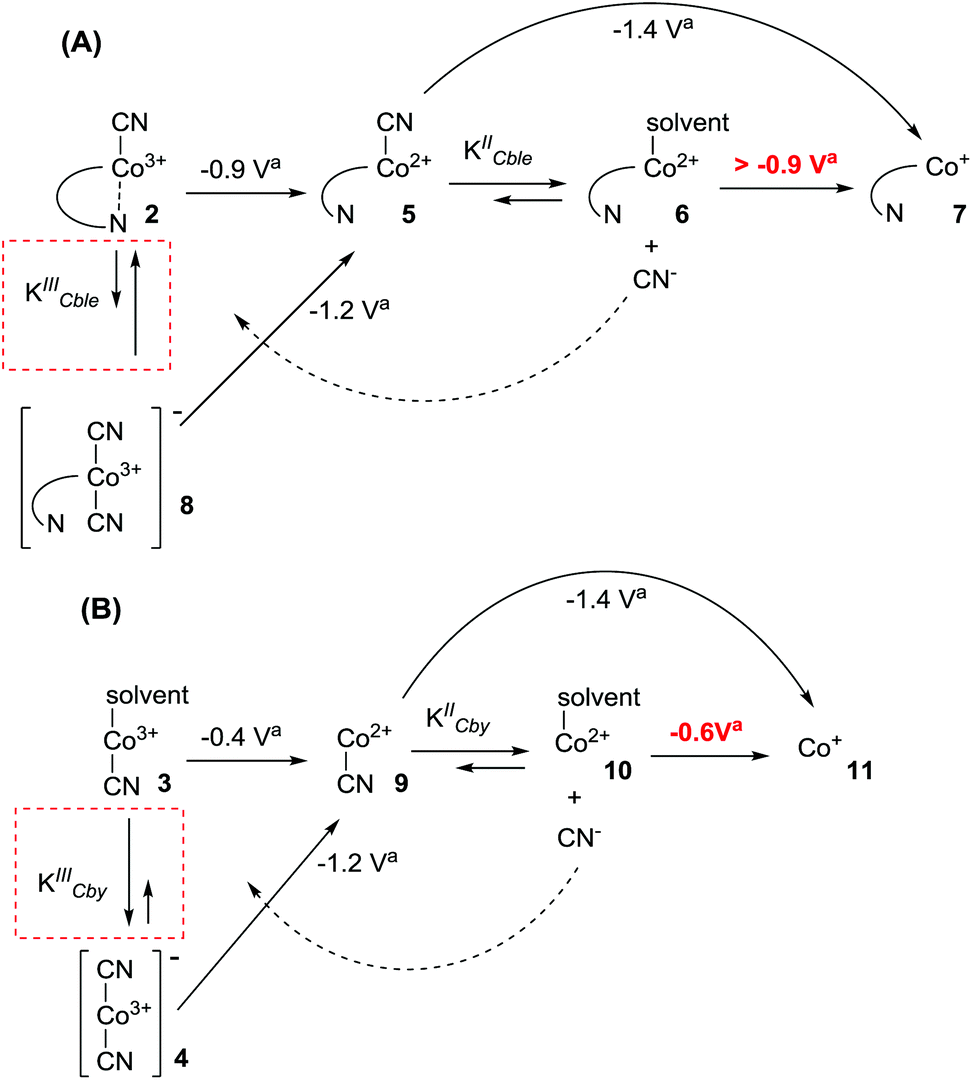 | ||
| Scheme 1 Proposed mechanism for the reduction of: (A) cobalester (2) and (B) monocyanocobyrinate (3). avs. Ag–AgCl. | ||
To further confirm the existence of (CN)Co(II) intermediate 5 and a key role of axial ligand exchange in the generation of Co(I) species 7, the reaction environment was modified in the following manner (Fig. 4). Firstly, the cyclic voltammogram was recorded using THF as a solvent. In contrast to the experiment in MeCN, a second reduction peak appeared at ca. −1.4 V vs. Ag–AgCl, indicating the direct reduction of cyano-form 5 to Co(I) complex 7. Such a behavior is consistent with our mechanistic hypothesis, as weaker coordination abilities (compared to MeCN) towards transition metals result in a lower KIICble value and afford stabilization of (CN)Co(II) complex 5 under the experimental conditions (Scheme 2). Secondly, hexabutylcobalamin (12) was synthesized by Bu4N+CN−-catalyzed aminolysis of cobalester (2) and examined using cyclic voltammetry. Due to changes at the periphery of the corrin ring, we expected cyanide ligand exchange to be altered. Indeed, a peak assigned to direct (CN)Co(II)/Co(I) reduction was observed (Fig. 4B), thus supporting our proposed mechanism in Scheme 1.
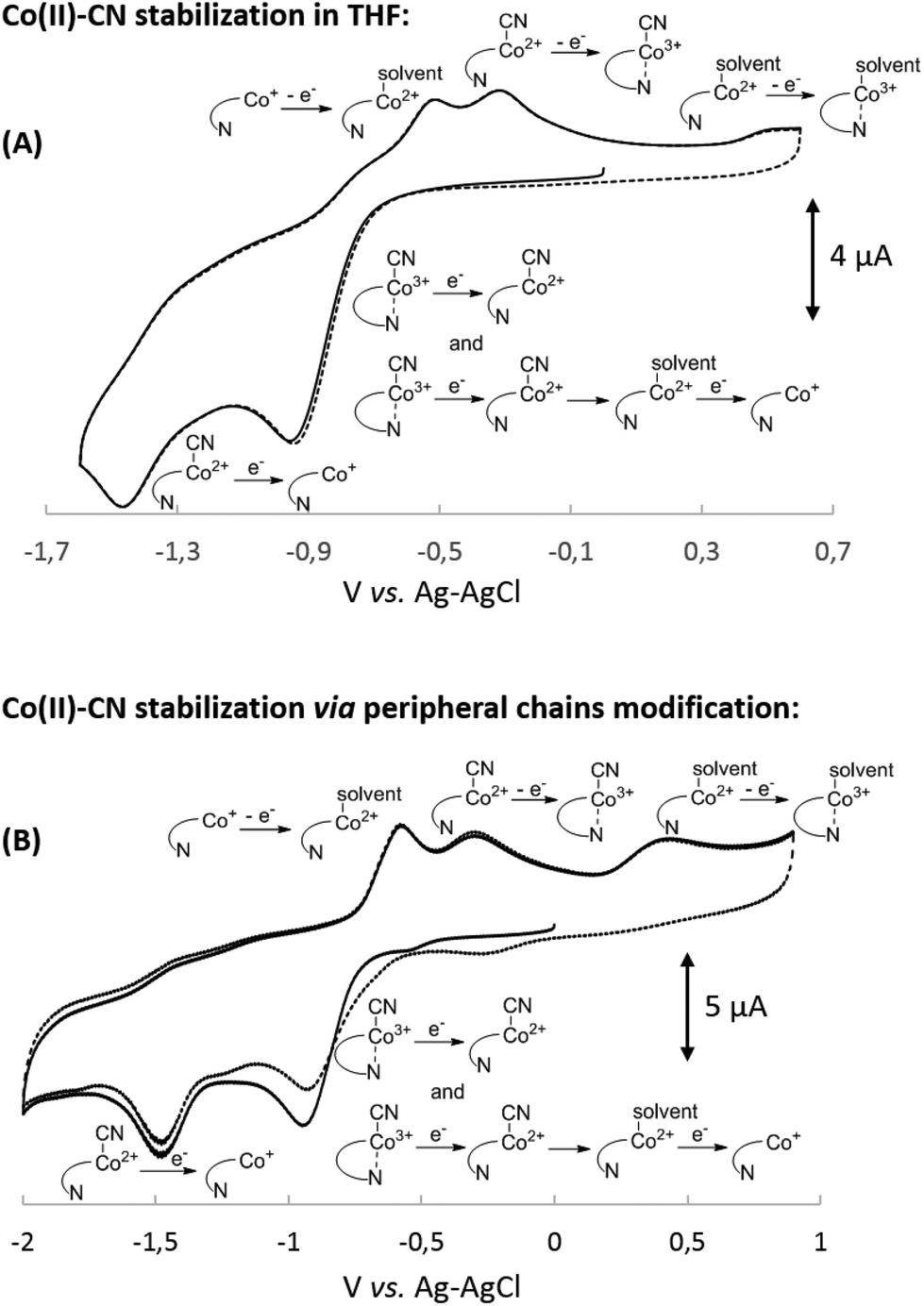 | ||
| Fig. 4 Cyclic voltammograms of 1.0 mM solutions of: (A) cyanocobalester (2) in THF and (B) hexabutylcobalamin (12) in MeCN. All solutions contained 0.1 M Bu4N+ClO4− and measurements were carried out at room temperature under a nitrogen atmosphere using the GC working electrode and Pt counter electrode; sweep rate: 0.1 V s−1. | ||
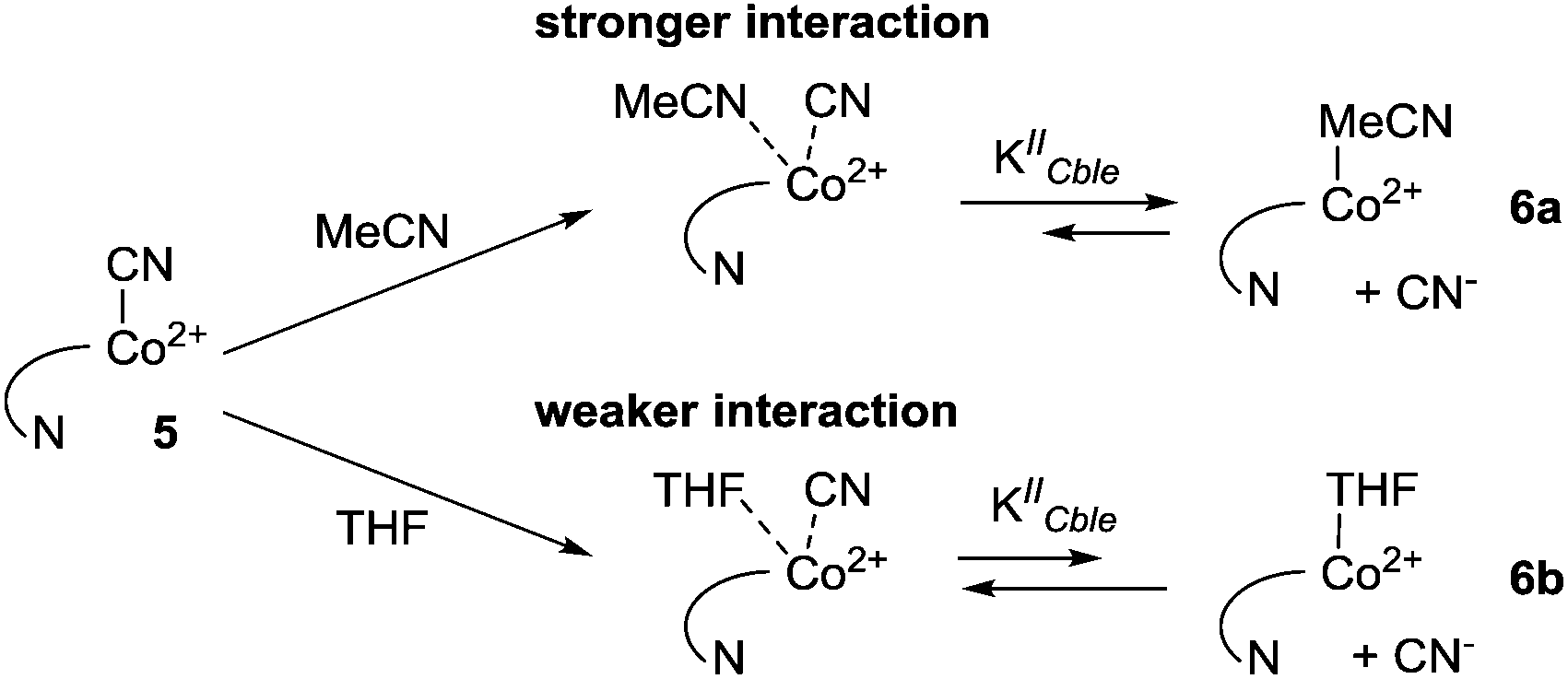 | ||
| Scheme 2 Interactions of solvent molecules with (CN)Co(II) complex 5. | ||
Redox behaviour of amphiphilic vitamin B12 in the presence of benzyl bromide
With our quest to understand the catalytic activity of vitamin B12 derivatives in a nonaqueous environment, we examined the redox behaviour of cobalester (2) in the presence of benzyl bromide. We have previously reported that cobalester (2) catalyzes dimerization of various benzyl bromides in the microwave-assisted reaction yielding substituted bibenzyl derivatives in high yields.18 A control measurement of benzyl bromide solution revealed a direct reduction peak at negative potential (Ered = −1.9 V vs. Ag–AgCl) (Fig. 5A). The addition of cobalester (2) (10 mol%) shifted the signal towards the anodic side by ca. 0.5 V marking a catalytic current (Fig. 5B). Moreover, when the scan was reversed at −0.95 V vs. Ag–AgCl, no reversible wave was observed (see the ESI†). This implies that once Co(I) species 7 is generated, it rapidly reacts with the electrophile forming complex 13 with the cobalt–carbon bond. The presence of a plateau between −0.9 and −1.0 V vs. Ag–AgCl indicated the stability of complex 13 in this range of the spectrum. Further negative sweeping of the potential might cause reduction of complex 13 to anion radical 14 that decomposed forming a benzyl radical and regenerating the active form of catalyst 7 (Scheme 3).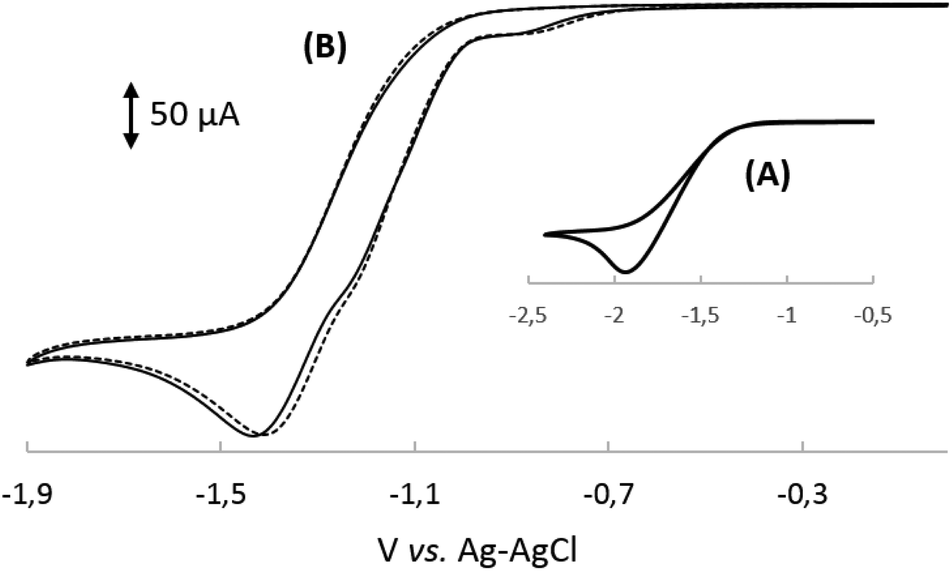 | ||
| Fig. 5 Cyclic voltammograms of MeCN solutions of: (A) benzyl bromide (10 mM) and (B) cyanocobalester 2 (1.0 mM) in the presence of BnBr (10 mM). All solutions contained 0.1 M Bu4N+ClO4− and measurements were carried out at room temperature under a nitrogen atmosphere using the GC working electrode and Pt counter electrode; sweep rate: 0.1 V s−1. | ||
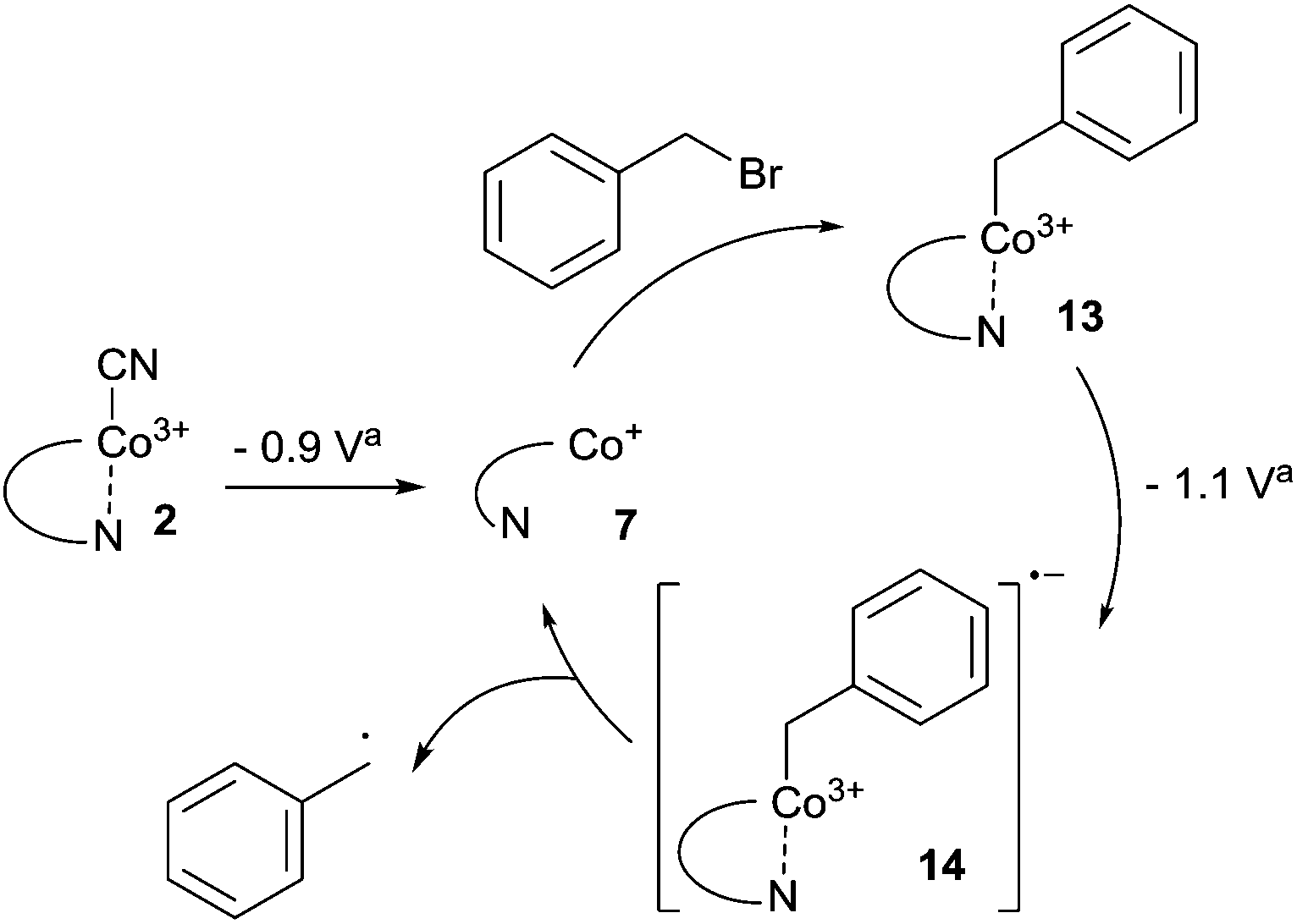 | ||
Scheme 3 Cobalester-catalyzed generation of the benzyl radical via intermediate Co–C species 13 and 14. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) vs. Ag–AgCl. vs. Ag–AgCl. | ||
Controlled-potential electrolysis of benzyl bromide
The controlled-potential electrolysis of benzyl bromide in the presence of (CN)Cble (2) gave bibenzyl (Table 1). ESI MS analysis of the reaction mixture revealed the presence of Co-benzyl intermediate 13 (see the ESI†).|
|
|||
|---|---|---|---|
| Entry | Working electrode | Potentialb [V] | Yieldc [%] |
| a Controlled-potential electrolyses were carried out in MeCN under a N2 atmosphere at rt using a divided cell system with the Zn counter-electrode. Initial concentration in the working-electrode compartment: BnBr: 0.25 M, (CN)Cble 2: 1.25 mM, Bu4N+ClO4−: 0.2 M. b vs. Ag–AgCl. c The quantity of the product is the average of at least two repeated experiments from the GC-MS results. d Reaction performed in THF. e 0.125 M solution of benzyl bromide was used. f No catalyst. | |||
| 1 | Pt | −1.1 | 69 |
| 2 | Carbon-felt | −1.1 | 76 |
| 3d | Carbon-felt | −1.1 | 13 |
| 4e | Carbon-felt | −1.1 | 71 |
| 5 | Carbon-felt | −1.0 | 67 |
| 6f | Carbon-felt | −1.1 | 13 |
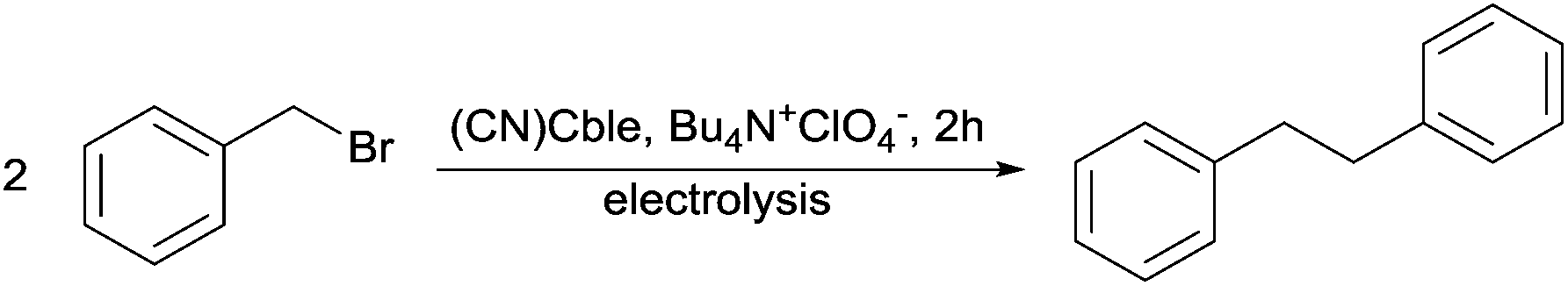
The use of carbon felt as a working electrode proved superior over Pt, which was consistent with the conditions of cyclic voltammetry measurements (compare entries 1 and 2). Among potentials examined, −1.1 V vs. Ag–AgCl was optimal giving the yield of 76% (entry 2). As expected, the use of THF as a solvent (entry 3) led to a dramatic decrease in the yield, due to impeded generation of active Co(I) species 7. Lowering the concentration of the starting material only slightly affected the yield (entry 4). As for the applied potential, −1.0 V vs. Ag–AgCl still allowed for an efficient reaction to occur (entry 5), however, no further decrease was possible (data not shown). The background reaction without the addition of the catalyst gave the desired product in low yield (entry 6).
Next we examined cobyrinate-derived complexes 3 and 4 as catalysts in bulk electrolysis (Table 2). Surprisingly, changing the catalyst to [(CN)Cby]+ClO4− (3) did not affect the reaction outcome (compare entries 1 and 2). This stays in contrast to the efficiency of Co(I) form generation which was proven to be higher for catalyst 2. Furthermore, the use of (CN)2Cby (4) afforded the product in comparable yield, suggesting a shift in Co(I) formation equilibria which is caused by the irreversible reaction of Co(I) species 7 with benzyl bromide. Moreover, once the active Co(I) complex is formed, it recovers in the catalytic cycle, masking initial differences.
| Entry | Catalyst | Conversionb [%] | Yieldb [%] |
|---|---|---|---|
| a Controlled-potential electrolyses were carried out at −1.1 V vs. Ag–AgCl in MeCN under a N2 atmosphere at rt using a divided cell system with the carbon-felt working electrode and Zn counter-electrode. Initial concentration in the working-electrode compartment: BnBr: 0.25 M, catalyst: 1.25 mM, Bu4N+ClO4−: 0.2 M. b The quantity of the product is the average of at least two repeated experiments from the GC-MS results. | |||
| 1 | (CN)Cble (2) | 95 | 76 |
| 2 | [(CN)Cby]+ClO4− (3) | 88 | 76 |
| 3 | (CN)2Cby (4) | 86 | 64 |
Conclusions
In conclusions, for the first time, the reduction pathway for cobalester (2) was determined in aprotic solvents and compared with mono- and dicyanocobyrinates 3 and 4. The reaction mechanism involves axial cyanide exchange which depends on the coordination abilities of a solvent, as well as the steric hindrance around the cobalt centre. Furthermore, it was found that the reduction process is influenced by the affinity of the cobalt cation to cyanide, which results in the formation of stable dicyano-species 4 and 8. The latter feature, however, does not determine the outcome of Co(I)-catalyzed bulk electrolysis of benzyl bromide, as the irreversible formation of the Co-benzyl complex facilitates the reduction process.Our findings provide a new insight into B12 derivatives redox chemistry in an organic, aprotic environment. We expect that the understanding of factors that influence the generation of active Co(I) will soon result in the development of new B12-catalyzed organic reactions.
Experimental
Materials
All solvents and chemicals were of reagent grade and used without further purification. Dry MeCN, THF and N,N-dimethylformamide (DMF) were purchased from Wako Chemicals. Tetra(n-butyl)ammonium perchlorate (n-Bu4N+ClO4−) was purchased from Sigma Aldrich. Cyanocobalester 2, monocyanocobyrinate 3 and dicyanocobyrinate 4 were synthesized according to reported procedures.12,18,26General analysis and measurements
The electronic absorption spectra were recorded using a Hitachi U-3000 spectrophotometer and a 10 mm cell.The cyclic voltammograms were obtained using a BAS ALS-630C electrochemical analyzer. The gas chromatography-mass spectra (GC-MS) were obtained using a Shimadzu GCMS-QP5050 equipped with a J&W Scientific DB-1 column (length: 30 m; ID: 0.25 mm, film: 0.25 μm) and helium as the carrier gas. For the measurement, the injector and detector temperatures were 250 °C, the oven temperature was initially held at 40 °C for 4 min, and then increased to 240 °C at a rate of 10 °C min−1. The ESI-MS was measured by using a JMS-T100LC AccuTOF from JEOL.
The spectroelectrochemical measurements for 2 and 3 were carried out in MeCN containing of 0.1 M n-Bu4N+ClO4− under N2 with a BAS SEC2000 spectrometer equipped with carbon-grid ITO glass as a working electrode, a Pt wire as an counter electrode, and an Ag/AgCl electrode (3 M aq. NaCl) as the reference.
Cyclic voltammetry
A cylindrical three-electrode cell that was used was equipped with a glassy carbon working electrode, a 25 mm platinum wire as the counter electrode and an Ag/AgCl (3.0 M NaCl) electrode as the reference electrode. The scan rate for a typical experiment was 100 mV s−1. The dry solution of the vitamin B12 derivatives (1.0 × 10−3 M) and n-Bu4N+ClO4− (1.0 × 10−1 M) was deaerated by N2 gas bubbling before the measurements, and the cyclic voltammetry was carried out under a N2 gas atmosphere at room temperature. The E1/2 value of the ferrocene–ferrocenium (Fc/Fc+) was 0.56 V vs. Ag/AgCl with this setup.Catalytic benzyl bromide dimerization reaction under electrochemical conditions
Controlled-potential electrolyses were carried out in dry MeCN at −1.1 V vs. Ag/AgCl under a N2 atmosphere in a divided electrolysis cell separated by a polypropylene membrane. The cathodic compartment was equipped with a Pt mesh or a carbon felt working electrode, an Ag/AgCl (3 M aq. NaCl) reference electrode and a stirring bar. The anodic compartment was equipped with a sacrificial Zn-plate electrode. The applied potential between the working and reference electrodes in the electrolysis was maintained constant with a Hokuto Denko HA BF-501A potentiostat, and the electrical quantity was also recorded on it. For a typical reaction, a MeCN solution of the vitamin B12 derivative (1.25 × 10−3 M), BnBr (2.5 × 10−1 M), n-Bu4N+ClO4− (2.0 × 10−1 M) was deaerated by N2 and subjected to electrolysis for 2 h at room temperature. Solutions from both compartments were then mixed together, passed through a silica pad and analyzed by GC-MS with biphenyl as the internal standard.Synthesis of hexa(n-butyl)cobalamin (12)
A sealed pressure tube was charged with cyanocobalester (2, 15 mg, 1.0 × 10−2 mmol), Bu4N+CN− (17 mg, 6 × 10−2 mmol), dry MeOH (0.1 mL) n-butylamine (0.5 mL) and diethyl cyanophosphonate (30 μL, 2.0 × 10−1 mmol). The reaction mixture was stirred for 4 days at 55 °C. It was then diluted with CH2Cl2 and washed with 10% HCl, water and saturated NaHCO3. The organic phase was dried over Na2SO4, filtered and concentrated in vacuo. The crude product was purified using reverse phase (C18) column chromatography (gradually from 50 to 75% MeOH in water) and subsequent chromatography on silica (gradually from 0 to 10% MeOH in toluene). Recrystallization from CH2Cl2/hexane gave a red solid (8 mg; 47%). HRMS ESI (m/z) calcd for C87H136N14O14PCoNa2 [M + 2Na]2+ 1736.92145, found 868.4607. UV/Vis (DMSO): λmax (ε [l mol−1 cm−1]) = 547 (8.65 × 103), 516 (7.59 × 103), 361 (2.56 × 104), 325 (8.75 × 103), 278 (1.67 × 104), 262 (1.66 × 104). Anal. calcd for C87H136CoN14O14P + CH2Cl2: C 59.48, H 7.83, N 11.04; found: C 59.50, H 7.83, N 10.40. 1H NMR (500 MHz, CD3OD): δ = 7.25 (s, 1H), 7.11 (s, 1H), 6.55 (s, 1H), 6.26 (d, J = 3.1 Hz, 1H), 6.03 (s, 1H), 4.70–4.63 (m, 1H), 4.57–4.51 (m, 1H), 4.37–4.28 (m, 1H), 4.19 (t, J = 3.5 Hz, 1H), 4.12–4.05 (m, 2H), 3.92–3.88 (m, 1H), 3.76–3.72 (m, 1H), 3.68 (d, J = 14.0 Hz, 1H), 3.63 (s, 1H), 3.49 (q, J = 5.2 Hz, 1H), 3.28–3.07 (m, 10H), 3.02–2.91 (m, 2H), 2.87–2.80 (m, 2H), 2.66–2.44 (m, 11H), 2.42–2.32 (m, 4H), 2.28–2.24 (m, 6H), 2.18–2.08 (m, 2H), 2.05–1.93 (m, 3H), 1.91–1.84 (m, 4H), 1.77–1.67 (m, 2H), 1.57–1.15 (m, 42H), 1.14–1.05 (m, 2H), 0.97–0.86 (m, 18H), 0.42 (s, 3H) ppm. 13C NMR (125 MHz, CD3OD): δ = 181.5, 180.2, 177.6, 175.6, 174.6, 174.4, 174.0, 173.6, 172.7, 172.5, 171.6, 167.2, 166.5, 143.4, 138.3, 135.4, 133.8, 131.5, 130.7, 117.9, 112.5, 109.0, 105.0, 95.6, 87.9, 86.5, 83.7, 76.2, 75.4, 73.7, 73.6, 71.5, 70.7, 62.7, 60.3, 57.8, 57.6, 55.0, 52.9, 46.85, 46.82, 45.2, 43.7, 40.6, 40.5, 40.4, 40.32, 40.28, 40.1, 36.9, 35.8, 33.6, 33.5, 33.0, 32.9, 32.7, 32.56, 32.55, 32.50, 32.4, 32.3, 30.7, 29.8, 27.7, 27.5, 23.7, 21.3, 21.24, 21.22, 21.17, 21.15, 21.1, 20.9, 20.43, 20.39, 20.3, 20.20, 20.17, 19.9, 17.6, 17.1, 16.3, 16.0, 14.10, 14.09, 14.08, 14.02 ppm.Acknowledgements
The authors acknowledge the generous support from the National Science Centre: grant OPUS no. 2012/07/B/ST5/02016, ETIUDA 2014/12/T/ST5/00113, a Grant-in-Aid for Scientific Research (C) (no. 26410122) from the Japan Society for the Promotion of Science (JSPS) and a Grant-in-Aid for Scientific Research on Innovative Areas “Stimuli-responsive Chemical Species for Creation of Functional Molecules” (no. l5H00952) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan.References
- K. L. Brown, Chem. Rev., 2005, 105, 2075 CrossRef CAS PubMed.
- B. Kraütler, B. T. Golding and D. Arigoni, Vitamin B12 and B12-Proteins, Wiley-VCH, 2008 Search PubMed.
- K. Gruber, B. Puffer and B. Kraütler, Chem. Soc. Rev., 2011, 40, 4346 RSC.
- R. Banerjee, Chemistry and Biochemistry of B12, John Wiley & Sons, 1999 Search PubMed.
- J. M. Pratt, Chem. Soc. Rev., 1985, 14, 161 RSC.
- M. Giedyk, K. Goliszewska and D. Gryko, Chem. Soc. Rev., 2015, 44, 3391 RSC.
- Y. Hisaeda, J. Synth. Org. Chem., Jpn., 1996, 54, 859 CrossRef CAS.
- Y. Hisaeda and H. Shimakoshi, in Handbook of Porphyrin Science, World Scientific Publishing Co., 2010 Search PubMed.
- R. Scheffold, S. Busato, E. Eichenberger, R. Härter, H. Su, O. Tinembart, L. Walder, C. Weymuth and Z.-D. Zhang, in Organic Free Radicals SE - 93, ed. H. Fischer and H. Heimgartner, Springer, Berlin, Heidelberg, 1988, p. 187 Search PubMed.
- I. A. Dereven'kov, D. S. Salnikov, R. Silaghi-Dumitrescu, S. V. Makarov and O. I. Koifman, Coord. Chem. Rev., 2016, 309, 68 CrossRef.
- K. O. Proinsias, M. Giedyk, D. Gryko and K. Ó. Proinsias, Chem. Soc. Rev., 2013, 42, 6605 RSC.
- Y. Murakami, Y. Hisaeda and A. Kajihara, Bull. Chem. Soc. Jpn., 1983, 56, 3642 CrossRef CAS.
- H. Shimakoshi and Y. Hisaeda, Angew. Chem., Int. Ed., 2015, 54, 15439 CrossRef CAS PubMed.
- J. Xu, H. Shimakoshi and Y. Hisaeda, J. Organomet. Chem., 2014, 782, 89 CrossRef.
- H. Shimakoshi and Y. Hisaeda, ChemPlusChem, 2014, 79, 1250 CrossRef CAS.
- W. Zhang, H. Shimakoshi, N. Houfuku, X.-M. Song and Y. Hisaeda, Dalton Trans., 2014, 43, 13972 RSC.
- Y. Hisaeda, T. Nishioka, Y. Inoue, K. Asada and T. Hayashi, Coord. Chem. Rev., 2000, 198, 21 CrossRef CAS.
- M. Giedyk, S. N. Fedosov and D. Gryko, Chem. Commun., 2014, 50, 4674 RSC.
- Y. Murakami, Y. Hisaeda, A. Kajihara and T. Ohno, Bull. Chem. Soc. Jpn., 1984, 57, 405 CrossRef CAS.
- Y. Murakami, Y. Hisaeda, T. Ozaki and Y. Matsuda, Chem. Lett., 1988, 469 CrossRef CAS.
- H. Shimakoshi, M. Tokunaga and Y. Hisaeda, Dalton Trans., 2004, 878 RSC.
- D. Lexa, J. M. Savéant and J. Zickler, J. Am. Chem. Soc., 1980, 102, 2654 CrossRef CAS.
- D. Lexa and J. M. Savéant, Acc. Chem. Res., 1983, 16, 235 CrossRef CAS.
- C. Männel-Croisé and F. Zelder, Inorg. Chem., 2009, 48, 1272 CrossRef PubMed.
- F. H. Zelder, Inorg. Chem., 2008, 47, 1264 CrossRef CAS PubMed.
- L. Werthemann, ETH Zurich, 1968.
Footnote |
| † Electronic supplementary information (ESI) available: Details of mechanistic studies, spectroscopic data of (CN)Cble (2), [(CN)Cby]+ClO4− (3), and hexa(n-butyl)cobalamin (12). See DOI: 10.1039/c6dt00355a |
| This journal is © The Royal Society of Chemistry 2016 |