
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transition metal (Rh and Fe) complexes and main-group (Se and B) adducts with N,N′-diphosphanyl NHC ligands: a study of stereoelectronic properties†
Pengfei
Ai
a,
Andreas A.
Danopoulos
*ab and
Pierre
Braunstein
*a
aLaboratoire de Chimie de Coordination, Institut de Chimie (UMR 7177 CNRS), Université de Strasbourg, 4 rue Blaise Pascal, 67081 Strasbourg Cedex, France. E-mail: danopoulos@unistra.fr; braunstein@unistra.fr
bInstitute for Advanced Study, USIAS, Université de Strasbourg, France
First published on 3rd February 2016
Abstract
Attempts to evaluate experimentally the donor characteristics of the N,N′-bis(di-tert-butylphosphanyl)-imidazole-2-ylidene (PCNHCP) hybrid ligand are described. Thus, reactions of PCNHCP with [Rh(μ-Cl)(COD)]2 and [Rh(μ-Cl)(CO)2]2 led to the formation of the mononuclear and dinuclear complexes, [Rh(PCNHCP,κP,κCNHC)2]Cl (PCNHCP-RhCl) and [Rh(CO)(PCNHC,κP,κCNHC,κN)]2 (PCNHC-RhCO), respectively, the latter resulting after in situ cleavage of one (t-Bu)2P–Nimid bond of PCNHCP. With ligands acting as a P,C-chelate, a straightforward evaluation of the Tolman electronic parameter (TEP) of the CNHC donor is problematical; the viability of dangling P- and bound CNHC-donors (i.e. κCNHC) has been observed in the trinuclear Fe(II) chain complex [Fe3Cl2(μ-Cl)4(THF)2(PCNHCP,κCNHC)2] (PCNHCP-Fe), obtained by the reaction of PCNHCP with [Fe4Cl8(THF)6] and, recently, established on CrII, CoII and AuI centres. Evaluation of the π-accepting properties of the PCNHCP (and the related Dipp-PCNHC) was based on the 77Se NMR chemical shifts of the corresponding NHC–Se adducts, PCNHCP-Se (and Dipp-PCNHC-Se), which were prepared from the free PCNHCP (and Dipp-PCNHC) and Se. The π-acidity of PCNHCP is found to be higher than that of Dipp-PCNHC but lower than that of SIPr. The donor ability of the CNHC in PCNHCP was explored by its reaction with the Lewis acids tris(pentafluorophenyl)borane (B(C6F5)3) and tris(pentafluorophenyl)boroxine ([(C6F5)BO]3), which resulted in stable donor–acceptor adducts with no FLP reactivity. The steric properties of PCNHCP and Dipp-PCNHC are conformation dependent, with the percent buried volume (%Vbur) of PCNHCP in the structurally characterised conformer calculated at 67.6, the largest value currently reported for NHC ligands.
Introduction
N-heterocyclic carbene (NHC) ligands have become ubiquitous in molecular chemistry and find extensive applications in e.g. catalytic transformations, medicinal chemistry and material sciences.1 Both experimental and theoretical studies on their electronic properties have established synergism resulting from their strong σ donor and weaker π acceptor properties. The most commonly used method for the experimental evaluation of the donor properties of NHCs is the Tolman electronic parameter (TEP), originally developed for phosphines by Tolman in 1970,2 which is based on the value of the A1ν(CO) infrared-stretching frequencies in [Ni(CO)3L], or the average ν(CO) frequencies in cis-[Ir(CO)2ClL] and cis-[Rh(CO)2ClL] (Chart 1), the latter two complexes being more widely used nowadays owing to the toxicity of [Ni(CO)4].2,3 However, the TEP quantifies the overall NHC electronic properties and does not separate the σ donation and π back-donation components.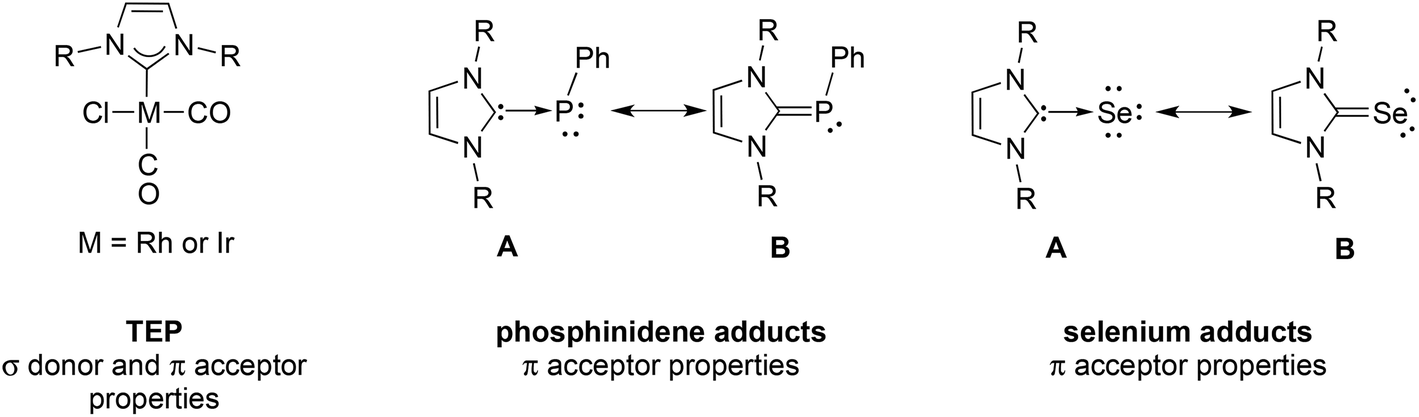 | ||
| Chart 1 Three experimental methods used to evaluate the electronic properties of NHC ligands. | ||
Recently, Bertrand and coll.4 and Ganter and coll.5 separately reported the use of phosphinidene (NHC)PPh and selenium adducts (NHC)Se (Chart 1) to probe the NHC π back-bonding ability, on the basis of the δp chemical shift in the 31P NMR and the δSe chemical shift in the 77Se NMR, respectively. These adducts can be represented by two limiting canonical structures: the polarized form A with a NHC–E (E = PPh or Se) single bond indicating little π accepting ability and the resonance form B with a NHC–E (E = PPh or Se) double bond consistent with a high π accepting ability of NHCs. In contrast to the narrow range of TEP values, the chemical shifts of the phosphinidene/selenium adducts are very sensitive to subtle changes of the electronic structures of NHCs and offer a finer grading of the NHCs.
Various groups have recently reported N-phosphanyl-functionalized NHC ligands, which feature the two adjacent, strong σ-donors groups linked by a direct P–N bond.6 Because of the rigidity and ease of tuning of their steric and electronic properties, they have not only been used as bridging ligands for d10 coinage metals but also as small bite angle (ca. 68.2°) chelating ligands for platinum group or late transition metals.6c–f,7 We have recently reported the synthesis of the new tridentate ligand N,N′-bis(di-tert-butylphosphanyl)-imidazole-2-ylidene (PCNHCP) (Scheme 1) and its behaviour as bridging ligand with coinage metals or palladium, resulting in polynuclear complexes,6h,i,8 and as PCNHC chelating ligand with chromium or palladium.8a,9 However, a more precise experimental estimate of the stereoelectronic properties of CNHC donor in the PCNHCP ligand is not straightforward, due to the high nucleophilicity of both donors and the propensity of formation of small ring chelates with the metals used as probes for the TEP. Herein, we describe Rh(I) carbonyl complexes with PCNHCP which display κP, κCNHC coordination, thus rendering the evaluation of the TEP cumbersome; in contrast, the selective access of NHC–Se adducts permits the evaluation of the π accepting ability of the CNHC in PCNHCP; furthermore we give an estimate of the steric bulk of PCNHCP, and describe adducts with B(C6F5)3 and FeCl2 in which CNHC coordination predominates.
 | ||
| Scheme 1 Reactions of PCNHCP with [Rh(μ-Cl)(COD)]2, [Rh(μ-Cl)(CO)2]2 and [Fe4Cl8(THF)6]. | ||
Results and discussion
Mono- and dinuclear rhodium complexes and trinuclear iron complex
Since no rhodium complex with the PCNHCP has yet been prepared, with the objective to prepare cis-[RhCl(CO)2(PCNHCP)] we firstly reacted PCNHCP with [Rh(μ-Cl)(COD)]2 in THF in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ligand/Rh ratio (Scheme 1). However, instead of the expected adduct [RhCl(COD)(PCNHCP,κCNHC)], the mononuclear complex [Rh(PCNHCP,κP,κCNHC)2]Cl (PCNHCP-RhCl) was isolated, the two PCNHCP ligands acting as P,C-chelates. This was confirmed by single-crystal X-ray diffraction analysis (Fig. 1).
:1 ligand/Rh ratio (Scheme 1). However, instead of the expected adduct [RhCl(COD)(PCNHCP,κCNHC)], the mononuclear complex [Rh(PCNHCP,κP,κCNHC)2]Cl (PCNHCP-RhCl) was isolated, the two PCNHCP ligands acting as P,C-chelates. This was confirmed by single-crystal X-ray diffraction analysis (Fig. 1).
 | ||
| Fig. 1 Thermal ellipsoid representation (30% probability level) of the structure of one cation in PCNHCP-RhCl. H atoms, the t-Bu methyl groups, the second complex cation and the chloride anions are omitted for clarity. Selected bond lengths (Å) and angles [°] (the two values are for the two independent cations in the unit cell): Rh1–C1 2.077(4)/2.082(4); Rh1–P1 2.300(1)/2.304(1), C1–Rh1–P1 67.26(11)/67.31(13). | ||
The unit cell of PCNHCP-RhCl contains two similar complex cations with slightly different metrical data and isolated chloride anions (Rh–Cl distance 7.418/7.359 Å). In each cation, the tetracoordinated Rh atom occupies a centre of symmetry and is chelated by two PCNHCP ligands through their CNHC and one phosphorus donor, with a bite angle of 67.26(11)/67.31(13)°. The solution 31P{1H} NMR spectrum of PCNHCP-RhCl contains a doublet at δ 113.1 (1J(P–Rh) = 146.9 Hz) and a singlet at δ 83.8, corresponding to the coordinated and the uncoordinated phosphines, respectively. The latter value is slightly different from that of the free PCNHCP (δ 98.7),6h indicating electronic communication between the P donors on coordination.
In view of the lability of the COD ligand that prevented the preparation of [RhCl(COD)(PCNHCP)], the complex [Rh(μ-Cl)(CO)2]2 was used as precursor and was directly reacted with PCNHCP in THF. However, instead of the expected cis-[RhCl(CO)2(PCNHCP,κCNHC)], the neutral, dinuclear complex [Rh(CO)(PCNHC,κP,κCNHC,κN)]2 (PCNHC-RhCO) containing two monoanionic PCNHC ligands was obtained after cleavage of one (t-Bu)2P–Nimid bond of PCNHCP (Scheme 1). The formation of (t-Bu)2PCl was evidenced in the reaction mixture as the only other P-containing product (δ 145 in 31P{1H} NMR). A similar P–N bond cleavage reaction has also been observed when PCNHCP was reacted with [AuCl(tht)] and rationalised by the reactivity of the P–N bond toward the nucleophilic chloride ligand.6i The 31P{1H} NMR spectrum of PCNHC-RhCO contains a doublet at δ 128.4 (1J(P–Rh) = 142.8 Hz). The structure of this complex exhibits an approximate C2 symmetry and the square-planar environment around each Rh atom contains one chelating PCNHC ligand, bonded through the P and CNHC 2 electron donors which form an angle of 67.3(3)/67.2(3)°, a negatively charged N atom from the other PCNHC ligand and one carbonyl ligand trans to CNHC (Fig. 2). Related dinuclear rhodium complexes with bridging arylimidazolide-N3,C2 ligands containing a central six-membered ring have been recently reported.10
 | ||
| Fig. 2 Thermal ellipsoid representation (30% probability level) of the structure of PCNHC-RhCO. H atoms and the t-Bu methyl groups are omitted for clarity. Selected bond lengths (Å) and angles [°]: Rh1–C1 2.071(10), Rh2–C12 2.080(9), Rh1–C23 1.833(10), Rh2–C24 1.842(11), Rh1–N4 2.080(8), Rh2–N2 2.065(8), Rh1–P1 2.249(3), Rh2–P2 2.243(3), C23–O1 1.166(12), C24–O2 1.152(12); C1–Rh1–P1 67.3(3), C12–Rh2–P2 67.2(3). | ||
Since PCNHC chelation was observed here with PCNHCP-RhCl and previously with Pd(II) and Cr(III) centres,8a,9 but not with Cr(II) or Co(II),9,11 we wondered whether the κCNHC coordination mode is more common with 3d metals. Thus, reaction of PCNHCP with [Fe4Cl8(THF)6] in THF inside a glovebox (Scheme 1) afforded colourless, paramagnetic, air-sensitive crystals after crystallisation by addition of pentane. Single-crystal X-ray diffraction established the formation of a centrosymmetric trinuclear Fe(II) chain complex [Fe3Cl2(μ-Cl)4(THF)2(PCNHCP,κCNHC)2] (PCNHCP-Fe) in which each outer Fe atom is coordinated to the CNHC donor of one PCNHCP ligand, with both P donors remaining uncoordinated (Fig. 3). The linear metal chain is assembled by two μ-chlorido ligands that connect the outer Fe atoms to the central Fe2. The outer Fe atoms adopt a tetrahedral coordination geometry consisting of three chloride ligands and one CNHC donor while the central iron is in an octahedral environment with four chloride ligands occupying equatorial positions and two THF molecules the axial positions. The higher occurrence of this coordination mode with 3d metals may be rationalised by the reduced M–P bond energies and the reduced size and strength of ligand fields of 3d rendering structures with dangling P donors energetically more accessible compared to 4d and 5d metals.
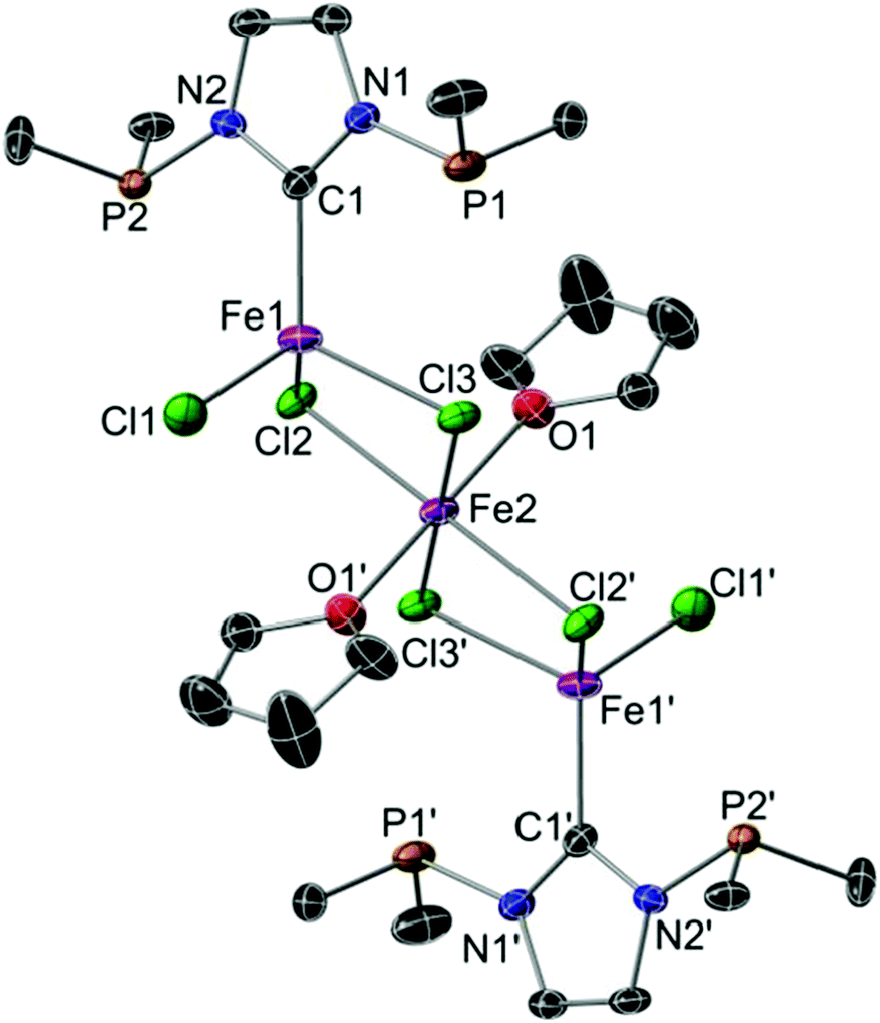 | ||
| Fig. 3 Thermal ellipsoid representation (30% probability level) of the structure of PCNHCP-Fe. H atoms and the t-Bu methyl groups are omitted for clarity. Selected bond lengths (Å) and angles [°]: Fe1–C1 2.103(6), Fe1–Cl1 2.2587(19), Fe1–Cl2 2.3807(17), Fe1–Cl3 2.4118(18), Fe2–Cl2 2.4781(14), Fe2–Cl3 2.4755(14), Fe2–O1 2.158(4); Fe1–Cl2–Fe2 91.98(5), Fe1–Cl3–Fe2 91.30(5), C1–Fe1–Cl1 112.98(16), C1–Fe1–Cl2 115.81(15), C1–Fe1–Cl3 115.31(16), Cl1–Fe1–Cl2 111.90(7), Cl1–Fe1–Cl3 109.59(6), Cl2–Fe1–Cl3 88.99(6), O1–Fe2–Cl3′ 88.33(11). | ||
NHC adducts with main group elements (Se, B)
Based on the results we shifted our attention to alternative adducts that may disclose information on the donor characteristics of the PCNHCP. The first attempt focused on the synthesis of phosphinidene adducts of NHCs, which carry information on the π-accepting strength of the NHC precursor.4 Two known synthetic routes were explored for the synthesis of the adducts (Scheme 2): (i) the direct reaction of PCNHCP with pentaphenylcyclopentaphosphane ((PPh)5); (ii) the reaction of PCNHCP with PhPCl2 followed by reduction with Mg. However, both failed: the former gave no reaction even after heating to 70 °C, and the latter led to the cleavage of a P–N bond of PCNHCP by PhPCl2 with formation of (t-Bu)2PCl.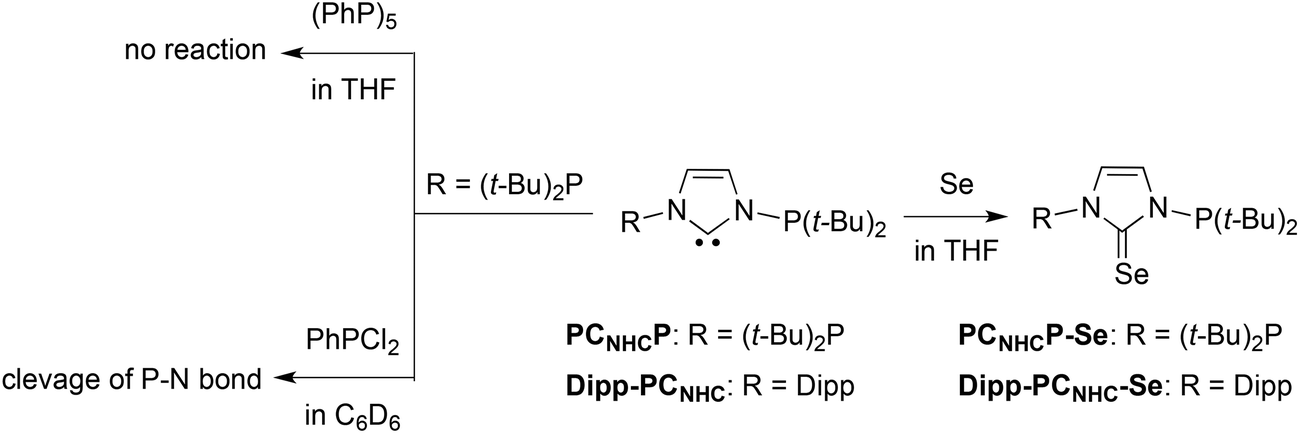 | ||
| Scheme 2 Reactions of PCNHCP and DIPP-PCNHC with PhPCl2, (PPh)5 and elemental selenium. | ||
The second attempt focused on the synthesis of selenium adducts; herein, the π-accepting properties of NHCs precursors can also be correlated with the 77Se NMR chemical shift observed.5 Thus, reaction of elemental selenium with PCNHCP in a 1:1 ratio in THF afforded selectively the desired adduct PCNHCP-Se, with intact phosphine groups (Scheme 2), as indicated by a 31P NMR shift (s, δ 91.0) (cf.PCNHCPδ s, 98.7). The structure of the product was further confirmed by an X-ray diffraction analysis (Fig. 4). The Se–CNHC bond distance 1.839(8) Å is intermediate between that of a Se–C single bond (aver. 1.94 Å) and of a typical Se![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond (aver. 1.74 Å).12 Its value is similar to that found in other selenium–NHC adducts.5b,12a–c Compared to the free carbene PCNHCP, the N–CNHC bond lengths (1.36(1)/1.370(9) Å, cf. 1.376(1)/1.378(1) Å in PCNHCP) are shortened and the N1–C1–N2 angle (108.2(6)°, cf. 102.5(1)° in PCNHCP) enlarged.
C double bond (aver. 1.74 Å).12 Its value is similar to that found in other selenium–NHC adducts.5b,12a–c Compared to the free carbene PCNHCP, the N–CNHC bond lengths (1.36(1)/1.370(9) Å, cf. 1.376(1)/1.378(1) Å in PCNHCP) are shortened and the N1–C1–N2 angle (108.2(6)°, cf. 102.5(1)° in PCNHCP) enlarged.
 | ||
| Fig. 4 Thermal ellipsoid representation (30% probability level) of the structure of PCNHCP-Se. H atoms and the t-Bu methyl groups are omitted for clarity. Selected bond lengths (Å) and angles [°]: C1–Se1 1.839(8), C1–N1 1.36(1), C1–N2 1.370(9), N1–P1 1.760(7), N2–P2 1.775(7); N1–C1–N2 108.2(6). | ||
For comparison with PCNHCP-Se, the selenium adduct Dipp-PCNHC-Se of a ligand containing only one phosphine group was prepared under similar reaction conditions (Scheme 2). The 77Se NMR spectra feature a triplet at δ 166.9 (2JSeP = 46.5 Hz) for PCNHCP-Se and a doublet at δ 131.1 (2JSeP = 40.0 Hz) for Dipp-PCNHC-Se. Accordingly, and by comparison with literature data, it can be concluded that the π-acidity of PCNHCP is stronger than that of Dipp-PCNHC and that both ligands are stronger π-acids than SIMes (δ 110 77Se NMR), but weaker π-acids than SIPr (δ 190 77Se NMR) (Scheme 3).13
 | ||
| Scheme 3 Comparative π-acidity of NHC ligands based on the 77Se NMR data (in CDCl3). | ||
To further explore the donor abilities of the CNHC and P donors in PCNHCP, the latter was reacted with tris(pentafluorophenyl)borane B(C6F5)3 in toluene (Scheme 4). The 11B NMR spectrum of the resulting product (PCNHCP-B1) shows a single resonance at δ −17.5, which is indicative of a four-coordinate boron centre and suggests the formation of a NHC–borane Lewis adduct with a stable B–CNHC bond.
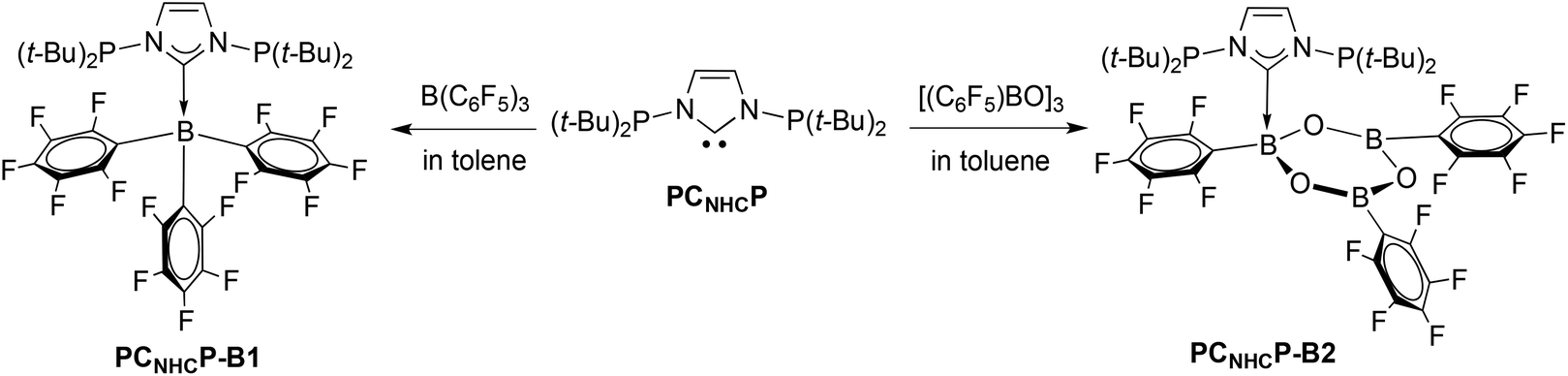 | ||
| Scheme 4 Synthesis of the adducts PCNHCP-B-1 and PCNHCP-B-2. | ||
Broad NMR resonances were not only observed for the three pentafluorophenyl groups in the 19F NMR and 13C NMR spectra but also for the phosphine groups (one broad singlet at δ 110.5) and CNHC (broad multiplet at δ 177.1). This, together with the singlet at δ 7.69 for the two protons on the imidazolylidene ring, indicates fast free rotation of the B(C6F5)3 moiety around the B–CNHC bond. A single-crystal X-ray diffraction analysis showed that the three C6F5 groups adopt a typical C3 propeller-type orientation (Fig. 5). For steric reasons, the lone pair on the P atoms is oriented toward the borane moiety.
 | ||
| Fig. 5 Thermal ellipsoid representation (30% probability level) of the structure of PCP-B1. H atoms, the t-Bu methyl groups, all the F atoms and one molecule of THF are omitted for clarity. Selected bond lengths (Å) and angles [°]: B1–C1 1.688(3), B1–C20 1.659(3), B1–C26 1.660(3), B1–C32 1.670(3); N1–C1–N2 104.98(15), C20–B1–C26 111.11(16), C20–B1–C32 112.03(16), C26–B1–C32 101.70(15), C20–B1–C1 106.09(15), C26–B1–C1 110.95(16), C32–B1–C1 115.06(15). | ||
Unlike other NHC–borane adducts,14PCNHCP-B1 exhibits no frustrated-Lewis-pair (FLP) reactivity toward H2 or THF, which is consistent with a rather strong donor–acceptor CNHC → B interaction, despite a moderate elongation of this bond (1.688(3) Å) when compared to that in the adduct IPr/B(C6F5)3 (1.663(5) Å)14a and in 1,3,4,5-tetramethyl-1,3-imidazole-2-ylidene/B(C6F5)3 (1.640(2) Å).15
Another donor–acceptor adduct was obtained by reaction of PCNHCP with a suspension of tris(pentafluorophenyl)boroxine [(C6F5)BO]3 in toluene (Scheme 4). An immediate reaction occurred with formation of a clear solution. A crystallographic analysis established again the formation of one CNHC → B bond in the boroxine adduct PCNHCP-B2, leaving the other two B atoms intact (Fig. 6). The B–CNHC bond distance 1.670(4) Å is slightly shorter than that in PCNHCP-B1. Obvious differences were observed between the three B–C6F5 moieties: (i) the B1–C32 bond (1.643(4) Å) is elongated compared to the other two (1.590(4) Å and 1.586(4) Å); (ii) the B2–C20 and B3–C26 bonds are coplanar with the boroxine ring while the B1–C32 bond bends out of the boroxine plane, away from the PCNHCP ligand with a distance of 1.393 Å between the C23 atom and the boroxine plane. This results from the coordination of B1 to CNHC forming a tetrahedral environment around the B1 atom.
 | ||
| Fig. 6 Thermal ellipsoid representation (30% probability level) of the structure of PCNHCP-B-2. H atoms, the t-Bu methyl groups, all the F atoms and one molecule of toluene are omitted for clarity. Selected bond lengths (Å) and angles [°]: C1–B1 1.670(4), C20–B2 1.590(4), C26–B3 1.586(4), C32–B1 1.643(4), N1–C1–N2 105.6(2), O1–B1–C1 108.6(2), O3–B1–C1 105.0(2), C32–B1–C1 113.7(2). | ||
The 31P NMR spectrum shows one singlet at δ 97.6 for the phosphine groups and a doublet is observed in the 1H NMR spectrum at δ 1.15 (3JHP = 12.9 Hz) for the t-Bu groups, which indicates free rotation of the boroxine moiety around the B–CNHC bond. The 11B NMR spectrum contains a broad signal at δ 26.4 and a singlet at δ −0.6 ppm corresponding to the three- and four-coordinate B atoms, respectively. Similarly to PCNHCP-B1, it exhibited no frustrated-Lewis-pair (FLP) reactivity toward H2 or THF.
Percent buried volume of the NHC ligands
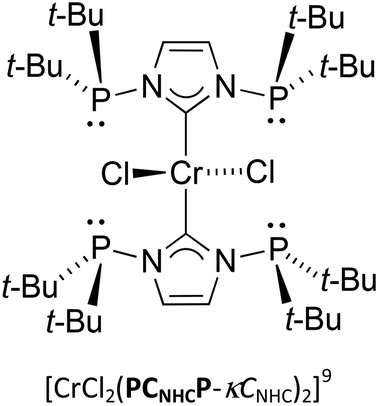
Lastly, the steric properties of PCNHCP and Dipp-PCNHC were evaluated from the percent buried volume (%Vbur) using the SambVca software,16 and metrical data from the crystal structures. The results are compiled in Table 1 assuming a 3.50 Å for the sphere radius, omitted hydrogen atoms and scaled bond radii by 1.17, as recommended. Rotation of the phosphine group around the P–N bond has dramatic effect on the steric environment at the carbene centre, especially for PCNHCP. The %Vbur of the free carbene PCNHCP reaches a value up to 67.6 (M–NHC 2.00 Å), for the conformation observed crystallographically, featuring anti lone pair arrangement; this value is the largest value among all reported NHC ligands (Table 1).17 In the conformation with syn arrangement of the lone pairs which is commonly found on κCNHC coordination or adduct formation the %Vbur decreases to 32.5 for PCNHCP-Se and 36.4 for [CrCl2(PCNHCP-κCNHC)2]9 (M–NHC 2.00 Å). Compared to the %Vbur values found in PCNHCP, that for Dipp-PCNHC is smaller (39.4, M–NHC 2.00 Å) but similar (35.8, M–NHC 2.00 Å) to that in the Cr(II) complexes.| Compounds | %Vbur for M–NHC length at | |
|---|---|---|
| 2.00 Å | 2.28 Å | |

|
67.6 | 63.1 |

|
32.5 | 28.3 |
|
36.4 | 31.7 |

|
39.4 | 36.2 |

|
35.8 | 31.0 |
Conclusions
Some stereoelectronic properties of the hybrid ligands PCNHCP and Dipp-PCNHC have been evaluated. Initial attempts to prepare cis-[RhCl(CO)2(PCNHCP-κCNHC)] by the reaction of PCNHCP with [Rh(μ-Cl)(COD)]2 or [Rh(μ-Cl)(CO)2]2 led instead to the formation of rhodium complexes with a chelating PCNHCP,κP,κCNHC or PCNHC,κP,κCNHC,κN ligand, and the additional coordination of the phosphine group hampered the evaluation of the TEP of the sole CNHC donor. However, the π-accepting properties of PCNHCP and Dipp-PCNHC were determined from the 77Se NMR chemical shift of the corresponding selenium adducts. The σ-donor ability of the carbene donor in PCNHCP was confirmed by the isolation of two stable donor–acceptor adducts with borane or boroxine. The free carbene donor of PCNHCP shows the largest value reported for percent buried volume in NHC ligands in the conformation of the free PCNHCP established crystallographically. Currently, it appears that a coordination mode through the CNHC donor (κCNHC) is more common with 3d metals.Experimental section
Synthesis and characterisation
20 and [Fe4Cl8(THF)6]21 were prepared according to the literature.
X-ray crystallography
Summary of the crystal data, data collection and refinement for the structures of PCP-RhCl·2CH2Cl2, PCNHC-RhCO, PCNHCP-Fe, PCNHCP-Se, PCNHCP-B-1·THF and PCNHCP-B-2·toluene are given in Table S1.†For PCNHCP-RhCl·2CH2Cl2, PCNHCP-Fe, PCNHCP-Se, PCNHCP-B-1·THF and PCNHCP-B-2·toluene, X-ray diffraction data collection was carried out on a Bruker APEX II DUO Kappa-CCD diffractometer equipped with an Oxford Cryosystem liquid N2 device, using Mo-Kα radiation (λ = 0.71073 Å). The crystal-detector distance was 38 mm. The cell parameters were determined (APEX2 software)22 from reflections taken from three sets of 12 frames, each at 10s exposure. The structure was solved by direct methods using the program SHELXS-97.23 The refinement and all further calculations were carried out using SHELXL-97.24 The H-atoms were included in calculated positions and treated as riding atoms using SHELXL default parameters. The non-H atoms were refined anisotropically, using weighted full-matrix least-squares on F2. A semi-empirical absorption correction was applied using SADABS in APEX2.22
For PCNHC-RhCO, X-ray diffraction data collection was carried out on a Nonius Kappa-CCD diffractometer equipped with an Oxford Cryosystem liquid N2 device, using Mo-Kα radiation (λ = 0.71073 Å). The crystal-detector distance was 36 mm. The cell parameters were determined (Denzo software)25 from reflections taken from one set of 10 frames (1.0° steps in phi angle), each at 20 s exposure. The structures were solved by direct methods using the program SHELXS-97.23 The refinement and all further calculations were carried out using SHELXL-97.24 The H-atoms were included in calculated positions and treated as riding atoms using SHELXL default parameters. The non-H atoms were refined anisotropically, using weighted full-matrix least-squares on F2. A semi-empirical absorption correction was applied using MULscanABS in PLATON.26
Conflict of interest
The authors declare no competing financial interest.Acknowledgements
The USIAS, CNRS, Région Alsace and Communauté Urbaine de Strasbourg are acknowledged for the award of fellowships and a Gutenberg Excellence Chair (2010–11) to AAD. We thank the CNRS and the MESR (Paris) for funding and Dr L. Karmazin and Miss C. Bailly (Service de Radiocristallographie, UdS) for the determination of the crystal structures. We are grateful to the China Scholarship Council for a PhD grant to P.A.References
- (a) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290 CrossRef CAS; (b) C. M. Crudden and D. P. Allen, Coord. Chem. Rev., 2004, 248, 2247 CrossRef CAS; (c) E. Peris and R. H. Crabtree, Coord. Chem. Rev., 2004, 248, 2239 CrossRef CAS; (d) F. Glorius, N-Heterocyclic Carbenes in Transition Metal Catalysis, Springer, Berlin Heidelberg, 2007 Search PubMed; (e) D. Pugh and A. A. Danopoulos, Coord. Chem. Rev., 2007, 251, 610 CrossRef CAS; (f) E. A. B. Kantchev, C. J. O'Brien and M. G. Organ, Angew. Chem., Int. Ed., 2007, 46, 2768 CrossRef CAS PubMed; (g) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612 CrossRef PubMed; (h) D. McGuinness, Dalton Trans., 2009, 6915 RSC; (i) K. Riener, S. Haslinger, A. Raba, M. P. Högerl, M. Cokoja, W. A. Herrmann and F. E. Kühn, Chem. Rev., 2014, 114, 5215 CrossRef CAS PubMed; (j) R. Visbal and M. C. Gimeno, Chem. Soc. Rev., 2014, 43, 3551 RSC; (k) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef CAS PubMed; (l) Z. Wang, L. Jiang, D. K. B. Mohamed, J. Zhao and T. S. A. Hor, Coord. Chem. Rev., 2015, 293–294, 292 CrossRef CAS; (m) D. Zhang and G. Zi, Chem. Soc. Rev., 2015, 44, 1898 RSC.
- C. A. Tolman, Chem. Rev., 1977, 77, 313 CrossRef CAS.
- D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723 RSC.
- O. Back, M. Henry-Ellinger, C. D. Martin, D. Martin and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 2939 CrossRef CAS PubMed.
- (a) A. Liske, K. Verlinden, H. Buhl, K. Schaper and C. Ganter, Organometallics, 2013, 32, 5269 CrossRef CAS; (b) K. Verlinden, H. Buhl, W. Frank and C. Ganter, Eur. J. Inorg. Chem., 2015, 2015, 2416 CrossRef CAS.
- (a) A. P. Marchenko, H. N. Koidan, A. N. Huryeva, E. V. Zarudnitskii, A. A. Yurchenko and A. N. Kostyuk, J. Org. Chem., 2010, 75, 7141 CrossRef CAS PubMed; (b) A. P. Marchenko, H. N. Koidan, I. I. Pervak, A. N. Huryeva, E. V. Zarudnitskii, A. A. Tolmachev and A. N. Kostyuk, Tetrahedron Lett., 2012, 53, 494 CrossRef CAS; (c) A. P. Marchenko, H. N. Koidan, A. N. Hurieva, I. I. Pervak, S. V. Shishkina, O. V. Shishkin and A. N. Kostyuk, Eur. J. Org. Chem., 2012, 4018 CrossRef CAS; (d) A. P. Marchenko, H. N. Koidan, E. V. Zarudnitskii, A. N. Hurieva, A. A. Kirilchuk, A. A. Yurchenko, A. Biffis and A. N. Kostyuk, Organometallics, 2012, 31, 8257 CrossRef CAS; (e) A. Marchenko, H. Koidan, A. Hurieva, O. Kurpiieva, Y. Vlasenko, A. Kostyuk, C. Tubaro, A. Lenarda, A. Biffis and C. Graiff, J. Organomet. Chem., 2014, 771, 14 CrossRef CAS; (f) P. Nägele, U. Herrlich, F. Rominger and P. Hofmann, Organometallics, 2013, 32, 181 CrossRef; (g) T. Wang and D. W. Stephan, Chem. – Eur. J., 2014, 20, 3036 CrossRef CAS PubMed; (h) P. Ai, A. A. Danopoulos, P. Braunstein and K. Y. Monakhov, Chem. Commun., 2014, 50, 103 RSC; (i) P. Ai, A. A. Danopoulos and P. Braunstein, Inorg. Chem., 2015, 54, 3722 CrossRef CAS PubMed.
- (a) A. P. Marchenko, H. N. Koidan, A. N. Hurieva, O. V. Gutov, A. N. Kostyuk, C. Tubaro, S. Lollo, A. Lanza, F. Nestola and A. Biffis, Organometallics, 2013, 32, 718 CrossRef CAS; (b) C. C. Brown, P. N. Plessow, F. Rominger, M. Limbach and P. Hofmann, Organometallics, 2014, 33, 6754 CrossRef CAS; (c) E. Kühnel, I. V. Shishkov, F. Rominger, T. Oeser and P. Hofmann, Organometallics, 2012, 31, 8000 CrossRef.
- (a) P. Ai, C. Gourlaouen, A. A. Danopoulos and P. Braunstein, Inorg. Chem., 2016, 55, 1219 CrossRef CAS PubMed; (b) P. Ai, M. Mauro, L. De Cola, A. A. Danopoulos and P. Braunstein, Angew. Chem., Int. Ed., 2016 DOI:10.1002/anie.201510150.
- P. Ai, A. A. Danopoulos and P. Braunstein, Organometallics, 2015, 34, 4109 CrossRef CAS.
- F. He, L. Ruhlmann, J.-P. Gisselbrecht, S. Choua, M. Orio, M. Wesolek, A. A. Danopoulos and P. Braunstein, Dalton Trans., 2015, 44, 17030 RSC.
- A. Massard, P. Braunstein, A. A. Danopoulos, S. Choua and P. Rabu, Organometallics, 2015, 34, 2429 CrossRef CAS.
- (a) G. Roy, M. Nethaji and G. Mugesh, J. Am. Chem. Soc., 2004, 126, 2712 CrossRef CAS PubMed; (b) G. Roy and G. Mugesh, J. Am. Chem. Soc., 2005, 127, 15207 CrossRef CAS PubMed; (c) G. Roy, P. N. Jayaram and G. Mugesh, Chem. – Asian J., 2013, 8, 1910 CrossRef CAS PubMed; (d) L. Pauling, The Nature of the Chemical Bond, Cornell University Press, New York, 3rd edn, 1960 Search PubMed.
- D. J. Nelson, F. Nahra, S. R. Patrick, D. B. Cordes, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2014, 33, 3640 CrossRef CAS.
- (a) P. A. Chase and D. W. Stephan, Angew. Chem., Int. Ed., 2008, 47, 7433 CrossRef CAS PubMed; (b) D. Holschumacher, T. Bannenberg, C. G. Hrib, P. G. Jones and M. Tamm, Angew. Chem., Int. Ed., 2008, 47, 7428 CrossRef CAS PubMed; (c) D. Holschumacher, C. Taouss, T. Bannenberg, C. G. Hrib, C. G. Daniliuc, P. G. Jones and M. Tamm, Dalton Trans., 2009, 6927 RSC; (d) S. Kronig, E. Theuergarten, D. Holschumacher, T. Bannenberg, C. G. Daniliuc, P. G. Jones and M. Tamm, Inorg. Chem., 2011, 50, 7344 CrossRef CAS PubMed; (e) E. L. Kolychev, T. Bannenberg, M. Freytag, C. G. Daniliuc, P. G. Jones and M. Tamm, Chem. – Eur. J., 2012, 18, 16938 CrossRef CAS PubMed.
- A. D. Phillips and P. P. Power, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2005, 61, o291 Search PubMed.
- (a) A. C. Hillier, W. J. Sommer, B. S. Yong, J. L. Petersen, L. Cavallo and S. P. Nolan, Organometallics, 2003, 22, 4322 CrossRef CAS; (b) A. Poater, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, Eur. J. Inorg. Chem., 2009, 2009, 1759 CrossRef.
- H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841 RSC.
- G. Giordano, R. H. Crabtree, R. M. Heintz, D. Forster and D. E. Morris, Inorg. Synth., 2007, 19, 88 Search PubMed.
- J. A. McCleverty, G. Wilkinson, L. G. Lipson, M. L. Maddox and H. D. Kaesz, Inorg. Synth., 1966, 8, 84 CrossRef.
- H. J. Frohn, N. Y. Adonin, V. V. Bardin and V. F. Starichenko, Z. Anorg. Allg. Chem., 2002, 628, 2827 CrossRef CAS.
- F. A. Cotton, R. L. Luck and K.-A. Son, Inorg. Chim. Acta, 1991, 179, 11 CrossRef CAS.
- Bruker AXS Inc, Madison USA, 2006.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1990, 46, 467 CrossRef.
- G. M. Sheldrick, Universität Göttingen, Göttingen Germany, 1999.
- B. V. Nonius, Delft, The Netherlands, 1997.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal data for PCP-RhCl·2CH2Cl2, PCNHC-RhCO, PCNHCP-Fe, PCNHCP-Se, PCNHCP-B1·THF and PCNHCP-B2·toluene. CCDC 1449234–1449239. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt00318d |
| This journal is © The Royal Society of Chemistry 2016 |