
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diphosphane 2,2′-binaphtho[1,8-de][1,3,2]dithiaphosphinine and the easy formation of a stable phosphorus radical cation†
Christin
Kirst
a,
Bela E.
Bode
b,
David B.
Cordes
b,
Phillip S.
Nejman
b,
Alexandra M. Z.
Slawin
b,
Konstantin
Karaghiosoff
*a and
J. Derek
Woollins
*b
aDepartment of Chemistry, Ludwig-Maximilians-University of Munich, Butenandtstr. 5-13 (D), 81377 Munich, Germany. E-mail: konstantin.karaghiosoff@cup.uni-muenchen.de
bEastCHEM School of Chemistry, University of St. Andrews, Fife, KY16 9ST, UK. E-mail: jdw3@st-andrews.ac.uk
First published on 14th March 2016
Abstract
A convenient synthesis route to 2,2′-binaphtho[1,8-de][1,3,2]di-thiaphosphinine (3) was found. Its stable radical cation 3˙+ was accessed easily through one-electron oxidation with NOBF4.
Main group radicals have been known for a long time, though those of the heavier group atoms have only been characterised in the 1970s.1 Having a lone pair and an unbound electron located at the phosphorus atom make phosphinyl radicals [R2P]˙ an interesting and reactive intermediate in phosphorus chemistry,2 useful in the addition of phosphines to olefins for example.3 Recently, persistent phosphinyl radicals have been published, either stabilised by kinetic or even resonance effects.4–12 Phosphinyl radicals can either be pure σ-radicals, with the unpaired electron being localised at the phosphorus atom, or π-based systems, localising the spin density over several nuclei.
Computational studies and EPR on these molecules can give more insight into the electronic structure and bonding situation of these compounds. A few of these π-delocalised and pure σ-radicals phosphorus radicals have been isolated and structurally characterised.1,5–13
It is also known, that dissociation of phosphorus dimers (R2PPR2) can be driven by release of steric strain between bulky substituents and thus favour the existence of the monomer.5–9 It is also possible to form diphosphene radical cations (R2PPR2)+˙, which can be stabilised by large carbenes, through one-electron oxidation of their dimer.14
Herein, we report the synthesis of the new diphosphane 2,2′-binaphtho[1,8-de][1,3,2]dithiaphosphinine (3) and an easy way to form the radical cation thereof by reaction of the diphosphane (3) with NOBF4. The products were characterised by mass spectrometry, elemental analysis and by single crystal X-ray diffraction. EPR studies and computational calculations were undertaken on 3 and its radical cation 3˙+.
We found that the direct reaction of lithiated naphtho[1,8-cd]-1,2-dithiole with PCl3 in THF does not lead to the preferred compound 2, thus lithiated naphtho[1,8-cd]-1,2-dithiole was reacted with one equivalent of (diethylamino)dichlorophosphine giving 2-(diethylamino)-naphtho[1,8-de][1,3,2]dithiaphosphinine (1) (95% yield).
In the next step the diethylamine group was removed by reacting 1 with PCl3 in THF to form 2-chloronaphtho[1,8-de][1,3,2]dithiaphosphinine (2) (93% yield).152 was then reacted with magnesium in THF under reflux.16
After workup, 2,2′-binaphtho[1,8-de][1,3,2]dithiaphosphinine (3) was obtained in a moderate yield (20%). The complete synthesis route is shown in Scheme 1.
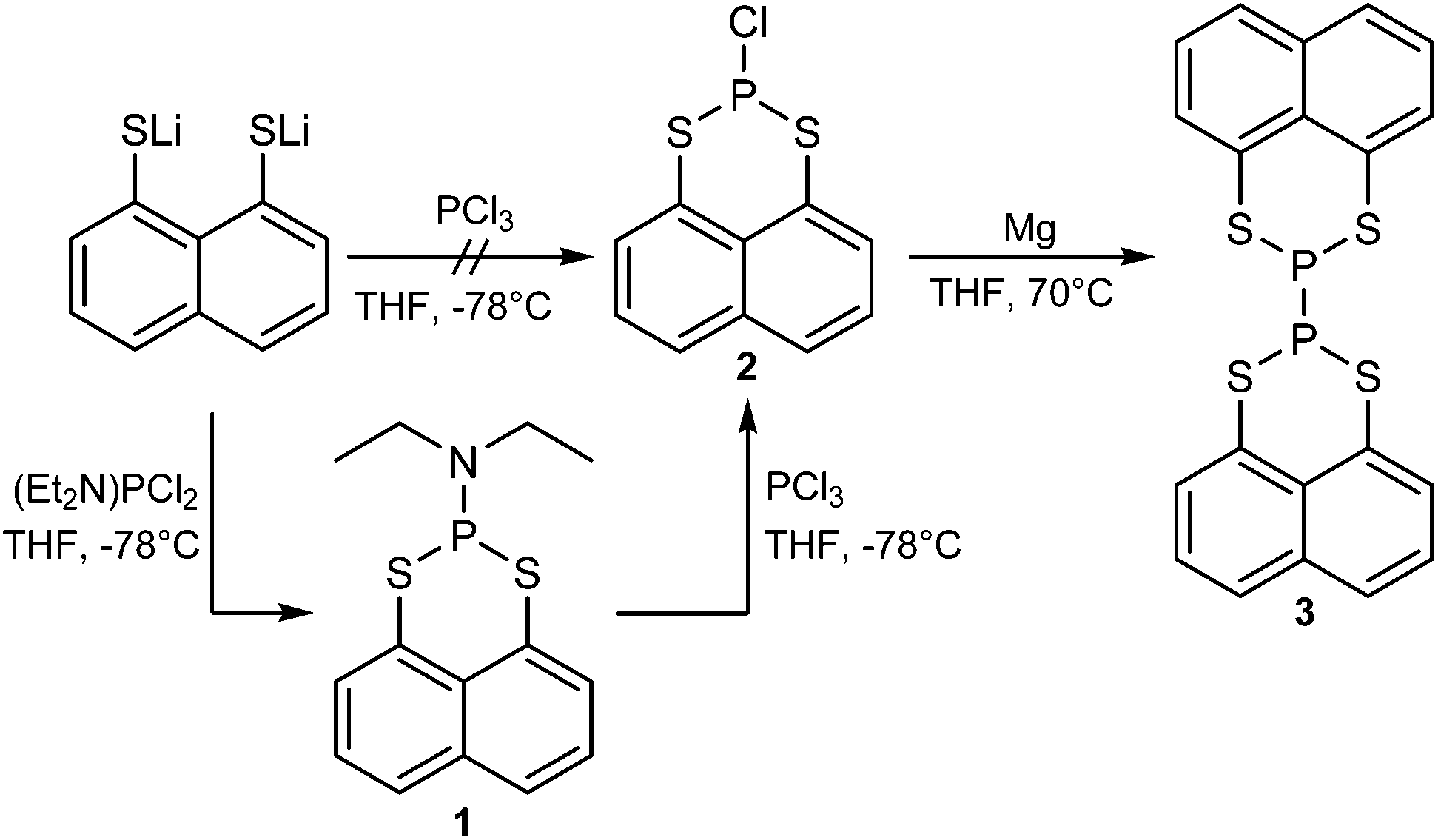 | ||
| Scheme 1 Synthesis route to 2,2′-binaphtho[1,8-de][1,3,2]di-thiaphosphinine (3). | ||
The intermediate 2-(diethylamino)naphtho[1,8-de][1,3,2]di-thiaphosphinine (1) crystallises in the orthorhombic space group P212121 with four formula units per unit cell (see Table A1, ESI†). In the 31P NMR spectrum 1 shows a signal at δ = 69.0 ppm. The APCI MS spectrum of 1 exhibits a molecular ion peak with the correct isotropic distribution pattern ([M + H]+ = 294).
We were unable to crystallise the 2-chloronaphtho[1,8-de][1,3,2]dithiaphosphinine (2) intermediate though the APCI MS spectrum exhibits a molecular ion peak with the correct isotropic distribution pattern ([M]+ = 256) and elemental analysis results agree with the C10H6ClPS2 formula of 2. The 31P NMR chemical shift of 2 (72.4 ppm) is low field shifted by only three ppm when compared to the precursor 1 (69.0 ppm). In the 1H NMR spectrum, only three signals in the aromatic region were observed and none in the alkyl group region, further proving the removal of the diethylamine group on the phosphorus atom.
The title compound 2,2′-binaphtho[1,8-de][1,3,2]dithiaphosphinine (3) crystallised in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with one formula unit per unit cell (see Table A1, ESI†). The APCI MS spectrum exhibits a molecular ion peak with the correct isotropic distribution pattern ([M + H]+ = 442). In the 31P NMR spectrum compound 3 shows a signal at δ = −15.1 ppm. The solid state structure of 1 and 3 are depicted in Fig. 1 and selected bond lengths and angles are listed in the caption of Fig. 1. The P–P bond distance in 3 (2.2306(13) Å) is indicative of a P–P single bond.17 The lengthening of the P–P bond adds to other structural distortions in order to minimise the steric strain between the C3S2 ring units and the naphthalene ring. Most obvious are the puckering of the C3S2P ring. The S⋯S peri-distance is 3.1972(13) Å which is longer than in normal chalcogenide substituted naphthalenes.18
with one formula unit per unit cell (see Table A1, ESI†). The APCI MS spectrum exhibits a molecular ion peak with the correct isotropic distribution pattern ([M + H]+ = 442). In the 31P NMR spectrum compound 3 shows a signal at δ = −15.1 ppm. The solid state structure of 1 and 3 are depicted in Fig. 1 and selected bond lengths and angles are listed in the caption of Fig. 1. The P–P bond distance in 3 (2.2306(13) Å) is indicative of a P–P single bond.17 The lengthening of the P–P bond adds to other structural distortions in order to minimise the steric strain between the C3S2 ring units and the naphthalene ring. Most obvious are the puckering of the C3S2P ring. The S⋯S peri-distance is 3.1972(13) Å which is longer than in normal chalcogenide substituted naphthalenes.18
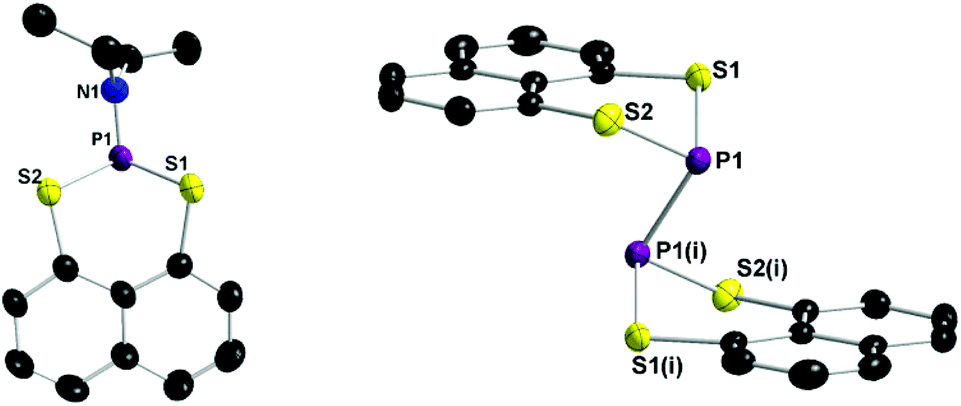 | ||
| Fig. 1 Solid-state molecular structures of 1 and 3. Thermal ellipsoids are set at 50% probability. H-atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): 1 P1–N1 1.652(6), P1–S1 2.122(2), P1–S2 2.108(2), S1–C1 1.789(6), S2–C8 1.769(6), N1–C9 1.482(9), N1–C11 1.465(8), S1⋯S2 3.1663(22), N1–P1–S1 103.8(2), N1–P1–S2 99.7(2), S1–P1–S2 96.91(9), P1–N1–C9 116.8(5), P1–N1–C11 125.6(4), P1–S1–C1 99.6(2), P1–S2–C8 101.1(2), C9–N1–C11 115.8(5); 3 P1–P1(i) 2.2306(13), P1–S1 2.0924(11), P1–S2 2.0903(11), S1–C1 1.782(2), S2–C8 1.770(2), S1⋯S2 3.1972(13), S1–P1–S2 99.70(6), P1–S1–C1 105.75(8), P1–S2–C8 106.53(8), S1–P1–P1(i) 99.48(5), S2–P1–P1(i) 99.53(5). Symmetry code: (i) −x, −y, 1 − z. | ||
Due to the presence of the P–P bond, 3 is EPR silent in toluene at room temperature, at 383 K,13 and even at 400 K in xylene though a change of colour of the solution from a pale yellow to yellow was observed. In contrast to the P–P dimer published by Edge et al.,9 the dimer (3) has a stronger P–P bond. The radical itself might not be as stabilised by the sulphur atoms in 3, which can favour the dimer over the monomer.
Yet in the APCI MS spectrum, which was done at min. 674 K, the molecular ion peak for the monomer of 3 was visible ([M/2]+ = 221).
Nevertheless, it was possible to create the stable radical cation 3˙+ by reacting 3 with 1.5 eq. equivalents of NOBF4 in DCM and prove its existence by mass spectrometry, IR, UV Vis and EPR at 80 K (Fig. 2).19 The positive ion ESI MS spectrum of 3˙+ exhibits a molecular ion peak with the correct isotropic distribution pattern ([M + 3H]+ = 445). In the IR spectrum the FBF deformation vibration is visible as a very strong signal at δas(FBF) = 1057 and the BF4 stretch vibration is visible as a medium signal at νas(BF4) = 528 which goes hand in hand with reported signals in the literature of the [Co(NH3)6](BF4)2 complex and NaBF4.20,21 The vibration at νas(BF4) = 528 is split-up into multiple peaks just like in NaBF4. The symmetry of the anion BF4− in 3˙+ differs from the expected Td-symmetry explaining the splitting.21
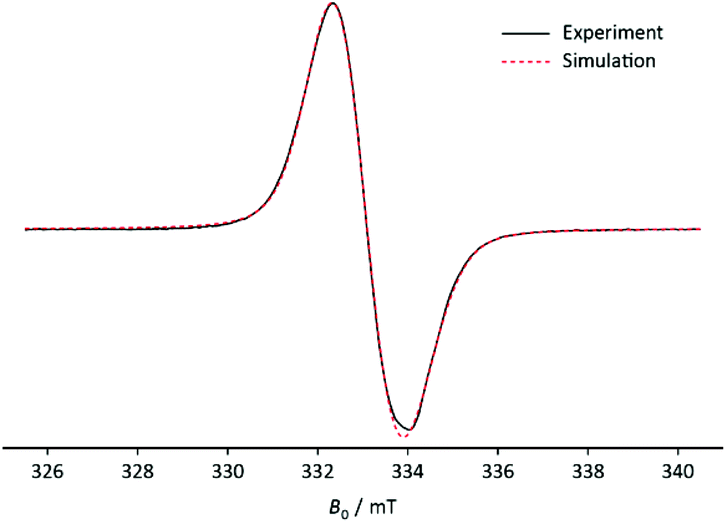 | ||
| Fig. 2 EPR spectrum of 3+˙ at 80 K (black) and the simulated spectrum at RT (red). Frequency: 9.3460 GHz; amplitude modulation: 0.05 mT. | ||
When reacting 3 with NOBF4 in DCM the solution turns orange and a very dark green/black precipitate forms. When measuring the EPR spectrum of the reaction mixture, the question arose, whether the signal came from the solution or from the precipitate. The result was that the latter was the case. The resulting EPR spectrum of the precipitate 3˙+ recorded at 80 K shows an intense and broad singlet (g = 2.007(3)). No hyperfine couplings were visible due to broad line width giving an upper limit of a(31P) = 1.7 mT. The small phosphorus coupling suggests that the unpaired electron is localised on the p-orbital.
Density functional theory (DFT) calculations were performed to gain insight into the nature of the frontier orbitals and to calculate the UV-Vis spectrum using the Gaussian 09 program package (see ESI† for details).22 Full geometry optimisations on the dimer 3 and the radical cation 3˙+ were performed at the B3LYP/6-31G(d) and UB3LYP/6-31G+(d) level, the obtained stationary points were confirmed by frequency calculations. Molecular orbitals, NBO values and spin densities were calculated using the same basis sets. The UV-Vis spectrum was calculated using TD-UB3LYP/6-31G+(d).
The bond order of 0.89 of the P–P bond in 3 indicates a relatively strong single bond, which explains why it is so difficult to split the bond through heating at high temperatures. The P–P bond in 3 and 3˙+ is a σ-bond combined with a trans-cofacial π*–π*-interaction similar to a 2c–2e− bond involving the p-orbitals of both phosphorus atoms explaining the geometry of the structure of 3 and 3˙+ (Fig. 3). In the corresponding HOMO−2(α) of 3˙+ an increase of electron density is visible in the p-orbitals of the P–P bond and a decrease in the p-orbitals of the lone pairs when compared to the HOMO of 3. The gain of electron density of the p-orbital of the P–P bond in 3˙+ further suggests that the unpaired electron is delocalised over the bond.
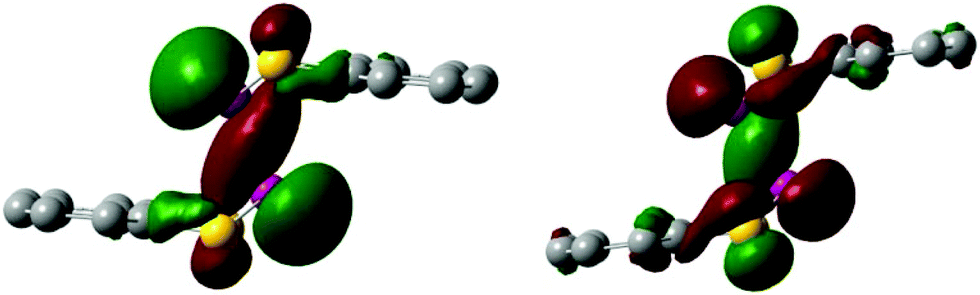 | ||
| Fig. 3 HOMO of the dimer 3 (left) and HOMO−2(α) of the radical cation 3˙+ (right). Isovalue set at 0.03. H-atoms are omitted for clarity. | ||
Attempts to recrystallize the radical salt from thf or acetonitrile or obtain a satisfactory elemental analysis proved unsuccessful because of sample decomposition though the sample appears thermally stable for days under nitrogen. The experimental powder UV Vis spectrum of the radical cation 3˙+ shows three absorption maxima in the visible spectral region, at λ = 876.9; 559.1 and 426.2 nm. These absorption maxima go hand in hand with the visually observed colour of a very dark green/black solid of the radical cation 3˙+.
TD-DFT calculations were performed on the optimised structure of the radical cation 3˙+. They result in three permitted transitions, at λ = 818.0; 612.6 and 486.1 nm, in the visible spectral region, which are in good agreement with the experiment (see ESI† for details). The longest-wave transition corresponds to a π → π*-transition from the HOMO−2(β) to LUMO(β). The transition at 612.6 nm describes the excitation of an electron from the lone pair of a sulfur atom, in detail a n → π*-transition from the HOMO−7(β) to the LUMO(β). The last one at 426.2 nm is the transition from the HOMO−1(β) to the LUMO+2(β) and from the HOMO(α) to the LUMO(α). The calculated UV Vis data explains the dark colour of the radical cation 3˙+, as well.
In conclusion we have developed a convenient synthesis to form a P–P dimer 3, including its structural characterisation. The radical cation 3˙+ was successfully formed by the easy one-electron oxidation in solution and could be isolated as a solid. The IR, MS and UV Vis results of 3˙+ indicate the success of the reaction and the corresponding DFT calculations circumstantiate these. The importance of radicals of heavier main-group elements means that the isolation of similar compounds to 3 can be expected using the same method as well as the exploration of the reactivities of their radicals and radical cations.
Notes and references
- P. P. Power, Chem. Rev., 2003, 103, 789 CrossRef CAS PubMed.
- S. Marque and P. Tordo, Top. Curr. Chem., 2005, 250, 43 CAS.
- D. Leca, L. Fensterbank, E. Lacúte and M. Malacria, Chem. Soc. Rev., 2005, 34, 858 RSC.
- P. Agarwal, N. A. Piro, K. Meyer, P. Mìller and C. C. Cummins, Angew. Chem., Int. Ed., 2007, 46, 3111 ( Angew. Chem. , 2007 , 119 , 3171 ) CrossRef CAS PubMed.
- O. Back, M. A. Celik, G. Frenking, M. Melaimi, B. Donnadieu and G. Bertrand, J. Am. Chem. Soc., 2010, 132, 10262 CrossRef CAS PubMed.
- O. Back, B. Donnadieu, P. Parameswaran, G. Frenking and G. Bertrand, Nat. Chem., 2010, 2, 369 CrossRef CAS PubMed.
- O. Back, B. Donnadieu, M. von Hopffgarten, S. Klein, R. Tonner, G. Frenking and G. Bertrand, Chem. Sci., 2011, 2, 858 RSC.
- A. Dumitrescu, V. L. Rudzevich, V. D. Romanenko, A. Mari, W. W. Schoeller, D. Bourissou and G. Bertrand, Inorg. Chem., 2004, 43, 6546 CrossRef CAS PubMed.
- R. Edge, R. J. Less, E. J. L. McInnes, K. Müther, V. Naseri, J. M. Rawson and D. S. Wright, Chem. Commun., 2009, 1691 RSC.
- S. L. Hinchley, C. A. Morrison, D. W. H. Rankin, C. L. B. Macdonald, R. J. Wiacek, A. H. Cowley, M. F. Lappert, G. Gundersen, J. A. C. Clyburnee and P. P. Power, Chem. Commun., 2000, 2045 RSC.
- S. Ito, M. Kikuchi, M. Yoshifuji, A. J. Arduengo, T. A. Konovalova and L. D. Kispert, Angew. Chem., Int. Ed., 2006, 45, 4341 ( Angew. Chem. , 2006 , 118 , 4447 ) CrossRef CAS PubMed.
- T. Sasamori, E. Mieda, N. Nagahora, K. Sato, D. Shiomi, T. Takui, Y. Hosoi, Y. Furukawa, N. Takagi and S. Nagase, et al. , J. Am. Chem. Soc., 2006, 128, 12582 CrossRef CAS PubMed.
- M. Scheer, C. Kuntz, M. Stubenhofer, M. Linseis, R. F. Winter and M. Sierka, Angew. Chem., Int. Ed., 2009, 48, 2600 ( Angew. Chem. , 2009 , 121 , 2638 ) CrossRef CAS PubMed.
- (a) O. Back, B. Donnadieu, P. Parameswaran, G. Frenking and G. Bertrand, Nat. Chem., 2010, 2, 369 CrossRef CAS PubMed; (b) R. Kinjo, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 5930 CrossRef CAS PubMed; (c) S. H. Strauss, Chem. Rev., 1993, 93, 927 CrossRef CAS; (d) C. Reed, Acc. Chem. Res., 1998, 31, 133 CrossRef CAS; (e) I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066 CrossRef CAS PubMed; (f) X. Pan, X. Chen, T. Li, Y. Li and X. Wang, J. Am. Chem. Soc., 2013, 135, 3414 CrossRef CAS PubMed; (g) X. Pan, Y. Su, X. Chen, Y. Zhao, Y. Li, J. Zuo and X. Wang, J. Am. Chem. Soc., 2013, 135, 5561 CrossRef CAS PubMed.
- D. W. Netzbandt, Aktivierung von Phosphanen durch Donor/Akzeptor Wechselwirkungen Untersuchungen zur Synthese und Struktur, dissertation, Rheinische Friedrich-Wilhelms-Universität, Bonn, 2008 Search PubMed.
- W. A. Henderson, M. Epstein and F. S. Seichter, J. Am. Chem. Soc., 1963, 5, 2462 CrossRef.
- A. F. Holleman, E. Wiberg and N. Wiberg, Lehrbuch der Anorganischen Chemie, Walter de Gruyter, Berlin, 102nd edn, 2007 Search PubMed.
- S. D. Robertson, A. M. Z. Slawin and J. D. Woollins, Heteroat. Chem., 2004, 15, 530 CrossRef.
- F. R. Knight, R. A. M. Randall, T. L. Roemmele, R. T. Boeré, B. E. Bode, L. Crawford, M. Bühl, A. M. Z. Slawin and J. D. Woollins, ChemPhysChem, 2013, 14, 3199 CrossRef CAS PubMed.
- S. Kummer and D. Babel, Z. Naturforsch., B: Anorg. Chem. Org. Chem., 1984, 39, 1118 Search PubMed.
- J. Weidlein, U. Müller and K. Dehnicke, Schwingungsfrequenzen I, Thieme Verlag, Stuttgart, 1981 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Norm, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, single crystal X-ray diffraction data, UV-Vis, EPR data and DFT calculation details. CCDC 1439936 and 1439937. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt00304d |
| This journal is © The Royal Society of Chemistry 2016 |