
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unusual dimer formation of cyclometalated ruthenium NHC p-cymene complexes†
David
Schleicher
a,
Alexander
Tronnier
a,
Hendrik
Leopold
a,
Horst
Borrmann
b and
Thomas
Strassner
*a
aPhysikalische Organische Chemie, Technische Universität Dresden, 01069 Dresden, Germany. E-mail: thomas.strassner@chemie.tu-dresden.de
bMax-Planck-Institut für Chemische Physik fester Stoffe, 01187 Dresden, Germany. E-mail: horst.borrmann@cpfs.mpg.de
First published on 3rd February 2016
Abstract
We present the synthesis and structural characterization of novel ruthenium complexes containing C^C* cyclometalated N-heterocyclic carbene ligands, η6-arene (p-cymene) ligands and one bridging chlorine ion. Complexes of the general formula [Ru(p-cymene)(C^C*)Cl] were prepared via a one-pot synthesis using in situ transmetalation from the correspondent silver NHC complexes. These complexes react with sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (NaBArF4) to form dinuclear complexes of the general structure [Ru(p-cymene)(C^C*)-μ-Cl-(p-cymene)(C^C*)Ru]+[BArF4]−. Solid-state structures confirm that the pseudo-tetrahedral coordination around the metal center with the η6-ligand aligned perpendicularly to the C^C* ligand and the i-Pr group “atop” is retained in the bimetallic complexes.
Over the last few years ruthenium complexes, especially with strong electron-donating ligands like N-heterocyclic carbenes (NHC) and/or cyclometalating moieties, have gained wide-spread interest for their diverse catalytic activity, e.g. in transfer hydrogenation1 metathesis,2 and water oxidation.3 Especially NHC complexes allow for a wide range of steric and electronic control at the metal center and are more stable than other widely used neutral donors like phosphine ligands.1a,4 Cyclometalated Ru(II) complexes have also become more and more interesting as sensitizers in dye-sensitized solar cells.5 Here the cyclometalating ligands have several advantages such as raising the HOMO/LUMO levels for more efficient electron transport into the TiO2 substrate as well as from the redox pair responsible for the regeneration of the dye. The chelating effect also increases their long term stability compared to complexes with monodentate thiocyanate ligands. Moreover, in the last few years ruthenium arene complexes with either cyclometalating or NHC ligands were used in cytotoxicity and anticancer studies.6
Since our group has a long-standing interest in alkyl–aryl-imidazolium salts7 and their respective transition metal (especially palladium8 and platinum9) complexes, we developed a synthetic one-pot route to ruthenium complexes featuring these C^C* ligand motifs. It should be noted that quite recently the group of Wang published similar complexes10 as intermediates in the synthesis of highly substituted imidazolium salts following up on their earlier report on sp2 and sp3 CH-activation in ruthenium NHC complexes.11 Furthermore Choudhury et al. reported cyclometalated Ru(II) NHC complexes based on a pyridyl ligand for studies of electronic properties of dinuclear complexes.12
To study the reactivity of the [Ru(p-cymene)(C^C*)Cl] complexes, we wanted to remove the chlorine ligand. Reaction with the sodium salt of the bulky non-coordinating anion [BArF4]− – to our surprise – did not lead to the cationic species [Ru(p-cymene)(C^C*)]+ [BArF4]− and sodium chloride, but rather gave rise to the formation of a ruthenium μ-chloro bridged structure consisting of two ruthenium centers with one C^C* ligand and p-cymene each and only one bridging chlorine ion. The single positive charge of the product complex is compensated by one [BArF4]− counter ion. This type of ruthenium dimers with only one bridging ion – to the best of our knowledge – has been described only twice in the literature before. More than 40 years ago Haines et al. reported the synthesis of halogen-bridged ruthenium piano stool complexes with η6-benzene and carbon monoxide ligands.13 And quite recently Oestereich described the formation of a ruthenium dinuclear species bearing a chelating η6-arene-sulfur and a phosphine ligand, which was catalytically active in hydrodefluorination,14 while the groups of Peris and Crabtree reported a dinuclear ruthenium complex with one bridging chlorine ion and an additional bridging bis-NHC ligand.15
We present the synthetic one-pot route to the [Ru(p-cymene)(C^C*)Cl] complexes depicted in Scheme 1. It starts from a mixture of the widely used [RuCl2(η6-p-cymene)]2 precursor and the corresponding 1-alkyl-3-aryl imidazolium iodide in dry dichloromethane. Addition of one equivalent of silver(I) oxide to the stirred suspension at room temperature under inert atmosphere and exclusion of light results in the formation of the desired ruthenium compounds. It should be noted that an isolation of the intermediate Ag(NHC) complexes and subsequent transmetalation was not found to give any improvement in overall yields.
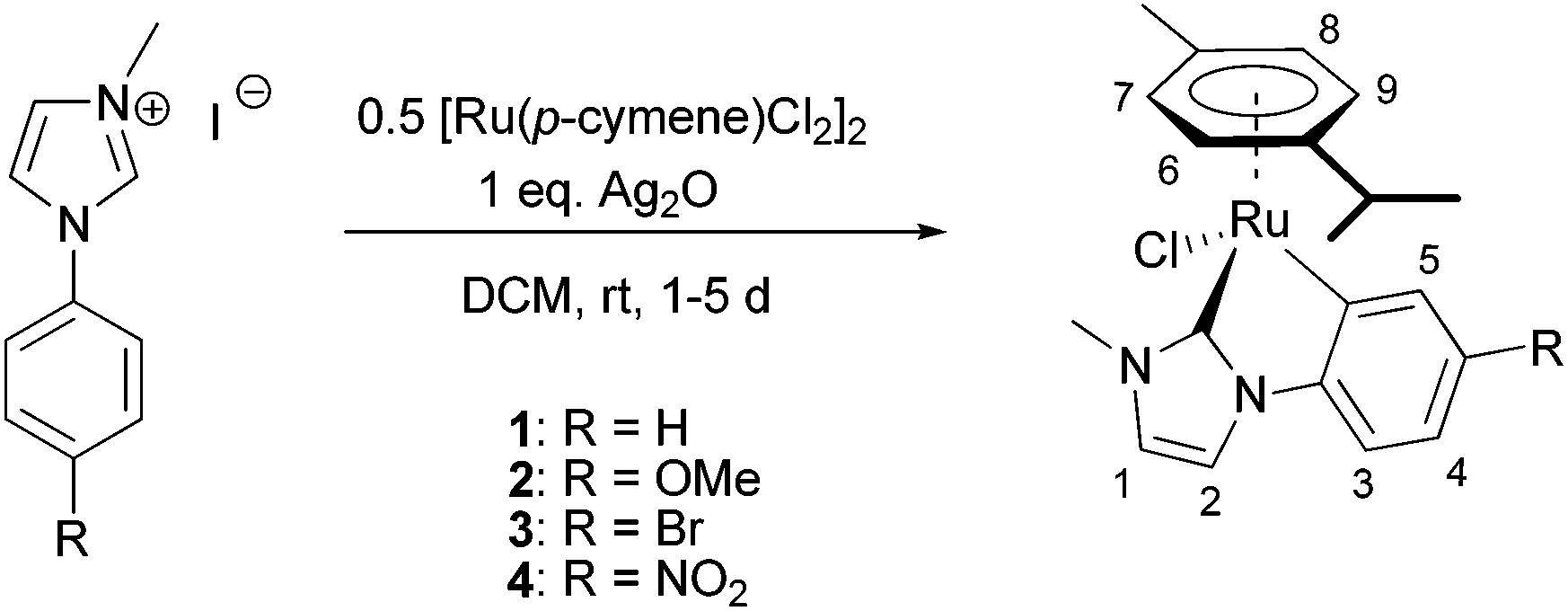 | ||
| Scheme 1 Synthetic route to cyclometalated Ru NHC complexes via transmetalation from silver carbene complexes; numbers correspond to NMR assignments. | ||
The crude product is (with the exception of 1) subjected to column chromatography (SiO2, DCM/MeOH 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1; yellow to orange band with Rf = 0.90–0.95) and the resulting yellow solid is dissolved in little amounts of THF, filtered over basic alumina, and slowly precipitated with pentane to give analytically pure samples.
:1; yellow to orange band with Rf = 0.90–0.95) and the resulting yellow solid is dissolved in little amounts of THF, filtered over basic alumina, and slowly precipitated with pentane to give analytically pure samples.
The isolated complexes are stable under ambient conditions. However, solutions in wet solvents (especially in slightly acidic ones like chloroform) slowly turn from yellow to green and ultimately to dark blue with complete decomposition of the complexes. These observations are in accordance to those reported by Albrecht et al.16 for similar dinuclear species.
Yellow-orange crystals of 1 and 2 were obtained by slow diffusion of pentane into a solution of the complex in a mixture of dichloromethane/diethyl ether. Their structures are shown in Fig. 1 and 2, crystallographic details can be found in the ESI (Tables S1 and S2†). The complexes show the typical “piano-stool” geometry with a pseudo-tetrahedral coordination sphere at the ruthenium center. The bite angle is significantly smaller than 90° (approx. 77°) resulting in a “yaw”-distortion17 at the carbene of 9.1° for 1 and 9.8° for 2.
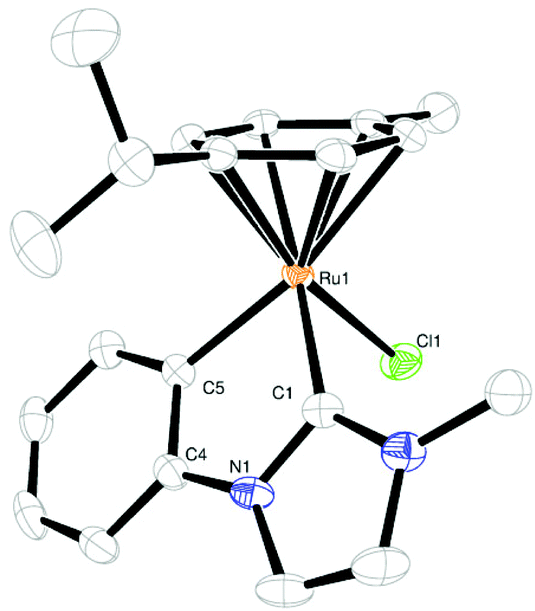 | ||
| Fig. 1 ORTEP of 1 in the solid state. Thermal ellipsoids are drawn at the 50% probability level, H atoms are omitted for clarity. Selected bond lengths [Å], angles and dihedral angles [°]: Ru(1)–Cl(1) 2.411(10); Ru(1)–C(1) 2.026(3); Ru(1)–C(5) 2.073(3); Ru(1)–Centroid (cymene) 1.727; C(1)–Ru(1)–C(5) 76.7(12); C(1)–N(1)–C(4)–C(5) −0.3(4). | ||
 | ||
| Fig. 2 ORTEP of 2 in the solid state. Thermal ellipsoids are drawn at the 50% probability level, H atoms are omitted for clarity. Selected bond lengths [Å], angles and dihedral angles [°]: Ru(1)–Cl(1) 2.4294(8); Ru(1)–C(1) 2.011(3); Ru(1)–C(5) 2.076(3); Ru(1)–Centroid 1.724; C(1)–Ru(1)–C(5) 77.25(11); C(1)–N(1)–C(4)–C(5) 0.7(3). | ||
Comparison of Ru–C bond lengths shows a slightly shorter distance to the carbene carbon atom than to the (anionic) phenyl carbon atom due to π-back donation. All these structural findings are in good agreement with previously reported similar complexes.11,16,18
The synthesis of the corresponding dimers (see Scheme 2) is again carried out in dry dichloromethane at room temperature using a slight excess (relative to 0.5 equivalents) of the BArF4 salt. Using 1 or 1.1 equivalents of the sodium salt gives the exact same complexes and still doesn't lead to the formation of the cationic ruthenium complexes. Prolonging the reaction time however causes slow decomposition of the complexes, which can be clearly observed by a darkening of the reaction mixture.
 | ||
| Scheme 2 Synthesis of the ruthenium dimers. | ||
The obtained complexes are readily soluble in diethyl ether, from which they can be crystallized by slow addition of pentane. Purification can be achieved by filtering the ether solutions consecutively through small pads of Celite and basic alumina. The dimers are air stable as solids, but solutions in solvents like chloroform, ethanol, and acetone turn blue quite rapidly. We confirmed the structure of the synthesized dinuclear compounds by 2D-NMR experiments, elemental analyses as well as solid state structure determination. NMR spectra of 5 and 6 (recorded in deuterated benzene) show that the signals for the C^C* ligand(s) don't shift significantly (<0.5 ppm) in comparison to the respective monomers. However, the signals for the aromatic protons of the p-cymene ring show a different pattern compared to the monomeric complexes. Two of the signals (two adjacent protons on “one side”) are shifted upfield by almost 2 ppm. This is evidence for their strong electronic shielding (in the “inner part” of the dinuclear complex) and clearly indicates that the dimers are also stable in solution. Complex 7 is barely soluble in benzene and we therefore measured the NMR spectra in deuterated acetonitrile. Interestingly, in this (coordinating) solvent different signal patterns are observed for the two metal centers (see ESI† for details), which is an indication of a slightly different arrangement of the η6 ligand. This effect can also be observed when dissolving complexes 5 or 6 in acetonitrile, but these solutions quite rapidly turn blue, showing decomposition.
The solid state structure of complex 6 (see Fig. 3, crystallographic details given in the ESI, Table S3†) shows that the geometry around both ruthenium centers is generally retained in regard to the monomeric structures. The complex still consists of two piano-stool “hemispheres” sharing the central chlorine ion. Bond distances and angles differ only slightly from the monomeric complexes. The bridging angle between both ruthenium atoms and the chlorine ion is very similar to the one reported by Peris (126.43(4)°)15 and significantly larger than the bridging angles in the [RuCl2(η6-p-cymene)]2 dimer, which are approx. 97–98°.19 One of the most interesting features about these dinuclear compounds is the opposing direction of the ligands around the two metal centers. In this way they can arrange in a sterically much more favorable position than with adjacent η6-arene ligands. Moreover, this leads to an almost ball-like geometry of the complex, which together with the perfectly spherical [BArF4] anions provides a good means for optimal packing in the crystal (see ESI, Fig. S1†).
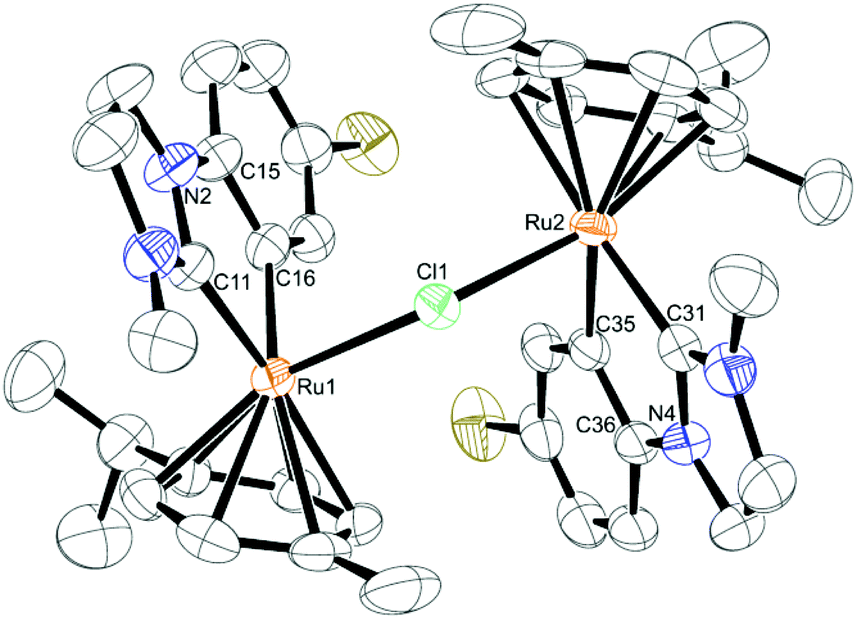 | ||
| Fig. 3 ORTEP of 6 in the solid state. Thermal ellipsoids are drawn at the 30% probability level, H atoms and the anion are omitted for clarity. Selected bond lengths [Å], angles and dihedral angles [°]: Ru(1)–Cl(1) 2.4490(9); Ru(2)–Cl(1) 2.4467(9); Ru(1)–C(11) 2.035(4); Ru(1)–C(16) 2.073(4); Ru(2)–C(31) 2.050(4); Ru(2)–C(35) 2.075(3); Ru(1)–Centroid (cymene) 1.735; Ru(2)–Centroid (cymene) 1.733; Ru(2)–Cl(1)–Ru(1) 129.28(4); C(11)–Ru(1)–C(16) 76.94(16); C(31)–Ru(2)–C(35) 76.30(16); C(11)–N(2)–C(15)–C(16) 2.2(6); C(31)–N(4)–C(36)–C(35) −0.4(5). | ||
In conclusion we prepared cyclometalated ruthenium NHC complexes via a one-pot transmetalation route. The [Ru(p-cymene)(C^C*)Cl] complexes are generally obtained in moderate to good yields and can be stored under ambient conditions.
Abstraction of the halide ligand proved to be difficult and several synthetic strategies were not successful. However, the reaction of these complexes with sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate lead to the formation of a novel class of ruthenium(II) dimers bridged only by a single chlorine ion. Their molecular structure, which was unambiguously confirmed by several analytical methods, shows a distorted pseudo-tetrahedral geometry around the ruthenium centers and weak CH–π interactions between the aromatic protons of the cymene ligand and the cyclometalated ring on the opposite ruthenium center.
Notes and references
- (a) V. Dragutan, I. Dragutan, L. Delaude and A. Demonceau, Coord. Chem. Rev., 2007, 251, 765–794 CrossRef CAS; (b) N. Pannetier, J.-B. Sortais, J.-T. Issenhuth, L. Barloy, C. Sirlin, A. Holuigue, L. Lefort, L. Panella, J. G. de Vries and M. Pfeffer, Adv. Synth. Catal., 2011, 353, 2844–2852 CrossRef CAS; (c) H. D. Velazquez and F. Verpoort, Chem. Soc. Rev., 2012, 41, 7032–7060 RSC; (d) W. B. Cross, C. G. Daly, Y. Boutadla and K. Singh, Dalton Trans., 2011, 40, 9722–9730 RSC; (e) M. Delgado-Rebollo, D. Canseco-Gonzalez, M. Hollering, H. Mueller-Bunz and M. Albrecht, Dalton Trans., 2014, 43, 4462–4473 RSC; (f) N. Ding and T. S. A. Hor, Dalton Trans., 2010, 39, 10179–10179 RSC; (g) H. Ohara, W. W. N. O, A. J. Lough and R. H. Morris, Dalton Trans., 2012, 41, 8797–8808 RSC; (h) X.-H. Zhu, L.-H. Cai, C.-X. Wang, Y.-N. Wang, X.-Q. Guo and X.-F. Hou, J. Mol. Catal. A: Chem., 2014, 393, 134–141 CrossRef CAS; (i) S. Enthaler, R. Jackstell, B. Hagemann, K. Junge, G. Erre and M. Beller, J. Organomet. Chem., 2006, 691, 4652–4659 CrossRef CAS; (j) J. DePasquale, M. Kumar, M. Zeller and E. T. Papish, Organometallics, 2013, 32, 966–979 CrossRef CAS; (k) F. E. Fernández, M. C. Puerta and P. Valerga, Organometallics, 2012, 31, 6868–6879 CrossRef; (l) D. Gnanamgari, E. L. O. Sauer, N. D. Schley, C. Butler, C. D. Incarvito and R. H. Crabtree, Organometallics, 2009, 28, 321–325 CrossRef CAS; (m) N. Pannetier, J.-B. Sortais, P. S. Dieng, L. Barloy, C. Sirlin and M. Pfeffer, Organometallics, 2008, 27, 5852–5859 CrossRef CAS; (n) Y.-B. Lai, C.-S. Lee, W.-J. Lin, A. R. Naziruddin and W.-S. Hwang, Polyhedron, 2013, 53, 243–248 CrossRef CAS.
- (a) R. Kadyrov, Chem. – Eur. J., 2013, 19, 1002–1012 CrossRef CAS PubMed; (b) X. Sauvage, Y. Borguet, A. F. Noels, L. Delaude and A. Demonceau, Adv. Synth. Catal., 2007, 349, 255–265 CrossRef CAS; (c) C. Lübbe, A. Dumrath, H. Neumann, M. Beller and R. Kadyrov, ChemCatChem, 2014, 6, 105–108 CrossRef; (d) Y. Kong, S. Xu, H. Song and B. Wang, Organometallics, 2012, 31, 5527–5532 CrossRef CAS.
- L. Bernet, R. Lalrempuia, W. Ghattas, H. Mueller-Bunz, L. Vigara, A. Llobet and M. Albrecht, Chem. Commun., 2011, 47, 8058–8060 RSC.
- (a) V. Dragutan and I. Dragutan, Platinum Metals Rev., 2005, 49, 123–137 CrossRef CAS; (b) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- (a) T. Funaki, H. Kusama, N. Onozawa-Komatsuzaki, K. Kasuga, K. Sayama and H. Sugihara, Eur. J. Inorg. Chem., 2014, 2014, 1303–1311 CrossRef CAS; (b) S. H. Wadman, J. M. Kroon, K. Bakker, M. Lutz, A. L. Spek, G. P. M. van Klink and G. van Koten, Chem. Commun., 2007, 1907–1909 RSC; (c) T. Bessho, E. Yoneda, J.-H. Yum, M. Guglielmi, I. Tavernelli, H. Imai, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2009, 131, 5930–5934 CrossRef CAS PubMed; (d) K. C. D. Robson, B. D. Koivisto, A. Yella, B. Sporinova, M. K. Nazeeruddin, T. Baumgartner, M. Grätzel and C. P. Berlinguette, Inorg. Chem., 2011, 50, 5494–5508 CrossRef CAS PubMed; (e) K. C. D. Robson, P. G. Bomben and C. P. Berlinguette, Dalton Trans., 2012, 41, 7814–7829 RSC; (f) P. G. Bomben, T. J. Gordon, E. Schott and C. P. Berlinguette, Angew. Chem., Int. Ed., 2011, 50, 10682–10685 CrossRef CAS PubMed; (g) P. G. Bomben, J. Borau-Garcia and C. P. Berlinguette, Chem. Commun., 2012, 48, 5599–5601 RSC; (h) P. G. Bomben, K. C. D. Robson, B. D. Koivisto and C. P. Berlinguette, Coord. Chem. Rev., 2012, 256, 1438–1450 CrossRef CAS; (i) P. G. Bomben, K. D. Thériault and C. P. Berlinguette, Eur. J. Inorg. Chem., 2011, 2011, 1806–1814 CrossRef; (j) T. Funaki, H. Funakoshi, O. Kitao, N. Onozawa-Komatsuzaki, K. Kasuga, K. Sayama and H. Sugihara, Angew. Chem., Int. Ed., 2012, 51, 7528–7531 CrossRef CAS PubMed; (k) B. Schulze, D. Escudero, C. Friebe, R. Siebert, H. Görls, S. Sinn, M. Thomas, S. Mai, J. Popp, B. Dietzek, L. González and U. S. Schubert, Chem. – Eur. J., 2012, 18, 4010–4025 CrossRef CAS PubMed; (l) C.-W. Hsu, S.-T. Ho, K.-L. Wu, Y. Chi, S.-H. Liu and P.-T. Chou, Energy Environ. Sci., 2012, 5, 7549–7554 RSC; (m) J.-J. Kim, H. Choi, S. Paek, C. Kim, K. Lim, M.-J. Ju, H. S. Kang, M.-S. Kang and J. Ko, Inorg. Chem., 2011, 50, 11340–11347 CrossRef CAS PubMed.
- (a) G. Lv, L. Guo, L. Qiu, H. Yang, T. Wang, H. Liu and J. Lin, Dalton Trans., 2015, 44, 7324–7331 RSC; (b) K. J. Kilpin, S. Crot, T. Riedel, J. A. Kitchen and P. J. Dyson, Dalton Trans., 2013, 43, 1443–1443 RSC; (c) L. Oehninger, M. Stefanopoulou, H. Alborzinia, J. Schur, S. Ludewig, K. Namikawa, A. Munoz-Castro, R. W. Koster, K. Baumann, S. Wolfl, W. S. Sheldrick and I. Ott, Dalton Trans., 2013, 1657–1666 RSC; (d) J. Dinda, S. D. Adhikary, G. Roymahapatra, K. K. Nakka and M. K. Santra, Inorg. Chim. Acta, 2014, 413, 23–31 CrossRef CAS; (e) F. Hackenberg, H. Müller-Bunz, R. Smith, W. Streciwilk, X. Zhu and M. Tacke, Organometallics, 2013, 32, 5551–5560 CrossRef CAS; (f) B. Pena, A. David, C. Pavani, M. S. Baptista, J.-P. Pellois, C. Turro and K. R. Dunbar, Organometallics, 2014, 33, 1100–1103 CrossRef CAS; (g) B. A. Albani, B. Peña, K. R. Dunbar and C. Turro, Photochem. Photobiol. Sci., 2014, 13, 272–272 RSC.
- S. Ahrens, A. Peritz and T. Strassner, Angew. Chem., Int. Ed., 2009, 48, 7908–7910 CrossRef CAS PubMed.
- D. Munz, A. Poethig, A. Tronnier and T. Strassner, Dalton Trans., 2013, 42, 7297–7297 RSC.
- Y. Unger, D. Meyer, O. Molt, C. Schildknecht, I. Münster, G. Wagenblast and T. Strassner, Angew. Chem., Int. Ed., 2010, 49, 10214–10216 CrossRef CAS PubMed.
- C. Ma, C. Ai, Z. Li, B. Li, H. Song, S. Xu and B. Wang, Organometallics, 2014, 33, 5164–5172 CrossRef CAS.
- C. Zhang, Y. Zhao, B. Li, H. Song, S. Xu and B. Wang, Dalton Trans., 2009, 5182–5189 RSC.
- M. Mondal, T. K. Ranjeesh, S. K. Gupta and J. Choudhury, Dalton Trans., 2014, 43, 9356–9356 RSC.
- R. J. Haines and A. L. Du Preez, J. Chem. Soc., Dalton Trans., 1972, 944–948 RSC.
- T. Stahl, H. F. T. Klare and M. Oestreich, J. Am. Chem. Soc., 2013, 135, 1248–1251 CrossRef CAS PubMed.
- M. Poyatos, W. McNamara, C. Incarvito, E. Clot, E. Peris and R. H. Crabtree, Organometallics, 2008, 27, 2128–2136 CrossRef CAS.
- L. Mercs, A. Neels, H. Stoeckli-Evans and M. Albrecht, Inorg. Chem., 2011, 50, 8188–8196 CrossRef CAS PubMed.
- C. H. Leung, C. D. Incarvito and R. H. Crabtree, Organometallics, 2006, 25, 6099–6107 CrossRef CAS.
- (a) D. Enders, H. Gielen, G. Raabe, J. Runsink and H. Henrique Teles, Chem. Ber., 1997, 130, 1253–1260 CrossRef CAS; (b) F. Simal, D. Jan, L. Delaude, A. Demonceau, M.-R. Spirlet and A. F. Noels, Can. J. Chem., 2001, 79, 529–535 CrossRef CAS; (c) K. Ogata, S. Inomata and S.-I. Fukuzawa, Dalton Trans., 2013, 42, 2362–2362 RSC.
- C. S. Allardyce, P. J. Dyson, D. J. Ellis, P. A. Salter and R. Scopelliti, J. Organomet. Chem., 2003, 668, 35–42 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental and crystallographic methods as well as NMR data of the complexes. CCDC 1441834, 1441835 and 1441914. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt00100a |
| This journal is © The Royal Society of Chemistry 2016 |