
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePincer-CNC mononuclear, dinuclear and heterodinuclear Au(III) and Pt(II) complexes supported by mono- and poly-N-heterocyclic carbenes: synthesis and photophysical properties†
S.
Gonell‡
,
M.
Poyatos
* and
E.
Peris
*
Institute of Advanced Materials (INAM), Universitat Jaume I, Av. Vicente Sos Baynat s/n, 12071-Castellón, Spain. E-mail: eperis@uji.es; poyatosd@uji.es
First published on 11th February 2016
Abstract
A family of cyclometallated Au(III) and Pt(II) complexes containing a CNC-pincer ligand (CNC = 2,6-diphenylpyridine) supported by pyrene-based mono- or bis-NHC ligands have been synthesized and characterized, together with the preparation of a Pt–Au hetero-dimetallic complex based on a Y-shaped tris-NHC ligand. The photophysical properties of all the new species and of two related Ru(II)–arene complexes were studied and compared. Whereas the pyrene-based complexes only exhibit emission in solution, those containing the Y-shaped tris-NHC ligand are only luminescent when dispersed in poly(methyl methacrylate) (PMMA). In particular, the pyrene-based complexes were found to be emissive in the range of 373–440 nm, with quantum yields ranging from 3.1 to 6.3%, and their emission spectra were found to be almost superimposable, pointing to the fluorescent pyrene-centered nature of the emission. This observation suggests that the emission properties of the pyrene fragment may be combined with some of the numerous applications of NHCs as supporting ligands allowing, for instance, the design of biological luminescent agents.
Introduction
During the last decade, the use of poly-N-heterocyclic carbene ligands has given access to a plethora of new polymetallic complexes whose properties often differ from their more closely related monometallic analogues.1 While most of the applications of these polymetallic species have been explored in the field of homogeneous catalysis, where the presence of two or more metals has given clear evidence of activity improvement,2 little has been done in other fields, such as the study of their potential photophysical applications, where the presence of two or more metals may also provide some benefits. Since the invention of luminescent organic devices in 1995,3 there has been increasing interest in the development of materials with emissive properties, mostly due to the current interest in the preparation of new optical devices and organic light emitting diodes (OLEDs). Most organic light emitters only show fluorescence,4 and if triplet excitons are formed, they are often deactivated by thermal processes. A breakthrough in the preparation of this type of materials was the incorporation of transition metal complexes in 1998,5 because the strong spin–orbit couplings introduced by the metal fragment allow fast energy transfer from the singlet to the triplet state to occur,6 thus improving the efficiency of the OLED material. Although many transition metals have been investigated as emitting systems, iridium(III) and platinum(II) are those that have provided the best results thus far,7 but ruthenium8 and gold9 have already shown very promising properties.10 Normally, the presence of a chelating group is desirable for the preparation of metal complexes with good emitting properties, and the presence of an organic chromophore is usually required to impart the desired luminescence. For the choice of the cyclometallated ligands those featuring a combination of aryl and N-heterocyclic rings that bind through a partnership of C and N atoms are those that have been most extensively used. The incorporation of N-heterocyclic carbene ligands (NHCs) into metal-based luminescent materials has also provided significant benefits,10 because NHCs are strong-field ligands that push the dσ* orbitals to higher energy levels, thus non-radiative deactivation from these states may be inhibited to an important extent, an effect that is particularly important for highly electrophilic metals like Au(III).11 Besides, NHCs provide stable complexes that facilitate the processability of the resulting emissive materials. In addition, pyrene has become one of the most widely studied organic materials in the field of photochemistry and photophysics,12 and for this reason it has been the chromophore of choice in the design of many coordination complexes used in fields such as optoelectronics, chemical sensors and even photodynamic therapeutic agents.13We recently reported the photophysical properties of a series of pyrene-containing azoliums and bisazoliums that showed emissions in the range of 370–420 nm, and quantum yields ranging from 0.29–0.75,14 although their coordination to rhodium(I), iridium(I) and platinum(II) to afford the related NHC complexes provided materials with negligible emissive properties due to non-radiative deactivation.
Based on these preliminary results, herein we describe the preparation of new platinum(II) and gold(III) complexes containing a pyrene-bis-N-heterocyclic carbene ligand and a CNC-pincer ligand (CNC = 2,6-diphenylpyridine).15 With the preparation of these complexes, we try to marry the benefits provided by the combined use of tridentate CNC ligands, NHCs and metal centers that can afford high emissive efficiencies. We also report the preparation of a Pt(II)–Au(III) hetero-dimetallic complex based on a Y-shaped tris-NHC ligand.16 We considered this ligand an excellent support for the preparation of NHC-based mixed-metal complexes,2 which may benefit from the combined properties of the metal fragments comprised in the heterometallic unit. An interesting example describing the preparation of a Au(I)–Ru(II) heterometallic complex was recently reported,17 in which the photophysical properties provided by the ruthenium fragment are combined with the biological activity afforded by the gold unit.
Results and discussion
Synthesis and characterization
As depicted in Scheme 1, the mono-gold complex 1 was obtained by reaction of the pyrene-imidazolium iodide A with [Au(CNC)Cl] (CNC = 2,6-diphenylpyridine) in refluxing acetonitrile in the presence of NaOAc in high yield (90%). The reaction of A with [Pt(CNC)(DMSO)] in THF in the presence of KOtBu led to the formation of the platinum complex 2 in 69% yield. | ||
| Scheme 1 Synthesis of complexes 1 and 2. | ||
By following a similar protocol, but starting from the pyrene-bis-imidazolium salt B, the related di-gold and di-platinum complexes 3 and 4 were obtained in 81 and 77% yield, respectively (Scheme 2). The new complexes were characterized by NMR spectroscopy, mass spectrometry and elemental analysis. The 1H NMR spectra of 1 and 2 confirm the twofold symmetry of both molecules due to the presence of a mirror plane, as exemplified by the equivalency of the protons of the methyl groups of the tert-butyl substituents, and the three resonances assigned to the six protons of the pyrene. The 1H NMR spectra of 3 and 4 are fully consistent with the pseudo-D2h symmetry of the complexes, as illustrated by the equivalence of the aromatic protons of the pyrene and of the resonances corresponding to the CNC ligands. The 13C NMR spectra of the complexes show the diagnostic signal assigned to the metallated carbene carbons, which appear at virtually identical chemical shifts for the two gold (164.0 ppm in 1, 163.9 ppm in 3) and platinum (179.5 ppm in 2, 181.1 ppm in 4) complexes.
 | ||
| Scheme 2 Synthesis of complexes 3 and 4. | ||
The molecular structure of the di-platinum complex 4 was unambiguously confirmed by X-ray diffraction studies (Fig. 1). The molecule consists of two Pt(CNC) fragments, bound by the pyrene-bis-NHC ligand. The CNC bite angle is 161.2(2)°. The coordination planes of the two metal fragments are parallel, and deviate from the plane established by the pyrene unit by 69.46(15)°. The distance between the two metal atoms is 13.12 Å, and the Pt–Ccarbene bond distance is 1.972(5) Å.
 | ||
| Fig. 1 Two perspectives of the molecular diagram of complex 4. Hydrogen atoms and solvent (CH2Cl2) have been omitted for clarity. Ellipsoids at the 50% probability level. Selected bond distances (Å) and bond angles (°): Pt(1)–C(1) 1.972(5), Pt(1)–N(1) 2.015(4), Pt(1)–C(3) 2.054(6), Pt(1)–C(2) 2.053(6), C(1)–Pt(1)–N(1) 178.5(2), C(1)–Pt(1)–C(3) 101.1(2), C(1)–Pt(1)–C(2) 97.7(2), N(1)–Pt(1)–C(3) 80.4(2), N(1)–Pt(1)–C(2) 80.8(2), C(2)–Pt(1)–C(3) 161.2(2). | ||
For the preparation of a hetero-dimetallic Pt(II)–Au(III) complex, we decided to use our previously reported tris-imidazolium salt C (Scheme 3).16b As we already reported,16a the ligand can bind to two different metals by a stepwise procedure in which the first step invariably implies the coordination via the chelating side of the ligand, therefore facilitating the rational design of hetero-dimetallic complexes. This type of stepwise coordination of a ditopic ligand has also been successfully used by other authors for other types of polytopic NHC-based ligands,18 although only in a few cases the stepwise metalation strategy is metal-selective,18c,e thus simplifying the access to heterometallic compounds. In this case we proceeded by the reaction of C with [Pt(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh)2(COD)] in refluxing acetonitrile, in the presence of NaOAc, which afforded the Pt(II) complex 5 in 74% yield. The second step of the synthesis involves the reaction of 5 with [Au(CNC)Cl] and NaOAc, in refluxing acetonitrile, providing the hetero-dimetallic complex 6 in 18% yield. Interestingly, complex 6 can also be obtained by a one-pot procedure, by the sequential addition of [Pt(CCPh)2(COD)] and [Au(CNC)Cl] over a solution of C in MeCN in the presence of NaOAc, as indicated in Scheme 3. In both synthetic procedures NH4BF4 was added at the end of the process in order to facilitate the formation of the tetrafluoroborate salt of 6. This stepwise synthesis of complex 6 is a new example that illustrates the easy access to hetero-dimetallic complexes by using the tris-imidazolium salt C. In this case, the preparation of 6 affords a good opportunity to study the photophysical properties of a dimetallic complex that combines the presence of two potentially active inorganic metal-based chromophores.
CPh)2(COD)] in refluxing acetonitrile, in the presence of NaOAc, which afforded the Pt(II) complex 5 in 74% yield. The second step of the synthesis involves the reaction of 5 with [Au(CNC)Cl] and NaOAc, in refluxing acetonitrile, providing the hetero-dimetallic complex 6 in 18% yield. Interestingly, complex 6 can also be obtained by a one-pot procedure, by the sequential addition of [Pt(CCPh)2(COD)] and [Au(CNC)Cl] over a solution of C in MeCN in the presence of NaOAc, as indicated in Scheme 3. In both synthetic procedures NH4BF4 was added at the end of the process in order to facilitate the formation of the tetrafluoroborate salt of 6. This stepwise synthesis of complex 6 is a new example that illustrates the easy access to hetero-dimetallic complexes by using the tris-imidazolium salt C. In this case, the preparation of 6 affords a good opportunity to study the photophysical properties of a dimetallic complex that combines the presence of two potentially active inorganic metal-based chromophores.
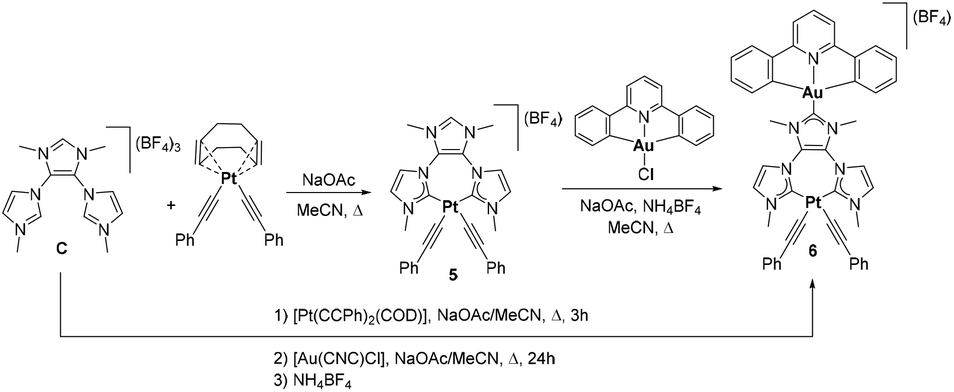 | ||
| Scheme 3 Synthesis of complexes 5 and 6. | ||
Compounds 5 and 6 were characterized by NMR spectroscopy and mass spectrometry. The electrospray MS spectrum of 5 displayed a peak at m/z 654.3 assigned to [5-BF4]. The heterometallic nature of 6 could be ascertained by its electrospray mass spectrum, which displayed a peak at m/z 1079.4, attributed to [6-BF4]. The 1H NMR of 5 shows a singlet at δ 8.67, assigned to the NCHN proton of the remaining imidazolium ring. The 13C NMR of 5 shows the resonance corresponding to the two equivalent carbene carbons at 175.9 ppm. The 13C NMR signals due to the distinct carbene carbons in 6 appear at 175.7 and 165.5 ppm, for the Pt–Ccarbene and Au–Ccarbene, respectively.
UV-Vis absorption and emission
The UV-visible absorption spectra of complexes 1–6 were studied in dichloromethane or acetonitrile at 298 K. We also included in our study the mono-ruthenium and di-ruthenium complexes 7 and 8 (Scheme 4),19 and the azolium and bisazolium salts A and B, for comparative purposes. The UV-Vis absorption spectra of the latter complexes were performed either in acetonitrile (A and B) or dichloromethane (7 and 8) at 298 K. All the photophysical data are summarized in Table 1. | ||
| Scheme 4 Ru(II) complexes 7 and 8. | ||
| Entry | Comp. | λ abs (nm) (log(ε))a |
λ
em![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) d (nm) d (nm) |
τ (ns) |
Φ
fd (%) |
|---|---|---|---|---|---|
| a Molar extinction coefficients (ε, in M−1 cm−1) were determined from Beer's law plots. b Measurements were performed in CH2Cl2 under ambient conditions. c Measurements were performed in MeCN under ambient conditions. d Emission quantum yields were measured in degassed MeCN, with recrystallized anthracene in degassed EtOH as standard (Φf = 0.278), λexc = 310 nm. e Measured in degassed MeCN, λexc = 304 nm. f Values in brackets correspond to emission quantum yields recorded in PMMA films. | |||||
| 1 | A | 251 (4.83), 277 (4.54), 307 (4.16), 321 (4.34), 336 (4.53)c | 373, 393, 416, 440 | 78.8 | 32 |
| 2 | B | 263 (4.79), 274 (4.63), 286 (4.69), 307 (4.05), 320 (4.19), 335 (4.34)c | 371, 391, 412, 436 | 43.1 | 41 |
| 3 | 1 | 246 (5.03), 281 (4.71), 325 (4.42), 341 (4.49)c | 373, 393, 415, 437 | 8.5 (15%) | 6.3 |
| 66.3 (85%) | |||||
| 4 | 2 | 255 (5.01), 304 (4.65), 352 (4.34)b | 372, 392, 414, 437 | 6.5 (8%) | 3.5 |
| 70.9 (92%) | |||||
| 5 | 3 | 236 (5.13), 281 (4.86), 293 (4.92), 344 (4.44), 379 (4.20)c | 372, 392, 413, 437 | 3.8 (21%) | 3.6 |
| 37.6 (79%) | |||||
| 6 | 4 | 257 (5.10), 305 (4.87), 348 (4.51), 394 (4.22)b | 372, 392, 413, 437 | 3.2 (25%) | 3.1 |
| 42.6 (75%) | |||||
| 7 | 5 | 257 (4.55), 305 (4.27)c | Not detected | — | [5.6]f |
| 8 | 6 | 246 (4.57), 305 (4.16)c | Not detected | — | [0.9]f |
| 9 | 7 | 255 (4.77), 293 (4.57), 302 (4.56), 348 (4.23)b | 374, 394, 415, 440 | 8.1 (17%) | 3.2 |
| 73.6 (83%) | |||||
| 10 | 8 | 262 (4.75), 300 (4.69), 356 (4.19), 385 (3.91)b | 380, 394 | 6.5 (13%) | 4.5 |
| 41.5 (87%) | |||||
The mono-NHC complexes 1, 2 and 7 exhibit an intense absorption band between 246–255 nm. The bis-NHC complexes 3, 4 and 8 show an intense absorption band between 236–262 nm. Complex 1, as well as compounds A and B, exhibits a less intense vibronic-structured band at 310–340 nm, with vibronic spacings of 1488 and 1443 cm−1. These bands are assigned to pyrene-centered π–π* transitions.
The photoluminescence properties of the synthesized complexes were studied in solution and dispersed in poly(methyl methacrylate) (PMMA) films at 298 K. Upon excitation at λ = 310 nm, degassed acetonitrile solutions of pyrene-based complexes 1–4, 7 and 8 exhibit strong luminescence at 373–440 nm. Their emission spectra featured vibronically resolved bands, with peak maxima at 393 nm (Fig. S3–S6, S9 and S10†). In fact, the emission spectra of all the pyrene-based NHC complexes and salts A and B were superimposable. These similarities suggest that the exciton that forms in all these molecules is largely localized on the pyrene linker, with minimum participation of the heterocyclic fragments or the metals. Under the same conditions, complexes 5 and 6, based on a Y-shaped tris-NHC ligand, were found to be non-emissive. These results may seem surprising, especially if we take into account the moderately intense luminescence (Φf = 3.9 × 10−3) shown by the gold(III) complex 9 (Scheme 5), which was originated from a metal perturbed 3[π → π*] (RC^N^CR) IL state, as previously published,20 although non-emissive systems were also obtained by slight modifications of the NHC substituents. Similarly, the Pt(II) complexes 10 (Scheme 5) are known to exhibit emission at room temperature in CH2Cl2, although with low quantum yields (Φf = 1–2 × 10−3).21
 | ||
| Scheme 5 Previously reported complexes 9 and 10. | ||
The carbene coordination reduces but does not suppress the pyrene-centered emission of complexes 1–4, 7 and 8 (compare entries 1 and 2 with 3–6, 9 and 10), suggesting that the carbene coordination to the Pt(II), Au(III) or Ru(II) center increases the proportion of non-radiative decay processes.22 All in all the quantum yields referred to the pyrene-based NHC metal complexes range between 3.1–6.3%, which are rather high values compared to those shown by other complexes with the same pyrene-based bis-NHC ligands.14b This result is interesting, because it indicates that the emission properties of the pyrene fragment may be combined with the well-known antitumoral activity displayed by several gold and platinum complexes,23 so that luminescent antitumoral agents may be accessible.
Emission lifetime measurements were performed using a nanosecond pulsed laser system in degassed acetonitrile. The excited-state lifetimes were found to be rather short, suggesting the fluorescent character of the luminescence for all metal complexes under study. The excited-state of complexes 1–8 exhibited bi-exponential decays, in which one long component (71–38 ns) with a larger contribution and a shorter one (3.2–8.5 ns) are present.
The emission quantum efficiencies were also recorded at 10 wt% (5 wt% for complexes 3 and 5 due to solubility issues) in PMMA films, exciting between 300 and 330 nm. Under these conditions, only the Y-shaped tris-NHC-based complexes 5 and 6 were found to be emissive, displaying quantum efficiencies of 5.6 and 0.9%, respectively (entries 7 and 8 in Table 1). In particular, complex 5 displays a vibronic-structured emission band between 415 and 600 nm with a band maximum at 430 nm (Fig. S11†). Complex 6 exhibits a less intense emission band between 475 and 575 nm with a band maximum at 517 nm (Fig. S12†), in line with the emission spectrum shown by 9.20
Electrochemistry
The electrochemical properties of complexes 1–6 were analysed by cyclic voltammetry (CV) and differential pulse voltammetry (DPV), in dichloromethane or acetonitrile using a 0.1 M solution of [NBu4][PF6] as the supporting electrolyte (Fig. S13–S19†). The cyclic voltammogram of the mono-gold complex 1 shows three irreversible reduction waves and no oxidation waves, which are assigned to the ligand-centered reduction of the CNC ligand.20,24 Unfortunately, the cyclic voltammogram of the di-gold complex 3 could not be recorded due to its high insolubility.The pyrene-based Pt(II) complexes 2 and 4 display a reversible and a quasi-reversible oxidation wave at 0.95 and 0.89 V, respectively, which can be attributed to a metal-centered redox process. In the case of the dimetallic complex 4, the DPV analysis of the oxidation wave indicates the absence of electronic coupling between the two platinum centers, in agreement with the results observed for other bimetallic complexes linked by the same NHC-based polyaromatic ligand.14a,19
Complexes 5 and 6 displayed irreversible platinum-centered oxidation waves at 1.3 and 1.2 V, respectively. These highly positive potentials may be attributed to the lower electron-donating character of the ligand, in combination with the cationic nature of the complexes (Fig. 2).
 | ||
| Fig. 2 CV plots of Pt(II)-based complexes 2, 4 and 5 (1 mM in dry dichloromethane or acetonitrile with 0.1 M [NBu4][PF6] as the supporting electrolyte, Fc+/Fc used as standard). 50 mV s−1 scan rate. | ||
Conclusions
In summary, in this work we prepared a series of pyrene-based monometallic and dimetallic complexes with pincer-CNC Au(III) and Pt(II) fragments. Additionally, based on a Y-shaped tris-NHC ligand, we prepared a Pt–Au hetero-dimetallic complex, together with a monometallic Pt(II) complex. These two complexes display the Pt(II) fragment at the chelating part of the ligand. The hetero-dimetallic complex contains a Au(III) fragment with a pincer CNC ligand bound to the mono-NHC part of the Y-shaped tris-NHC. The preparation of the Pt(II)/Au(III) hetero-dimetallic complex constitutes a novel example of the easy access to heterometallic complexes using our previously reported Y-shaped tris-NHC. While our initial interest in obtaining heterometallic complexes was based on their use for homogeneously catalyzed tandem processes, in this case we aimed to determine the influence of combining with potentially emissive fragments on the luminescence properties of the resulting complex.The study of the emissive properties of the pyrene-based metal complexes in solution showed that they displayed moderate pyrene-centered emissions, although with significant lower efficiencies compared to their related mother ligands. The same complexes are non-emissive in the solid state, probably due to molecular aggregation (such as π–π stacking interactions).
On the other hand, the two complexes based on the Y-shaped ligand are emissive in the solid state, but non-emissive in solution. By comparing the solid state emissions, it can be concluded that for the two complexes the emission is centered on the Pt(II) fragment.
Our results did not fully accomplish our initial objectives, which aimed to provide homo- and hetero-dimetallic complexes with enhanced luminescence properties compared to their monometallic analogues, but we believe that we established a wide-open pathway for the exploration of new families of multimetallic complexes with great photophysical potential. Our lab is currently searching for the preparation of heterodimetallic complexes in which the properties of the two different metal fragments are combined in order to afford new materials with enhanced luminescence properties.
Experimental
General methods
The metal complexes [Au(CNC)Cl],25 [Pt(CNC)DMSO],26 and [Pt(CCPh)2(COD)]27 were prepared according to literature methods. The imidazolium salts A,19B14a and C16b were previously reported by us, as well as the ruthenium based complexes 7 and 8.19 All operations were carried out by using standard Schlenk tube techniques under a nitrogen atmosphere unless otherwise stated. Anhydrous solvents were dried using a solvent purification system (SPS M BRAUN) or purchased from Aldrich and degassed prior to use by purging with nitrogen and kept over molecular sieves. All other reagents were used as received from commercial suppliers. NMR spectra were recorded on a Varian Inova 300 and 500 MHz, using CDCl3, CD3CN or DMSO-d6 as solvents. Electrospray mass spectra (ESI-MS) were recorded on a Micromass Quattro LC instrument; nitrogen was employed as drying and nebulizing gas. Exact mass analysis was realized using a Q-TOF Premier mass spectrometer with an electrospray source (Waters, Manchester, UK) operating at a resolution of ca. 16000 (FWHM). Elemental analyses were carried out on a EuroEA3000 EuroVector Analyzer. Infrared spectra (FTIR) were obtained on a Bruker EQUINOX 55 spectrometer with a spectral window of 4000–600 cm−1.
General spectroscopic considerations
UV-visible absorption spectra were recorded on a Varian Cary 300 BIO spectrophotometer using CH3CN or CH2Cl2 as a solvent, under ambient conditions. Emission spectra were recorded on a modular Horiba FluoroLog®-3 spectrofluorometer employing degassed CH3CN. Extinction coefficients (ε) were determined from Beer's law measurements using solutions with the concentration of the analyte of 10, 20, 30 and 40 μM or 10, 15, 20 and 25 μM. Quantum yields in solution were determined relative to recrystallized anthracene in degassed EtOH as a standard (Φf = 0.27),28 exciting at 317 nm. All solutions were prepared in air and de-aerated by sparging with nitrogen for 20 min prior to performing emission and quantum yield measurements. Time-resolved fluorescence measurements were performed using time-correlated single-photon counting (TCSPC) on the Horiba Jobin-Yvon IBH-5000U. The samples were excited using a 304 nm NanoLED with a FWHM of 1.3 ns at a repetition ratio of 100 kHz. Signals were collected by using an IBH Data Station Hub photon-counting module. The quality of the fit was assessed by minimizing the reduced chi (χ2) squared function and visual inspection of the weighted residuals. Direct quantum yield measurements were performed for solid samples at room temperature in a Hamamatsu absolute PL quantum yield spectrometer C11347.Electrochemistry
Electrochemical studies were carried out by using an Autolab Potentiostat, Model PGSTAT101 using a three-electrode cell. The cell was equipped with platinum working and counter electrodes, as well as a silver wire reference electrode. In all experiments, [NBu4][PF6] (0.1 M in dry CH2Cl2 or MeCN) was used as the supporting electrolyte with an analyte concentration of approximately 1 mM. Measurements were performed at 50 mV s−1 scan rate. All redox potentials were referenced to the Fc+/Fc couple as an internal standard with E1/2 (Fc/Fc+) vs. SCE = +0.46 V.29C), 106.1 (Pt–CC), 39.4 (NCH3), 35.8 (NCH3). Anal. Calc. for PtN6C29H27BF4 (741.20): C, 46.97; H, 3.67; N, 11.34. Found: C, 46.53; H, 3.63; N, 11.29. Electrospray MS (20 V, m/z): 654.3 [M]+.
:3 to 1:1) afforded the separation of an orange band which contained compound 6. Precipitation with a mixture of acetonitrile/diethyl ether gave the desired product as a brownish solid. Yield: 20 mg, 18%. 1H NMR (500 MHz, CD3CN): δ 8.16 (t, 3JH–H = 8.0 Hz, 1H, CHpyridyl), 7.86–7.81 (m, 4H, CHpyridyl, CHphenyl), 7.64 (d, 3JH–H = 2.1 Hz, 2H, CHimid), 7.44–7.31 (m, 9H, CHphenyl, CHimid), 7.23–7.17 (m, 7H, CHphenyl), 7.11–7.07 (m, 2H, CHphenyl), 4.10 (s, 6H, NCH3), 3.87 (s, 6H, NCH3). 13C NMR (126 MHz, CD3CN): δ 175.7 (Pt–Ccarbene), 165.5 (Au–Ccarbene), 165.5 (Cq CNC), 164.8 (Cq CNC), 164.6 (Cq CNC), 153.5 (Cq CNC), 150.8 (Cq CNC), 150.8 (Cq CNC), 145.5 (CH), 137.4 (CH), 136.9 (CH), 133.4 (CH), 133.3 (CH), 131.6 (CH), 130.1 (Cq), 129.1 (CH), 129.0 (CH), 127.7 (CH), 127.6 (CH), 126.5 (CH), 126.0 (CHimid), 125.7 (CH), 121.5 (CHimid), 119.5 (CH), 119.4 (CH), 118.0 (Cq), 107.9 (Pt–CC), 106.5 (Pt–CC), 39.3 (NCH3), 37.8 (NCH3). Slow decomposition of the complex prevented a correct elemental analysis. Electrospray MS (20 V, m/z): 1079.4 [M]+.
Acknowledgements
We gratefully acknowledge financial support from MINECO of Spain (CTQ2014-51999-P) and the Universitat Jaume I (P11B2014-02). The authors are grateful to the Serveis Centrals d'Instrumentació Científica (SCIC) of the Universitat Jaume I for providing spectroscopic and X-ray facilities. We are very grateful to Prof. M. Concepción Gimeno (CSIC-Universidad de Zaragoza) for the measurement of the photophysical properties of the samples in PMMA films. We are also grateful to Prof. Francisco Galindo (Universitat Jaume I) for his help in the emission lifetime measurements.References
- (a) M. Poyatos, J. A. Mata and E. Peris, Chem. Rev., 2009, 109, 3677–3707 CrossRef CAS PubMed; (b) J. A. Mata, M. Poyatos and E. Peris, Coord. Chem. Rev., 2007, 251, 841–859 CrossRef CAS.
- J. A. Mata, F. E. Hahn and E. Peris, Chem. Sci., 2014, 5, 1723–1732 RSC.
- J. Kido, M. Kimura and K. Nagai, Science, 1995, 267, 1332–1334 CrossRef CAS PubMed.
- (a) L. S. Hung and C. H. Chen, Mater. Sci. Eng., R, 2002, 39, 143–222 CrossRef; (b) S.-H. Hwang, C. N. Moorefield and G. R. Newkome, Chem. Soc. Rev., 2008, 37, 2543–2557 RSC; (c) T. W. Kelley, P. F. Baude, C. Gerlach, D. E. Ender, D. Muyres, M. A. Haase, D. E. Vogel and S. D. Theiss, Chem. Mater., 2004, 16, 4413–4422 CrossRef CAS; (d) Z. Wang, P. Lu, S. Chen, Z. Gao, F. Shen, W. Zhang, Y. Xu, H. S. Kwok and Y. Ma, J. Mater. Chem., 2011, 21, 5451–5456 RSC.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS.
- (a) H. Yersin, Transition Metal and Rare Earth Compounds III, 2004, vol. 241, pp. 1–26 Search PubMed; (b) H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622–2652 CrossRef CAS.
- (a) L. X. Xiao, Z. J. Chen, B. Qu, J. X. Luo, S. Kong, Q. H. Gong and J. J. Kido, Adv. Mater., 2011, 23, 926–952 CrossRef CAS PubMed; (b) J. Kalinowski, V. Fattori, M. Cocchi and J. A. G. Williams, Coord. Chem. Rev., 2011, 255, 2401–2425 CrossRef CAS; (c) K. Binnemans, Chem. Rev., 2005, 105, 4148–4204 CrossRef CAS PubMed; (d) S. Reineke, F. Lindner, G. Schwartz, N. Seidler, K. Walzer, B. Luessem and K. Leo, Nature, 2009, 459, 234–U116 CrossRef CAS PubMed; (e) S. Fantacci and F. De Angelis, Coord. Chem. Rev., 2011, 255, 2704–2726 CrossRef CAS; (f) Y. You and S. Y. Park, Dalton Trans., 2009, 1267–1282 RSC; (g) J. A. G. Williams, Chem. Soc. Rev., 2009, 38, 1783–1801 RSC; (h) R. C. Evans, P. Douglas and C. J. Winscom, Coord. Chem. Rev., 2006, 250, 2093–2126 CrossRef CAS; (i) M. Mauro, A. Aliprandi, D. Septiadi, N. S. Kehra and L. De Cola, Chem. Soc. Rev., 2014, 43, 4144–4166 RSC.
- (a) Q. Sun, S. Mosquera-Vazquez, Y. Suffren, J. Hankache, N. Amstutz, L. M. L. Daku, E. Vauthey and A. Hauser, Coord. Chem. Rev., 2015, 282, 87–99 CrossRef; (b) A. O. Adeloye and P. A. Ajibade, Molecules, 2014, 19, 12421–12460 CrossRef PubMed; (c) V. Marin, E. Holder, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 618–635 RSC; (d) E. A. Medlycott and G. S. Hanan, Chem. Soc. Rev., 2005, 34, 133–142 RSC.
- (a) V. W.-W. Yam and E. C.-C. Cheng, Chem. Soc. Rev., 2008, 37, 1806–1813 RSC; (b) A. Barbieri, G. Accorsi and N. Armaroli, Chem. Commun., 2008, 2185–2193 RSC; (c) V. W.-W. Yam and K. K. W. Lo, Chem. Soc. Rev., 1999, 28, 323–334 RSC.
- R. Visbal and M. Concepcion Gimeno, Chem. Soc. Rev., 2014, 43, 3551–3574 RSC.
- C. Bronner and O. S. Wenger, Dalton Trans., 2011, 40, 12409–12420 RSC.
- (a) T. M. Figueira-Duarte and K. Muellen, Chem. Rev., 2011, 111, 7260–7314 CrossRef CAS PubMed; (b) F. M. Winnik, Chem. Rev., 1993, 93, 587–614 CrossRef CAS.
- A. J. Howarth, M. B. Majewski and M. O. Wolf, Coord. Chem. Rev., 2015, 282, 139–149 CrossRef.
- (a) S. Gonell, M. Poyatos and E. Peris, Chem. – Eur. J., 2014, 20, 9716–9724 CrossRef CAS PubMed; (b) S. Ibanez, A. Guerrero, M. Poyatos and E. Peris, Chem. – Eur. J., 2015, 21, 10566–10575 CrossRef CAS PubMed.
- C. K.-L. Li, R. W.-Y. Sun, S. C.-F. Kui, N. Zhu and C.-M. Che, Chem. – Eur. J., 2006, 12, 5253–5266 CrossRef CAS PubMed.
- (a) S. Gonell, M. Poyatos, J. A. Mata and E. Peris, Organometallics, 2012, 31, 5606–5614 CrossRef CAS; (b) S. Gonell, M. Poyatos, J. A. Mata and E. Peris, Organometallics, 2011, 30, 5985–5990 CrossRef CAS.
- L. Boselli, M. Carraz, S. Mazeres, L. Paloque, G. Gonzalez, F. Benoit-Vical, A. Valentin, C. Hemmert and H. Gornitzka, Organometallics, 2015, 34, 1046–1055 CrossRef CAS.
- (a) M. T. Zamora, M. J. Ferguson and M. Cowie, Organometallics, 2012, 31, 5384–5395 CrossRef CAS; (b) M. T. Zamora, M. J. Ferguson, R. McDonald and M. Cowie, Dalton Trans., 2009, 7269–7287 RSC; (c) R. Maity, A. Bit, C. Schulte to Brinke, J. Koesters and F. E. Hahn, Organometallics, 2013, 32, 6174–6177 CrossRef CAS; (d) R. Maity, A. Rit, C. Schulte to Brinke, C. G. Daniliuc and F. E. Hahn, Chem. Commun., 2013, 49, 1011–1013 RSC; (e) R. Maity, H. Koppetz, A. Hepp and F. E. Hahn, J. Am. Chem. Soc., 2013, 4966–4969 CrossRef CAS PubMed; (f) R. Maity, T. Tichter, M. van der Meer and B. Sarkar, Dalton Trans., 2015, 44, 18311–18315 RSC; (g) R. Maity, C. Schulte to Brinke and F. E. Hahn, Dalton Trans., 2013, 42, 12857–12860 RSC; (h) M. T. Zamora, M. J. Ferguson, R. McDonald and M. Cowie, Organometallics, 2012, 31, 5463–5477 CrossRef CAS.
- S. Gonell and E. Peris, ACS Catal., 2014, 4, 2811–2817 CrossRef CAS.
- V. K.-M. Au, K. M.-C. Wong, N. Zhu and V. W.-W. Yam, J. Am. Chem. Soc., 2009, 131, 9076–9085 CrossRef CAS PubMed.
- Y. Zhang, J. A. Garg, C. Michelin, T. Fox, O. Blacque and K. Venkatesan, Inorg. Chem., 2011, 50, 1220–1228 CrossRef CAS PubMed.
- J. J. Hu, S.-Q. Bai, H. H. Yeh, D. J. Young, Y. Chi and T. S. A. Hor, Dalton Trans., 2011, 40, 4402–4406 RSC.
- (a) S.-W. L. C.-M. Che, in Gold Chemistry. Applications and Future Directions in the Life Science, ed. F. Mohr, Wiley-VCH Verlag, 2008 Search PubMed; (b) D. L. Ma and C. M. Che, Chem. – Eur. J., 2003, 9, 6133–6144 CrossRef CAS PubMed.
- K. M.-C. Wong, L.-L. Hung, W. H. Lam, N. Zhu and V. W.-W. Yam, J. Am. Chem. Soc., 2007, 129, 4350–4365 CrossRef CAS PubMed.
- K. H. Wong, K. K. Cheung, M. C. W. Chan and C. M. Che, Organometallics, 1998, 17, 3505–3511 CrossRef CAS.
- U. Belio, S. Fuertes and A. Martin, Dalton Trans., 2014, 43, 10828–10843 RSC.
- A. Klein, J. van Slageren and S. Zalis, J. Organomet. Chem., 2001, 620, 202–210 CrossRef CAS.
- W. R. Dawson and M. W. Windsor, J. Phys. Chem., 1968, 72, 3251–3260 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR, UV-Vis absorption and emission spectra of the new complexes, cyclic voltammograms of the complexes, CIF file of the molecular structure of complex 4, and a summary of the crystal data, data collection and refinement details. CCDC 1446681. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt00198j |
| ‡ Current address: Homogeneous, Supramolecular and Bio-Inspired Catalysis, Van't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098XH Amsterdam (The Netherlands). |
| This journal is © The Royal Society of Chemistry 2016 |