
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reaction of a 2,4,6-triphenylphosphinine ferrate anion with electrophiles: a new route to phosphacyclohexadienyl complexes†
Christian M.
Hoidn
and
Robert
Wolf
*
University of Regensburg, Institute of Inorganic Chemistry, D-93040 Regensburg, Germany. E-mail: robert.wolf@ur.de; Web: http://www.uni-regensburg.de/chemistry-pharmacy/inorganic-chemistry-wolf/index.html
First published on 22nd April 2016
Abstract
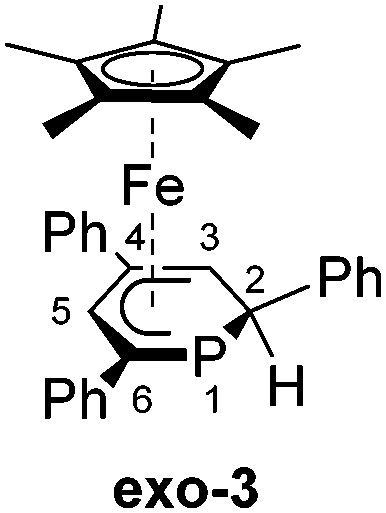
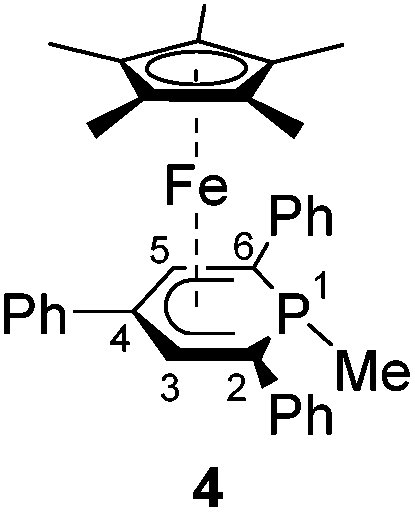
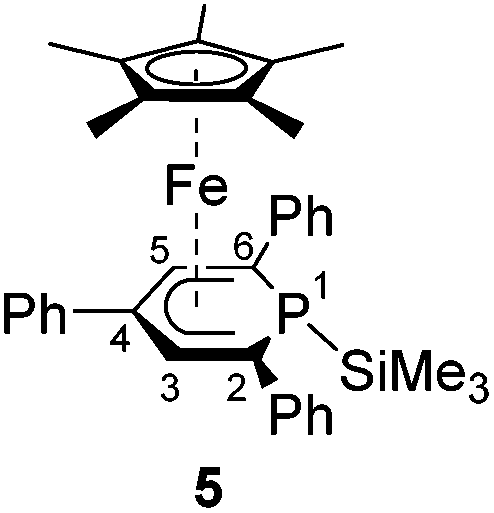
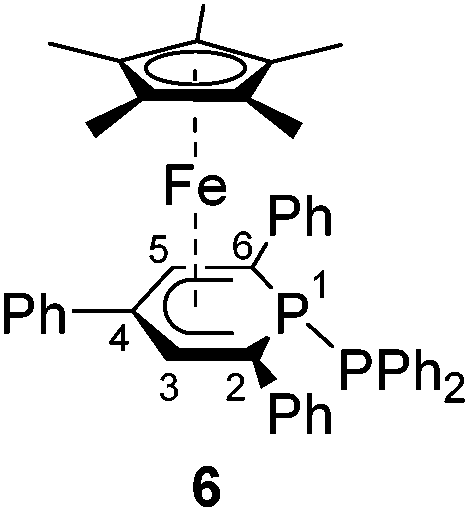
A novel, versatile route to phosphorus- and carbon-substituted η5-phosphacyclohexadienyl complexes was developed. Reaction of the anionic 2,4,6-triphenylphosphinine iron complex [K([18]crown-6)(thf)2][Cp*Fe(PC5Ph3H2)] (1) with selected main group element electrophiles afforded the new complexes [Cp*Fe(2-endo-H-PC5Ph3H2)] (endo-3), [Cp*Fe(2-exo-H-PC5Ph3H2)] (exo-3), [Cp*Fe(1-Me-PC5Ph3H2)] (4), [Cp*Fe(1-Me3Si-PC5Ph3H2)] (5), [Cp*Fe(1-PPh2-PC5Ph3H2)] (6) and [Cp*Fe(2-BCat-PC5Ph3H2)] (7, BCat = 2-benzo[d][1,3,2]dioxaborol-2-yl). Initial attack of the electrophile at phosphorus was observed, leading to a P-substitued phosphinine ligand. A subsequent rearragement occured in some cases, resulting in C-substituted phosphinine complexes endo-3, exo-3 and 7. The new complexes were characterized by 1H, 31P{1H}, and 13C{1H} NMR spectroscopy, UV-vis spectroscopy and elemental analysis; their molecular structures were determined by X-ray crystallography.
Introduction
Phosphacyclohexadienyls are six-membered heterocycles, which can be distinguished from the related purely carbon-containing cyclohexadienyls by their ambidentate donor properties and versatile coordination behaviour (Fig. 1).1 The donation of six π-electrons through an η5-coordinated anionic carbocyclic backbone of the phosphacyclohexadienyl ligand (C5 mode) is the most common motif.2–7 Additionally, the phosphorus atom may also become part of the coordinated π-system (C4P unit and CPC3 mode, Fig. 1).8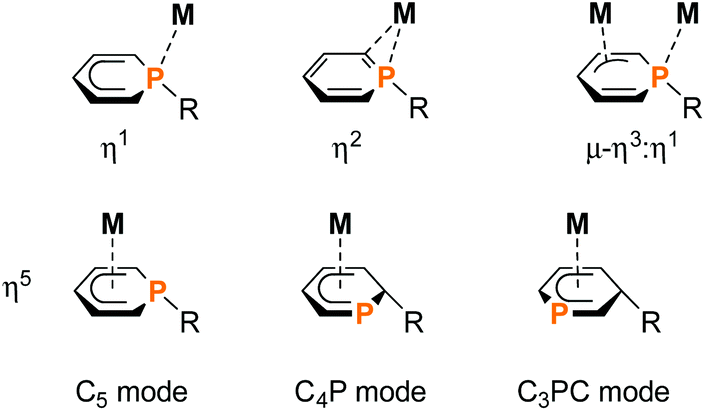 | ||
| Fig. 1 Overview of the versatile coordination modes of the ambidentate 1-substituted, 2-substituted and 3-substituted phosphacyclohexadienyl complexes; M = transition metal; R = organic residue; further substituents on the phosphinine rings are omitted for clarity. | ||
While η5-coordination is the most frequent coordination mode, η1-coordination can be found less often. The two electron donation of the P lone pair may be induced by additional chelating donor moieties such as pyridyl and phosphasulfide in the periphery of 1-substituted phosphacyclohexadienyl complexes.1,2,9–16 Rare η2-coordination was found in PdII and PtII complexes,17 while a bridging η3:η1-mode was observed for dinuclear Ni and Zr complexes.1,18
Phosphacyclohexadienyls thus are versatile ligands that bind to a range of transition metals and have successfully been applied in homogeneous catalysis, e.g. catalytic olefin polymerization and hydroformylation.1,5 Nevertheless, preparative methods are limited to merely three routes (Fig. 2). The conventional method (exemplified in Fig. 2a) is based on the initial synthesis of phosphacyclohexadienyl anions by reacting a phosphinine with an organometallic nucleophile and subsequent salt metathesis.2,9,11,17 This method was first established by Märkl in 1974.3 Nief and Fischer developed the reduction of the phosphinine oxide complex C with HSiCl3 as a more specialized approach (Fig. 2b) to the synthesis of hydrophosphacyclohexadienyl complexes.8 The P–H functionalized complex D was formed as the kinetic product with an excess of HSiCl3 at room temperature. The thermodynamically more stable carbon-protonated isomer exo-F was obtained by refluxing a solution of C with an excess of HSiCl3 in toluene for three hours. When the reaction was stopped after 55 min, complex E was identified as the main product. Isolated E undergoes a quantitative isomerization to endo-F by refluxing overnight.
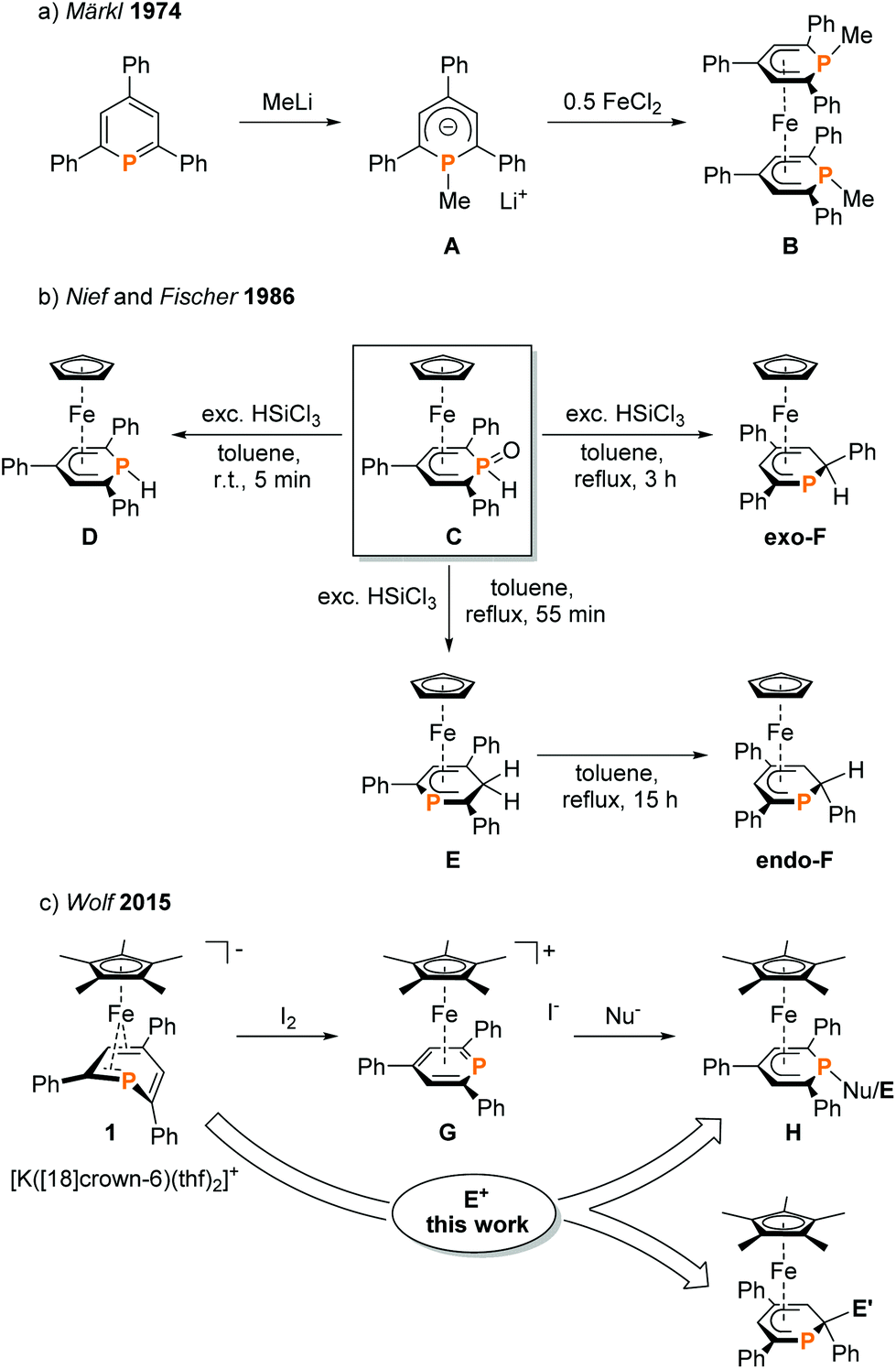 | ||
| Fig. 2 Synthesis of phosphacyclohexadienyl complexes; Nu = H, NMe2, Cp*;7 E = H, Me, SiMe3, PPh2; E′ = H, BCat. | ||
To our knowledge, endo-F is the only crystallographically characterized example of a 2-substituted phosphacyclohexadienyl complex with the C4P coordination mode so far. Using a different approach, we synthesized η5-phosphacyclohexadienyl iron complexes of type H by oxidizing the anionic pentamethylcyclopentadienyl complex 1 with iodine, followed by the conversion of the resulting cationic complex G with nucleopiles (Fig. 2c).7 Nucleophilic attack occurs at the phosphorus atom, giving 1-substituted λ3σ3-phosphacyclohexadienyl complexes. While this route can in principle give access to a large family of complexes, a disadvantage is the required two-step reaction sequence. In this paper, we report a new, complementary route to phosphacyclohexadienyl complexes that is based on the direct reaction of the anion 1 with electrophiles. The application of this new method resulted in the synthesis and structural characterization of six new phosphacyclohexadienyl complexes endo-3, exo-3, and 4–7. The formation of 1-phosphacyclohexadienyls (where the substituents are attached to phosphorus) and 2-substituted phosphacyclohexadienyls (where the substituent is connected to an adjacent carbon atom) is observed. Consequently, the molecular structures of the complexes display the C5 and the C4P coordination mode, respectively (Fig. 1).
Results and discussion
Synthesis of novel phosphacyclohexadienyl complexes
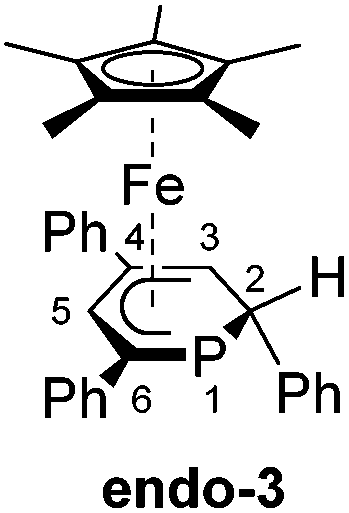
We recently reported that the 1-hydrophosphacyclohexadienyl complex 2 can be synthesized by reacting the cationic phosphinine complex G with one equivalent LiBHEt3 (Fig. 2c). Assuming that the protonation of the anionic complex 1 might give the same product, 1 was treated with one equivalent of HCl(OEt2) in THF. The reaction affords a mixture of compounds, including 2 and the new compounds endo-3 and exo-3. The latter are isomers of 2 and display 2-hydrophosphacyclohexadienyl ligands. In the case of endo-3, the hydrogen atom in the 2-position of the phosphinine is attached to the metal-coordinated face, causing the phenyl substituent to point to the bottom. The diastereomer exo-3 formally results from protonation of the phosphinine ring at the remote face to the iron center. Isomers 2, endo-3 and exo-3 are analogous to D, endo-F and exo-F previously prepared by Nief and Fisher via a completely different route (Fig. 2b).831P{1H} NMR monitoring ([D8]THF, Fig. 3) revealed the signal of 2 (−80 ppm) at −80 °C. Two additional signals at 10 ppm and −64 ppm arise from unknown intermediates, which disappear at higher temperature. The signal of the starting material 1 (−49 ppm) continuously decreased on slow warming to 0 °C, whereas the signal of 2 increased. The signals of the 2-H-substituted species exo-3 (−162 ppm) and endo–3 (−137 ppm) were observed in low intensity at −30 °C; their intensity increased significantly at 0 °C, whereas the signal of 2 decreased. An additional signal corresponding to an unidentified species became apparent at −14 ppm at −40 °C. This signal could plausibly arise from a by-product similar to complex E (−20.4 ppm)8 or a decomposition product. The 31P{1H} NMR spectrum of the reaction mixture recorded at room temperature displays the signals of 2, exo-3, endo-3 as well as a few weak singlets of further unidentified species. The signal intensities did not change further after one day. Stirring the raw product mixtures of 2, endo-3 and exo-3 at 50 °C for several days (31P{1H} NMR monitoring) also did not lead to a further change of the integral ratios.
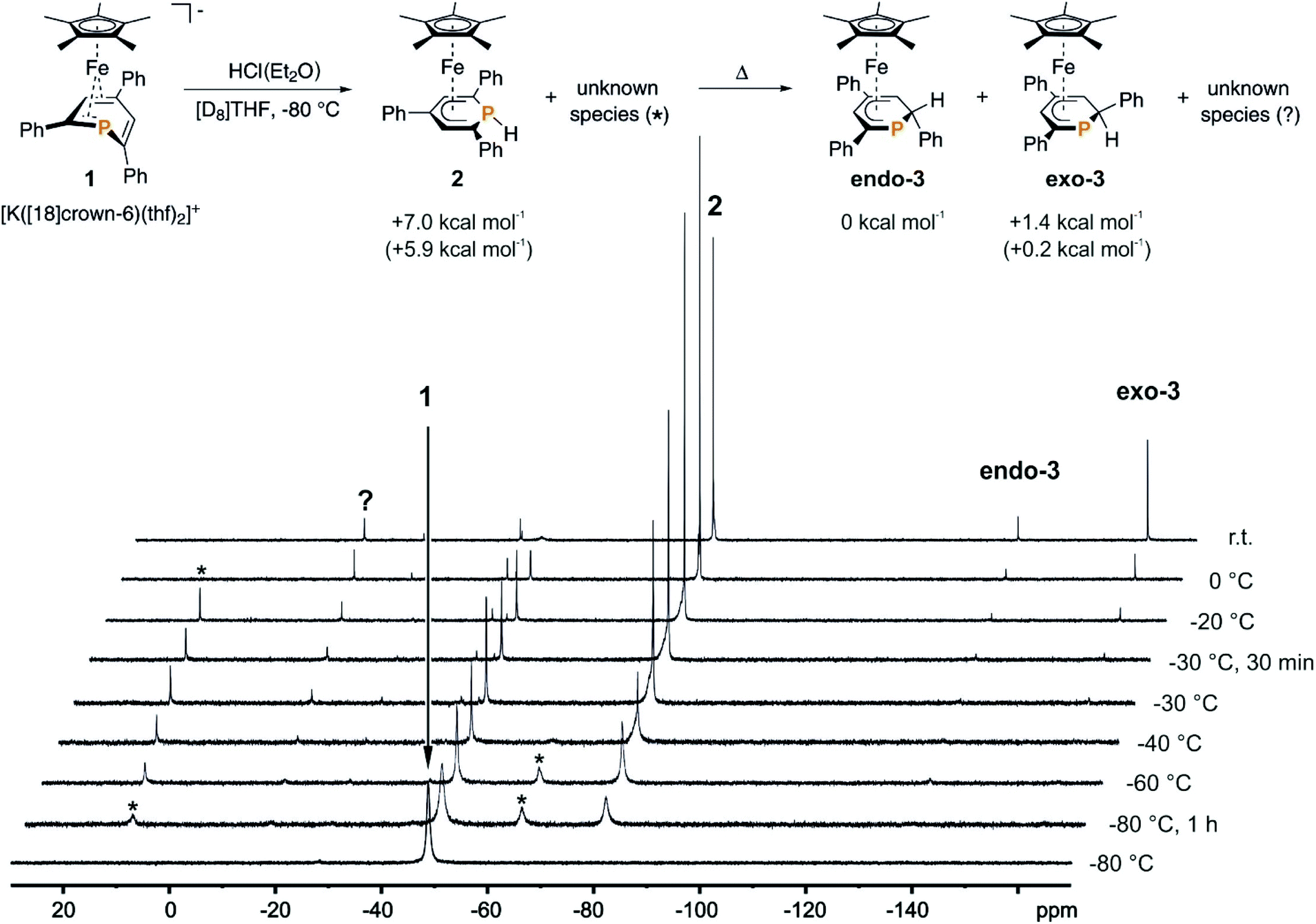 | ||
| Fig. 3 31P{1H} NMR monitoring of the reaction of 1 with HCl(Et2O) in [D8]THF; * and ? designate unknown intermediates or products; the scheme represents the proposed reaction pathway for the protonation of 1; DFT-calculated (BP86/def2-TZVP level) relative thermal enthalpies (ΔH at 298 K) are given of 2, endo-3 and exo-3; the relative free enthalpies (ΔG at 298 K) are given in parentheses; see the ESI† for details. | ||
Even though 2 appears to be formed selectively at low temperature, we were not able to isolate it as a pure material from reactions performed at −40 °C. However, 2 slowly converts to exo-3 upon treatment with HCl(OEt2) (10 mol%) at room temperature in [D8]THF. This indicates the rearrangement to be acid-catalysed. Attempts to optimise the reaction gave poorly reproducible product mixtures. Thus, it appears difficult to access 2, exo-3 and endo-3 as pure compounds by protonationg 1 with HCl(OEt2).
The results of the monitoring experiment indicate that the 1-hydrophosphacyclohexadienyl complex 2 is formed as the main kinetic product along with two unidentified species (marked with an asterisk in Fig. 3). The 2-hydrophosphacyclohexadienyl complexes endo-3 and exo-3 appear to be thermodynamic products that form at higher temperatures. Indeed, gas-phase DFT calculations performed at the BP86/def2-TZVP level (see the Experimental section for details) indicate that endo-3 and exo-3 are close in energy, while 2 was calculated to be +7.0 kcal mol−1 less stable than endo-3 (Fig. 3, see the Experimental section for details).
Gratifyingly, the reaction of 1 with one equiv. isopropyl chloride in THF at room temperature (Scheme 1) proceeded cleanly, reproducibly affording a mixture of endo-3 and exo-3 in a 65![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :35 ratio (NMR integration). The formation of 2 as an intermediate was not observed by 31P{1H} NMR in this case, which indicates that the reaction proceeds via a different mechanism. Purification by column chromatography gave NMR-spectroscopically pure exo-3 and endo-3 after crystallization.
:35 ratio (NMR integration). The formation of 2 as an intermediate was not observed by 31P{1H} NMR in this case, which indicates that the reaction proceeds via a different mechanism. Purification by column chromatography gave NMR-spectroscopically pure exo-3 and endo-3 after crystallization.
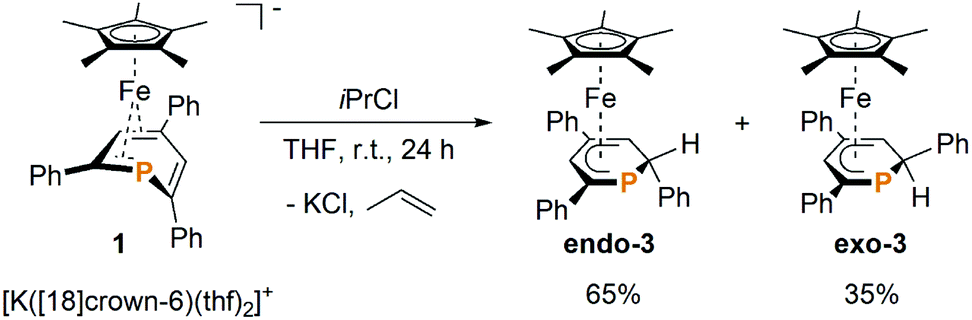 | ||
| Scheme 1 Preparation of endo-3 and exo-3. | ||
Exo-3 was isolated as orange rods in 25% yield, whereas pure endo-3 crystallized as orange plates in 41% yield. Both compounds are air-sensitive and dissolve well in n-hexane, diethyl ether, toluene and THF.
Complexes 4–6 are accessible in a similar fashion in moderate yields by reacting 1 with one equiv. of MeI, Me3SiCl, and Ph2PCl (Scheme 2a–c).‡ The compounds are deeply coloured crystalline solids that dissolve well in polar and apolar solvents such as n-pentane, n-hexane, diethyl ether, toluene and THF.
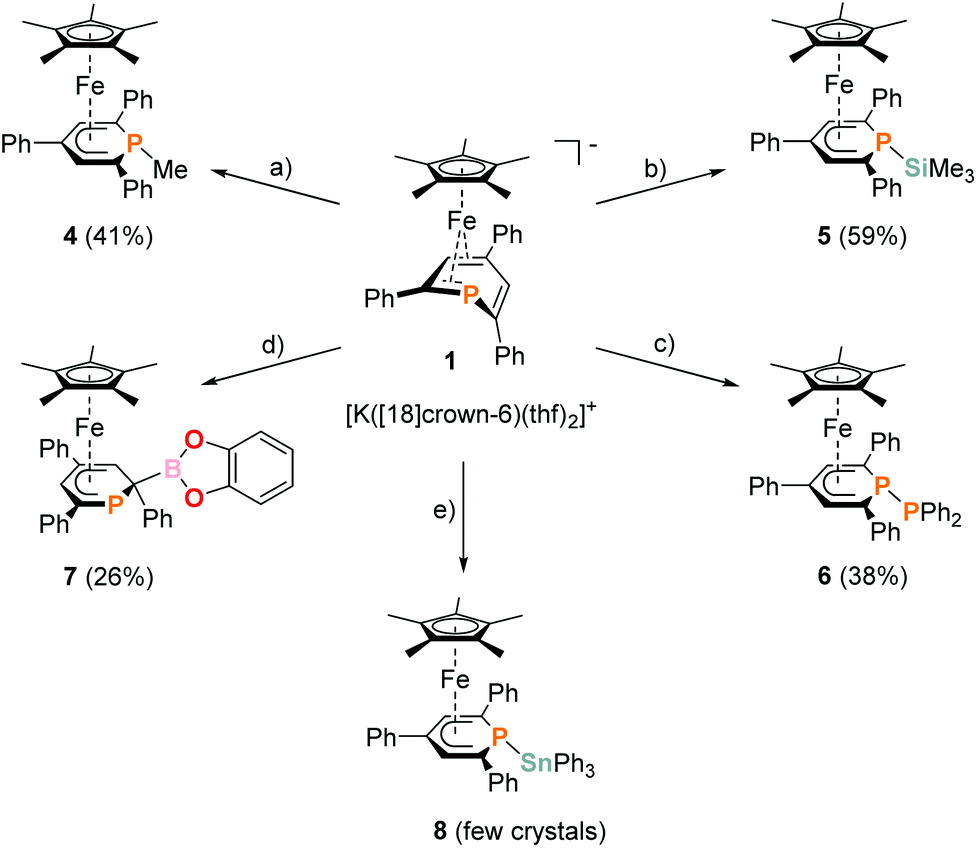 | ||
| Scheme 2 Synthesis of novel η5-phosphacyclohexadienyl complexes 4–8; reagents and by-products: (a) +MeI/−KI, −[18]crown-6; (b) +Me3SiCl/−KCl, −[18]crown-6; (c) +Ph2PCl/−KCl, −[18]crown-6; (d) +chlorocatecholborane/−KCl, −[18]crown-6; (e) +Ph3SnCl/−KCl, −[18]crown-6; isolated yields of the products are given in parentheses. | ||
The 2-substituted phosphacyclohexadienyl complex 7 was obtained as bright orange crystals by a similar reaction with one equiv. of chlorocatecholborane (Scheme 2d). Compound 7 is moderately soluble in n-pentane and n-hexane, but dissolves well in more polar solvents such as diethyl ether, toluene and THF. 31P{1H} NMR monitoring ([D8]THF, see ESI, Fig. S23†) at −100 °C revealed a signal at −26 ppm, which we tentatively assign to the intermediate [Cp*Fe(1-BCat-PC5Ph3H2)] (7-P, Scheme 3) containing a direct P–B bond. The signal is broad, therefore the 31P–10/11B coupling constant cannot be precisely determined, but the characteristic 1:1:1:1 quartet structure is clearly visible. The resonance of intermediate 7–B decreased upon warming and completely disappeared at −40 °C. The signal of 7 simultaneously appeared above −60 °C.
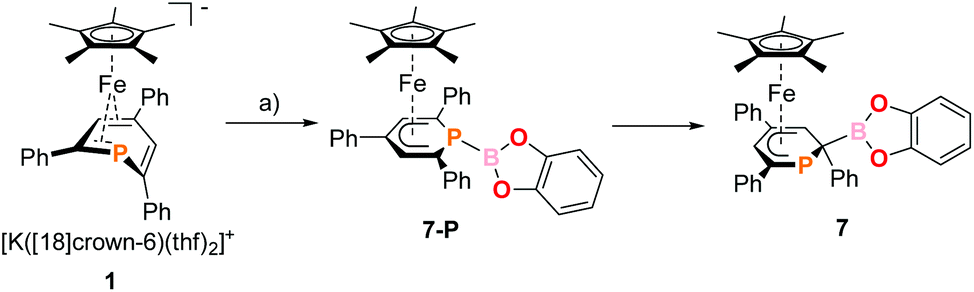 | ||
| Scheme 3 Presumed formation of 2-substituted 7via the 1-substituted intermediate 7-P; reagents and by-products: (a) +chlorocatecholborane/−KCl, −[18]crown-6. | ||
The 31P{1H} NMR spectrum recorded at room temperature shows the presence of 7, the diphoshinine complex [Cp*2Fe2(μ-{PC5Ph3H2}2)], the hydrophosphinine complex endo-3 and a small signal for an unidentified by-product (singlet at −50 ppm). The observation of this mixture shows that other processes than borylation may also occur, explaining the modest isolated yield (26%).
An analogous reaction with Ph3SnCl in THF produced the P–Sn functionalized complex 8 (Scheme 2e), but the reaction was unselective. According to 31P{1H} NMR integration complex 8 is only present in a low amount (26% of the total P content) in the reaction mixture after stirring for 17 h at room temperature. Several attempts to isolate it as a pure compound were not successful due to its low stability. Diphospinine [Cp*2Fe2(μ-{PC5Ph3H2}2)] and hexaphenyldistannane were identified as decomposition products by 31P{1H} and 119Sn{1H} NMR, suggesting decomposition by a radical pathway.
Crystallographic characterization of exo-3, endo-3, and 4–7
Single-crystal X-ray structure determinations of exo-3, endo-3 and 4–7 (Fig. 4 and Table 1) revealed η5-Cp* and η5-phosphacyclohexadienyl ligands. As a consequence, the phosphacyclohexadienyl units are not planar. The P atom points away from iron in complexes 4–6, and the heterocycle is folded along the C1–C5 axis. The dihedral angles between the carbocyclic mean plane and the plane defined by C1/P1/C5 (39.4° for 4, 37.0° for 5) are close to the values in the related complexes 2 (38.2°) and [Cp*Fe(1-Cp*-PC5Ph3H2)] (39.7°) previously reported by us (Fig. 2c, vide supra).7 The corresponding fold angle for 6 (27.2°) is over 10° shorter and similar to that of [Cp*Fe(1-NMe2-PC5Ph3H2)] (28.1°, vide supra).7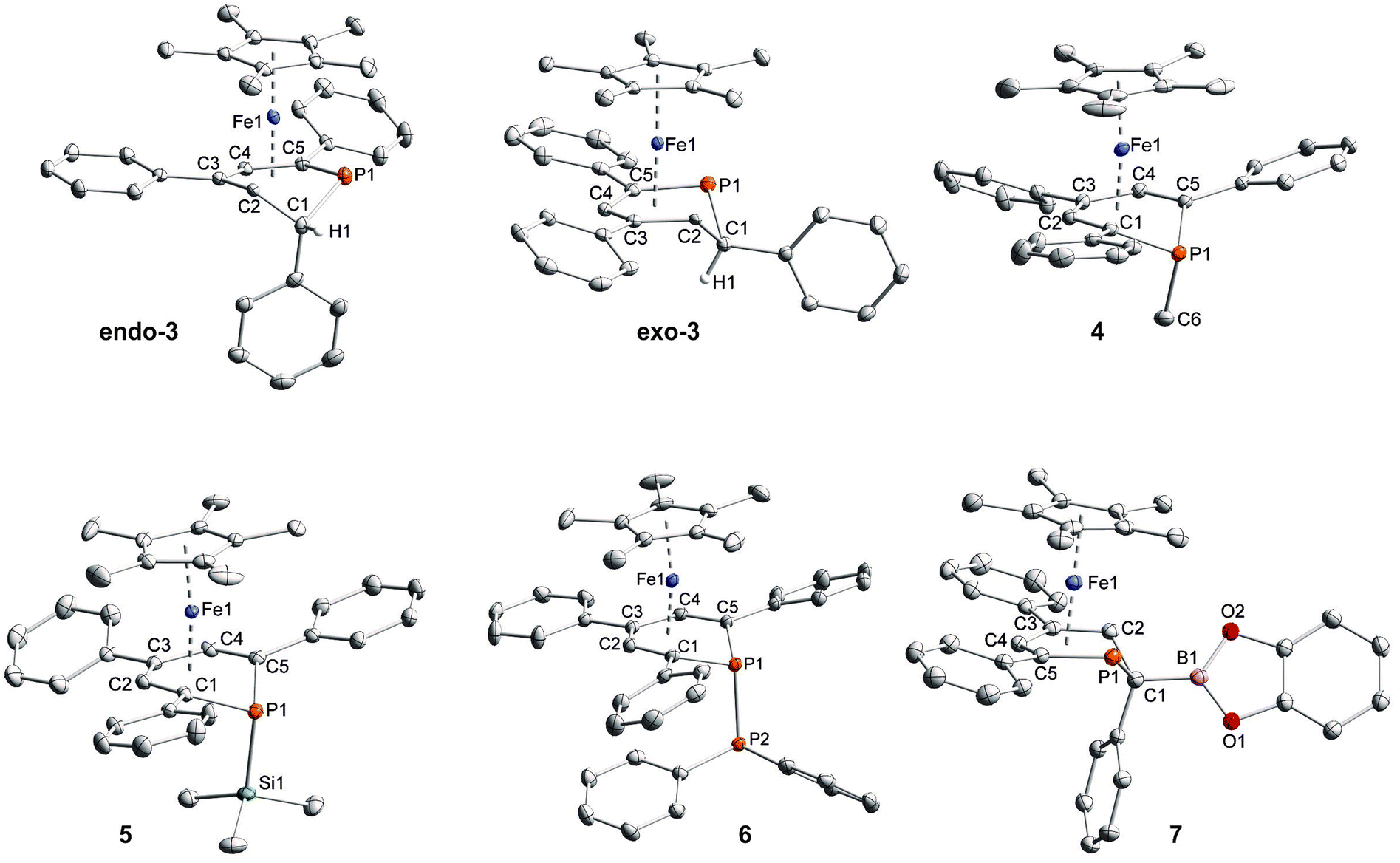 | ||
| Fig. 4 Solid-state molecular structures of complexes endo-3, exo-3 and 4–7. Ellipsoids are drawn at the 40% probability level; H atoms except H1 in endo-3 and exo-3, and [18]crown-6 in 7 are omitted for clarity, key bond lengths (Å) and angles (°) for 4: P1–C6 1.853(2), C1–P1–C6 103.94(7), C5–P1–C6 102.72(7); for 5: P1–Si1 2.270(2), C1–P1–Si1 104.4(1), C5–P1–Si1 106.6(1); for 6: P1–P2 2.3062(7), C1–P1–P2 107.54(7), C5–P1–P2 105.82(7); for 7: C1–B1 1.563(2), B1–O1 1.385(2), B1–O2 1.388(2), P1–C1–B1 104.11(1), C2–C1–B1 113.0(1), O1–B1–C1 123.3(1), O2–B1–C1 125.5(1), O1–B1–O2 111.2(1); see Table 1 for additional structural data. | ||
| endo-3 | exo-3 | 4 | 5 | 6 | 7 | |
|---|---|---|---|---|---|---|
| a Cp*(c) = centroid of the cyclopentadienyl ring. | ||||||
| Fe1–P1 | 2.3125(5) | 2.2946(6) | 2.8786(4) | 2.871(1) | 2.7785(5) | 2.2783(4) |
| Fe1–C1 | 2.881(2) | 2.933(2) | 2.160(1) | 2.168(4) | 2.170(2) | 2.901(2) |
| Fe1–C2 | 2.103(2) | 2.133(2) | 2.059(1) | 2.052(4) | 2.048(2) | 2.087(2) |
| Fe1–C3 | 2.059(1) | 2.061(2) | 2.091(1) | 2.088(4) | 2.086(2) | 2.061(2) |
| Fe1–C4 | 2.090(2) | 2.085(2) | 2.049(1) | 2.050(4) | 2.057(2) | 2.092(2) |
| Fe1–C5 | 2.098(2) | 2.096(2) | 2.150(1) | 2.151(4) | 2.161(2) | 2.096(2) |
| Fe1–Cp*(c)a | 1.700(1) | 1.703(1) | 1.698(1) | 1.699(2) | 1.693(1) | 1.701(1) |
| P1–C1 | 1.851(2) | 1.870(2) | 1.818(1) | 1.828(5) | 1.795(2) | 1.891(2) |
| C1–C2 | 1.521(2) | 1.540(3) | 1.418(2) | 1.404(6) | 1.414(3) | 1.526(2) |
| C2–C3 | 1.459(2) | 1.456(3) | 1.422(2) | 1.435(6) | 1.428(3) | 1.432(2) |
| C3–C4 | 1.433(2) | 1.421(3) | 1.428(2) | 1.430(6) | 1.423(3) | 1.4272) |
| C4–C5 | 1.419(2) | 1.427(3) | 1.423(2) | 1.429(6) | 1.414(3) | 1.416(2) |
| P1–C5 | 1.786(2) | 1.792(2) | 1.809(1) | 1.819(4) | 1.801(2) | 1.785(2) |
| P1–C1–C2 | 98.3(1) | 96.0(1) | 119.5(1) | 120.9(3) | 123.6(2) | 95.50(9) |
| C1–P1–C5 | 98.86(7) | 98.35(9) | 93.46(6) | 93.5(2) | 95.07(8) | 99.20(7) |
In exo-3, endo-3 and 7 the C1 atom adjacent to phosphorus is bent away from the iron center; consequently the six-membered phosphinine ring is folded along the P1–C2 axis. The corresponding plane to plane angles are larger than in 4–6 (60.6° for endo-3, 63.4° for exo-3 and 59.7° for 7). Complexes endo-3, exo-3 and 7 are rare phosphinine-type complexes, which show η5-coordination through a C4P-unit. To the best of our knowledge, the sole example comprising the same structural motif is endo-F (Fig. 2b, vide supra).8
The C–C distances of the η5-coordinated C5 and C4P-units exo-3, endo-3 and 4–7 (Table 1) are in between typical single and double bond distances.19 Similar bond lengths were observed for the η6-coordinated phosphinine ring in complex G.7 In addition, it is noteworthy that the C1–C2 distances of endo-3 (1.521(2) Å), exo-3 (1.540(3) Å) and 7 (1.526(2) Å) correspond to the value for a normal single bond.20 The P–C bond lengths in 4–6 are typical for single bonds and similar to those in B and H (Fig. 2, vide supra).4,7 The P–C bond lengths are distinct in endo-3, exo-3 and 7: the P1–C1 distances (1.891(2)–1.851(2) Å) are in the typical range for P–C single bonds,20 whereas the P1–C5 (1.792(2)–1.785(2) Å) bonds are shorter and close to those found in the η6-coordinated ring in G.7
While the P1–Si1 bond length (2.270(2) Å) of 5 is typical for a P–Si single bond,19 the P1–P2 bond (2.3062(7) Å) of 6 is elon-gated compared to that in unsymmetrically-substituted diphosphanes such as 9-diphenylphosphanyl-9-phosphabicyclo-[3.3.1]nonane (2.229(1) Å).21 An analogous observation was made by Gudat et al. for P-phosphanyldiazaphospholenes, e.g. 2-diphenylphosphanyl-1,3-dimesityldiazaphospholene, which displays a similarly elongated P–P bond (2.334(1) Å).22
In 7, the B1–C1 distance (1.563(2) Å) is in the range of normal boron–carbon single bonds (1.597 Å). The boron centre comprises a trigonal planar environment (angular sum = 360°).20 It seems noteworthy that Mathey and co-workers synthesized related phosphinine borates, e.g. Li[2-BEt3-PC5H4] by reaction of 2-bromophosphinines with two equiv. LiBHEt3.23 These anionic molecules contain a tetrahedral boron atom in the 2-position; they can be converted into 2-ethylated phosphinines by reaction with iodine. Braunschweig et al. prepared a series of (dimethoxyborylmethyl)dimethylphosphane complexes where a P–C–B(OMe)2 unit coordinates to chromium or iron via the P atom.24 An example is the compound [FeH(CO)3(SiPh3){Me2PCH2B(OMe)2}]. Different from these σ-coordinated complexes, the phosphacyclohexadienyl ligand of 7 acts as a π-ligand to iron through the planar C4P-unit. Thus, the phosphorus lone pair remains uncoordinated and should be able to act as a Lewis base. The trivalent boron center might function as a Lewis acid in related complexes with less strongly electron-donating substituents at boron, enabling the formation of a new frustrated Lewis pair type system.
NMR and UV-Vis spectroscopic characterization
Table 2 summarizes 1H, 13C{1H} and 31P{1H} NMR data of endo-3, exo-3 and 4–7 recorded in [D8]THF. The 31P NMR signals of exo-3 (−160.7 ppm, 2JPH not detected) and endo-3 (−136.3 ppm, 2JPH = 15.2 Hz) are slightly downfield shifted in comparison with those of the cyclopentadienyl analogues exo-F (−173.0 ppm) and endo-F (−150.1 ppm) synthesised by Nief and Fischer,8 which otherwise feature similar 1H and 13C NMR shifts of the phosphacyclohexadienyl units as their Cp*-substituted equivalents. Notably, the simple change of the configuration of a carbon atom of the phosphinine ring causes an upfield shift of the 31P NMR doublet of exo-3 by almost 25 ppm with respect to endo-3. The aliphatic hydrogen atom of the phosphacyclohexadienyl ring resonates at 2.76 ppm for endo-3 and at 1.66 ppm for exo-3. The spectrum of exo-3 thus displays a pronounced upfield shift for the exo-hydrogen atom comparable to that observed for the related cyclohexadienyl complex [CpFe(η5-C6H7)].25| 31P{1H} NMR | 1H NMR | 13C{1H} NMR | ||||
|---|---|---|---|---|---|---|
| Complex | P1 (ppm) | C5(CH3)5 (ppm) | C2–H, C4–H (ppm) | 3 J HP (Hz) | C1, C5 (ppm) | 1 J C1P, 1JC5P (Hz) |
| a Overlapping with meta-H of C5-Ph. b Overlapping with Ar-H of C1-Ph and meta/para-H of C3-Ph/C5-Ph. c Overlapping with ortho-H of C1-Ph. n.d. = not detected. | ||||||
| endo-3 | −136.3 (s) | 1.36 (s) | 2.98 (dd), 7.20–7.24 (m)a | 2.4 | 34.7 (d), 95.2 (d) | 23.2, 69.9 |
| exo-3 | −160.7 (s) | 1.30 (s) | 2.44 (s), 7.13–7.40 (m)b | n.d. | 31.7 (d), 97.7 (d) | 19.7, 67.5 |
| 4 | −57.4 (s) | 1.02 (s) | 6.07 (d) | 2.6 | 41.1 (d) | 1.1 |
| 5 | −77.7 (s) | 1.02 (s) | 6.21 (d) | 2.5 | 29.2 (d) | 0.9 |
| 6 | −38.8 (d) | 0.98 (s) | 6.02 (d) | 2.9 | 41.0 (dd) | 12.5 |
| 7 | −126.7 (s) | 1.34 (s) | 3.51 (s), 6.03–6.05 (m)c | n.d. | n.d., 95.2 (d) | n.d., 74.5 |
The spectra of 1-substituted 4–6 overall resemble those of related complexes of type H (Fig. 2c).7 Characteristic 1H NMR features of 4 and 5 are the doublets at −0.13 ppm (2JHP = 5.5 Hz) for the methyl group of 4 and the Me3Si group of 5 (−0.41 ppm, 3JHP = 3.4 Hz). The 31P{1H} NMR signal of the trimethylsilyl-substituted complex 5 is upfield shifted by 20.3 ppm compared to the methyl-substituted analogue 4. Two 31P{1H} NMR doublets are observed for 6 at 12.8 and −38.8 ppm with a large 1JPP coupling constant (293 Hz) in the typical range for a covalent P–P single bond.26 The signal at 12.8 ppm is assigned to the PPh2 group, because it splits into a doublet of quintets in the 31P NMR spectrum (3JPH = 6.5 Hz).
Complex 7, which features a 2-substituted phosphacyclohexadienyl moiety, gives rise to a similar high-field 31P{1H} NMR singlet (−126.7 ppm) as endo-3 (−136.3 ppm); the 1H NMR data (Table 2) are also similar in agreement with the similar structures.§
The UV/vis spectra of endo-3–7 were recorded in n-hexane. The spectra of 2-H-substituted endo-3 and exo-3 are similar and display a weak shoulder at 450 nm; three stronger bands are found in the UV range (endo-3 220, 260 and 320 nm; exo-3 230, 275 and 325 nm). The spectrum of the structurally related complex 7 is analogous, showing slightly bathochromically shifted bands at 260, 290sh, 360sh and 460sh nm. The UV/vis spectra of the 1-substituted species 4–6 are distinct from those of the aforementioned complexes and feature two visible absorptions each with moderate intensities in the ranges λmax = 550–580 nm and λmax = 480–580 nm, respectively. Similar spectra were observed for other complexes of this type (type H, Fig. 2c).7 Previous TD-DFT calculations indicated that these bands predominantly arise from excitations from filled metal-centered MOs into the ligand-based unoccupied MOs (MLCT).7
Conclusions
The reaction of the anionic phosphinine complex 1 with diverse electrophiles represents a novel and straightforward synthetic pathway to phosphacyclohexadienyl iron complexes. Protonation of 1 using HCl(OEt2) initially affords the 1-substituted complex 2 at low temperature, which appears to undergo an acid catalyzed rearrangement and converts to a mixture of isomers, including the 2-H-substituted compounds endo-3 and exo-3. The latter complexes were conveniently isolated in good yields from the reaction of 1 with isopropyl chloride. An analogous 2-substitued complex 7 formed in the reaction with chlorocatecholborane. Similar to the hydrophosphinine complexes, an initial formation of a phosphorus-substituted complex followed by a subsequent 1,2-shift of the substituent was observed. Using MeI, Me3SiCl, Ph2PhCl and Ph3SnCl, 1-substituted complexes 4–6 and 8 were obtained. Thus, HCl(OEt2), isopropyl chloride and chlorocatecholborane result in products substituted at the 2-carbon atom, whereas MeI, Me3SiCl, Ph3SnCl and Ph2PhCl provide phosphorus-substituted products.An extensive family of related compounds could become accessible via this route. In addition, the reactivity and possible catalytic activity of the new complexes presented here needs to be examined, where the unusually long P–P bond in 6 and the FLP type motif in 7 will be of particular interest. Investigations in these directions are underway in our laboratory.
Experimental
General considerations
All experiments were performed under an atmosphere of dry argon, by using standard Schlenk and glovebox techniques. Solvents were purified, dried, and degassed with an MBraun SPS800 solvent purification system. NMR spectra were recorded on Bruker Avance 300 and Avance 400 spectrometers at 300 K and internally referenced to residual solvent resonances. The assignment of the 1H and 13C NMR signals was confirmed by two-dimensional (COSY, HSQC, and HMBC) experiments. Melting points were measured on samples in sealed capillaries on a Stuart SMP10 melting point apparatus. UV/vis spectra were recorded on a Varian Cary 50 spectrometer. Elemental analyses were determined by the analytical department of Regensburg University. The starting material [K([18]crown-6)(thf)2][Cp*Fe(η4-PC5Ph3H2)] (1)7 was prepared according to literature procedures. HCl(Et2O) solution, methyl iodide, trimethylsilyl chloride, chlorocatecholborane and chlorodiphenylphosphane were purchased from Sigma-Aldrich and TCI and were used as received.M.p. 196 °C. UV/vis: (n-hexane, λmax/nm, εmax/L mol−1 cm−1): 220sh (37
 000), 260 (29600), 320sh (9300), 450sh (670). 1H NMR (400.13 MHz, 300 K, [D8]THF): δ = 1.36 (s, 15H, C5(CH3)5), 2.76 (dd, 2JHP = 15.2 Hz, 3JHH = 8.1 Hz, 1H, C2–H of TPP), 2.98 (dd, 3JHP = 2.4 Hz, 3JHH = 8.1 Hz, 1H, C3–H of TPP), 6.77–6.81 (m, 3H, C2,4,6–H of C2–Ph), 6.90–6.94 (m, 2H, C3,5–H of C2–Ph), 7.16 (t, 3JHH = 7.2 Hz, 1H, C4–H of C6–Ph), 7.20–7.24 (m, 3H, C3,5–H of C6–Ph overlapping with C5–H of TPP), 7.31 (t, 3JHH = 7.3 Hz, 1H, C4–H of C4–Ph), 7.37–7.42 (m, 2H, C3,5–H of C4–Ph), 7.80 (d, 2H, C2,6–H of C6–Ph), 7.93 (d, 3JHH = 7.8 Hz, 2H, C2,6–H of C4–Ph). 13C{1H} NMR (100.61 MHz, 300 K, [D8]THF): δ = 10.0 (d, 3JCP = 3.4 Hz, C5(CH3)5), 26.3 (s, C3–H of TPP), 34.7 (d, 1JCP = 23.2 Hz, C2 of TPP), 87.9 (s, C5(CH3)5), 88.8 (d, 2JCP = 7.9 Hz, C5–H of TPP), 91.8 (s, C4 of TPP), 95.2 (d, 1JCP = 69.9 Hz, C6 of TPP), 125.2 (s, C4–H of C2–Ph), 126.3 (d, 3JCP = 3.5 Hz, C2–H of C2–Ph), 127.1 (d, 5JCP = 1.1 Hz, C4–H of C6–Ph), 127.7 (s, C4–H of C4–Ph), 127.5 (s, C2,6–H of C4–Ph), 127.9 (s, C2,6–H of C4–Ph), 128.1 (s, C3,5–H of C2–Ph), 128.6 (d, 4JCP = 1.0 Hz, C3,5–H of C6–Ph), 129.0 (s, C3,5–H of C4–Ph), 141.8 (s, C1 of C4–Ph), 143.8 (d, 2JCP = 17.8 Hz, C1 of C6–Ph), 147.1 (d, 2JCP = 1.8 Hz, C1 of C2–Ph). 31P{1H} NMR (161.98 MHz, 300 K, [D8]THF): δ = –136.3 (s). 31P NMR (161.98 MHz, 300 K, [D8]THF): δ = –136.3 (d, 2JPH = 15.3 Hz). Elemental analysis calcd for C33H33FeP (Mw = 516.45 g mol−1) C 76.75, H 6.44; found C 76.11, H 6.55.
000), 260 (29600), 320sh (9300), 450sh (670). 1H NMR (400.13 MHz, 300 K, [D8]THF): δ = 1.36 (s, 15H, C5(CH3)5), 2.76 (dd, 2JHP = 15.2 Hz, 3JHH = 8.1 Hz, 1H, C2–H of TPP), 2.98 (dd, 3JHP = 2.4 Hz, 3JHH = 8.1 Hz, 1H, C3–H of TPP), 6.77–6.81 (m, 3H, C2,4,6–H of C2–Ph), 6.90–6.94 (m, 2H, C3,5–H of C2–Ph), 7.16 (t, 3JHH = 7.2 Hz, 1H, C4–H of C6–Ph), 7.20–7.24 (m, 3H, C3,5–H of C6–Ph overlapping with C5–H of TPP), 7.31 (t, 3JHH = 7.3 Hz, 1H, C4–H of C4–Ph), 7.37–7.42 (m, 2H, C3,5–H of C4–Ph), 7.80 (d, 2H, C2,6–H of C6–Ph), 7.93 (d, 3JHH = 7.8 Hz, 2H, C2,6–H of C4–Ph). 13C{1H} NMR (100.61 MHz, 300 K, [D8]THF): δ = 10.0 (d, 3JCP = 3.4 Hz, C5(CH3)5), 26.3 (s, C3–H of TPP), 34.7 (d, 1JCP = 23.2 Hz, C2 of TPP), 87.9 (s, C5(CH3)5), 88.8 (d, 2JCP = 7.9 Hz, C5–H of TPP), 91.8 (s, C4 of TPP), 95.2 (d, 1JCP = 69.9 Hz, C6 of TPP), 125.2 (s, C4–H of C2–Ph), 126.3 (d, 3JCP = 3.5 Hz, C2–H of C2–Ph), 127.1 (d, 5JCP = 1.1 Hz, C4–H of C6–Ph), 127.7 (s, C4–H of C4–Ph), 127.5 (s, C2,6–H of C4–Ph), 127.9 (s, C2,6–H of C4–Ph), 128.1 (s, C3,5–H of C2–Ph), 128.6 (d, 4JCP = 1.0 Hz, C3,5–H of C6–Ph), 129.0 (s, C3,5–H of C4–Ph), 141.8 (s, C1 of C4–Ph), 143.8 (d, 2JCP = 17.8 Hz, C1 of C6–Ph), 147.1 (d, 2JCP = 1.8 Hz, C1 of C2–Ph). 31P{1H} NMR (161.98 MHz, 300 K, [D8]THF): δ = –136.3 (s). 31P NMR (161.98 MHz, 300 K, [D8]THF): δ = –136.3 (d, 2JPH = 15.3 Hz). Elemental analysis calcd for C33H33FeP (Mw = 516.45 g mol−1) C 76.75, H 6.44; found C 76.11, H 6.55.
M.p. 177 °C. UV/vis: (n-hexane, λmax nm−1, εmax/L mol−1 cm−1): 230sh (72
 000), 275 (59000), 325sh (17000), 450sh (1100). 1H NMR (400.13 MHz, 300 K, [D8]THF): δ = 1.30 (s, 15H, C5(CH3)5), 1.66 (s br, 1H, C2–H of TPP), 2.44 (s br, 1H, C3–H of TPP), 7.13–7.40 (overlapping m, 12H, Ar–H of C2–Ph + C5–H of TPP + C3,4,5–H of C4–Ph + C3,4,5–H of C6–Ph), 7.86 (d, 3JHH = 7.9 Hz, 2H, C2,6–H of C4–Ph), 7.90 (d, 3JHH = 7.9 Hz, 2H, C2,6–H of C6–Ph). 13C{1H} NMR (100.61 MHz, 300 K, [D8]THF): δ = 9.9 (d, 3JCP = 2.8 Hz, C5(CH3)5), 21.4 (d, 2JCP = 5.1 Hz, C3–H of TPP), 31.7 (d, 1JCP = 19.7 Hz, C2 of TPP), 86.9 (d, 2JCP = 7.3 Hz, C5–H of TPP), 88.1 (s, C5(CH3)5), 92.3 (d, 3JCP = 1.8 Hz, C4 of TPP), 97.7 (d, 1JCP = 67.5 Hz, C6 of TPP), 125.9, 127.2, 127.3, 127.4, 127.5, 127.6, 127.7, 128.7, 128.9, 129.0 (C2,3,4,5,6 of C2–Ph, C2,3,4,5,6 of C4–Ph C2,3,4,5,6 of C6–Ph), 140.8 (s, C1 of C4–Ph), 143.6 (d, 2JCP = 16.7 Hz, C1 of C6–Ph), 145.9 (d, 2JCP = 12.9 Hz, C1 of C2–Ph). 31P{1H} NMR (161.98 MHz, 300 K, [D8]THF): δ = −160.7 (s). 31P NMR (161.98 MHz, 300 K, [D8]THF): δ = −160.7 (s br). Elemental analysis calcd for C33H33FeP (Mw = 516.45 g mol−1) C 76.75, H 6.44; found C 77.15, H 6.50.
000), 275 (59000), 325sh (17000), 450sh (1100). 1H NMR (400.13 MHz, 300 K, [D8]THF): δ = 1.30 (s, 15H, C5(CH3)5), 1.66 (s br, 1H, C2–H of TPP), 2.44 (s br, 1H, C3–H of TPP), 7.13–7.40 (overlapping m, 12H, Ar–H of C2–Ph + C5–H of TPP + C3,4,5–H of C4–Ph + C3,4,5–H of C6–Ph), 7.86 (d, 3JHH = 7.9 Hz, 2H, C2,6–H of C4–Ph), 7.90 (d, 3JHH = 7.9 Hz, 2H, C2,6–H of C6–Ph). 13C{1H} NMR (100.61 MHz, 300 K, [D8]THF): δ = 9.9 (d, 3JCP = 2.8 Hz, C5(CH3)5), 21.4 (d, 2JCP = 5.1 Hz, C3–H of TPP), 31.7 (d, 1JCP = 19.7 Hz, C2 of TPP), 86.9 (d, 2JCP = 7.3 Hz, C5–H of TPP), 88.1 (s, C5(CH3)5), 92.3 (d, 3JCP = 1.8 Hz, C4 of TPP), 97.7 (d, 1JCP = 67.5 Hz, C6 of TPP), 125.9, 127.2, 127.3, 127.4, 127.5, 127.6, 127.7, 128.7, 128.9, 129.0 (C2,3,4,5,6 of C2–Ph, C2,3,4,5,6 of C4–Ph C2,3,4,5,6 of C6–Ph), 140.8 (s, C1 of C4–Ph), 143.6 (d, 2JCP = 16.7 Hz, C1 of C6–Ph), 145.9 (d, 2JCP = 12.9 Hz, C1 of C2–Ph). 31P{1H} NMR (161.98 MHz, 300 K, [D8]THF): δ = −160.7 (s). 31P NMR (161.98 MHz, 300 K, [D8]THF): δ = −160.7 (s br). Elemental analysis calcd for C33H33FeP (Mw = 516.45 g mol−1) C 76.75, H 6.44; found C 77.15, H 6.50.
A solution of methyl iodide in THF (1 mL, c = 0.106 mol L−1) was added to a dark orange solution of 1 (102 mg, 0.106 mmol) in THF (5 mL) at room temperature. The reaction mixture turned burgundy red immediately, and was stirred for four hours at room temperature. After removing the solvent in vacuo, the remaining dark red residue was extracted with n-hexane (10 × 1 mL). The burgundy red extracts were combined and the solution was concentrated to 5 mL. Dark red crystals of 4 formed during storage at −30 °C for three days. Yield: 23 mg (41%). M.p. 239 °C. UV/vis: (n-hexane, λmax nm−1, εmax L−1 mol−1 cm−1): 380sh (3150), 480 (1440), 550 (1000). 1H NMR (400.13 MHz, 300 K, [D8]THF): δ = −0.13 (d, 2JHP = 5.5 Hz, 3H, P–CH3), 1.02 (s, 15H, C5(CH3)5), 6.07 (d, 3JHP = 2.6 Hz, 2H, C3,5–H of TPP), 7.12 (t, 3JHH = 7.3 Hz, 2H, C4–H of C2,6–Ph), 7.27–7.31 (m, 4H, C3,5–H of C2,6–Ph), 7.46 (t, 3JHH = 7.3 Hz, 1H, C4–H of C4–Ph), 7.54–7.58 (m, 2H, C3,5–H of C4–Ph), 8.11–8.13 (m, 4H, C2,6–H of C2,6–Ph), 8.32 (d, 3JHH = 7.7 Hz, 2H, C2,6–H of C4–Ph). 13C{1H} NMR (100.61 MHz, 300 K, [D8]THF): δ = 8.9 (s, C5(CH3)5), 18.2 (d, 1JCP = 39.0 Hz, P–CH3), 41.1 (d, 1JCP = 1.1 Hz, C2,6 of TPP), 79.1 (d, 2JCP = 7.3 Hz, C3,5–H of TPP), 85.3 (s, C5(CH3)5), 95.9 (d, 3JCP = 2.4 Hz, C4 of TPP), 124.7 (d, 5JCP = 2.8 Hz, C4–H of C2,6–Ph), 128.1 (C4–H of C4–Ph overlapping with C2,6–H of C4–Ph), 128.7 (s, C3,5–H of C2,6–Ph), 128.8 (d, 3JCP = 19.5 Hz, C2,6–H of C2,6–Ph), 129.4 (s, C3,5–H of C4–Ph), 141.6 (s, C1 of C4–Ph), 145.5 (d, 2JCP = 25.2 Hz, C1 of C2,6–Ph). 31P{1H} NMR (161.98 MHz, 300 K, [D8]THF): δ = –57.4 (s). 31P NMR (161.98 MHz, 300 K, [D8]THF): δ = –57.4 (s, br). Elemental analysis calcd for C34H35FeP (Mw = 530.47 g mol−1) C 76.98, H 6.65; found C 77.22, H 6.40.
A solution of trimethylsilyl chloride in toluene (0.9 mL, c = 0.152 mol L−1) was added to a dark orange solution of 1 (132 mg, 0.137 mmol) in THF (7 mL) and stirred at room temperature for 16 h. The resulting dark greenish brown mixture was dried in vacuo, and the residue was extracted with n-pentane (16 × 0.5 mL). The fractions were combined and dried in vacuo. [18]crown-6 was sublimed at 60 °C and <1.0 × 10−3 mbar. The remaining residue was dissolved in n–hexane (8 mL). The greenish black solution was filtered and concentrated to 5 mL. 5 was isolated as dark green to black crystals after storage at −30 °C for three days. Yield: 48 mg (59%). M.p. 213 °C. UV/vis: (n-hexane, λmax nm−1, εmax L−1 mol−1 cm−1): 250 (36
 300), 300 (26000), 500 (2300), 580 (2100). 1H NMR (400.13 MHz, 300 K, [D8]THF): δ = −0.41 (d, 3JPH = 3.4 Hz, 9H, P–Si(CH3)3), 1.02 (s, 15H, C5(CH3)5), 6.21 (d, 3JPH = 2.5 Hz, 2H, C3,5–H of TPP), 7.10 (t, 3JHH = 7.2 Hz, 2H, C4–H of C2,6–Ph), 7.22–7.27 (m, 4H, C3,5–H of C2,6–Ph), 7.47 (t, 3JHH = 7.3 Hz, 1H, C4–H of C4–Ph), 7.56–7.60 (m, 2H, C3,5–H of C4–Ph), 8.08–8.11 (m, 4H, C2,6–H of C2,6–Ph), 8.34 (d, 3JHH = 7.8 Hz, 2H, C2,6–H of C4–Ph). 13C{1H} NMR (100.61 MHz, 300 K, [D8]THF): δ = −0.9 (d, 2JCP = 8.5 Hz, P–Si(CH3)3), 8.8 (s, C5(CH3)5), 29.2 (d, 1JCP = 0.9 Hz, C2,6 of TPP), 82.2 (d, 2JCP = 8.3 Hz, C3,5–H of TPP), 85.7 (s, C5(CH3)5), 97.9 (d, 3JCP = 1.2 Hz, C4 of TPP), 124.6 (d, 5JCP = 3.1 Hz, C4–H of C2,6–Ph), 127.9 (s, C4–H of C4–Ph), 128.3 (s, C2,6–H of C4–Ph), 128.7 (s, C3,5–H of C2,6–Ph and d, 3JCP = 19.4 Hz, C2,6–H of C2,6–Ph), 129.5 (s, C3,5–H of C4–Ph), 142.5 (s, C1 of C4–Ph), 146.5 (d, 2JCP = 24.2 Hz, C1 of C2,6–Ph). 29Si DEPT NMR (79.49 MHz, 300 K, [D8]THF): δ = −3.3 (d, 1JSiP = 62.4 Hz). 31P{1H} NMR (161.98 MHz, 300 K, [D8]THF): δ = −77.7 (s, 29Si-satellites: 1JPSi = 62.1 Hz). 31P NMR (161.98 MHz, 300 K, [D8]THF): δ = −77.7 (s, br). Elemental analysis calcd for C36H41FePSi (Mw = 588.63 g mol−1) C 73.46, H 7.02; found C 73.81, H 6.97.
300), 300 (26000), 500 (2300), 580 (2100). 1H NMR (400.13 MHz, 300 K, [D8]THF): δ = −0.41 (d, 3JPH = 3.4 Hz, 9H, P–Si(CH3)3), 1.02 (s, 15H, C5(CH3)5), 6.21 (d, 3JPH = 2.5 Hz, 2H, C3,5–H of TPP), 7.10 (t, 3JHH = 7.2 Hz, 2H, C4–H of C2,6–Ph), 7.22–7.27 (m, 4H, C3,5–H of C2,6–Ph), 7.47 (t, 3JHH = 7.3 Hz, 1H, C4–H of C4–Ph), 7.56–7.60 (m, 2H, C3,5–H of C4–Ph), 8.08–8.11 (m, 4H, C2,6–H of C2,6–Ph), 8.34 (d, 3JHH = 7.8 Hz, 2H, C2,6–H of C4–Ph). 13C{1H} NMR (100.61 MHz, 300 K, [D8]THF): δ = −0.9 (d, 2JCP = 8.5 Hz, P–Si(CH3)3), 8.8 (s, C5(CH3)5), 29.2 (d, 1JCP = 0.9 Hz, C2,6 of TPP), 82.2 (d, 2JCP = 8.3 Hz, C3,5–H of TPP), 85.7 (s, C5(CH3)5), 97.9 (d, 3JCP = 1.2 Hz, C4 of TPP), 124.6 (d, 5JCP = 3.1 Hz, C4–H of C2,6–Ph), 127.9 (s, C4–H of C4–Ph), 128.3 (s, C2,6–H of C4–Ph), 128.7 (s, C3,5–H of C2,6–Ph and d, 3JCP = 19.4 Hz, C2,6–H of C2,6–Ph), 129.5 (s, C3,5–H of C4–Ph), 142.5 (s, C1 of C4–Ph), 146.5 (d, 2JCP = 24.2 Hz, C1 of C2,6–Ph). 29Si DEPT NMR (79.49 MHz, 300 K, [D8]THF): δ = −3.3 (d, 1JSiP = 62.4 Hz). 31P{1H} NMR (161.98 MHz, 300 K, [D8]THF): δ = −77.7 (s, 29Si-satellites: 1JPSi = 62.1 Hz). 31P NMR (161.98 MHz, 300 K, [D8]THF): δ = −77.7 (s, br). Elemental analysis calcd for C36H41FePSi (Mw = 588.63 g mol−1) C 73.46, H 7.02; found C 73.81, H 6.97.
A solution of chlorodiphenylphosphane (35 mg, 0.159 mmol) in THF (5 mL) at −95 °C was added dropwise to a dark orange solution of 1 (150 mg, 0.156 mmol) in THF (10 mL) at −95 °C. The reaction mixture turned deep red to violet and was slowly warmed to room temperature. The solvent was removed in vacuo after stirring at room temperature for 16 h. The remaining residue was extracted with n-pentane (8 × 1 mL). The extracted fractions were combined and concentrated to 4 mL. 6 was isolated as dark violet crystals after storage at −30 °C for three days. Yield: 48 mg (38%). M.p. 192 °C. UV/vis: (n-hexane, λmax nm−1, εmax L−1 mol−1 cm−1): 240sh (42
 000), 295 (30800), 485 (2400), 550 (1900). 1H NMR (400.13 MHz, 300 K, [D8]THF): δ = 0.98 (s, 15H, C5(CH3)5), 6.02 (d, 3JHP = 2.9 Hz, 2H, C3–H of TPP), 6.84–6.93 (m, 6H, C3,4,5–H of PPh2), 6.96–7.00 (m, 4H, C2,6–H of PPh2), 7.10 (t, 3JHH = 7.2 Hz, 2H, C4–H of C2,6–Ph), 7.21–7.25 (m, 4H, C3,5–H of C2,6–Ph), 7.47 (t, 3JHH = 7.2 Hz, 1H, C4–H of C4–Ph), 7.52–7.56 (m, 2H, C3,5–H of C4–Ph), 7.99–8.02 (m, 4H, C2,6–H of C2,6–Ph), 8.13 (d, 3JHH = 7.8 Hz, 2H, C2,6–H of C4–Ph). 13C{1H} NMR (100.61 MHz, 300 K, [D8]THF): δ = 8.79 (s, C5(CH3)5), 41.0 (dd, 1JCP = 12.5 Hz, 2JCP = 10.6 Hz, C2,6 of TPP), 80.9 (dd, 2JCP = 12.6 Hz, 3JCP = 10.5 Hz, C3–H of TPP), 85.8 (s, C5(CH3)5), 97.3 (d, 3JCP = 2.2 Hz, C4 of TPP), 125.0 (d, 5JCP = 3.3 Hz, C4–H of C2,6–Ph), 127.4 (s, C4–H of PPh2), 128.0 (d, 3JCP = 6.0 Hz, C3,5 of PPh2), 128.1 (s, C4–H of C4–Ph), 128.4 (s, C2,6–H of C4–Ph), 128.6 (s, C3,5–H of C2,6–Ph), 129.1 (s, C3,5–H of C4–Ph), 129.2 (d, 3JCP = 18.1 Hz, C2–H of C2,6–Ph), 134.7 (dd, 2JCP = 17.2 Hz, 3JCP = 4.1 Hz, C2,6–H of PPh2), 138.7 (dd, 1JCP = 25.5 Hz, 2JCP = 6.9 Hz, C1 of PPh2), 141.0 (s, C1 of C4–Ph), 144.9 (d, 2JCP = 24.5 Hz, C1 of C2,6–Ph). 31P{1H} NMR (161.98 MHz, 300 K, [D8]THF): δ = 12.8 (d, 1JPP = 293 Hz, PPh2), −38.8 (d, 1JPP = 293 Hz, P of TPP). 31P NMR (161.98 MHz, 300 K, [D8]THF): δ = 12.8 (dquint, 1JPP = 293 Hz, 3JPH = 6.5 Hz, PPh2), −38.8 (d, 1JPP = 293 Hz, P of TPP). Elemental analysis calcd for C45H42FeP2 (Mw = 700.62 g mol−1) C 77.14, H 6.04; found C 77.55, H 6.36.
000), 295 (30800), 485 (2400), 550 (1900). 1H NMR (400.13 MHz, 300 K, [D8]THF): δ = 0.98 (s, 15H, C5(CH3)5), 6.02 (d, 3JHP = 2.9 Hz, 2H, C3–H of TPP), 6.84–6.93 (m, 6H, C3,4,5–H of PPh2), 6.96–7.00 (m, 4H, C2,6–H of PPh2), 7.10 (t, 3JHH = 7.2 Hz, 2H, C4–H of C2,6–Ph), 7.21–7.25 (m, 4H, C3,5–H of C2,6–Ph), 7.47 (t, 3JHH = 7.2 Hz, 1H, C4–H of C4–Ph), 7.52–7.56 (m, 2H, C3,5–H of C4–Ph), 7.99–8.02 (m, 4H, C2,6–H of C2,6–Ph), 8.13 (d, 3JHH = 7.8 Hz, 2H, C2,6–H of C4–Ph). 13C{1H} NMR (100.61 MHz, 300 K, [D8]THF): δ = 8.79 (s, C5(CH3)5), 41.0 (dd, 1JCP = 12.5 Hz, 2JCP = 10.6 Hz, C2,6 of TPP), 80.9 (dd, 2JCP = 12.6 Hz, 3JCP = 10.5 Hz, C3–H of TPP), 85.8 (s, C5(CH3)5), 97.3 (d, 3JCP = 2.2 Hz, C4 of TPP), 125.0 (d, 5JCP = 3.3 Hz, C4–H of C2,6–Ph), 127.4 (s, C4–H of PPh2), 128.0 (d, 3JCP = 6.0 Hz, C3,5 of PPh2), 128.1 (s, C4–H of C4–Ph), 128.4 (s, C2,6–H of C4–Ph), 128.6 (s, C3,5–H of C2,6–Ph), 129.1 (s, C3,5–H of C4–Ph), 129.2 (d, 3JCP = 18.1 Hz, C2–H of C2,6–Ph), 134.7 (dd, 2JCP = 17.2 Hz, 3JCP = 4.1 Hz, C2,6–H of PPh2), 138.7 (dd, 1JCP = 25.5 Hz, 2JCP = 6.9 Hz, C1 of PPh2), 141.0 (s, C1 of C4–Ph), 144.9 (d, 2JCP = 24.5 Hz, C1 of C2,6–Ph). 31P{1H} NMR (161.98 MHz, 300 K, [D8]THF): δ = 12.8 (d, 1JPP = 293 Hz, PPh2), −38.8 (d, 1JPP = 293 Hz, P of TPP). 31P NMR (161.98 MHz, 300 K, [D8]THF): δ = 12.8 (dquint, 1JPP = 293 Hz, 3JPH = 6.5 Hz, PPh2), −38.8 (d, 1JPP = 293 Hz, P of TPP). Elemental analysis calcd for C45H42FeP2 (Mw = 700.62 g mol−1) C 77.14, H 6.04; found C 77.55, H 6.36.
A solution of chlorocatecholborane (24 mg, 0.155 mmol) in THF (5 mL) was added to a dark orange solution of 1 (150 mg, 0.156 mmol) in THF (10 mL) at −35 °C. The mixture was stirred and warmed to room temperature overnight. All volatiles were removed in vacuo and the dark orange-green residue was washed with n–hexane (5 × 1 mL) and extracted with diethyl ether (10 × 0.5 mL). The deep orange diethyl ether fractions were combined and the major impurities including [18]crown-6 were crystallized by storage at room temperature for five days. The deep orange mother liquor was decanted and concentrated to 3 mL. Deep orange crystals of 7 formed during storage at room temperature for two days. Yield: 25 mg (25%). M.p. 196 °C (decomposition to a dark green solid). UV/vis: (n–hexane, λmax nm−1, εmax L−1 mol−1 cm−1): 260 (15
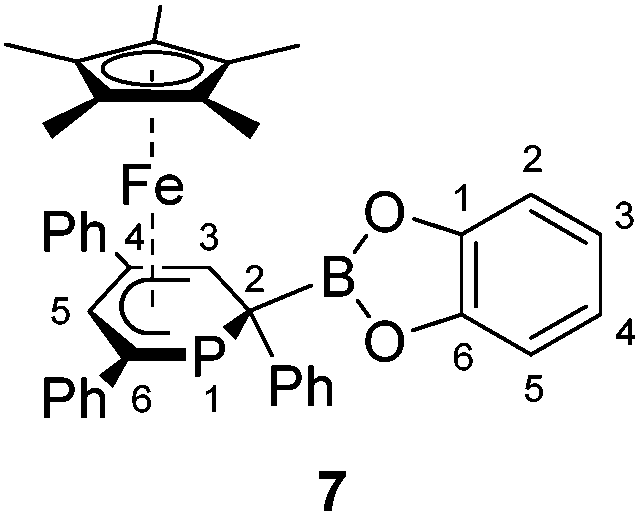 000), 290sh (10000), 360sh (1700), 460sh (300). 1H NMR (400.13 MHz, 300 K, [D8]THF): δ = 1.34 (s, 15H, C5(CH3)5), 3.51 (s, 1H, C3–H of TPP), 6.68–6.72 (m, 1H, C4–H of C2–Ph), 6.89–6.91 (m, 2H, C3,5–H of C2–Ph), 6.03–6.05 (overlapping m, 3H, C2,6–H of C2–Ph overlapping with C5–H of TPP), 7.07–7.11 (m, 2H, C2,5–H of catecholboryl), 7.17–7.21 (m, 1H, C4–H of C6–Ph), 7.23–7.26 (m, 2H, C3,5–H of C6–Ph), 7.28–7.30 (m, 2H, C3,4–H of catecholboryl), 7.32–7.36 (m, 1H, C4–H of C4–Ph), 7.44–7.48 (m, 2H, C3,5–H of C4–Ph), 7.85 (d, 3JHH = 7.9 Hz, 2H, C2,6–H of C6–Ph), 8.04 (d, 3JHH = 7.9 Hz, 2H, C2,6–H of C4–Ph). 13C{1H} NMR (100.61 MHz, 300 K, [D8]THF): δ = 9.7 (d, 3JCP = 3.2 Hz, C5(CH3)5), 26.1 (s, C3–H of TPP), 88.4 (s, C5(CH3)5), 88.6 (d, 2JCP = 7.3 Hz, C5–H of TPP), 90.9 (s, C4 of TPP), 95.2 (d, 1JCP = 74.5 Hz, C6 of TPP), 112.9 (s, C3,4–H of catecholboryl), 123.3 (s, C2,5–H of catecholboryl), 124.3 (s, C4–H of C2–Ph), 127.3 (s, C2,6–H of C4–Ph overlapping with C4 of C6–Ph), 127.4 (d, 3JCP = 4.6 Hz, C2,6 of C2–Ph), 127.6 (s, C4–H of C4–Ph), 127.7 (s, C3,5–H of C2–Ph), 127.9 (d, 3JCP = 15.0 Hz, C2,6 of C6–Ph), 128.7 (s, C3,5–H of C6–Ph), 129.2 (s, C3,5–H of C4–Ph), 141.6 (s, C1 of C4–Ph),143.5 (d, 2JCP = 18.0 Hz, C1 of C6–Ph), 147.8 (s, C1 of C2–Ph), 149.5 (s, C1,6 of catecholboryl), the signal for C2 of TPP was not observed. 11B{1H} NMR (128.38 MHz, 300 K, [D8]THF): δ = 34.3 (s br). 11B NMR (128.38 MHz, 300 K, [D8]THF): δ = 34.3 (s br). 31P{1H} NMR (161.98 MHz, 300 K, [D8]THF): δ = −126.7 (s). 31P NMR (161.98 MHz, 300 K, [D8]THF): δ = −126.7 (s br). Elemental analysis calcd for C39H36BFeO2P (Mw = 634.34 g mol−1) C 73.84, H 5.72; found C 73.45, H 5.73.
000), 290sh (10000), 360sh (1700), 460sh (300). 1H NMR (400.13 MHz, 300 K, [D8]THF): δ = 1.34 (s, 15H, C5(CH3)5), 3.51 (s, 1H, C3–H of TPP), 6.68–6.72 (m, 1H, C4–H of C2–Ph), 6.89–6.91 (m, 2H, C3,5–H of C2–Ph), 6.03–6.05 (overlapping m, 3H, C2,6–H of C2–Ph overlapping with C5–H of TPP), 7.07–7.11 (m, 2H, C2,5–H of catecholboryl), 7.17–7.21 (m, 1H, C4–H of C6–Ph), 7.23–7.26 (m, 2H, C3,5–H of C6–Ph), 7.28–7.30 (m, 2H, C3,4–H of catecholboryl), 7.32–7.36 (m, 1H, C4–H of C4–Ph), 7.44–7.48 (m, 2H, C3,5–H of C4–Ph), 7.85 (d, 3JHH = 7.9 Hz, 2H, C2,6–H of C6–Ph), 8.04 (d, 3JHH = 7.9 Hz, 2H, C2,6–H of C4–Ph). 13C{1H} NMR (100.61 MHz, 300 K, [D8]THF): δ = 9.7 (d, 3JCP = 3.2 Hz, C5(CH3)5), 26.1 (s, C3–H of TPP), 88.4 (s, C5(CH3)5), 88.6 (d, 2JCP = 7.3 Hz, C5–H of TPP), 90.9 (s, C4 of TPP), 95.2 (d, 1JCP = 74.5 Hz, C6 of TPP), 112.9 (s, C3,4–H of catecholboryl), 123.3 (s, C2,5–H of catecholboryl), 124.3 (s, C4–H of C2–Ph), 127.3 (s, C2,6–H of C4–Ph overlapping with C4 of C6–Ph), 127.4 (d, 3JCP = 4.6 Hz, C2,6 of C2–Ph), 127.6 (s, C4–H of C4–Ph), 127.7 (s, C3,5–H of C2–Ph), 127.9 (d, 3JCP = 15.0 Hz, C2,6 of C6–Ph), 128.7 (s, C3,5–H of C6–Ph), 129.2 (s, C3,5–H of C4–Ph), 141.6 (s, C1 of C4–Ph),143.5 (d, 2JCP = 18.0 Hz, C1 of C6–Ph), 147.8 (s, C1 of C2–Ph), 149.5 (s, C1,6 of catecholboryl), the signal for C2 of TPP was not observed. 11B{1H} NMR (128.38 MHz, 300 K, [D8]THF): δ = 34.3 (s br). 11B NMR (128.38 MHz, 300 K, [D8]THF): δ = 34.3 (s br). 31P{1H} NMR (161.98 MHz, 300 K, [D8]THF): δ = −126.7 (s). 31P NMR (161.98 MHz, 300 K, [D8]THF): δ = −126.7 (s br). Elemental analysis calcd for C39H36BFeO2P (Mw = 634.34 g mol−1) C 73.84, H 5.72; found C 73.45, H 5.73.
X-Ray crystallography
Crystals of endo-3, exo-3, 4, 5, and 8 suitable for X-ray diffraction were obtained from n-hexane. Crystals of 6 were obtained from diethyl ether. X-ray quality crystals of 7 were obtained from concentrated diethyl ether solutions of the crude reaction mixture resulting in crystallization along with half a molecule of [18]crown-6. The single crystal X-ray diffraction data were recorded on an Agilent Technologies Gemini Ultra R diffractometer (exo-3 and 5) and an Agilent Technologies SuperNova (endo-3, 4, and 6–8) with Cu Kα radiation (λ = 1.54184 Å). Semi-empirical multi-scan absorption corrections27 and analytical ones28 were applied to the data. The structures were solved with SHELXT29 and least-square refinements on F2 were carried out with SHELXL.30CCDC 1448678–1448684 contain the supplementary crystallographic data for this paper.
DFT calculations
The calculations on 2, endo-3 and exo-3 were performed using the ORCA program package (version 3.0.2).31 The BP86 density functional and the Ahlrichs def2-TZVP basis set were employed for all atoms.32–36 The RI approximation was used.37,38 The Ahlrichs Coulomb fitting basis for the TZVP basis for all atoms (TZV/J) and the atom-pair-wise dispersion correction to the DFT energy with Becke-Johnson damping (d3bj) were applied.39,40 The nature of the stationary points was verified by numerical frequency analysis.Acknowledgements
Funding by the Deutsche Forschungsgemeinschaft is gratefully acknowledged.Notes and references
- C. Müller, L. E. E. Broeckx, I. de Krom and J. J. M. Weemers, Eur. J. Inorg. Chem., 2013, 187–202 CrossRef.
- M. Bruce, G. Meissner, M. Weber, J. Wiecko and C. Müller, Eur. J. Inorg. Chem., 2014, 1719–1726 CrossRef CAS.
- G. Märkl and C. Martin, Angew. Chem., 1974, 86, 445–446 CrossRef.
- G. Baum and W. Massa, Organometallics, 1985, 4, 1572–1574 CrossRef CAS.
- A. Moores, N. Mézailles, L. Ricard and P. Le Floch, Organometallics, 2005, 24, 508–513 CrossRef CAS.
- A. Moores, L. Ricard, P. Le Floch and N. Mézailles, Organometallics, 2003, 22, 1960–1966 CrossRef CAS.
- B. Rezaei Rad, U. Chakraborty, B. Mühldorf, J. A. W. Sklorz, M. Bodensteiner, C. Müller and R. Wolf, Organometallics, 2015, 34, 622–635 CrossRef CAS.
- F. Nief and J. Fischer, Organometallics, 1986, 5, 877–883 CrossRef CAS.
- M. Doux, N. Mézailles, L. Ricard and P. Le Floch, Organometallics, 2003, 22, 4624–4626 CrossRef CAS.
- H. Lehmkuhl, J. Elsäßer, R. Benn, B. Gabor, A. Rufińska, R. Goddard and C. Krüger, Z. Naturforsch., B: Anorg. Chem. Org. Chem., 1985, 40, 171–181 Search PubMed.
- B. Nuber and M. L. Ziegler, Acta Crystallogr. C: Cryst. Struct. Commun., 1987, 43, 59–61 CrossRef.
- M. Doux, L. Ricard, P. Le Floch and Y. Jean, Organometallics, 2005, 24, 1608–1613 CrossRef CAS.
- T. Arliguie, M. Doux, N. Mézailles, P. Thuéry, P. Le Floch and M. Ephritikhine, Inorg. Chem., 2006, 45, 9907–9913 CrossRef CAS PubMed.
- M. Doux, P. Thuéry, M. Blug, L. Ricard, P. Le Floch, T. Arliguie and N. Mézailles, Organometallics, 2007, 26, 5643–5653 CrossRef CAS.
- T. Arliguie, M. Blug, P. Le Floch, N. Mézailles, P. Thuéry and M. Ephritikhine, Organometallics, 2008, 27, 4158–4165 CrossRef CAS.
- T. Arliguie, P. Thuéry, P. L. Floch, N. Mézailles and M. Ephritikhine, Polyhedron, 2009, 28, 1578–1582 CrossRef CAS.
- A. Moores, N. Mézailles, L. Ricard, Y. Jean and P. le Floch, Organometallics, 2004, 23, 2870–2875 CrossRef CAS.
- H. Lehmkuhl, R. Paul, C. Krüger, Y.-H. Tsay, R. Benn and R. Mynott, Liebigs Ann. Chem., 1981, 1147–1161 CrossRef CAS.
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 12770–12779 CrossRef PubMed.
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 186–197 CrossRef PubMed.
- D. L. Dodds, J. Floure, M. Garland, M. F. Haddow, T. R. Leonard, C. L. McMullin, A. G. Orpen and P. G. Pringle, Dalton Trans., 2011, 40, 7137–7146 RSC.
- S. Burck, D. Gudat and M. Nieger, Angew. Chem., 2004, 116, 4905–4908 CrossRef.
- D. Carmichael, P. L. Floch, H. G. Trauner and F. Mathey, Chem. Commun., 1996, 971 RSC.
- H. Braunschweig, R. Dirk and B. Ganter, J. Organomet. Chem., 1997, 545–546, 257–266 CrossRef CAS.
- S. D. Ittel, J. Organomet. Chem., 1980, 195, 331–335 CrossRef CAS.
- J. Emsley and D. Hall, The Chemistry of Phosphorus, Harper & Row Ltd, London, 1976 Search PubMed.
- (a) SCALE3ABS, CrysAlisPro, Aglient Technologies Inc., Oxford, UK, 2015 Search PubMed; (b) G. M. Sheldrick, SADABS, Bruker AXS, Madison, USA, 2007 Search PubMed.
- (a) R. C. Clark and J. S. Reid, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1995, 51, 887–897 CrossRef; (b) CrysAlisPro, Agilent Technologies Inc., Oxford, UK, 2015 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986, 33, 8822–8824 CrossRef.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- D. L. Dodds, M. F. Haddow, A. G. Orpen, P. G. Pringle and G. Woodward, Organometallics, 2006, 25, 5937–5945 CrossRef CAS.
- R. A. Oliveira, R. O. Silva, G. A. Molander and P. H. Menezes, Magn. Reson. Chem., 2009, 47, 873–878 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H, 13C{1H}, 31P{1H} and UV-vis spectra of complexes endo-3–7, crystallographic data of endo-3–7, and details of the DFT calculations (relative thermal and free enthalpies of 2, endo-3 and exo–3). Full experimental details and crystallographic data of [Cp*Fe(1-Ph3Sn-PC5Ph3H2)] (8). CCDC 1448678–1448684. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt00336b |
| ‡ The synthesis of 6 needs to be performed at −95 °C by slow addition of Ph2PCl to a solution of complex 1. In this case, 6 was isolated in 38% yield. Addition of Ph2PCl at room temperature led to an almost quantitative formation of tetraphenyldiphosphane and the dimeric complex [Cp*2Fe2(μ-{PC5Ph3H2}2)].7,41 |
| § The 13C{1H} NMR resonance for sp3-hybridized carbon atom of 7 attached to phosphorus and boron was not observed. Coupling in the 31P–13C–10/11B spin system and the quadrupole relaxation mechanism of the 11B nucleus presumably lead to substantial broadening of this signal.42 |
| This journal is © The Royal Society of Chemistry 2016 |