
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Extraction and coordination studies of a carbonyl–phosphine oxide scorpionate ligand with uranyl and lanthanide(III) nitrates: structural, spectroscopic and DFT characterization of the complexes†
Anna G.
Matveeva
*a,
Anna V.
Vologzhanina
a,
Evgenii I.
Goryunov
a,
Rinat R.
Aysin
a,
Margarita P.
Pasechnik
a,
Sergey V.
Matveev
a,
Ivan A.
Godovikov
a,
Alfiya M.
Safiulina
b and
Valery K.
Brel
a
aNesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, 28 Vavilov Str., Moscow 119991, Russia
bOJSC United Chemical Company Uralchem, Presnenskaya nab. 6/2, Moscow, 123317 Russia. E-mail: matveeva@ineos.ac.ru; Fax: +7-4991355085; Tel: +7-4991359366
First published on 18th February 2016
Abstract
Hybrid scorpionate ligand (OPPh2)2CHCH2C(O)Me (L) was synthesized and characterized by spectroscopic methods and X-ray diffraction. The selected coordination chemistry of L with UO2(NO3)2 and Ln(NO3)3 (Ln = La, Nd, Lu) has been evaluated. The isolated mono- and binuclear complexes, namely, [UO2(NO3)2L] (1), [{UO2(NO3)L}2(μ2-O2)]·EtOH (2), [La(NO3)3L2]·2.33MeCN (3), [Nd(NO3)3L2]·3MeCN (4), [Nd(NO3)2L2]+·(NO3)−·EtOH (5) and [Lu(NO3)3L2] (6) have been characterized by IR spectroscopy and elemental analysis. Single-crystal X-ray structures have been determined for complexes 1–5. Intramolecular intraligand π-stacking interactions between two phenyl fragments of the coordinated ligand(s) were observed in all complexes 1–5. The π-stacking interaction energy was estimated from Bader's AIM theory calculations performed at the DFT level. Solution properties have been examined using IR and multinuclear (1H, 13C, and 31P) NMR spectroscopy in CD3CN and CDCl3. Coordination modes of L vary with the coordination polyhedron of the metal and solvent nature showing many coordination modes: P(O),P(O), P(O),P(O),C(O), P(O),C(O), and P(O). Preliminary extraction studies of U(VI) and Ln(III) (Ln = La, Nd, Ho, Yb) from 3.75 M HNO3 into CHCl3 show that scorpionate L extracts f-block elements (especially uranium) better than its unmodified prototype (OPPh2)2CH2.
1. Introduction
Bidentate neutral organophosphorus extractants, first of all phosphine oxides (carbamoylphosphine oxides, alkylenediphosphine dioxides), are the most efficient extractants for the recovery of transplutonium, rare earth, and other elements from the waste of spent nuclear fuel reprocessing, to recover different metals from processing solutions of hydrometallurgy and to design analytical test objects for the same metals.1–4 These compounds are also important in medicine for the diagnosis and treatment of different pathologies, mainly affecting the locomotor apparatus. The design of novel functionalized phosphine oxides showing higher efficiency and selectivity is one of the major and topical fields of extractive and synthetic chemistry.Thus, highly efficient extractants for the recovery of actinides and rare earth elements from nitric acid solutions were found among derivatives of methylenediphosphine dioxide2–5 [Scheme 1(a) and (b)]. This fact favored the development of the coordination chemistry of these ligands, in particular their ability to coordinate ions of f-block elements.6–8
 | ||
| Scheme 1 Structures of different types of ligands bearing >P(O)CH2P(O)<. | ||
The introduction of Ar2P(O)CH2 and Ar2PCH2 functional groups showing coordination ability into the methylene bridge of dioxide [Scheme 1, (a)] leads to new scorpionate ligands [Scheme 1, (c)–(d),)] that are promising extractants for nuclear fuel reprocessing.9 All strongly donating Ar2P(O) and Ar2P groups of these scorpionates, as expected, participate in coordination to lanthanide-like Y(III).9 The coordination properties of such a scorpionate ligand change considerably if the donor phosphorus-containing group in substituent Ar2P(O)CH2–, Ar2PCH2– is replaced by a less basic AlkC(O)– group [Scheme 1, (e)]. Less basic carbonyl groups may not form coordination bonds with cations or form weaker bonds than phosphorus functionalities, as well as participate in other weak interactions. Consequently, such a modification can considerably change not only coordination, but also the extractive properties of the ligand.
In this paper, we report the modified synthesis of a scorpionate ligand [Ph2P(O)]2CHCH2C(O)Me (L) and its new complexes with uranyl and lanthanide(III) nitrates, the structural characterization of all compounds in the solid state (X-ray crystallography for L, 1–5) and in solution by IR and multinuclear NMR (1H, 13C, 31P) spectroscopy, and extraction studies towards the f-block elements. Furthermore, we report herein the results of AIM analysis (Bader's “Atoms in molecules” approach) for the π-stacking interactions in the U(VI), La(III), and Nd(III) complexes. Extraction ability of ligand L for the recovery of U(VI) and Ln(III) from nitric acid solution into chloroform in comparison with the Ph2P(O)CH2P(O)Ph2 (L′) prototype was evaluated.
2. Results and discussion
2.1. Synthesis and characterization of the ligand L
We prepared compound Lvia a modified variant10 of the Conant reaction11 that consists of combining Ph2PCl with (E)-4-(diphenylphosphoryl)but-3-en-2-one, we developed the synthesis of the latter earlier.12 The reaction was conducted in anhydrous acetonitrile solution at ambient temperature with the addition of acetic acid (Scheme 2). Under these conditions, the reaction was completed over 48 h to give 4,4-bis(diphenylphosphoryl)butan-2-one (L) in 90% yield (according to 31P{1H} NMR spectra of the reaction mixture). It should be noted that this is the first example of the successful use of the Conant reaction for the synthesis of gem-diphosphoryl-substituted alkanones. | ||
| Scheme 2 Synthesis of 4,4-bis(diphenylphosphoryl)butan-2-one (L). | ||
The ligand L has been characterized by elemental analysis, IR, 1H, 31P{1H}, and 13C NMR spectroscopy. Thus, in particular, the 1H NMR spectrum of this compound, whose molecule contains two identical diphenylphosphoryl groups, shows the proton signals of the CH2 and CH groups as a doublet of triplets and triplet of triplets, respectively, which transform into a doublet and triplet under broad band 1H–31P decoupling (1H{31P} NMR spectrum). The 13C{1H} NMR spectrum also shows triplets of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CH carbon atoms due to spin–spin coupling with the phosphorus nuclei of two Ph2P(O) groups. The IR spectrum of a crystalline sample of L exhibits ν(PO) bands at 1202 and 1182 cm−1 and a ν(CO) band at 1720 cm−1. The DFT computational data for the normal vibration frequencies of ligand L agree well with the experimental values without any scaling.13
O and CH carbon atoms due to spin–spin coupling with the phosphorus nuclei of two Ph2P(O) groups. The IR spectrum of a crystalline sample of L exhibits ν(PO) bands at 1202 and 1182 cm−1 and a ν(CO) band at 1720 cm−1. The DFT computational data for the normal vibration frequencies of ligand L agree well with the experimental values without any scaling.13
In addition to spectral experiments, compound L was characterized in the solid state using single-crystal X-ray diffraction. Scorpionate ligand L displays typical bond lengths and angles, with the oxygen atoms of the phosphoryl groups trans-situated with respect to the C13 atom (Fig. 1). Selected bond distances are given in Table 1. Such a conformation of L is additionally stabilized with π-stacking between the phenyl groups of different phosphorus functionalities (the centroid–centroid distance is equal to 3.681(2) Å and the dihedral angle between the ring planes is equal to 10.81(8)°). Thus, the realization of a bi- and tridentate coordination mode for this ligand requires the rotation of donor arms as compared with its conformation in the solid state. All three donor groups are involved in weak intra- and intermolecular C–H⋯O interactions. Intermolecular CPh–H⋯O contacts are the most significant. The shortest r(H⋯O) distances are 2.60 Å for the carbonyl group, 2.28, 2.38 Å (for P1–O1) and 2.46, 2.62 Å (for P2–O2) for the phosphoryl groups (Fig. S1, ESI†).
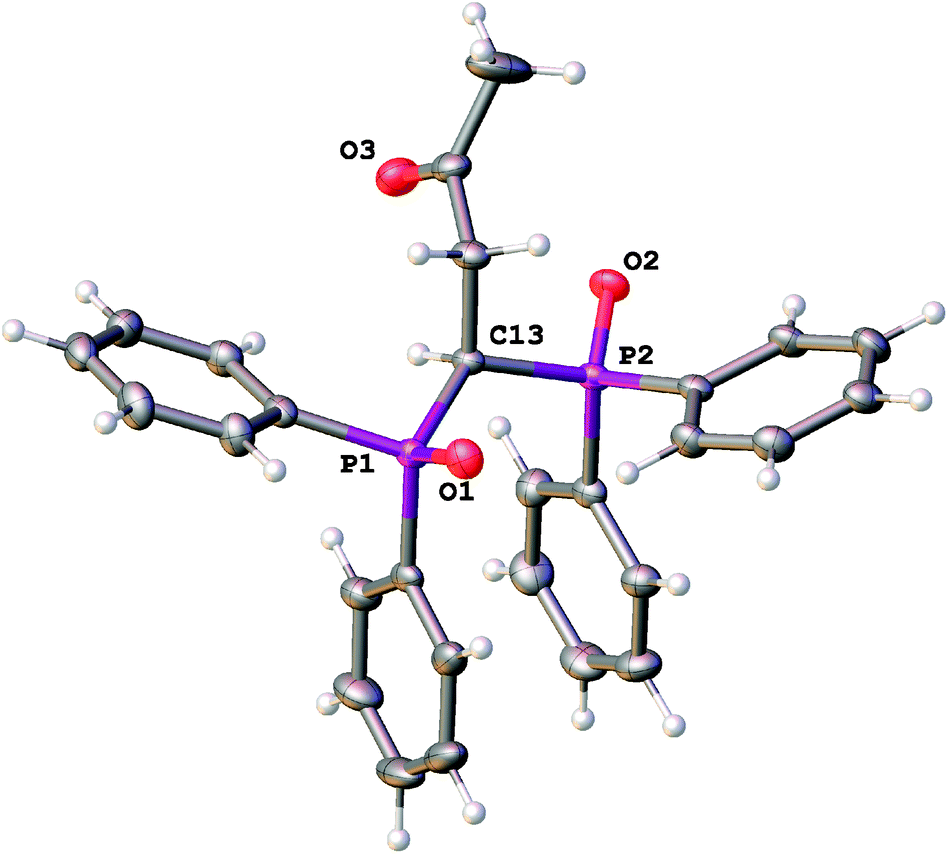 | ||
| Fig. 1 General view of L with the atoms represented as thermal ellipsoids drawn at p = 50%. | ||
| Bond | L | 1 | 2 |
|---|---|---|---|
| P1O1 |
1.485(1) | 1.500(2) | 1.487(6) |
| P2O2 |
1.493(1) | 1.503(2) | 1.495(6) |
| CO |
1.208(2) | 1.213(4) | 1.202(12) |
| U1O1 |
1.761(2) | 1.877(11) | |
| U1O2 |
1.760(2) | 1.658(12) | |
| U1–O3(L)/O9(L) | 2.393(2) | 2.397(6) | |
| U1–O4(L)/O8(L) | 2.398(2) | 2.463(6) | |
| U1–O(nitrate) | 2.506(3)–2.540(3) | 2.520(7)–2.549(7) | |
| U1–O(peroxo) | 2.361(12)–2.407(12) |
As far as we know, compound L is the first example of a hybrid scorpionate ligand combining two P(O) and one C(O) side arms that has ever been characterized in the solid state.
2.2. Synthesis and solid state characterization of the complexes
Compound L is a hybrid scorpionate ligand with a combination of two phosphoryl and one carbonyl groups. Therefore the coordination behavior of this ligand is interesting since the free rotation of donor arms can give a different possible combination of coordination modes. Various possible coordination modes of L, as depicted in Scheme 3, can be observed for mononuclear complexes.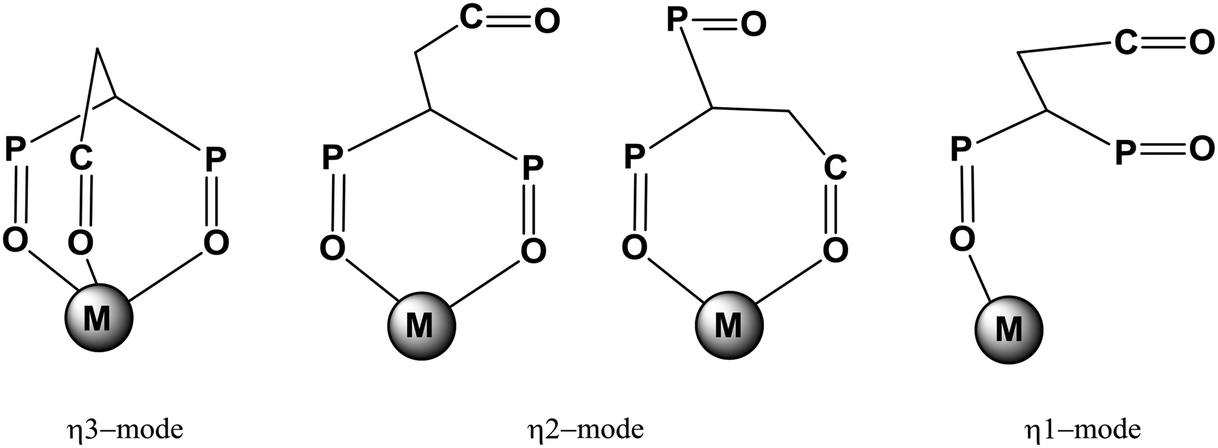 | ||
| Scheme 3 Possible coordination modes of ligand L in mononuclear complexes. | ||
C(O)-Monodentate coordination is the least probable (not shown in the Scheme). Obviously, the metal and the composition of the complex affect the choice of coordination mode. The f-block element coordination chemistry of this scorpionate ligand was studied in order to understand its coordination behavior.
Mononuclear complex [UO2(L)(NO3)2] (1) was prepared by addition of 1 equivalent of UO2(NO3)2·(H2O)6 in acetonitrile to 1 equivalent of compound L in chloroform to form a bright yellow microcrystalline powder. Crystallization of an ethanolic solution of 1 when exposed to sunlight yielded a trace amount of a yellow product with structural analyses consistent with the formula [{UO2(NO3)L}2(μ2-O2)]·EtOH (2). The appearance of the bidentate peroxo [O2]2− anion in uranyl nitrate solutions in the presence of atmospheric dioxygen is possible due to the sunlight photolysis of EtOH.14 The mechanism of this photolysis has been previously reported.15
Mononuclear bisligand complexes [La(NO3)3L2]·2.33MeCN (3), [Nd(NO3)3L2]·3MeCN (4), [Nd(NO3)2L2]·(NO3)·EtOH (5) and [Lu(NO3)3L2] (6) isolated in a pure state were obtained by combining stoichiometric amounts of the ligand and the salts in a mixture of aprotic solvents followed by crystallization from the corresponding solvent.
The composition and structures of the complexes in the solid state were studied using elemental analysis, and IR spectroscopy. The structures of the crystal complexes 1–5 were also elucidated by X-ray diffraction.
O group is oriented in the opposite direction with respect to the uranium atom).
 | ||
| Fig. 2 General view of 1 with the atoms represented as thermal ellipsoids drawn at p = 50%. | ||
The absence of strong intermolecular interactions that involve the oxygen atoms of the uranyl group, results in its linearity (the O1–U1–O2 angle is equal to 179.9(1)°) and similarity of the UO distances (1.760(2) and 1.761(2)Å) in 1. In complex [UO2(NO3)2L′] (7)7 (where L′ is Ph2P(O)CH2P(O)Ph2), which has a similar molecular structure to 1, the UO distances differ rather considerably, 1.781(11) and 1.768(11) Å, due to the presence of intermolecular C–H⋯O1 bonding. Complex 1 also involves intermolecular contacts C–H⋯OC, with r(H⋯O) distances in them being virtually the same as in the structure of the ligand (Fig. S2, ESI†).
Neutral dinuclear complex 2 (Fig. 3) contains only half of the complex in the asymmetric unit. L acts as a bidentate chelate P(O),P(O)-ligand and the other equatorial positions of the uranium(VI) atom in 2 are occupied by the oxygen atoms of the bidentate chelate nitrate anion and the bridging bidentate peroxo anion.
 | ||
| Fig. 3 General view of 2 with the atoms represented as thermal ellipsoids drawn at p = 50%. Uranyl oxygen atoms, the peroxo group and an oxygen atom of the nitrate anion are disordered over two sites, and only one of the disordered components is depicted. | ||
The resulting coordination polyhedron UO8 adopts a hexagonal bipyramidal geometry with the uranyl oxygen atoms in apical positions. Two nitrates (and two L ligands) are trans-situated to each other with respect to the UO2U species as it was previously observed in complexes with a similar [{UO2(NO3)X}2(μ2-O2)] composition, where X is the bidentate chelate neutral ligand (X = tetraethylsuccinamide,16 2,2′-bipyridyl17 or 5,5′-dimethyl-2,2′-bipyridine18). The severe disorder of the peroxo, NO3 and uranyl groups in 2 provides no possibility to analyze its molecular geometry in detail, but the mutual disposition of the constituting moieties can still be assessed. Particularly, the equatorial planes of two UO8 polyhedra connected through the peroxo anion are not parallel (the corresponding dihedral angle is ca. 21°), and deviate from planarity. Although this deviation is a rare case, it was previously reported for several peroxo complexes of uranium.19
Compounds 3 and 4 are isostructural, although the number of solvate molecules in their structures obtained from X-ray diffraction is not equal, probably due to the loss of solvent molecules in air. These contain only half of the molecule in the asymmetric unit with Ln, N1 and O5 atoms situated on a two-fold rotation axis. The Ln atom in 3 and 4 coordinates three nitrate anions and two L ligands in a bidentate chelate mode to form a LnO10 coordination polyhedron (the molecular view of 3 is given in Fig. 4 as an example). The nitrate anions are situated in a T-shape according to the metal atom with the NLnN angles slightly deviating from 90° (the angle value is equal to 83.2(2) and 83.0(2)°) due to the electronic and steric effects of the L ligands.
 | ||
| Fig. 4 General view of molecule 3 with the atoms represented as thermal ellipsoids drawn at p = 50%. | ||
The resulting polyhedron forms a pseudo-capped trigonal prism with the N1 atom in the capped position, if the nitrate anions are regarded as one polyhedron vertex (Fig. 5).
 | ||
| Fig. 5 Visualization of the pseudo-capped trigonal prismatic environment around La(III) in 3. | ||
In the closely related family of lanthanide nitrate complexes with L′, the compounds were characterized to be of the compositions [Ln(NO3)3(L′)2]·solv (Ln = La, solv = EtOH; Ln = Ce, solv = acetone), [Ln(NO3)2(H2O)(L′)2][Ln(NO3)4L′]·MeCN (Ln = Pr, Eu), [Ln(NO3)2(H2O)(L′)2](NO3)·solv (Ln = Nd, Gd, solv = H2O; Ln = Ho, solv = EtOH) and [Ln(NO3)(L′)3](NO3)2·solv (Ln = Gd, Yb; Ln = Gd, solv = EtOH).6 Thus, although a lanthanide cation can coordinate up to four bis(diphenylphosphino)methane dioxide ligands (as in the structure of [Eu(L′)4](ClO4)3·2 H2O),20 nitrate anions compete with those for a place in the lanthanide coordination sphere. The ratio Ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NO3:L′ (or L) = 1:3:2 is expected only for light elements (La, Ce); lanthanide contraction is accompanied by a decrease in this ratio as 1:2:2 (Ln = Pr–Ho) or as 1:1:3 (Ln = Gd–Yb). Thus, coordination isomerism is possible in this series.
:NO3:L′ (or L) = 1:3:2 is expected only for light elements (La, Ce); lanthanide contraction is accompanied by a decrease in this ratio as 1:2:2 (Ln = Pr–Ho) or as 1:1:3 (Ln = Gd–Yb). Thus, coordination isomerism is possible in this series.
Indeed, we succeeded to obtain complex 5 from ethanol for which the ratio is Nd:NO3:L = 1:2:2 (Fig. 6).
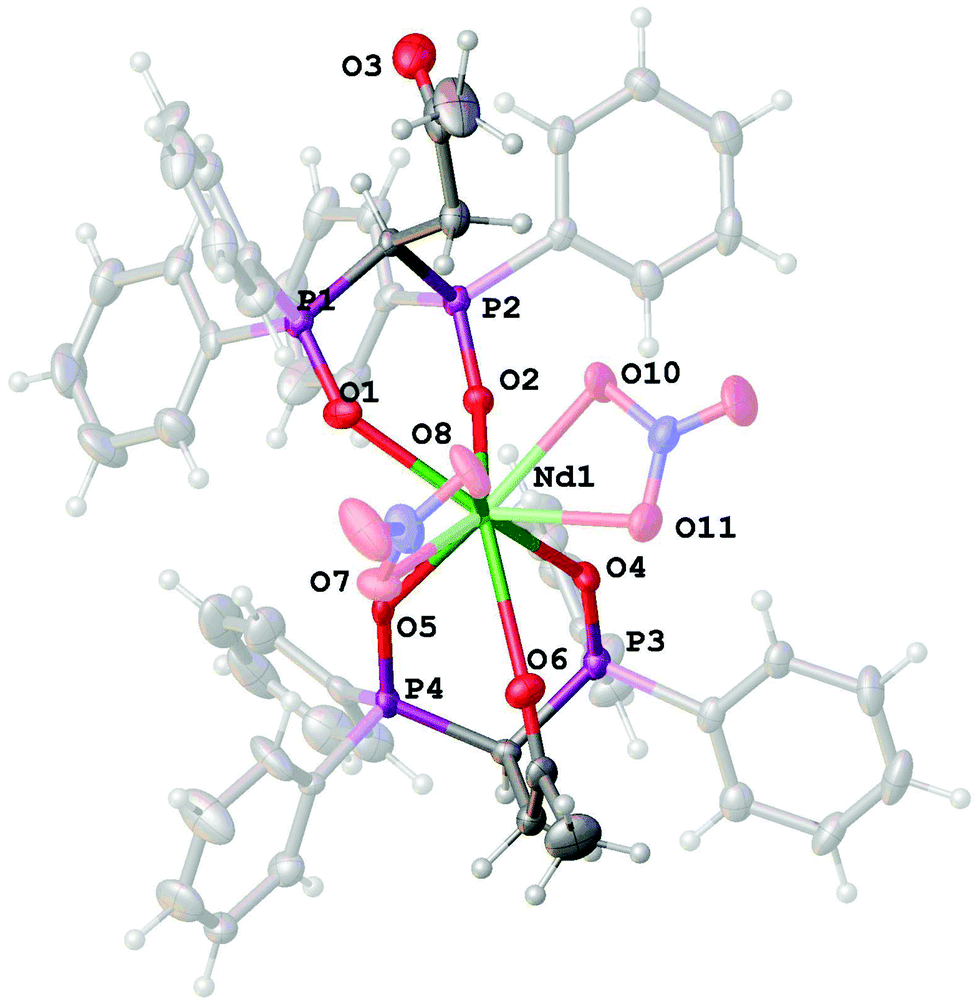 | ||
| Fig. 6 General view of the complex cation [Nd(NO3)2L2]+ in the structure of 5 with the atoms represented as thermal ellipsoids drawn at p = 50%. | ||
It contains two L ligands in both the P(O),P(O)-bidentate and tridentate coordination modes, and only two nitrate anions coordinated by the neodymium atom. The NdO9 polyhedron adopts a tricapped trigonal prismatic geometry (Fig. 7).
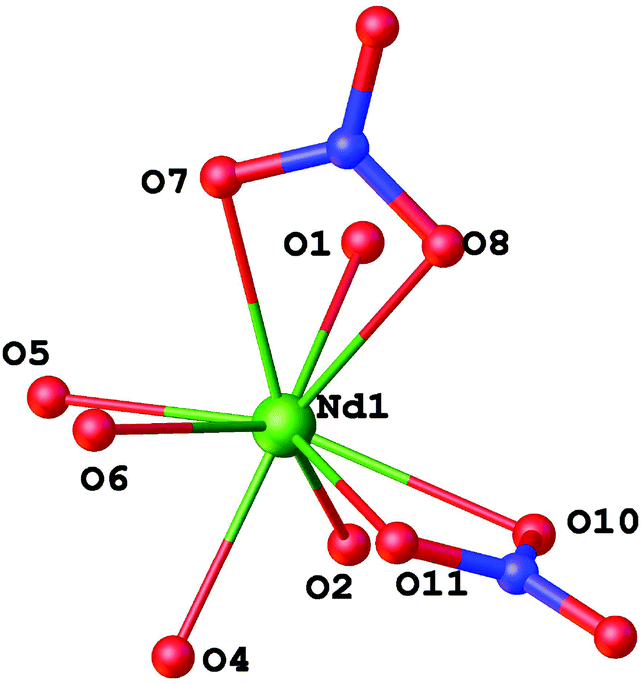 | ||
| Fig. 7 Visualization of the tricapped trigonal prismatic environment around Nd(III) in 5. | ||
For compounds 3–5, the Ln–O(L) bond distances are shorter than those for the Ln–O(nitrate) bonds (Table 2); and the latter bonds are typically alternated. In 5, the Nd–O(C) bond is the longest, and, hence, the weakest among the coordination bonds. The lanthanide contraction for isostructural compounds 3 and 4 is expressed as a shortening of the respective coordination bonds.
| Bond | 3 (La) | 4 (Nd) | 5 (Nd) |
|---|---|---|---|
| Ln–O1(L) | 2.541(6) | 2.5016(9) | 2.462(5) |
| Ln–O2(L) | 2.501(7) | 2.4573(9) | 2.410(4) |
| Ln–O4(L) | 2.389(4) | ||
| Ln–O5(L) | 2.405(5) | ||
| Ln–O(C) | 2.656(5) | ||
| CO (free) |
1.202(13) | 1.214(2) | 1.217(9) |
| CO (coordinated) |
1.222(7) | ||
| Ln–O (nitrate) | 2.599(8)–2.653(6) | 2.555(1)–2.609(1) | 2.536(5)–2.551(4) |
| P1–O1 | 1.491(7) | 1.4957(9) | 1.514(5) |
| P2–O2 | 1.504(7) | 1.5010(9) | 1.492(4) |
| P3–O4 | 1.496(4) | ||
| P4–O5 | 1.505(4) |
The coordination of phosphoryl and acetyl groups does not affect the lengths of the PO and CO bonds, although the oxygen atom of the longest PO bond in 5 is involved in O–H⋯O hydrogen bonding with ethanol molecules (r(O⋯O) = 3.844(8) Å, ∠(OHO) = 165.9°) (Fig. S4, ESI†). With the exception of one ligand in 5, the acetyl group of L in complexes 1–5 is involved in the CH⋯OC intra- and intermolecular bonding (Fig. S1–S4, ESI†). The most significant noncovalent CH⋯OC interactions are observed in complexes 3 and 4 with the shortest O⋯H distances 2.53–2.55 Å and corresponding value of the CHO angle ∼145° (Fig. S3, ESI†). In our opinion, the presence of an uncoordinated acetyl group available for complex⋯solvate hydrogen bonding could be the reason for the enhanced extraction ability of L compared with L′. To reveal the crystal packing effects on the IR spectra of 1 and 3–5 in the solid state, we analyzed the closest environment of these complexes. The O–H⋯O bond in 5 should affect the PO vibrations. Moreover, one can expect the effect of weak intermolecular C–H⋯OC interactions (for 3 and 4) on acetyl group vibration.
Along with numerous C–H⋯O intramolecular bonds, intramolecular π-bonding can be suggested for 1–5 that could affect the stability of complexes. These are analyzed in the next section.
To evaluate the π-stacking energy, topological analysis of the ED for complexes 1–5 was performed using X-ray geometry data at the DFT level of theory. Unlike the neutral compounds 1–4, complex 5 is both ionic and paramagnetic, for which convergence of SCF equations has not been achieved in spite of our efforts to change the basis set for the neodymium atom or convergence algorithms. We calculated this value for model free ligands in geometrical configuration as in the X-ray structure of complex 5.
The AIM results are presented in Fig. 8 and in Table 3. Molecular graphs of complexes 1–5 exhibit various sets of bond critical points (BCP) and bond paths corresponding to different types of π-stacking interactions.
 | ||
| Fig. 8 The fragments of QTAIM graphs for complexes 1–5 exhibiting π-stacking interactions in these complexes (a, b – tri- and bidentate ligand coordination in complex 5). Hydrogen atoms are omitted for clarity. Color codes for the atoms: orange (P), grey (C). The π-stacking bond paths are shown as green dotted lines; BCPs (3;−1) are red, ring (3;+1) critical points are yellow, and cage (3;+3) critical points are blue. | ||
| Complex | BCP (3;−1) | ρ(r), a.u. | ∇2ρ(r), a.u. | V(r), a.u. | E cont, kcal/mol | ∑Econt, kcal/mol |
|---|---|---|---|---|---|---|
| a For tridentate ligand coordination. b For bidentate ligand coordination. | ||||||
| 1 | C38–C60 | +0.008252 | +0.022976 | −0.003955 | 1.3 | 2.3 |
| C45–C63 | +0.003848 | +0.010428 | −0.001669 | 0.5 | ||
| C43–C65 | +0.004001 | +0.010002 | −0.001642 | 0.5 | ||
| 2 | C44–C57 | +0.008598 | +0.024180 | −0.003988 | 1.3 | 2.0 |
| C42–C60 | +0.005467 | +0.014297 | −0.002330 | 0.7 | ||
| 3 | C35–C46 | +0.006458 | +0.018423 | −0.002927 | 0.9 | 1.4 |
| C40–C51 | +0.001889 | +0.005211 | −0.000794 | 0.3 | ||
| C42–C55 | +0.001672 | +0.004601 | −0.000699 | 0.2 | ||
| 4 | C35–C47 | +0.006593 | +0.018433 | −0.002946 | 0.9 | 1.6 |
| C39–C49 | +0.003703 | +0.009296 | −0.001505 | 0.5 | ||
| C42–C52 | +0.001324 | +0.003886 | −0.000592 | 0.2 | ||
| 5 | C27–C58a | +0.005527a | +0.015985a | −0.002511a | 0.8a | 0.8a |
| C28–C49b | +0.007119 | +0.019434 | −0.003138 | 1.0b | 1.5b | |
| C30–C52b | +0.003728 | +0.009775 | −0.001713 | 0.5b | ||
The change in the parameters of the intramolecular π-stacking interactions (Table 3) for complexes 1–5 depends on the mutual geometric configuration of the benzene rings (Table 4). The larger the distance between the rings and corresponding interplanar angle the smaller the interaction energy value. The topology of molecular graphs is affected by the parallel shift of two benzene rings (e.g. for complexes 2 and 3), hence, the number of bond paths and overall interaction energy decrease. The most pronounced stacking interaction is observed in complex 1 (interplane distances are 3.505 Å). The total energy of the π-stackings in 1–5 amounts to 2.3, 4.0, 2.8, 3.2 and 2.3 kcal mol−1, respectively. Interestingly, the ligands in 5 exhibit different coordinations: bi- and tridentate fashion. This leads to different molecular graphs in the π-stacking interaction fragment (5a and 5b in Fig. 8), containing one and two bond paths for tridentate and bidentate ligands, respectively. Thus, the π-stacking interaction in the tridentate ligand (5a) is the weakest (only 0.8 kcal mol−1) in the series studied.
| 1 | 2 | 3 | 4 | 5 | 5 | |
|---|---|---|---|---|---|---|
| a d is the average distance between the contacting planes; l is the centroid–centroid distance; β is the average dihedral angle between the planes of contacting fragments. b For tridentate ligand coordination. c For bidentate ligand coordination. | ||||||
| M | U | U | La | Nd | Nd | Nd |
| d, Å | 3.505 | 3.710 | 3.816 | 3.849 | 4.644 | 3.831 |
| l, Å | 3.505 | 3.760 | 3.827 | 3.841 | 4.123 | 3.825 |
| β, ° | 9.25 | 8.03 | 23.15 | 24.21 | 29.82 | 13.76 |
The obtained π-stacking interaction energy values are close to those for intramolecular π-stacking in a Co(III) complex24 of about 1–3 kcal mol−1 and to those for intramolecular interligand π-stacking interactions in Lu(III) complexes25 of about 1.9–3.3 kcal mol−1.
The analysis of literature data on the structure of crystalline complexes of methylenediphosphine dioxide L′ with uranyl nitrate [UO2(NO3)2L′] (7)7 and lanthanum nitrate [La(NO3)3(L′)2]·EtOH (8)6 also indicates π-stacking in the molecules of coordinated ligand L′. However, the stacking interactions in the complexes of unmodified ligand L′ are weaker than in complexes of L of the same chelate coordination. Thus, the dihedral angles between contacting planes in uranyl complexes 1 and 7 are 9.25° and 13.17°, respectively, while the distances between centroids are 3.505 and 3.736 Å, respectively. In the lanthanum nitrate complexes with L and L′, 3 and 8, the noted angles are 23.1 and 12.17, 12.66°, while the distances are 3.816 and 3.940, 4.079 Å (the contacting fragments in complex 8 are skewed toward each other).
Thus, the π-stacking interaction between the two Ph substituents at the phosphorus atom is observed in molecule(s) of coordinated ligand L for all of the crystalline complexes 1–5. The highest energy of the π-stacking interaction was found for bisligand uranyl complex 2. The change of ligand denticity from P(O),P(O)-bidentate to P(O),P(O),C(O)-tridentate almost doubly decreases the energy of the π-stacking interaction (see Table 3).
The above discourse deals with the stabilization of complexes in a crystal, but the situation may change in solution. Solvent nature is known to considerably affect the aromatic π-stacking interaction. However, π-stacking interactions are retained as a rule in dipolar aprotic solvents.26
O → M coordination bond results in the shift of the ν(PO) band in the IR spectra of crystalline complexes 1 and 3–6 by ∼40 cm−1 to the low frequency region with respect to the band of the free ligand (Table 5), which is slightly lower than in similar complexes of phosphoryl-containing ligands (Δν(PO) ∼ 65 cm−1 for UO2 complexes,7,27 and ∼50 cm−1 for Ln complexes28). The spectrum of complex 5, along with the ν(PO) band at 1160 cm−1, also displays a band at 1150 cm−1 responsible for vibrations of the coordinated PO group participating in the formation of a supplementary weak H bond with a solvate EtOH molecule. The formation of the CO → M coordination bond in complex 5 causes a shift of the ν(CO) band to the low-frequency region but only by 9 cm−1 relative to the band of the free ligand, whereas usually coordination of CO to a cation leads to a shift of 20–25 cm−1 for lanthanide complexes and 20–60 cm−1 for UO2 complexes.16,27b,29 The band of the uncoordinated CO group in the spectrum of crystalline complex 1 is observed30 at the same frequency as in the spectrum of the free ligand (Table 5). In the isostructural complexes of La and Nd (3 and 4, respectively), the shift of the ν(CO) band by 4–3 cm−1 corresponds to vibrations of the CO group involved in weak intra- and intermolecular CH⋯OC interactions (Table 5). The spectrum of crystalline complex 5 exhibits a band of the free CO group, that does not become involved in supplementary interactions, at 1722 cm−1.
| Compound | Sample |
ν(PO) |
ν(CO) |
ν(NO) |
ν as(NO2) | δ P(W1/2)a |
|---|---|---|---|---|---|---|
|
a The band width at half-height (in ppm).
b
c = 0.02 M.
c Saturated solution, c ∼ 0.003 M.
d Non-covalent CH⋯OC interaction (see section 2.2.1).
e A wide absorption in the region 1300–1400 cm−1 (weak CH⋯ON interaction of “free” nitrate – see section 2.2.3).
f Weak OH⋯O bonding between coordinated PO and EtOH (see section 2.2.1).
g The strong band at 1356 cm−1 – νE(NO3) (see section 2.3.1).
|
||||||
| L | Cryst. | 1202, 1182 | 1720 | |||
| In CD3CNb | 1207, 1200 | 1720 | 29.8 (0.01) | |||
| In CDCl3b |
1199sh, 1181 | 1719 | 31.2 (0.03) | |||
| 1 | Cryst. | 1166 | 1720 | 1518 | 1308, 1282 | |
| In CD3CNc | 1195, 1165 | 1719 | 1523 | 1290, 1273 | 46.2 (0.02) | |
| In CDCl3c |
1171 | 1718 | 1526 | 1283 | 44.4 (0.03) | |
| 3 | Cryst. | 1166 | 1716d | 1458 | 1313 | |
| In CD3CN | 1170 | 1720 | 1455 | 1318 | 37.0 (0.08) | |
| In CDCl3 | 1173 | 1713 | 1450 | 1319 | 36.0 (0.7) | |
| 4 | Cryst. | 1164 br | 1717d | 1465 | 1306 | |
| In CD3CN | 1162 | 1709, 1720 | 1469 | 1308 | 86 (2.5) | |
| In CDCl3 | 1173, 1162 | 1712 | 1460 | 1313 | 86(5), 70(6) | |
| 5 | Cryst.e | 1160, 1150f | 1711, 1722sh | 1503, 1465 | 1285, 1300 | |
| In CD3CN | 1162 | 1709, 1720 | 1469 | 1308 | 86 (2.5) | |
| In CDCl3 | 1174, 1162 | 1712 | 1460 | 1313 | 86(5), 70(6) | |
| 6 | Solid | 1185, 1161 | 1721 | 1494, 1518sh | 1310 | |
| In CD3CNg | 1189, 1159 | 1723 | 1527, 1512 | 1293 | 42.4 (0.5) | |
| In CDCl3 | 1195, 1174, 1162 | 1715 | 1490 | 1310 | 40.3 (0.5) | |
The IR spectra of complexes 1 and 3–6 show absorption bands of bidentate nitrate ions at ∼1500 cm−1 for ν(NO), and ∼1300 cm−1 for νas(NO2) (Table 5). Full details of the nitrate bands are shown in Table S1 (ESI†). In contrast to other complexes, X-ray structure 5 includes, along with bidentate coordinated nitrate ions, outer-sphere “free” nitrate ions involved in many weak CH⋯ON interactions. The symmetry of an uncoordinated nitrate ion is known to be violated on weak interactions in crystal or contact ion pairs (CIPs) on account of cation–anion interactions that cause strong splitting of the νE(NO3) vibration.31 As should be expected, the spectrum of crystalline compound 5 shows no absorption for the free nitrate ion, which usually appears as a narrow intense band at ∼1370 cm−1,31 but displays a wide absorption in the region 1300–1500 cm−1 with several submaxima at 1338, 1384, and 1396 cm−1.
According to elemental analysis and X-ray crystallographic data, complexes 3 and 4 contain solvate acetonitrile, but the IR spectra of 3 and 4 exhibit no absorption for the CN group.32 The IR spectra of 5 show bands typical for outer-sphere ethanol molecules in the region of ∼3400 cm−1.
We failed to prepare complex 6 in a crystalline state. But, elemental analysis and IR spectra allow us to suppose unambiguously that one ligand molecule in bisligand complex 6 is coordinated in a P(O),P(O)-bidentate mode, while another molecule has a P(O)-monodentate coordination. The IR spectrum of solid complex 6 (Table 5) shows bands of free PO and CO groups at 1185 and 1721 cm−1 along with the band of the coordinated PO group at 1161 cm−1. The strong broad IR bands of bidentate NO3 groups are detected at 1494, 1310 and 1030 cm−1. In the region of 3200–3400 cm−1 the band of metal-coordinated water (typically at ∼3200 cm−1) is absent. In accordance with these data, one can suppose that compound 6 most likely has the structure of neutral mononuclear complex [Lu{P(O),P(O)-L}{P(O)-L}(O,O-NO3)3], and that the coordination number of lutetium is nine.
Thus, lanthanide contraction is observed for the structure of the studied neutral complexes of ligand L. The coordination number (CN) of light lanthanides (La and Nd) in complexes 3 and 4, [Ln{P(O),P(O)-L}2(NO3)3], equals ten, while the CN of lutetium in complex 6, [Lu{P(O),P(O)-L}{P(O)-L}(O,O-NO3)3], is nine.
2.3. Solution state characterization
The structure of the complexes in acetonitrile (AN) and chloroform solutions was studied by IR and multinuclear NMR spectroscopy. We were interested to study the effect of solvent nature on the structure of complexes and ligand L coordination mode. The parameters of IR and 31P, 1H, and 13C NMR spectra for the complexes 1, 3–6 in comparison with the data for the free ligand L are given in Tables 5–7 (see also Fig. S5–S12, ESI†).| Compound | δ 1H | δ 13C | |||||
|---|---|---|---|---|---|---|---|
C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3 3 |
C2 |
C |
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H3 H3 |
H |
H2 |
CO |
|
| a Spectra of complexes 4 and 5 are identical. b 0.02 M solution. c Saturated solution, c ∼ 0.003 M. d Not observed. | |||||||
| L | 1.55 s | 2.98 dt | 4.46 tt | 28.28 s | 35.61 t | 38.13 s | 204.04 t |
| 1 | 1.30 br s | 2.98 dt | 5.19 br t | 28.15 s | 32.64 t | 36.94 s | 202.6 br s |
| 3 | 1.47 s | 3.19 dt | 4.67 tt | 28.18 s | 32.00 s | 37.81 s | 203.3 br s |
| 4 | 1.10 s | 4.1 br s | —d | 28.30 s | 29.9 br s | 39.3 br s | 205.4 br s |
| 6 | 1.44 s | 3.10 dt | 4.93 tt | 28.10 s | 31.72 t | 37.66 s | 202.8 br s |
| Compound | δ 1H | δ 13C | |||||
|---|---|---|---|---|---|---|---|
| C3 |
C2 |
C |
H3 |
H |
H2 |
O |
|
| a 0.02 M solution. b Saturated solution, c ∼ 0.003 M. | |||||||
| L | 1.54 s | 2.94 dt | 4.45 tt | 28.81 s | 36.73 t | 38.70 s | 203.84 t |
| 1 | 1.38 s | 2.84 dt | 4.95 br t | 28.83 s | 31.93 br s | 37.72 s | 203.4 br s |
| 3 | 1.62 s | 3.36 t | 4.63 br s | 29.17 s | 32.43 br s | 39.15 br s | 206.2 br s |
| 6 | 1.61 s | 3.28 t | 5.04 br s | 28.98 s | 33.05 t | 39.81 s | 205.7 br s |
The coordination of the PO groups can be reliably determined from the NMR spectra of compounds 1 and 3–6. The signals of the phosphorus nuclei as well as the protons and carbon nuclei of neighboring groups exhibit expected shifts (Tables 5–7) close to those for known complexes of akin phosphoryl-containing ligands.8a,c,27a,b,28 The coordination of phosphoryl-containing ligands with UO2(II), La(III), Lu(III), and Nd(III) cations causes a downfield shift in the signals of the phosphorus nuclei by 5–60 ppm, while signals of the carbon nuclei of neighboring groups are shifted upfield; the signals of the paramagnetic neodymium complex show considerable broadening. The participation of the CO group in coordination appears in the 13C and 1H NMR spectra as a downfield shift of the carbon signals of the CO group as well as the carbon and proton nuclei signals of neighboring CH3 and CH2 groups relative to the free ligand signals (Tables 6 and 7), however, the value of these shifts in the spectra of complexes of ligand L is lesser than those of corresponding complexes for the majority of carbonyl ligands (for example16,27b,29b).
O group at 1165 cm−1 as well as the bands of free PO and CO groups at 1195 and 1719 cm−1. The bands of the nitrato groups are virtually retained when compared with the spectrum of the crystalline sample (Table 5). One can suppose that the ligand adopts a P(O)-monodentate coordination in AN solution, and the coordination sphere of the cation is supplemented by solvent molecules, while the neutral complex has the structure [UO2{P(O)-L}(OO-NO3)2·MeCN].
The NMR spectra of compound 1 (Tables 5 and 6) agree well with the proposed structure. Certain signals in the spectra are broadened. The 31P NMR spectrum displays only one slightly broadened signal, which seems to be explained by fast exchange processes. The signal is shifted downfield relative to the signal of the free ligand by 16.4 ppm. In the 13C NMR spectrum, the carbon signals of the CO, H3, and H2 groups are upfield shifted (Table 6). The largest upfield shift (−2.97 ppm) is observed for the broadened signal of the H group, which is typical when the PO group is involved in the coordination. The corresponding effects are observed in the 1H NMR spectrum (Table 6).
The lanthanide complexes have good solubility in AN, chloroform, and methanol. According to the IR spectral data, the structure of the crystalline lanthanum complex 3 is retained in AN solution (Scheme 4). The bands of the PO, CO groups and nitrate ions have expected frequencies (Table 5), the complicated pattern in the spectrum of the crystalline sample caused by supplementary weak interactions disappears. Bisligand complex 3 in AN solution, like in the crystalline state, seems to remain neutral [La{P(O),P(O)-L}2(OO-NO3)3], where the CN of lanthanum is ten.
 | ||
| Scheme 4 Complex 3 (Ln = La) in the crystalline state and in AN solution, and complex 4 (Ln = Nd) in the crystalline state. | ||
NMR spectral data for compound 3 (Tables 5 and 6) agree well with the proposed structure (Scheme 4). 31P NMR spectra display a sole narrow signal (W1/2 = 0.08 ppm) of the phosphorus nuclei downfield shifted by 7.2 ppm from its position in the free ligand. The signals of indicator groups (CO, H3, and H2) in the 13C NMR are shifted upfield, which indicate the lack of coordination of the CO group. The largest shift in the 13C NMR spectrum is observed for the signals of the CH groups (−3.61 ppm). The shifts in the 1H NMR spectrum are less considerable (Table 6), the protons of the CH3 group show the least shift (−0.08 ppm), while the protons of CH and CH2 groups exhibit the largest shift (0.21 ppm). A fine signal structure is observed in the spectrum.
IR and NMR spectra of neodymium complexes 4 and 5 in AN solution are identical (Tables 5 and 6). The complications in the IR spectrum of the crystalline sample of 5 are caused by weak interactions in the crystal that disappear in the solution spectrum. The main analytical bands have close positions to those in the spectrum of crystalline 5 (Table 5). We observed no band for the vibrations of free nitrate ions, expected at ∼1360 cm−1. Both complexes seem to be present in AN solution as a contact ion pair [Nd{P(O),P(O)-L}{P(O),P(O),C(O)-L}(OO-NO3)2]+·(NO3)−, where the CN of neodymium is nine (Scheme 5).
 | ||
| Scheme 5 Complex cation of compounds 4 and 5 in AN solution. | ||
The sole signal in the 31P NMR spectrum of neodymium complex 4 in AN at 86 ppm (analysis of NMR spectra is given for one complex because the spectra of solutions of 4 and 5 are identical) is considerably broadened (W1/2 = 2.5 ppm). In the 1H NMR spectrum, the proton signals of all groups except for CH3 and CH2 are also broadened, no signal for the CH protons is observed probably due to both paramagnetic properties of the neodymium cation and dynamic equilibria in solution. The signals of the CH3 and CH2 groups are shifted from their positions in the free ligand by −0.14 and 1.12 ppm, respectively. The 13C NMR spectrum exhibits one set of signals, which seem to correspond to the fast dynamic equilibrium of several complex species. Alterations of the chemical shifts in the 13C NMR spectrum agree well with the suggested complex structure (Scheme 5).
According to IR spectroscopic measurements, the structures of the lutetium complex 6 in AN solution and in the solid state differ. The main difference is the emergence of a strong vibrational band of the free nitrate ion at 1356 cm−1. Furthermore, strong bands of bidentate coordinated nitrato groups are detected at 1527, 1512, and 1293 cm−1. Vibration bands of the PO and CO groups are observed at almost the same frequencies as in the spectrum of the solid sample (Table 5). Obviously, ligand molecules retain the same coordination as in the solid complex. In accordance with these data, one can suppose that complex 6 in AN solution exists as either a solvent-separated ion pair (SSIP) or cationic complex [Lu{P(O),P(O)-L}{P(O)-L}(OO-NO3)2]+ and free nitrate ion. In this case, the CN of lutetium should be equal to seven. Both the forms, most probably, are in equilibrium. Since CNs of lutetium of eight and nine33 are more typical, one can suppose that the remaining sites in the lutetium coordination sphere will be occupied by solvent molecules [Lu{P(O),P(O)-L}{P(O)-L}(OO-NO3)2(MeCN)m]+·(NO3)− (Scheme 6). Complex species with coordinated solvent molecules are most likely to be involved in dynamic equilibria with species containing no coordinated solvent molecules to cause signal broadening in the 31P NMR spectrum.
 | ||
| Scheme 6 Complex 6 in the solid state (n = 3, m = 0), and complex cation of compound 6 (n = 2, m = 1 or 2) in AN solution. | ||
The NMR spectra (Tables 5 and 6) agree well with the supposed structure (Scheme 6). The 31P NMR shows a sole broadened signal for the phosphorus nuclei, which seems to be explained by fast exchange processes. The signal is downfield shifted by 12.6 ppm relative to the free ligand signal. The 1H and 13C NMR spectra display expected changes.
Thus, in AN solutions, the lanthanum (3) and neodymium (5) complexes retain the structure revealed in the crystal. The structures of the uranyl (1), neodymium (4), and lutetium (6) complexes change in solution. In the studied complexes in AN solutions, the scorpionate ligand L shows three coordination modes: P(O)-monodentate, chelate P(O),P(O)-bidentate, and P(O),P(O),C(O)-tridentate. The uranyl (1) and lanthanum (3) complexes in AN solution are neutral, whereas the neodymium (4 and 5) and lutetium (6) complexes are cationic.
Uranyl complex 1 is relatively poorly soluble in chloroform (∼0.003 M), however, it has much better solubility (≥0.02 M) in solution containing 3 equiv. of ligand L. The 31P NMR spectra of the 3:1 mixture showed at least 3 broad peaks at 45.9, 45.3 and 45.0 and one peak at 31.6 ppm with an integral intensity ratio ∼2:3, which indicates the presence of complex species of different stoichiometry containing both P(O)- and P(O),P(O)-coordinated ligand molecules. The position of the analytical bands in the IR spectrum of a solution of compound 1 slightly differs compared with the spectrum of the crystalline sample (Table 5). The 31P NMR spectrum shows a singlet at 44.4 ppm. The 1H NMR spectrum of complex 1 shows that the CH resonance is shifted downfield by ca. −0.5 ppm with respect to the free ligand, and the CH3 resonance is shifted downfield by ca. −0.10 and −0.25 ppm (Table 7). The proton signal of the CH group is broadened. The carbon signals of the indicator groups CO, H, and H2 in the 13C NMR spectrum are shifted upfield relative to the free ligand signals (Table 7). These changes agree well with a chelating P(O),P(O)-mode of ligand coordination. The neutral uranyl complex in chloroform solution has the structure [UO2{P(O),P(O)-L}(OO-NO3)2].
The structure of lanthanum complex 3 in chloroform solution according to IR spectroscopic data (Table 5) differs from that in the crystal and AN solution. Both ligand molecules in complex 3 are coordinated in a P(O),P(O),C(O)-tridentate mode. The bands of coordinated PO and CO groups are observed at 1173 and 1713 cm−1. The bands at 1450 and 1319 cm−1 correspond to bidentately coordinated nitrato groups. Taking into account the typical lanthanum coordination number of ten, one can suppose that complex 3 in chloroform solution is present as a contact ion pair [La{P(O),P(O),C(O)-L}2(OO-NO3)2]+·(NO3)− (Scheme 7).
 | ||
| Scheme 7 Complex cations compounds 3 (Ln = La), 4 and 5 (Ln = Nd) in CDCl3 solutions. | ||
The NMR spectra (Tables 5–7) agree well with the proposed structure. The 31P NMR spectrum shows a sole broadened phosphorus signal at 36.0 ppm shifted from the free ligand signal by 4.8 ppm. The 1H NMR spectrum displays the signals of the CH3, CH2, and CH groups downfield shifted relative to the free ligand signal, the signals of the two latter groups are broadened. The largest shift is observed for the protons of the CH2 group (0.42 ppm), whereas the signals of CH3 and CH groups are shifted by 0.08 and 0.18 ppm. The 13C NMR spectrum of complex 3 in chloroform solution exhibits considerably broadened signals for the H2, H, and O groups. The signals of the H and O groups have the largest shift relative to the free ligand signal: −4.3 and 2.4 ppm, respectively. The signals of the H3 and H2 groups are shifted much less: 0.36 and 0.5 ppm. Signal broadening observed for all of the NMR spectra of chloroform solutions of 3 indicate relatively fast on the NMR time scale equilibria with participation of other types of complexes (for example [La{P(O),P(O)-L}2(OO-NO3)3] and similar species).
The spectra of neodymium complexes 4 and 5 in chloroform are identical except for the bands of solvate ethanol at 3683 and 3622 cm−1 in the spectrum of 5. According to IR data, the structures of complexes 4 and 5 in chloroform solutions differ from those revealed in the crystalline forms and in AN solution. In chloroform solution, both ligand molecules are coordinated in a tridentate mode. The bands of coordinated PO groups are detected at 1173 and 1162 cm−1, those of coordinated CO groups are observed at 1712 cm−1. The bands at 1460 and 1313 cm−1 correspond to nitrato group vibrations. One can suppose that the neodymium bisligand complex in chloroform solution has the same structure as lanthanum complex 3: [Nd{P(O),P(O),C(O)-L}2(OO-NO3)2]+·(NO3)− (Scheme 7). The CN of neodymium is ten.
Paramagnetic properties of neodymium hamper the use of NMR spectroscopy to study the structure of Nd complexes. However, signal broadening in NMR spectra of complexes 4 and 5 is much larger than that observed for neodymium nitrate complexes with other phosphoryl-containing ligands.28b–e,34 Thus, the 1H NMR spectrum of complex 4, the signals of all protons either considerably broadened or not detected at all. The 31P NMR spectrum shows two broad signals at 86 and 70 ppm (W1/2 = 5 and 6 ppm, respectively) with approximate ratio 8:1 (Table 5), which indicates the presence of equilibrium in solution of the studied complex. One can suppose that dynamic equilibria involve structural isomers with different ligand coordination modes, species differing in the number of coordinated nitrato groups or ligand molecules, intermolecular exchange processes, etc. 13C NMR spectrum could not be interpreted correctly because of the presence of additional signals.
The IR spectrum of lutetium complex 6 in chloroform solution shows the bands of coordinated PO groups at 1162–1173 cm−1 and a shoulder at 1195 cm−1 due to the vibrations of the free PO group. The band at 1715 cm−1 slightly shifted relative to the free ligand band (1719 cm−1) corresponds to vibrations of the CO groups. A broad absorption at 3200 cm−1 corresponds to vibrations of coordinated water. The bands at 1490 and 1310 cm−1 are consistent with bidentately coordinated nitrato groups. In accordance with these data, one can suppose that complex 6 in chloroform solution exists as an ion pair. Both ligand molecules are coordinated through one and two phosphoryl groups, however, it is rather difficult to determine the interaction mode for the CO groups. One can suppose that both CO groups form H-bonds with coordinated water molecules, the CN of lutetium will be eight (Scheme 8).
 | ||
| Scheme 8 Complex cation of compound 6 in CDCl3 solution. | ||
This type of ligand coordination is not unique. A number of complexes are known where ligands form H-bonds with coordinated water molecules rather than coordination bonds with a metal.35 The NMR spectroscopic data agree well with the suggested structure (Scheme 8). Thus, the 31P NMR spectrum shows a sole signal for the phosphorus nuclei shifted by 9.1 ppm relative to the free ligand signal (W1/2 = 0.5 ppm) (Table 5). On cooling to −50 °C, a second broadened resonance at about 35 ppm, which may be related to an uncoordinated P(O) group,36 appears along with the main broadened signal at ∼40 ppm. Fast exchange processes with participation of coordinated and uncoordinated P(O) groups seem to take place in the coordination sphere of Lu in complex 6 at ambient temperature.
The 1H and 13C NMR spectra of complex 6 (Tables 6 and 7) are as expected for the structure of the complex cation in Scheme 8. Thus for 13C, the change in the chemical shifts are ΔδC(H3) 0.17 ppm, ΔδC(H2) 1.11 ppm, ΔδC(H) −3.68 ppm, and ΔδC(CO) 1.9 ppm. The signal of the CO group is broad. These changes are close to those observed for the spectrum of complex 3 and agree well with the conclusion of the participation of the CO group in certain interactions. The changes in chemical shifts (ΔδH) for the C3 and C2 groups also confirm the conclusion of the involvement of the CO group in “coordination”. The water signal in the spectrum of 6 is at 5.4 ppm, whereas it is usually detected in chloroform at ∼1.6 ppm.
It is possible that this complex is in equilibrium with other complexes, for example, [Lu{P(O),P(O),C(O)-L}{P(O),C(O)-L}(H2O)(OO-NO3)2]+·(NO3)−, where both CO groups are weakly coordinated to the metal (CN = 10), neutral complex [Lu{P(O),P(O)-L}{P(O)-L}(H2O)(OO-NO3)3] (CN = 10), etc. However, CN = 10 is less typical for lutetium cations.
Thus, scorpionate ligand L in complexes with f-block element nitrates in chloroform show variable denticity. In the complex with uranyl nitrate 1, this ligand is coordinated in a P(O),P(O) bidentate mode like in the crystal. This kind of chelate coordination is not realized in chloroform solutions of complexes with lanthanide nitrates 3–6. The main coordination is P(O),P(O)C(O)-tridentate, while P(O)C(O)-bidentate coordination, however through H-bond formation, is observed for the first time in the lutetium complex.
It should be noted that almost all studied complexes are labile in solution. Except for uranyl complex 1 in chloroform and AN, all other complexes have one or two broadened resonances in the 31P NMR spectra (Table 5). Although we did not conduct a detailed study, we suppose that the structures shown in Schemes 4–8 and noted in the text are only the main species present in solution. Virtually all of the studied complexes exhibit fluxional behavior in solution.
Let us note that only monoligand uranyl complex [UO2(NO3)2L] is neutral in chloroform solution, all of the studied bisligand lanthanide complexes are cationic [Ln(NO3)2(L)2]+·(NO3)−. This fact should be taken into account in analyzing the data on the extraction of f-block elements from nitric acid solutions into chloroform.
Thus, the coordination mode of the scorpionate ligand L varies with not only the requirements of the metal coordination polyhedron and complex composition, but also depending on the solvent, and shows large variation.
2.4. Extraction studies
The extraction ability of ligand L towards the f-block elements was studied by the example of the extraction of a group of lanthanides(III), as well as uranium(VI) from nitric acid solutions into CHCl3. To compare the efficiency and selectivity of the studied ligand L and well-known extractant L′, we compared the distribution ratios of the f-block elements (D = [M]org/[M]aq) for both extractants under the same experimental conditions (Fig. 9, Table S2, ESI†). | ||
| Fig. 9 Comparison of the distribution ratios of U(VI), La(III), Nd(III), Ho(III), and Yb(III) for the extraction with ligands L and L′ (0.01 M solutions in CHCl3) from 3.75 M HNO3; the initial concentration of lanthanide and uranyl nitrates in the aqueous phase is 2.5 × 10−4 M. | ||
Fig. 9 shows that both compounds extract U(VI) much more efficiently than lanthanides. However, scorpionate ligand L extracts uranium by a factor of 6 more efficiently than its prototype L′. The fraction extracted for uranium over one step is 98%. Let us note that the values of DU for common extractants – (BuO)3P(O), (C8H17)3P(O), and Ph2P(O)CH2C(O)NBu2 – is lower by 1.0 under the same experimental conditions. Compound L is also better (about 3 times) at extracting lanthanides than ligand L′. The extraction selectivity U(VI)/Ln(III) of ligand L is higher than that of ligand L′.
As noted above, both ligands, L and L′, form crystalline complexes of the same composition with uranyl and lanthanide nitrates with the same chelate coordination of the phosphoryl groups. The π-stacking interaction between the Ph fragments of coordinated ligand molecules is inherent in not only ligand L but also to a lesser extent in ligand L′ (section 2.2.2). At the same time, the CO group of scorpionate L in chloroform solutions of all of the lanthanide complexes participates in coordination along with two phosphoryl groups (section 2.3.2). Judging from the spectral and X-ray diffraction data, the CO → Ln bond in the studied complexes is rather weak; however, we believe that it is its formation that leads to the better extraction of lanthanides with ligand L. It should also be noted that lanthanide complexes 3–6 in chloroform solution are ionic in contrast to neutral uranyl complex 1, which also favors the better extraction of the latter. In uranyl complex 1, the CO group is not coordinated, however, it is available for other interactions. In the crystal, the uncoordinated CO group participates in various CH⋯OC contacts and one can expect that its availability for interaction with diluent will favor better extraction of the corresponding compound.
Thus, the modification of methylene dioxide L′ by the introduction of a MeC(O)CH2– substituent leads to a considerable improvement in the extraction properties of the hybrid scorpionate ligand L toward f-block elements.
3. Conclusion
Coordination properties of the neutral organophosphorus scorpionate ligand L toward f-block elements were examined. Mono- and binuclear complexes of L with uranyl and lanthanide(III) nitrates were studied in the solid state (X-ray, IR) and solution (IR, 31P NMR, 13C NMR, and 1H NMR). In the studied complexes, ligand L exhibits variable denticity: PO,PO-, PO,PO,CO-, PO,CO-, and PO-. The CO → Ln coordination bond is rather weak; at the same time, the noncoordinated CO group is involved in weak noncovalent interactions in both solution and in the crystalline form. Ligand coordination mode and complex structure are readily variable depending on both the nature of the metal and interactions in the second coordination sphere. The extraction experiments revealed an expected increase in the affinity of scorpionate ligand L over its unmodified prototype L′ for studied metal ions.
4. Experimental
4.1 General
Solvents were purified and dried using standard procedures. Deuterated solvents, CD3CN (99.8% D, Sigma-Aldrich) and CDCl3 (99.8% D, Sigma-Aldrich), were used as received. Multinuclear NMR spectra were recorded on a Bruker Avance 400 spectrometer, operating frequency 400.23 MHz (1H and 1H{31P}), 100.61 MHz (13C) and 161.98 MHz (31P and 31P{1H}) at ambient temperature using CD3CN or CDCl3 solution (0.01 M), unless otherwise stated. Chemical shifts (ppm) refer to the residual protic solvent peaks (for 1H and 13C), and 85% H3PO4 (for 31P) as external standards and coupling constants are expressed in hertz (Hz), the band width at half-height (W1/2) in ppm (for 31P{1H} NMR spectra). IR spectra in the region 400–4000 cm−1 were obtained on a Bruker Tensor 37 FTIR spectrometer. The samples were KBr pellets, mulls in Nujol and hexachlorobutadiene as well as 0.01 M solutions (CD3CN, CDCl3) in CaF2 cuvettes, unless otherwise stated. Raman spectra of the solid samples were obtained in the region 100–3500 cm−1 using a Jobin-Yvon LabRAM 300 laser Raman spectrometer with 632.8 nm excitation of 2 mW output power. Elemental analyses were performed at the Laboratory of Microanalysis, INEOS RAS.The reagents (E)-4-(Diphenylphosphoryl)but-3-en-2-one12 and methylenediphosphine dioxide (L′)37 were prepared according to literature procedures. Chlorodiphenylphosphine (Acros) was purified by vacuum distillation immediately prior to use. Glacial acetic acid (reagent grade) was distilled before reaction. Basic Brockmann activity grade I Al2O3, 50–200 μm (Acros) and silica gel 130–270 mesh, 60 Å (Aldrich) were used. Acetonitrile was dried by distillation over P2O5 prior to use. All manipulations with chlorodiphenylphosphine were carried out under an argon atmosphere.
The following reagents were used for the preparation of solutions in the extraction study: bidistilled water, CHCl3 (reagent grade), arsenazo III (analytical grade), HNO3 (high purity grade), UO2(NO3)2·6H2O (reagent grade), La(NO3)3·6H2O (reagent grade), Nd(NO3)3·6H2O (reagent grade), Ho(NO3)3·6H2O (pure grade), and Yb(NO3)3·6H2O (pure grade). Solutions for spectral and extraction studies were prepared by volumetric/gravimetric method.
O), 1202s and 1182s (PO). 1H NMR (400 MHz, CDCl3, 0.1 M): δ 1.54 (3H, s, CH3,), 2.94 (2H, dt, 3JHH 5.3, 3JHP 14.5, CH2), 4.45 (1H, tt, 3JHH 5.2, 2JHP 14.3, CH), 7.23–7.31 (8H, m, m-Ph), 7.31–7.38 (4H, m, p-Ph), 7.64–7.73 (4H, m, o-Ph), 7.76–7.85 (4H, m, o-Ph). 1H{31P} NMR (400 MHz, CDCl3, 0.1 M): δ 1.55 (3H, s, CH3), 2.94 (2H, d, 3JHH 5.0, CH2), 4.46 (1H, t, 3JHH 5.1, CH), 7.23–7.31 (8H, m, m-Ph), 7.31–7.39 (4H, m, p-Ph), 7.68 (4H, d, 3JHH 7.7, o-Ph), 7.81 (4H, d, 3JHH 7.3, o-Ph). 13C{1H} NMR (100.61 MHz, CDCl3, 0.1 M): δ 28.81 (s, CH3), 36.73 (t, 1JCP 59.4, CH), 38.70 (s, CH2), 128.10 (d, 3JCP 12.5, m-Ph), 128.12 (d, 3JCP 12.5, m-Ph), 130.82 (d, 1JCP 103.0, ipso-Ph), 131.33 (dd, 1JCP 100.6, 3JCP = 3.0, ipso-Ph), 131.45–131.53 (m, p-Ph), 131.48 (d, 2JCP 9.8, o-Ph), 131.52 (d, 2JCP 10.1, o-Ph), 131.63–131.71 (m, p-Ph), 203.84 (t, 3JCP 4.9, CO). 31P{1H} NMR (161.98 MHz, CDCl3, 0.1 M): δ 31.25 (s).
:1 or 1:2. The yields were 50–90%, but no attempts were made to optimize the yield for each individual complex.
(b) A solution of UO2(NO3)2·6H2O (35.1 mg, 0.070 mmol) in acetonitrile (1.5 mL) was added dropwise to a stirred solution of ligand L (100.0 mg, 0.212 mmol) in chloroform (1 mL) at room temperature. The transparent light yellow solution was concentrated at ∼60 °C in vacuo (∼5 Torr) down to volume of ∼1 mL. Light yellow transparent crystals of 1, including those suitable for X-ray diffraction studies, were obtained under cooling to room temperature. The resulting yellow crystals were collected by filtration, washed with diethyl ether, and dried in air at r.t. to give 1 (30.0 mg, 50%). Mp 235–236 °C. Found: C, 38.56; H, 3.05; N, 3.01; P, 7.31; U, 27.89. Calc. for C28H26N2O11P2U: C, 38.81; H, 3.02; N, 3.23; P, 7.15; U, 27.47%. IR (KBr disk): νmax/cm−1 1720s (CO), 1166s (PO), 1518s (NO), 1308s and 1282s (NO2)as, 1032w (NO2)s, 939s (OUO)as.
Raman: ν/cm−1 858 (OUO)as, 1037 (NO2)s. 1H NMR (400.13 MHz, CDCl3, ∼0.003 M): δ 1.38 (3H, s, Me), 2.85 (2H, d.t., 3JHH 5.0, 3JHP 14.7, CH2), 4.96 (1H, br m., CH), 7.18–7.22 (4H, m, m–Ph); 7.34–7.38 (2H, m, p-Ph); 7.63–7.68 (6H, m, m + p-Ph); 7.84–7.89 (4H, m, o-Ph); 8.14–8.19 (4H, m, o-Ph). 13C{1H} NMR (100.61 MHz, CDCl3, ∼0.003 M): δ 28.84 (s, CH3), 31.93 (s, CH), 37.72 (s, CH2), 129.10–129.40 (m, Ph), 129.71–129.95 (m, Ph), 130.95–131.05 (m, Ph), 131.10–131.28 (m, Ph), 133.44 (s, Ph), 134.14 (s, Ph), 202.42s (s, CO). No signals of ipso-Ph were observed. 31P{1H} (161.98 MHz, CDCl3, ∼0.003 M): δ 44.4 (s, W1/2 0.03).
O), 1166s (PO), 1458s (NO), 1313s (NO2)as, 1031w (NO2)s. 1H NMR (400.13 MHz, CDCl3): δ 1.62 (3H, s, CH3), 3.36 (2H, v br t, CH2), 4.63 (1H, v br s, CH), 7.16–7.23 (4H, m, m-Ph), 7.24–7.30 (2H, m, p-Ph), 7.42–7.56 (6H, m, m + p-Ph), 7.82–8.00 (8H, br m, o-Ph), and 2.03 (6H, s, CH3CN). 13C{1H} NMR (100.61 MHz, CDCl3): δ 29.17 (s, CH3), 32.4 (br s, CH), 39.2 (br s, CH2), 128.65 (d, 1JCP 92.0, ipso-Ph), 128.86 (d, 1JCP 91.0, ipso-Ph), 128.70–129.70 (m, m-Ph), 131.12 (br s, o-Ph), 132.49 (s, p-Ph), 133.02 (s, p-Ph), 206.2 (br s, CO), and 1.92 (s, CH3CN), 116.4 (s, CH3CN). 31P{1H} NMR (161.98 MHz, CDCl3): δ 36.0 (br s, W1/2 0.7).
O), 1164s br (PO), 1465(NO), 1306s (NO2)as, 1030w (NO2)s. 1H NMR (400.13 MHz, CD3CN): δ 1.10 (3H, br s, CH3), 4.1 (2H, v br s, CH2), 7.04 (5H, br s, Ph), 7.31 (3H, br s, Ph), 7.67–7.83 (8H, br m, Ph), 8.82 (4H, v br s, Ph). No signal of CH was observed. 13C{1H} NMR (100.61 MHz, CD3CN): δ 28.30 (s, CH3), 29.9 (br s, CH), 39.3 (br s, CH2), 128.90 (d, 3JCP 12.5, Ph), 129.79 (d, 3JCP 12.5, Ph), 131.62–131.90 (br m, Ph), 132.58 (br d, 2JCP 8.8, Ph), 133.01 (s, Ph), 133.66 (s, Ph), 205.4 (br s, CO). No signals of ipso–Ph were observed.
31P{1H} (161.98 MHz, CD3CN): δ 86 (br s, W1/2 2.5).
O), 1160s and 1150 m (PO), 1503, 1465(NO), 1285, 1306 (NO2)as, 1030w (NO2)s, 1300–1500 wide absorption (NO3, see section 2.2.3), 3440 br (OH). NMR spectra of complex 5 are identical to those of complex 4 except for the signals of solvent of crystallization in the 1H and 13C NMR spectra.
O), 1161s and 1185 m (PO), 1494s and 1518sh (NO), 1310s (NO2)as, 1030w (NO2)s, 3200 br (OH). 1H NMR (400.13 MHz, CDCl3): δ 1.61 (3H, s, CH3), 3.28 (2H, t, 2JHP 14.0, CH2), 5.0 (1H, v br s, CH), 7.24–7.25 (4H, m, Ph), 7.34–7.41 (2H, t, Ph), 7.43–7.50 (4H, m, Ph), 7.52–7.59 (2H, m, Ph), 7.84–7.99 (8H, br m, Ph). 13C{1H} NMR (100.61 MHz, CDCl3): δ 28.98 (s, CH3), 33.05 (t, 1JCP 54.7, CH), 39.81 (s, CH2), 127.51 (d, 1JCP 108.6, ipso–Ph), 127.85 (d, 1JCP 107.9, ipso–Ph), 128.80–129.15 (m, Ph), 129.20–129.15 (m, Ph), 131.00–131.42 (m, Ph), 133.09 (s, Ph), 133.42 (s, Ph), 205.7 (s, CO). 31P{1H} NMR (161.98 MHz, CDCl3): δ 40.3 (br s, W1/2 0.5).
4.2. Extraction of f-block elements
The distribution of U(VI), La(III), Nd(III), Ho(III), and Yb(III) in the extraction systems was studied in model solutions of 3.75 M nitric acid at metal concentrations of 0.25 mM. Extractant solutions (0.01 M) in CHCl3 were prepared from precisely weighed amounts of the reagents. The experiments were carried out in ampoules with ground stoppers at 20 ± 1 °C. The volumes of both organic and aqueous phases were equal to 2 mL. The solution was stirred for 30 min at 80 rpm to achieve constant values of the distribution ratio (D = [M]org/[M]aq). After the extraction, 0.5 mL of the aqueous solution was taken for further analysis. Metal concentrations in the initial and equilibrated aqueous solutions were determined by spectrophotometry.384.3. X-ray crystallography
The X-ray diffraction data were collected on a Bruker Apex II CCD diffractometer using Mo-Kα (λ = 0.71073 Å) radiation. The structures were solved by direct methods and refined by full-matrix least squares against F2. Non-hydrogen atoms were refined in anisotropic approximation with an exception of the disordered atoms. The oxygen atoms of the uranyl cation in 2 are equally disordered over two sites and were refined isotropically. Occupation of solvate molecules in 2–5 was refined as a free variable giving full occupation with an exception of one MeCN solvent molecule in 3 which was further fixed at 1/3. Complex 3 crystallizes as a twinned single crystal; twin components were separated using PLATON39 and refined using the HKLF 5 instruction and additional BASF factor. Hydrogen atoms were included in the refinement by the riding model with Uiso(H) = nUeq(C), where n = 1.5 for methyl and OH groups and 1.2 for the other atoms. All calculations were performed using SHELXL201340 and OLEX2.041 program packages.CCDC 1442097–1442102 for compounds L and 1–5 contain the supplementary crystallographic data for this study (Table 8).
| Compound | L 0.5C6H6 | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|---|
| a R = ∑||Fo| − |Fc||/∑|Fo|. b R w = [∑(w(Fo2 – Fc2)2)/∑(w(Fo2))]1/2. c GOF = [∑w(Fo2 − Fc2)2/(Nobs − Nparam)]1/2. | ||||||
| Empirical formula | C31H29O3P2 | C28H26N2O11P2U | C58H58N2O19P4U2 | C60.67H59LaN5.33O15P4 | C62H61N6NdO15P4 | C58H58N3NdO16P4 |
| F w | 511.48 | 866.48 | 1687.00 | 1365.59 | 1398.28 | 1321.19 |
| Color, habit | Colorless, prism | Yellow, plate | Yellow, needle | Colorless, needle | Pink, needle | Pink, prism |
| Crystal size (mm3) | 0.23 × 0.18 × 0.15 | 0.23 × 0.18 × 0.10 | 0.29 × 0.08 × 0.08 | 0.29 × 0.08 × 0.08 | 0.31 × 0.08 × 0.06 | 0.23 × 0.19 × 0.11 |
| F(000) | 538 | 1672 | 3264 | 2789 | 2860 | 1350 |
| T, K | 148 | 100 | 100 | 120 | 120 | 120 |
| Space group, Z | Triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , 2 , 2 |
Monoclinic, P21/c, 4 | Orthorhombic, Pbca, 4 | Monoclinic, C2/c, 4 | Monoclinic, C2/c, 4 | Triclinic, P, 2 |
| a (Å) | 9.8274(12) | 11.3280(7) | 18.2266(7) | 13.444(8) | 13.4706(8) | 12.590(7) |
| b (Å) | 11.8989(15) | 13.8253(9) | 17.8372(7) | 17.849(9) | 17.8919(10) | 13.510(10) |
| c (Å) | 13.0901(16) | 19.7227(13) | 19.0939(8) | 26.571(15) | 26.4711(15) | 19.148(15) |
| α (°) | 70.615(2) | 90 | 90 | 90 | 90 | 90.574(16) |
| β (°) | 68.839(2) | 104.445(1) | 90 | 90.709(14) | 91.378(1) | 96.836(16) |
| γ (°) | 87.156(2) | 90 | 90 | 90 | 90 | 111.923(17) |
| V (Å3) | 1342.3(3) | 2991.2(3) | 6207.6(4) | 6375(6) | 6378.1(6) | 2994(4) |
| d c (g/cm3) | 1.266 | 1.924 | 1.805 | 1.423 | 1.456 | 1.465 |
| μ (MoKα) (cm−1) | 0.193 | 5.597 | 5.387 | 0.839 | 0.984 | 1.043 |
| θ max (°) | 29.00 | 31.77 | 27.00 | 25.96 | 31.62 | 29.906 |
| I hkl coll/uniq | 16085/7124 |
34749/7917 |
62107/6743 |
11231/8846 |
44834/10755 |
38088/17037 |
| R int | 0.042 | 0.055 | 0.052 | — | 0.027 | 0.145 |
| Obs. refl./N/restraints | 5432/327/0 | 6105/398/0 | 4803/406/11 | 4827/394/14 | 10078/403/2 |
8453/752/41 |
| R,a % [I > 2σ(I)] | 0.047 | 0.028 | 0.065 | 0.099 | 0.023 | 0.078 |
| R w,b % | 0.125 | 0.057 | 0.158 | 0.245 | 0.060 | 0.152 |
| GOFc | 1.02 | 1.04 | 1.06 | 1.03 | 1.06 | 1.03 |
4.4. Computational details
Topological analysis of electron density according to Bader's “Atoms in Molecules” theory (AIM)21 was performed using the AIMAll42 program. The electron density ρ(r) (ED) in wfx-format for AIM calculations was generated for complexes 1–5 in the GAUSSIAN 0943 software suite at the DFT level of theory. The hybrid PBE044 functional and all-electron scalar relativistic basis set45 for U and La atoms and 6-311+G** basis set46 for other atoms were utilized. The geometry parameters were used as those obtained from X-ray diffraction experiments. Interaction energies were estimated with Espinosa's correlation scheme Econt = −1/2V(r).22 For paramagnetic ionic complex 5, a simplified procedure was applied by reason of a non-convergence SCF equation. π-Stacking energy evaluation was performed for “frozen” ligands in conformations that they adopt in complex 5. This technique was checked by computation for complexes 1 and 4. The difference in energies obtained by simplified and complete procedures was not larger than 0.02 kcal mol−1.Acknowledgements
This work was supported by the Russian Foundation for Basic Research (Grant No. 14-03-00695-a).Notes and references
- For reviews, see: (a) A. E. V. Gorden, M. A. DeVore and B. A. Maynard, Inorg. Chem., 2013, 52, 3445 CrossRef CAS PubMed; (b) E. V. Sharova, O. I. Artyushin and I. L. Odinets, Russ. Chem. Rev., 2014, 83, 95 CrossRef; (c) D. Rosario-Amorin, S. Ouizem, D. A. Dickie, Y. Wen, R. T. Paine, J. Gao, J. K. Grey, A. de Bettencourt-Dias, B. P. Hay and L. H. Delmau, Inorg. Chem., 2013, 52, 3063 CrossRef CAS PubMed; (d) P. Tkac, G. F. Vandegrift, G. J. Lumetta and A. V. Gelis, Ind. Eng. Chem. Res., 2012, 51, 10433 CrossRef CAS; (e) S. Tachimori and Y. Morita, in Ion exchange and solvent extraction, ed. B.A. Moyer, CRC Press, 2010, vol. 19, pp. 1–64 Search PubMed; (f) V. Manchanda, P.N. Pathak and P.K. Mohapatra, in Ion exchange and solvent extraction, ed. B.A. Moyer, CRC Press, 2010, vol. 19, pp. 66–118 Search PubMed; (g) C. Hill, in Ion exchange and solvent extraction, ed. B.A. Moyer, CRC Press, 2010, vol. 19, pp. 119–194 Search PubMed; (h) V. A. Babain, in Ion exchange and solvent extraction, ed. B.A. Moyer, CRC Press, 2010, vol. 19, pp. 359–380 Search PubMed.
- A. M. Rozen and B. V. Krupnov, Russ. Chem. Rev., 1996, 65, 973 CrossRef.
- H. J. Cristau, D. Virieux, J. F. Dozol and H. Rouquette, J. Radioanalyt. Nucl. Chem., 1999, 241, 543 CrossRef CAS.
- I. V. Smirnov, Radiokhimiya (in Russian), 2007, 49, 44 CAS.
- (a) A. N. Turanov, V. K. Karandashev and N. A. Bondarenko, Radiokhimiya (in Russian), 2007, 49, 55 CAS; (b) A. N. Turanov, V. K. Karandashev, A. N. Yarkevich and Z. V. Safronova, Solvent Extr. Ion Exch., 2004, 22, 391 CrossRef CAS; (c) T. Yaita and S. Tachimori, Radiochim. Acta, 1996, 73, 27 CrossRef CAS; (d) A. N. Turanov, V. K. Karandashev, V. E. Baulin, A. N. Yarkevich and S. V. Nosenko, Radiokhimiya (in Russian), 2012, 54, 363 CAS; (e) A. N. Turanov, V. K. Karandashev, A. N. Yarkevich and Z. V. Safronova, Radiokhimiya (in Russian), 2011, 53, 264 CAS; (f) N. E. Kochetkova, O. E. Kojro, N. P. Nesterova, T. Ya. Medved’, M. K. Chmutova, B. F. Myasoedov and M. I. Kabachnik, Radiokhimiya (in Russian), 1986, 28, 338 CAS; (g) A. Yu. Shadrin, I. V. Smirnov, R. N. Kiseleva, N. P. Nesterova, Yu. M. Polikarpov and M. I. Kabachnik, Radiokhimiya (in Russian), 1993, 35, 50 CAS.
- A. M. J. Lees and A. W. G. Platt, Inorg. Chem., 2003, 42, 4673 CrossRef CAS PubMed.
- S. Kannan, N. Rajalakshmi, K. V. Chetty, V. Venugopal and M. G. B. Drew, Polyhedron, 2004, 23, 1527 CrossRef CAS.
- (a) S. M. Cornet, I. May, M. P. Redmond, A. J. Selvage, C. A. Sharrad and O. Rosnel, Polyhedron, 2009, 28, 363 CrossRef CAS; (b) N. J. Hill, W. Levason, M. C. Popham, G. Reid and M. Webster, Polyhedron, 2002, 21, 445 CrossRef CAS; (c) A. D. Sutton, G. H. John, M. J. Sarsfield, J. C. Renshaw, I. May, L. R. Martin, A. J. Selvage, D. Collison and M. Helliwell, Inorg. Chem., 2004, 43, 5480 CrossRef CAS PubMed.
- P. E. Sues, A. J. Lough and R. H. Morris, Inorg. Chem., 2012, 51, 9322 CrossRef CAS PubMed.
- A. N. Turanov, V. K. Karandashev, G. V. Bodrin, E. I. Goryunov and V. K. Brel, Radiokhimiya (in Russian), 2015, 57, 509 Search PubMed.
- J. B. Conant, J. B. S. Braverman and R. E. Hussey, J. Am. Chem. Soc., 1923, 45, 165 CrossRef CAS.
- E. I. Goryunov, G. V. Bodrin, I. B. Goryunova, Yu. V. Nelyubina, P. V. Petrovskii, T. V. Strelkova, A. S. Peregudov, A. G. Matveeva, M. P. Passechnik, S. V. Matveev and E. E. NifantҐev, Russ. Chem. Bull., Int. Ed., 2013, 62, 780 CrossRef CAS.
- Geometry optimization and normal coordinate analysis for the X-ray conformation of L were carried out using DFT at PBE level by Priroda 6 program package (2006.08.20) [ D.N. Laikov, Chem. Phys. Lett., 1997, 281, 151 CrossRef CAS; D.N. Laikov and Yu. A. Ustynyuk, Russ. Chem. Bull., Int. Ed., 2005, 54, 820 CrossRef; J. P. Perdew, K. Bruke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef PubMed ].
- (a) G. A. Doyle, D. M. L. Goodgame, A. Sinden and D. J. Williams, Chem. Commun., 1993, 1170 RSC; (b) G. H. John, I. May, M. J. Sarsfield, H. M. Steele, D. Collison, M. Helliwell and J. D. McKinney, Dalton Trans., 2004, 734 RSC; (c) B. T. McGrail, L. S. Pianowski and P. C. Burns, J. Am. Chem. Soc., 2014, 136, 4797 CrossRef CAS PubMed.
- W. D. Wang, A. Bakas and J. H. Espenson, Inorg. Chem., 1995, 34, 6034 CrossRef CAS.
- S. Wahu, J.-C. Berthet, P. Thuéry, D. Guillaumont, M. Ephritikhine, R. Guillot, G. Cote and C. Bresson, Eur. J. Inorg. Chem., 2012, 3747 CrossRef CAS.
- I. A. Charushnikova and C. Den Auwer, Russ. J. Coord. Chem., 2004, 30, 546 CrossRef.
- N. Akbarzadeh-T and T. Kondori, Res. Chem. Intermed., 2015, 41, 845 CrossRef CAS.
- See for example: (a) P. Charpin, G. Folcher, M. Lance, M. Nierlich and D. Vigner, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1985, 41, 1302 CrossRef; (b) B. Masci and P. Thuery, Polyhedron, 2005, 24, 229 CrossRef CAS; (c) G. A. Doyle, D. M. L. Goodgame, A. Sinden and D. J. Williams, Chem. Commun., 1993, 1170 RSC; (d) I. M. Aladzheva, O. V. Bykhovskaya, Y. V. Nelyubina, Z. S. Klemenkova, P. V. Petrovskii and I. L. Odinets, Inorg. Chim. Acta, 2011, 373, 130 CrossRef CAS.
- L. Huang, B.-Q. Ma, C.-H. Huang, T. C. W. Mak, G.-Q. Yao and G.-X. Xu, J. Coord. Chem., 2001, 54, 95 CrossRef CAS.
- R. F. W. Bader, Atoms in Molecules. A Quantum Theory, Clarendron Press, Oxford, 1990 Search PubMed.
- (a) E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170 CrossRef CAS; (b) E. Espinosa, I. Alkorta, I. Rozas, J. Elguero and E. Molins, Chem. Phys. Lett., 2001, 336, 457 CrossRef CAS.
- For example: (a) A. A. Korlyukov and M. Yu Antipin, Russ. Chem. Rev., 2012, 81, 105 CrossRef CAS; (b) A. A. Korlyukov, Russ. Chem. Rev., 2015, 84, 422 CrossRef CAS; (c) C. Matta and R. J. Boyd, The Quantum Theory of Atoms in Molecules, Wiley-VCH, Weinheim, Germany, 2007 Search PubMed; (d) I. V. Glukhov, K. A. Lyssenko, A. A. Korlyukov and M. Yu. Antipin, Faraday Discuss., 2007, 135, 203 RSC.
- J. Overgaard, M. P. Waller, R. Piltz, J. A. Platts, P. Emseis, P. Leverett, P. A. Williams and D. E. Hibbs, J. Phys. Chem. A, 2007, 111, 10123 CrossRef CAS PubMed.
- A. G. Matveeva, Z. A. Starikova, R. R. Aysin, R. S. Skazov, S. V. Matveev, G. I. Timofeeva, M. P. Passechnik and E. E. Nifant'ev, Polyhedron, 2013, 61, 172 CrossRef CAS.
- R. K. Castellano, F. Diederch and E. A. Mayer, Angew. Chem., Int. Ed., 2003, 42, 1210 CrossRef PubMed.
- See for example: (a) M. B. Jones and A. J. Gaunt, Chem. Rev., 2013, 113, 1137 CrossRef CAS PubMed; (b) B. G. Vats, S. Kannan, K. Parvathi, D. K. Maity and M. G. B. Drew, Polyhedron, 2015, 89, 116 CrossRef CAS; A. G. Matveeva, M. S. Grigoriev, T. K. Dvoryanchikova, S. V. Matveev, A. M. Safiulina, O. A. Sinegribova, M. P. Passechnik, I. A. Godovikov, D. A. Tatarinov, V. F. Mironov and I. G. Tananaev, Russ. Chem. Bull., Int. Ed., 2012, 61, 399 Search PubMed.
- (a) A. G. Matveeva, A. S. Peregudov, E. I. Matrosov, Z.A. Starikova, S. V. Matveev and E. E. Nifant'ev, Inorg. Chim. Acta, 2009, 362, 3607 CrossRef CAS; (b) W. Levason, E. H. Newman and M. Webster, Polyhedron, 2000, 19, 2697 CrossRef CAS; (c) M. Bosson, W. Levason, T. Patel, M. C. Popham and M. Webster, Polyhedron, 2001, 20, 2055 CrossRef CAS; (d) A. Bowden, S. J. Coles, M. B. Pitak and A. W. G. Platt, Polyhedron, 2014, 68, 258 CrossRef CAS; (e) A. Bowden, P. N. Horton and A. W. G. Platt, Inorg. Chem., 2011, 50, 2553 CrossRef CAS PubMed; (f) N.J. Hill, W. Levason, M.C. Popham, G. Reid and M. Webster, Polyhedron, 2002, 21, 445 CrossRef CAS.
- See for example: (a) R. Babecki, A. W. G. Platt and J. Fawcett, J. Chem. Soc., Dalton Trans., 1992, 675 RSC; (b) B.G. Vats, S. Kannan, I. C. Pius, D. M. Noronha, D. K. Maity and M. G. B. Drew, Polyhedron, 2014, 75, 81 CrossRef CAS.
- CH⋯OC contacts were revealed in the ligand L (r(H⋯O) = 2.4/2.6–2.8 Å) for intra- and intermolecular interactions) and uranyl complex 1 (r(H⋯O) = 2.3–3.0/2.6–2.7 Å), however, they are much weaker than in complexes 3 and 4 (Fig. S1–S3, ESI†). Accordingly, the IR spectra of L and complex 1 display ν(CO) at 1720 cm−1.
- K. Nakamoto, IR and Raman Spectra of Inorganic and Coordination Compounds, Wiley, New York, 5th edn, 1997, Part B, 432 pp Search PubMed.
- Vibrations of the C
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N group have a low intensity. Moreover, solvate acetonitrile molecules are poorly retained in the crystal lattice of the studied complexes. Thus, freshly prepared finely crystalline complex 4 when stored in air becomes amorphous over time according to X-ray powder diffraction measurements. The content of MeCN in 3 and 4 according to elemental analysis data and NMR is lower than that determined from X-ray crystallography.
N group have a low intensity. Moreover, solvate acetonitrile molecules are poorly retained in the crystal lattice of the studied complexes. Thus, freshly prepared finely crystalline complex 4 when stored in air becomes amorphous over time according to X-ray powder diffraction measurements. The content of MeCN in 3 and 4 according to elemental analysis data and NMR is lower than that determined from X-ray crystallography. - For example, CN of ytterbium, whose radius is close to that of lutetium, equals to eight in the crystalline complex of akin ligand [Yb(NO3)(L′)3](NO3)2·2H2O.6.
- (a) A. Bowden, K. Singh and A. W. G. Platt, Polyhedron, 2012, 42, 30 CrossRef CAS; (b) A. G. Matveeva, T. V. Baulina, Z. A. Starikova, M. S. Grigor'ev, Z. S. Klemenkova, S. V. Matveev, L. A. Leites, R. R. Aysin and E. E. Nifant'ev, Inorg. Chim. Acta, 2012, 384, 266 CrossRef CAS.
- (a) D. Das, S. Kannan, D. K. Maity and M. G. B. Drew, Inorg. Chem., 2012, 51, 4869 CrossRef CAS PubMed; (b) C. Villiers, P. Thuéry and M. Ephritikhine, Polyhedron, 2004, 23, 1613 CrossRef CAS; (c) D. J. McCabe, E. N. Duesler and R. T. Paine, Inorg. Chem., 1985, 24, 4626 CrossRef CAS; (d) S. M. Bowen, E. N. Duesler and R. T. Paine, Inorg. Chim. Acta, 1982, 61, 155 CrossRef CAS.
- In the spectra of lanthanide nitrate complexes with polydentate bisphosphoryl-containing ligands, the signal of the uncoordinated P(O) group is usually broadened and slightly shifted downfield relative to the free ligand signal.28a,34b.
- E. N. Tsvetkov, N. A. Bondarenko, I. G. Malakhova and M. I. Kabachnik, Zh. Obshch. Khim. (in Russian), 1985, 55, 11 CAS.
- S. B. Savin, Arsenazo III, Atomizdat, Moscow, 1966, 256 pp. (in Russian) Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- Todd A. Keith, AIMAll (Version 10.05.04), TK Gristmill Software, Overland Park, KS, USA, 2012 <http://aim.tkgristmill.com> Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- (a) D. A. Pantazis and F. Neese, J. Chem. Theory Comput., 2009, 5, 2229 CrossRef CAS PubMed; (b) D. A. Pantazis and F. Neese, J. Chem. Theory Comput., 2011, 7, 677 CrossRef CAS.
- (a) R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS; (b) A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Selected IR data for complexes 1, 3–6, X-ray data for compounds L, 1–5, selected NMR spectra for compounds L, 1, 3, 6, and extraction data for L and L′. CCDC 1442097–1442102. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt04963f |
| This journal is © The Royal Society of Chemistry 2016 |