
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Donor–acceptor organo-imido polyoxometalates: high transparency, high activity redox-active NLO chromophores†
Ahmed
Al-Yasari‡
ac,
Nick
Van Steerteghem‡
b,
Hani
El Moll
a,
Koen
Clays
*b and
John
Fielden
*a
aSchool of Chemistry, University of East Anglia, Norwich, NR4 7TJ, UK. E-mail: John.Fielden@uea.ac.uk
bDepartment of Chemistry, University of Leuven, Celestijnenlaan 200D, B-3001 Leuven, Belgium. E-mail: Koen.Clays@fys.kuleuven.be
cCollege of Pharmacy, University of Kerbala, Kerbala, Iraq
First published on 19th January 2016
Abstract
We show that polyoxometalates (POMs) are an excellent redox-active acceptor on which to base high performance 2nd order non-linear optical (NLO) chromophores. This is demonstrated through three new organoimido-Lindqvist derivatives with HRS β0-values exceeding those of any dipolar organic system with comparable donor, π-system and absorption profile. Thus, organoimido POMs may provide a new generation of high performance, high transparency, and potentially redox-switchable NLO materials.
Non-linear optical (NLO) materials can manipulate laser light through 2nd order phenomena such as second harmonic generation (SHG) and 3rd order effects like multiphoton absorption.1 As such, they are essential to current and future technologies including telecommunications, optical and electro-optical computing, optical power limiting and imaging. The need for materials with fast responses and tunable properties, driven by advanced applications, has led to development of many organic donor–acceptor compounds with strong 2nd order non-linearities, β, which are now finding technological use (e.g. dimethylaminostilbazolium tosylate, DAST).1–3 Meanwhile, metallo-organic species, with responses based on metal-to-ligand or ligand-to-metal charge transfer (CT) absorptions, have attracted attention as a means to facilitate construction of 2- and 3-D chromophores with multi-dimensional responses,4 or introduce additional properties such as reversible redox chemistry4–6 that could be exploited in materials with switchable responses.6,7 An important challenge for both organic and metallo-organic materials, however, is overcoming transparency/non-linearity trade-offs whereby modifications that increase β also increase absorption of visible light. This leads to lower device efficiency, and often reduced photostability.
So far, this challenge has been tackled either through octupolar and other multi-dimensional chromophores,4,8 which are often synthetically complex, or through dipolar organic materials with unusual electronic structures. These unusual electronic structures have originated either from unconventional twisted (“tictoid”) π-bridges,9 or unusual donor sets.10 A complementary strategy is to explore new acceptors. To this end, we have begun to study polyoxometalates (POMs), a major class of molecular materials that are so far little investigated in NLO and photonics. These anionic molecular metal oxide clusters are based on low-cost earth-abundant elements (e.g. Mo, W),11 and many are good electron acceptors featuring fast, stable, often multi-electron redox chemistry.12 POMs also offer a high density of heavy atoms that can be beneficial for both 2nd and 3rd order NLO,13 and can be derivatized with organic groups,14 making them a highly attractive platform for redox-active hybrid donor–acceptor chromophores. However, the molecular 2nd order NLO properties of POMs have so far only been addressed by calculation,15 with experimental reports limited to bulk SHG materials containing underivatized POMs.16
Herein, we present the first experimental study of POM derivatives as molecular NLO chromophores, focusing on the synthesis, structure and 2nd order NLO properties of three new arylimido Lindqvist clusters ([RNMo6O18]2−, Scheme 1) with organic donor groups. Such Lindqvist arylimido derivatives show the strong CT transitions that are a pre-requisite for 2nd order NLO properties, and have an increasingly well-developed synthetic chemistry enabling post-synthetic modification of the organic group.17 The results show that such materials attain much better transparency/non-linearity trade-offs than purely organic dipolar chromophores with comparable donors and π-systems. Furthermore, the compounds have reversible electrochemistry and can incorporate electropolymerizable pyrroles, which may eventually open a new, convenient route to microfabrication18 of redox-switchable thin films with covalently derivatized POMs.
 | ||
| Scheme 1 Organoimido-Lindqvist anion based chromophores 1 to 3. | ||
Divalent anions 1 to 3 (Scheme 1) were obtained as [NBu4]+ salts using adapted procedures for the synthesis and post-functionalization of organoimido hexamolybdates,17,19 and have been unambiguously characterized (see ESI†). By comparison with the parent anilines, the 1H-NMR spectra of 1 to 3 clearly demonstrate their donor–acceptor nature: the protons ortho- to the aniline group shift downfield ca. 1 ppm upon formation of the Mo-imido bond, due to the electron withdrawing {Mo6} cluster. The pyrrole protons of 1 and 2, however, show relatively little change in δ upon attachment of the POM, implying a certain degree of electronic isolation between donor and acceptor. Crystal structures (Fig. 1) show characteristic bond lengths and angles for {Mo6}-organoimido species (Table S2, ESI†),17a,20 including ca. 180° Mo–N–C bond angles indicative of Mo–N triple bond character and a formal positive charge on N. The relative electronic isolation of the donors suggested by 1H-NMR is supported by observation of non-zero twist angles between the pyrrole and phenyl rings, of ca. 19.3° (in 1) and 13° (in 2).
 | ||
| Fig. 1 ORTEP representations of anions 1 to 3. Thermal ellipsoids are at the 30% probability level. Color scheme: Mo is green; O, red; C, gray; N, blue; H atoms are represented by green circles of arbitrary radii. | ||
The electronic spectra (Table 1, Fig. 2a) also show classic donor–acceptor behaviour. High energy spectral features can be ascribed to O → Mo charge transfer (CT) in the hexamolybdate acceptor unit and intra-ligand (π → π*) processes in the appended organic. At lower energy, however, an intense band is observed, absent from the spectra of both [Mo6O19]2− and the parent anilines. Tentative assignment of these bands to ligand-to-polyoxometalate charge transfer (LPCT)20b is supported by the red shift (371 to 386 nm) and increase (ca. 50%) in ε observed upon extending the conjugated system from 1 to 2. Replacing pyrrole with more strongly donating –NH2 produces a further red shift, to 406 nm, albeit with a decrease in ε. Such behaviour is typical of charge transfer dyes.1–7 Cyclic voltammetry (CV) confirms donation of electron density from the organic groups to the POM (Fig. 2b), through a ca. 180 mV negative shift in E1/2 from [Mo6O19]2− to the derivatives. Furthermore, as expected for POM species,12 near-ideal reversibility is seen on the CV timescale. However, consistent with the NMR and X-ray data, a degree of electronic isolation between N-donor and POM acceptor is implied by the minimal difference in reduction potential (±7 mV) between the derivatives. Therefore, while we use LPCT as shorthand, we cannot exclude that the observed electronic transitions may in fact be between the donor and electron deficient imido-N, rather than the POM.
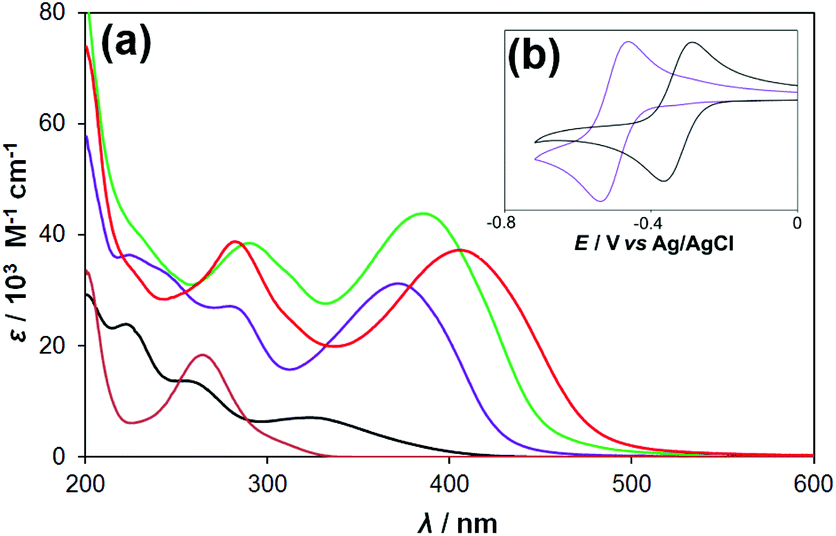 | ||
| Fig. 2 (a) Electronic spectra of 4-(1H-pyrrol-1-yl)aniline (brown) and [NBu4]+ salts of [Mo6O19] (black), 1 (purple), 2 (green) and 3 (red) at 298 K. (b) Inset – cyclic voltammograms of 10−3 M [NBu4]2[Mo6O19] (black) and [NBu4]2[1] (purple) in 0.1 M [NBu4][BF4] in MeCN, showing the cathodic shift in the [XMo6O18]2−/3− potential upon derivatization. Scan rate 100 mV s−1, Fc/Fc+ internal reference E1/2 = 0.46 V vs. Ag/AgCl. | ||
| Compound | λ max/nma (ε, 103 M−1 cm−1) | E max (eV) | Assignment | E 1/2, V vs. Ag/AgClb (ΔEp, mV) |
|---|---|---|---|---|
| a Concentrations ca. 10−5 M in MeCN. b Solutions ca. 10−3 M in analyte and 0.1 M in [NBu4][BF4] at a glassy carbon working electrode with a scan rate of 100 mV s−1. Ferrocene internal reference E1/2 = 0.46 V, ΔEp = 80 mV. | ||||
| [NBu4]2[Mo6O19] | 223 (24.9) | 5.56 | O → Mo | −0.315 (69) |
| 261 (13.4) | 4.75 | O → Mo | ||
| 323 (7.1) | 3.84 | O → Mo | ||
| [NBu4]2 [1] | 223 (39.6) | 5.56 | O → Mo/π → π* | −0.500 (63) |
| 281 (28.2) | 4.41 | O → Mo/π → π* | ||
| 371 (32.3) | 3.34 | LPCT | ||
| [NBu4]2 [2] | 290 (40.1) | 4.28 | O → Mo/π → π* | −0.496 (61) |
| 386 (45.1) | 3.21 | LPCT | ||
| [NBu4]2 [3] | 282 (38.8) | 4.40 | O → Mo/π → π* | −0.493 (67) |
| 406 (37.3) | 3.05 | LPCT | ||
Hyper-Rayleigh scattering (HRS) measurements21 (Table 2) suggest great potential for these organoimido-Lindqvist based materials in NLO. This is because although headline β values are around an order of magnitude lower than HRS-determined records, comparison of static first hyperpolarizabilities β0 (corrected for any one or two photon resonance enhancement) indicates that these compounds outperform many dipolar organic systems with similar donors and π-systems. Moreover, to our knowledge no dipolar organic compounds having both similar transparency to 1 and 3 and comparable donors/bridges exceed their non-resonant β0 (determined at 1064 nm). Our results, and comparisons are detailed below.
| Compound | β zzz, 800 | β 0,800b | β zzz, 1064 | β 0,1064 |
|---|---|---|---|---|
| (10−30 esu) | ||||
| a β zzz calculated assuming a single dominant tensor component, measured using 800 nm and 1064 nm fundamental laser beams. The quoted units (esu) can be converted into SI units (C3 m3 J−2) by dividing by a factor of 2.693 × 1020. b Non-resonant, static β estimated from βzzz using the two state model.22 c Underestimated due to proximity of LPCT maximum to the SH wavelength at 400 nm. | ||||
| [NBu4]2[1] | 292 ± 34 | (32 ± 4)c | 123 ± 10 | 56 ± 5 |
| [NBu4]2[2] | 557 ± 56 | (29 ± 3)c | 143 ± 10 | 59 ± 5 |
| [NBu4]2[3] | 716 ± 63 | (16 ± 1)c | 372 ± 22 | 133 ± 8 |
Firstly, large (up to 716 × 10−30 esu) dynamic 2nd order non-linearities (βzzz, assuming a single dominant tensor component for linear systems) were obtained at 800 nm. Substantial increases in activity from 1 to 2 (90%), and 2 to 3 (30%) show that modifications used to enhance β in purely organic chromophores (increased conjugation and donor strength) also apply to organoimido-POMs. At 1064 nm βzzz are lower, most likely because resonance enhancement is effectively eliminated, and the activity gain from 1 to 2 is much smaller, while that from 2 to 3 is larger. This may indicate wavelength dependence in the effects of extended conjugation and increased donor strength, with the caveat that analysis of 2 was complicated by the need to subtract signal from two-photon fluorescence, at both measured wavelengths.
While the dynamic βzzz values compare well to many purely organic systems measured under the same conditions,2d,3a,b to fairly assess the efficacy of the POM acceptor versus other materials we must use the resonance-corrected static first hyperpolarizability, β0, and limit the comparison to dipolar systems with similar donors and π-bridges. This is possible at 1064 nm, because λmax for 1 to 3 are distant from (≥126 nm shorter than) the 532 nm second harmonic, and have only minimal residual absorption at this wavelength. Considering the weak to moderate donor strength, modest π-systems and high transition energies, the β0 values obtained (from 56 × 10−30 esu for 1 to 133 × 10−30 esu for 3) are unusually high. They comfortably exceed that of the technologically valuable DAS+ cation (Fig. 3, 4) under non-resonant conditions,2b while the β0 for 3 also compares well to those of many other, more active stilbazolium chromophores with stronger donors and much lower energy transitions (i.e. poorer transparency).2b,c Restricting comparison to reasonable structural and spectral analogues of 1 to 3 (Fig. 3) indicates a consistent and significant advantage for the POM compounds, which in all cases have higher β0 values.§ Only p-nitroaniline shows better transparency, for a large sacrifice in β. While far higher β-values have been obtained with other organic acceptors (e.g. TCF), these materials are a poor comparison due to much lower transparency – the closest TCF analogue to 3 has a solvent-dependent λmax of between 540 and 590 nm.2e Indeed, taking transparency into account such materials do not seem to perform better than other organic acceptors.5d Comparison with a range of metal and main-group based acceptors4f also reveals an advantage for the POMs.
 | ||
| Fig. 3 Organic chromophores with comparable donors, π-systems and absorption profiles to 1 to 3, and non-resonant β0 values.2b,c,3c,d,21a,e | ||
Performance is further put in context by considering electron number adjusted β (β0/N3/2; N = no. bridge π-conjugated electrons) and λmax, an analysis that suggests an apparent limit for most dipolar organic materials of ca. β0/N3/2 = 1.5 when λmax = 370 nm (for 1) or 2 when λmax = 406 nm (for 3).23 Compound 1 (β0/N3/2 = 3.81) and compound 3 (β0/N3/2 = 2.54) both breach this limit (Fig. S4, ESI†). Certain dipolar organic materials (e.g. TICT chromophores9 and a pyridinium tetrazolate10) appear to breach these performance limits more spectacularly than our POMs, which has been ascribed to their unusual electronic structures. Thus future work must address how the electronic structure of organoimido-POMs enables them to act as unusually efficient acceptors, and whether combining POMs with more state-of-the-art organic components could lead to record performance.
In summary, we have shown that organo-imido POM derivatives are an excellent new class of NLO materials, with much better transparency/non-linearity trade-offs than comparable organic systems and reversible electrochemistry. Tailoring the organic component of these materials, through donor, bridge, or octupoles, could therefore lead to extremely high performance, switchable NLO chromophores. Such studies are already underway, as are preliminary investigations of electropolymer films, and efforts to deepen understanding of the LPCT transitions and electronic structure.
We thank the UK EPSRC National Crystallography Service in Southampton for X-ray data for [NBu4]2[2] and [NBu4]2[3], and the UK EPSRC National Mass Spectrometry Facility (Swansea) for MS. This work was supported by: the Iraqi Government (HCED scholarship to AAY), Royal Society of Chemistry, EPSRC (EP/M00452X/1), EU FP7 (Marie Curie IOF POMHYDCAT contract 254339 to JF) and Fonds voor Wetenschappelijk Onderzoek-Vlaanderen (FWO-V, PhD fellowship for NVS).
Notes and references
- (a) Nonlinear Optics of Organic Molecules and Polymers, ed. H. S. Nalwa and S. Miyata, CRC Press, Boca Raton, FL, 1997 Search PubMed; (b) S. R. Marder, Chem. Commun., 2006, 131 RSC; (c) M. G. Kuzyk, J. Mater. Chem., 2009, 19, 7444 RSC; (d) M. Pawlicki, H. A. Collins, R. G. Denning and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 3244 CrossRef CAS PubMed.
- (a) S. R. Marder, J. W. Perry and W. P. Schaefer, Science, 1989, 245, 626 CAS; (b) B. J. Coe, J. A. Harris, I. Asselberghs, K. Clays, G. Olbrechts, A. Persoons, J. T. Hupp, R. C. Johnson, S. J. Coles, M. B. Hursthouse and K. Nakatani, Adv. Funct. Mater., 2002, 12, 110 CrossRef CAS; (c) B. J. Coe, J. A. Harris, I. Asselberghs, K. Wostyn, K. Clays, A. Persoons, B. S. Brunschwig, S. J. Coles, T. Gelbrich, M. E. Light, M. B. Hursthouse and K. Nakatani, Adv. Funct. Mater., 2003, 13, 347 CrossRef CAS; (d) B. J. Coe, J. Fielden, S. P. Foxon, M. Helliwell, I. Asselberghs, K. Clays, K. De Mey and B. S. Brunschwig, J. Org. Chem., 2010, 75, 8550 CrossRef CAS PubMed; (e) S.-H. Jang, J. Luo, N. M. Tucker, A. Leclercq, E. Zojer, M. A. Haller, T.-D. Kim, J.-W. Kang, K. Firestone, D. Bale, D. Lao, J. B. Benedict, D. Cohen, W. Kaminsky, B. Kahr, J.-L. Brédas, P. Reid, L. R. Dalton and A. K.-Y. Jen, Chem. Mater., 2006, 18, 2982 CrossRef CAS.
- (a) B. J. Coe, J. Fielden, S. P. Foxon, J. A. Harris, M. Helliwell, B. S. Brunschwig, I. Asselberghs, K. Clays, J. Garín and J. Orduna, J. Am. Chem. Soc., 2010, 132, 10498 CrossRef CAS PubMed; (b) B. J. Coe, J. Fielden, S. P. Foxon, M. Helliwell, B. S. Brunschwig, I. Asselberghs, K. Clays, J. Olesiak, K. Matczyszyn and M. Samoc, J. Phys. Chem. A, 2010, 114, 12028 CrossRef CAS; (c) L. T. Cheng, W. Tam, S. H. Stevenson, G. R. Meredith, G. Rikken and S. R. Marder, J. Phys. Chem., 1991, 95, 10631 CrossRef CAS; (d) L. T. Cheng, W. Tam, S. R. Marder, A. E. Steigman, G. Rikken and C. W. Spangler, J. Phys. Chem., 1991, 95, 10643 CrossRef CAS.
- (a) C. Dhenaut, I. Ledoux, I. D. W. Samuel, J. Zyss, M. Bourgault and H. Le Bozec, Nature, 1995, 374, 339 CrossRef CAS; (b) A. M. McDonagh, M. G. Humphrey, M. Samoc, B. Luther-Davies, S. Houbrechts, T. Wada, H. Sasabe and A. Persoons, J. Am. Chem. Soc., 1999, 121, 1405 CrossRef CAS; (c) S. Di Bella, Chem. Soc. Rev., 2001, 30, 355 RSC; (d) B. J. Coe, S. P. Foxon, M. Helliwell, D. Rusanova, B. S. Brunschwig, K. Clays, G. Depotter, M. Nyk, M. Samoc, D. Wawrzynczyk, J. Garín and J. Orduna, Chem. – Eur. J., 2013, 19, 6613 CrossRef CAS PubMed; (e) O. Maury and H. Le Bozec, Acc. Chem. Res., 2005, 38, 691 CrossRef CAS PubMed; (f) H. Le Bozec and T. Renouard, Eur. J. Inorg. Chem., 2000, 229 CrossRef CAS.
- (a) M. Malaun, Z. R. Reeves, R. L. Paul, J. C. Jeffery, J. A. McCleverty, M. D. Ward, I. Asselberghs, K. Clays and A. Persoons, Chem. Commun., 2001, 49 RSC; (b) G. T. Dalton, M. P. Cifuentes, S. Petrie, R. Stranger, M. G. Humphrey and M. Samoc, J. Am. Chem. Soc., 2007, 129, 11882 CrossRef CAS PubMed; (c) B. J. Coe, J. Fielden, S. P. Foxon, I. Asselberghs, K. Clays, S. Van Cleuvenbergen and B. S. Brunschwig, Organometallics, 2011, 30, 5731 CrossRef CAS; (d) P. Kaur, M. Kaur, G. Depotter, S. Van Cleuvenbergen, I. Asselberghs, K. Clays and K. Singh, J. Mater. Chem., 2012, 22, 10597 RSC.
- (a) I. Asselberghs, K. Clays, A. Persoons, M. D. Ward and J. McCleverty, J. Mater. Chem., 2004, 14, 2831 RSC; (b) K. A. Green, M. P. Cifuentes, M. Samoc and M. G. Humphrey, Coord. Chem. Rev., 2011, 255, 2530 CrossRef CAS; (c) L. Boubekeur-Lecaque, B. J. Coe, K. Clays, S. Foerier, T. Verbiest and I. Asselberghs, J. Am. Chem. Soc., 2008, 130, 3286 CrossRef CAS; (d) S. Di Bella, I. P. Oliveri, A. Colombo, C. Dragonetti, S. Righetto and D. Roberto, Dalton Trans., 2012, 41, 7013 RSC.
- (a) J. Boixel, V. Guerchais, H. Le Bozec, D. Jacquemin, A. Amar, A. Boucekkine, A. Colombo, C. Dragonetti, D. Marinotto, D. Roberto, S. Righetto and R. De Angelis, J. Am. Chem. Soc., 2014, 136, 5367 CrossRef CAS PubMed; (b) V. Guerchais, L. Ordronneau and H. Le Bozec, Coord. Chem. Rev., 2010, 254, 2533 CrossRef CAS.
- (a) G. Alcaraz, L. Euzenat, O. Mongin, C. Katan, I. Ledoux, J. Zyss, M. Blanchard-Desce and M. Vaultier, Chem. Commun., 2003, 2766 RSC; (b) H. Kang, P. Zu, Y. Yang, A. Fachetti and T. J. Marks, J. Am. Chem. Soc., 2004, 126, 15974 CrossRef CAS PubMed.
- (a) H. Kang, A. Facchetti, H. Jiang, E. Cariati, S. Righetto, R. Ugo, C. Zuccaccia, A. Macchioni, C. L. Stern, Z. Liu, S. T. Ho, E. C. Brown, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 3267 CrossRef CAS PubMed; (b) Y. Shi, D. Frattarelli, N. Watanabe, A. Facchetti, E. Cariati, S. Righetto, E. Tordin, C. Zuccaccia, A. Macchioni, S. L. Wegener, C. L. Stern, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2015, 137, 12521 CrossRef CAS PubMed.
- L. Beverina, A. Sanguineti, G. Battagliarin, R. Ruffo, D. Roberto, S. Righetto, R. Soave, L. L. Presti, R. Ugo and G. A. Pagani, Chem. Commun., 2011, 47, 292 RSC.
- (a) C. L. Hill, Chem. Rev., 1998, 98, 1 CrossRef CAS PubMed; (b) D.-L. Long and L. Cronin, Dalton Trans., 2012, 41, 9815 RSC; (c) A. Al-Yasari and J. Fielden, Rev. Adv. Sci. Eng., 2014, 3, 304 CrossRef.
- M. Sadakane and E. Steckhan, Chem. Rev., 1998, 98, 219 CrossRef CAS PubMed.
- (a) B. F. Milne, F. Nogueira and C. Cardoso, Dalton Trans., 2013, 42, 3695 RSC; (b) J. Li, J. Zhang, M. G. Humphrey and C. Zhang, Eur. J. Inorg. Chem., 2013, 328 CrossRef.
- (a) A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009 CrossRef CAS PubMed; (b) A. Proust, B. Matt, R. Villanneau, G. Guillemot, P. Gouzerh and G. Izzet, Chem. Soc. Rev., 2012, 41, 7605 RSC.
- (a) G. Yang, W. Guan, L. Yan, Z. Su, L. Xu and E. B. Wang, J. Phys. Chem. B, 2006, 110, 23092 CrossRef CAS PubMed; (b) M. R. S. A. Janjua, M. Amin, M. Ali, B. Bashir, M. U. Khan, M. A. Iqbal, W. Guan, L. Yan and Z.-M. Su, Eur. J. Inorg. Chem., 2012, 705 CrossRef CAS; (c) W. Guan, G. Yang, C. Liu, P. Song, L. Fang, L. Yan and Z. Su, Inorg. Chem., 2008, 47, 5245 CrossRef CAS PubMed.
- (a) H. Murakami, T. Kozeki, Y. Suzuki, S. Ono, H. Ohtake, N. Surukura, E. Ishikawa and T. Yamase, Appl. Phys. Lett., 2001, 79, 3564 CrossRef CAS; (b) J.-D. Compain, P. Mialane, A. Dolbecq, J. Marrot, A. Proust, K. Nakatani, Y. Pei and F. Sécheresse, Inorg. Chem., 2009, 48, 6222 CrossRef CAS PubMed; (c) X.-M. Zhang, B.-Z. Shan, C.-Y. Duan and X.-Z. You, Chem. Commun., 1997, 1131 RSC; (d) Y. M. Xie, Q.-S. Zhang, Z.-G. Zhao, X.-Y. Wu, S.-C. Chen and C.-Z. Lu, Inorg. Chem., 2008, 47, 8086 CrossRef CAS.
- (a) Y. Wei, B. Xu, C. L. Barnes and Z. Peng, J. Am. Chem. Soc., 2001, 123, 4083 CrossRef CAS PubMed; (b) J. Zhang, F. Xiao, J. Hao and Y. Wei, Dalton Trans., 2012, 41, 3599 RSC; (c) C. Lv, R. N. N. Kahn, J. Zhang, J. Hu, J. Hao and Y. Wei, Chem. – Eur. J., 2013, 19, 1174 CrossRef CAS PubMed.
- (a) P. Kim, A. K. Epstein, M. Khan, L. D. Zarzar, D. J. Lipomi, G. M. Whitesides and J. Aizenberg, Nano Lett., 2012, 12, 527 CrossRef CAS PubMed; (b) L. Soleymani, Z. Fang, X. Sun, H. Yang, B. J. Taft, E. H. Sargent and S. O. Kelley, Angew. Chem., Int. Ed., 2009, 48, 8457 CrossRef CAS PubMed.
- (a) I. Bar-Nahum, K. V. Narasimhulu, L. Weiner and R. Neumann, Inorg. Chem., 2005, 44, 4900 CrossRef CAS PubMed; (b) M. Araghi, V. Mirkhani, M. Moghadam, S. Tangestaninejad and I. Mohammdpoor-Baltork, Dalton Trans., 2012, 41, 3087 RSC.
- (a) B. Xu, Y. Wei, C. L. Barnes and Z. Peng, Angew. Chem., Int. Ed., 2001, 40, 2290 CrossRef CAS; (b) J. B. Strong, G. P. A. Yap, R. Ostrander, L. M. Liable-Sands, A. L. Rheingold, R. Thouvenot, P. Gouzerh and E. A. Maatta, J. Am. Chem. Soc., 2000, 122, 639 CrossRef CAS.
- (a) K. Clays and A. Persoons, Phys. Rev. Lett., 1991, 66, 2980 CrossRef CAS PubMed; (b) K. Clays and A. Persoons, Rev. Sci. Instrum., 1992, 63, 3285 CrossRef CAS; (c) E. Hendrickx, K. Clays and A. Persoons, Acc. Chem. Res., 1998, 31, 675 CrossRef CAS; (d) G. Olbrechts, R. Strobbe, K. Clays and A. Persoons, Rev. Sci. Instrum., 1998, 69, 2233 CrossRef CAS; (e) G. Olbrechts, K. Wostyn, K. Clays and A. Persoons, Opt. Lett., 1999, 24, 403 CrossRef CAS PubMed; (f) K. Clays, K. Wostyn, G. Olbrechts, A. Persoons, A. Watanabe, K. Nogi, X.-M. Duan, S. Okada, H. Oikawa, H. Nakanishi, H. Vogel, D. Beljonne and J.-L. Brédas, J. Opt. Soc. Am. B, 2000, 17, 256 CrossRef CAS.
- (a) J. L. Oudar and D. S. Chemla, J. Chem. Phys., 1977, 66, 2664 CrossRef CAS; (b) J. L. Oudar, J. Chem. Phys., 1977, 67, 446 CrossRef CAS.
- (a) K. Tripathy, J. Pérez Moreno, M. G. Kuzyk, B. J. Coe, K. Clays and A. M. Kelley, J. Chem. Phys., 2004, 121, 7932 CrossRef CAS PubMed; (b) K. Clays and B. J. Coe, Chem. Mater., 2003, 15, 642 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic, crystallographic and other experimental details, CIF files. CCDC 1428590–1428592. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt00115g |
| ‡ AAY and NVS contributed equally to this manuscript. |
| § While comparison of β from different techniques/wavelengths must be made cautiously, by definition β0 from non-resonant measurements should be frequency independent. Moreover, there is generally good agreement between non-resonant, non-fluorescent/fluorescence corrected βzzz from HRS, and from EFISHG. See for example ref. 21e. |
| This journal is © The Royal Society of Chemistry 2016 |