
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiamidophosphines with six-membered chelates and their coordination chemistry with group 4 metals: development of a trimethylene-methane-tethered [PN2]-type “molecular claw”†
S.
Batke‡
,
T.
Kothe‡
,
M.
Haas
,
H.
Wadepohl
and
J.
Ballmann
 *
*
Anorganisch-Chemisches Institut, Ruprecht-Karls-Universität Heidelberg, Im Neuenheimer Feld 276, 69120 Heidelberg, Germany. E-mail: Joachim.ballmann@uni-heidelberg.de
First published on 25th January 2016
Abstract
The coordination chemistry of the phosphine-tethered diamidophosphine ligands PhP(CH2CH2CH2NHPh)2 (pr[NPN]H2) and PhP(1,2-CH2-C6H4-NHSiMe3)2 (bn[NPN]H2) featuring six-membered N–C3–P chelates was explored with group 4 metals, which allowed for the consecutive development of a new trimethylene-methane-tethered [PN2] scaffold. In the case of the propylene-linked system pr[NPN]H2, access to the sparingly soluble dibenzyl derivative pr[NPN]ZrBn2 (3-Zr) was gained, while thermally sensitive zirconium and hafnium diiodo complexes bn[NPN]MI2 (5-M, M = Zr, Hf) were isolated in the case of the benzylene-linked derivative bn[NPN]H2. Despite the related phosphine-tethered backbone architectures of both of these ligands, their group 4 complexes were found to exhibit either C1-symmetric (bn[NPN]MX2) or averaged CS-symmetric (pr[NPN]MX2) structures in solution. To restrain the overall flexibility of these systems and thereby control the properties of the resulting complexes without disrupting the six-membered chelates, the new trimethylene-methane-tethered N,N′-di-(tert-butyl)-substituted [PN2]H2 protioligand was designed. This tripodal ligand system was prepared on a gram scale and its CS-symmetric dichloro complexes [PN2]MCl2 (6-M, M = Ti, Zr, Hf) were isolated subsequently. The benzene-soluble dibenzyl derivative [PN2]ZrBn2 (7-Zr) was synthesised as well and characterised by X-ray diffraction. These results are discussed not only in conjunction with the known [NPN]-coordinated group 4 complexes incorporating five-membered chelates, but also in the context of “molecular claws” that are related to the new [PN2] tripod.
Introduction
Monoanionic amidodiphosphines (commonly abbreviated as [PNP] ligands) are particularly well-studied hybrid ligands1–4 and numerous amidodiphosphine complexes with the central metals embedded in five-5–15 or six-membered chelates16–18 have been prepared over the past decades.19 Novel stoichiometric transformations and applications in catalysis have been discovered for these complexes, not only for late20–29 but also for early transition metals.30–35 For the related dianionic diamidophosphines (commonly abbreviated as [NPN] ligands), complexes of the early transition metals are dominating the field,36 possibly due to their unique reactivities towards small molecules.37–43 Zirconium halides in an [NPN] coordination environment, for example, were shown to react with gaseous dinitrogen upon reduction, although particularly fine-tuned ligands are required for these challenging transformations.44–47 Chronologically, the first silyl-substituted and silylmethylene-linked [NPN] systems A and B (see Chart 1) have been prepared almost simultaneously by Schrock48 and Fryzuk49 more than 15 years ago. Considering that related silylmethylene- and ethylene-linked [PNP] ligands were already well-established at the time,3,50,51 it is not surprising that [NPN] ligands are still not as well developed as their [PNP] counterparts. Over the past years, however, the original systems A and B have been optimised by Fryzuk and co-workers in order to circumvent silylamide bond cleavage reactions and other undesired decomposition pathways.52,53 Following an iterative approach, the phenylene-, thiophenylene- and cyclopentenylene-linked systems C, D and E (see Chart 1) were prepared and their zirconium complexes isolated and examined subsequently.54–56 | ||
| Chart 1 Monomeric [NPN] complexes of the group 4 metals incorporating five-membered chelates (highlighted in blue). A: R = tBu, Ph, 2,6-Me2C6H3; C: R = Mes, p-Tol, Et; E: Ar = 2,6-Me2C6H3, 2,6-iPr2C6H3; A–F: M = Zr (Ti or Hf only in case of A, C and F); X = monoanionic ligand (e.g. Cl, I, NMe2, Bn, Me). | ||
System C was found to be well-suited for the preparation of dinitrogen complexes46 and for the stabilization of a reactive dimethyl zirconium species.56 A derivative of system C, the 2-methyl-tetrahydroquinoline-derived system F (see Chart 1),57 seems noteworthy as well, as it might open up an avenue for the introduction of chirality. What emerges from the inspection of systems A–F is that each ligand was designed to form five-membered chelates upon complexation of a metal ion. Indeed, diamidophosphines with six-membered chelates have not been explored so far, while [PNP] scaffolds with six-membered chelates are known since 1988.4
Thus, an exploration of [NPN] systems with six-membered chelates seemed essential to close the “development gap” between [PNP] and [NPN] systems. In view of the above mentioned iterative optimization that was required to fine-tune [NPN] scaffolds with five-membered chelates, a similar approach was planned to access and optimise the corresponding systems with six-membered chelates.
Expecting that the propylene- and the benzylene-linked ligands, pr[NPN] and bn[NPN] (see Chart 2), can be prepared easily, an exploration of their group 4 chemistry was initially pursued. The findings that we made with these complexes allowed for a consecutive ligand optimization, which led to the development of a new tripodal trimethylene-methane-tethered diamidophosphine scaffold. This system was then investigated with respect to its coordination chemistry with group 4 metals. The results, which were attained in this two-step development process are discussed in the following.
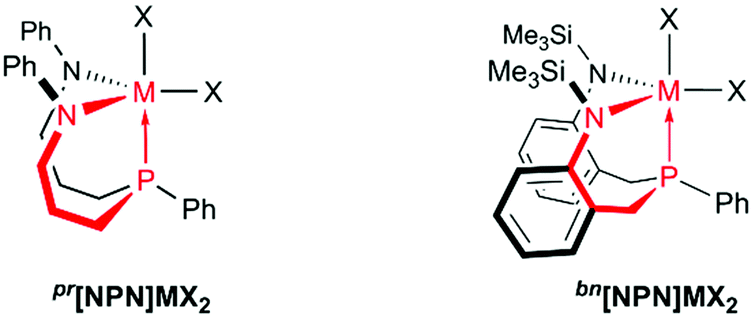 | ||
| Chart 2 Diamidophosphine complexes of the group 4 metals incorporating six-membered chelates (highlighted in red). | ||
Results and discussion
Step 1: group 4 complexes of pr[NPN]H2 and bn[NPN]H2
In agreement with the above mentioned expectation, the propylene- and benzylene-linked ligands, pr[NPN]H2 and bn[NPN]H2, were readily synthesised following an adaption of our previously reported experimental procedure for the preparation of related triamidophosphine scaffolds (see Scheme S1 in the ESI†).58,59 Both ligands were obtained in gram quantities and high purities, which allowed for their direct use to prepare the envisioned group 4 complexes shown in Chart 2.In the case of pr[NPN]H2, the bis-dimethylamido complexes pr[NPN]M(NMe2)2 (1-M, M = Zr, Hf, see Scheme 1) were obtained via protonolysis with M(NMe2)4 (M = Zr, Hf) and isolated in good yields (1-Zr: 70% with δ(31P) = −28.0 ppm; 1-Hf: 67% with δ(31P) = −20.2 ppm).
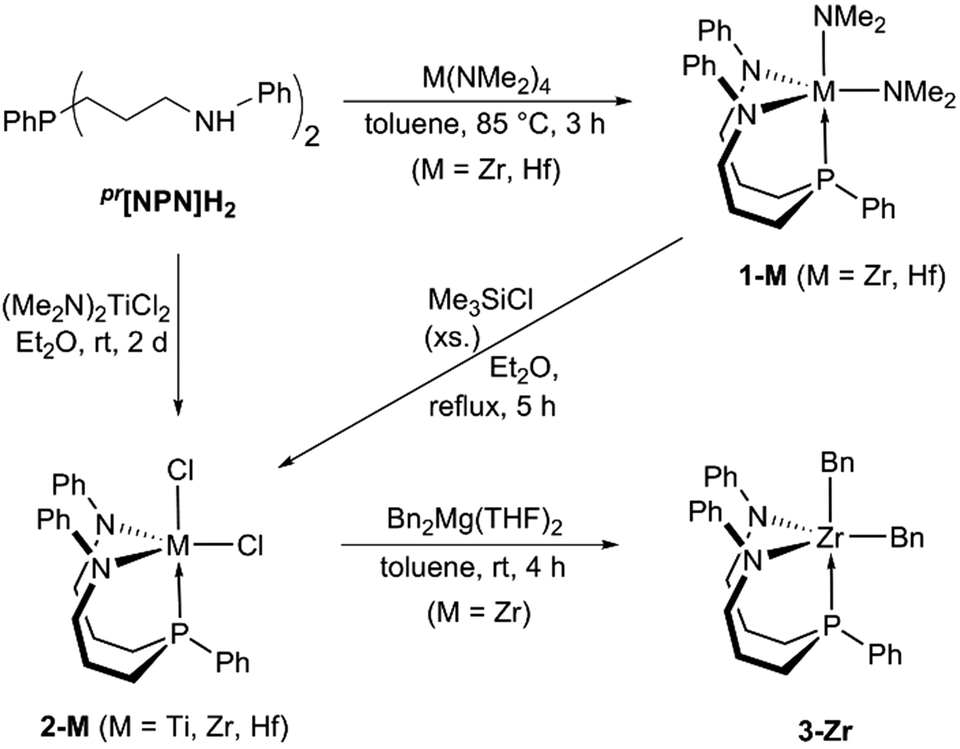 | ||
| Scheme 1 Synthesis of complexes 1-M, 2-M and 3-Zr. | ||
In the 1H NMR spectra of complexes 1-M (M = Zr, Hf), two signals each were detected for the dimethylamides, indicative of trigonal bipyramidal complex geometries. At room temperature, only one set of signals was detected for the equivalent ligand sidearms and significant signal broadening was observed at −80 °C. Whether the fac- or the mer-isomers of complexes 1-M (M = Zr, Hf) were present in solution could not be distinguished by NMR spectroscopy, although the facial coordination mode shown in Scheme 1 seems more likely in view of the solution structure of 3-Zr (vide infra).
Following a frequently used synthetic pathway, complexes 1-M were converted to the corresponding dichlorides pr[NPN]MCl2 (2-M, M = Zr, Hf) via reaction with Me3SiCl. In the present case, these reactions were only successful when carried out in boiling diethylether, which led to the precipitation of the desired products in yields of 62% (2-Zr) and 51% (2-Hf), respectively. A similar procedure relying on the precipitation of the product was found to be effective for titanium as well. Upon reaction of pr[NPN]H2 with (Me2N)2TiCl2 in diethylether, a brick-red precipitate of pr[NPN]TiCl2 (2-Ti) formed over the course of two days at room temperature and was isolated in modest yields of 35% (see Scheme 1).
In the 31P{1H} NMR spectra of 2-M (M = Ti, Zr, Hf), uninformative singlets were observed at 8.0 ppm (2-Ti), −13.1 ppm (2-Zr) and −4.0 ppm (2-Hf) in dichloromethane-d2, while rapid decomposition was noticed in THF-d8. In the respective 1H NMR spectra of these complexes, partially overlapping methylene resonances were detected at room temperature in addition to three well-resolved signals for the N-phenyl substituents. On basis of these NMR data, an overall CS-symmetry was deduced for all three complexes 2-M (M = Ti, Zr, Hf) in solution. To elucidate their corresponding solid state structures, single crystals suitable for X-ray diffraction were grown from pentane-layered dichloromethane solutions (2-Ti: see Fig. 1, 2-Zr, 2-Hf: see Fig. S21 and S22 in the ESI,†cf.Table 1 for selected metrical parameters). In each case, a trigonal bipyramidal complex geometry with the ligand binding in a facial coordination mode was found, but the expected CS-symmetry was not observed in the solid state molecular structures.
 | ||
| Fig. 1 ORTEP diagram of the molecular structure of 2-Ti (hydrogen atoms omitted for clarity, thermal ellipsoids set at 50% probability). Selected bond lengths and angles are summarised in Table 1. | ||
| 2-Ti | 2-Zr | 2-Hf | |
|---|---|---|---|
| M–P (Å) | 2.5431(10) | 2.7481(11) | 2.7262(6) |
| M–N (Å) | 1.9132(11) | 2.0503(12) | 2.0432(18) |
| 1.9181(11) | 2.0654(12) | 2.0545(18) | |
| M–X (Å) | 2.3345(9) | 2.4555(9) | 2.4293(6) |
| 2.3476(11) | 2.4038(7) | 2.3835(6) | |
| P–M–X (°) | 169.895(14) | 174.441(11) | 174.694(18) |
| 82.802(17) | 87.20(2) | 87.41(2) | |
| N–M–N (°) | 111.90(5) | 115.61(4) | 115.79(8) |
| δ(31P) (ppm) | 8.0 | −13.1 | −4.0 |
Instead, both ligand sidearms were found in a mutually twisted arrangement with the N-phenyl substituents pointing in opposite directions. In solution, these N-phenyl substituents were found to be equivalent, although broadened resonances were recorded in the respective proton NMR spectra at −80 °C. In the 1H NMR spectrum of 2-Ti, for example, two sets of geminally coupled signals centred at approximately 4.3 ppm were detected at −80 °C and assigned to the N-methylene protons (αH and βH, see Scheme 2). Thus, these protons were identified as symmetry-related even at low temperatures, indicating that the averaged CS-symmetry was retained in solution over a wide temperature range. The virtually temperature independent 31P{1H} NMR chemical shifts implied that the facial coordination mode of the ligand (analogous to 3-Zr) is unaffected over this temperature range, although a meridional coordination mode cannot be ruled out merely by NMR spectroscopy (precipitation of the complexes from dichloromethane-d2 hampered NMR experiments at lower temperatures). Based on these findings, the averaged CS-symmetry in solution was interpreted as a consequence of a dynamic twist of the ligand sidearms, which renders them identical on the NMR timescale. For complexes 1-M (M = Zr, Hf) practically identical observations were made upon analysis of their variable temperature NMR spectra, but no single crystals suitable for X-ray diffraction were obtained for these derivatives.
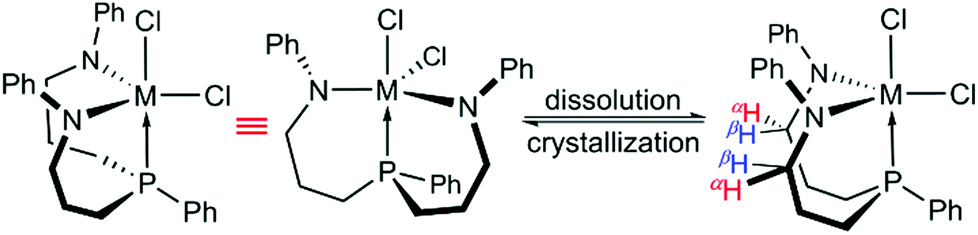 | ||
| Scheme 2 Structures of complexes 2-M (M = Ti, Zr, Hf) in the solid state (left) and in solution (right). | ||
With the dichlorides 2-M (M = Ti, Zr, Hf) in hand, the synthesis of alkyl complexes was pursued and 3-Zr was generated upon reaction of 2-Zr with Bn2Mg(THF)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 60 in toluene suspension (see Scheme 1). Over the course of this reaction, gradual dissolution of 2-Zr was observed along with precipitation of the product as a bright yellow powder, which was separated from magnesium salt by-products by extraction with dichloromethane. In case of titanium and hafnium, similar reactions met with failure, although it remains unclear whether sparingly soluble dibenzyl derivatives were actually formed. This uncertainty is mainly due to the required extraction with polar chlorinated solvents, which might result in decomposition of the products prior to analysis by NMR spectroscopy.
60 in toluene suspension (see Scheme 1). Over the course of this reaction, gradual dissolution of 2-Zr was observed along with precipitation of the product as a bright yellow powder, which was separated from magnesium salt by-products by extraction with dichloromethane. In case of titanium and hafnium, similar reactions met with failure, although it remains unclear whether sparingly soluble dibenzyl derivatives were actually formed. This uncertainty is mainly due to the required extraction with polar chlorinated solvents, which might result in decomposition of the products prior to analysis by NMR spectroscopy.
In the proton NMR spectrum of 3-Zr, two sets of signals were observed for the inequivalent benzyl groups and significantly different 3JH,P coupling constants of 2.6 and 7.9 Hz were determined. Thus, a facial coordination mode of the ligand with one of the benzyl substituents located in trans- and the other in cis-position to the phosphine is in agreement48 with the solution structure proposed for 3-Zr (see Scheme 1). The 31P{1H} NMR signal was found at −19.1 ppm and only one set of 1H NMR signals was detected for the ligand sidearms, indicative of a close structural relationship between 3-Zr and complexes 2-M in solution.
As the low solubility of 3-Zr hampered further conversions (e.g. in attempted hydrogenations or ethylene polymerization reactions), we turned our attention to the N,N′-bis-(trimethylsilyl)-substituted benzylene-linked system bn[NPN]H2 expecting that this ligand scaffold might form adequately soluble group 4 complexes. Following a synthetic strategy similar to the one used for the propylene-linked ligand, the tetrakis-(dimethylamido) precursors M(NMe2)4 (M = Ti, Zr, Hf) were reacted with bn[NPN]H2 and clean conversions were observed in the case of zirconium and hafnium. The resulting dimethylamido complexes bn[NPN]M(NMe2)2 (M = Zr, Hf) (4-M, see Scheme 3) were isolated (4-Zr: 52% yield with δ(31P) = 8.2 ppm; 4-Hf: 72% yield with δ(31P) = 15.6 ppm), but only ill-defined product mixtures were obtained in the case of titanium. In contrast to the protioligand bn[NPN]H2, inequivalent sets of 1H and 13C{1H} NMR signals for the ligand sidearms were detected for each complex 4-M (M = Zr, Hf). In the proton NMR spectra of 4-Zr, for example, two singlet resonances for the different trimethylsilyl units (δ = −0.06 ppm and 0.33 ppm) and four sets of signals for the different methylene protons (δ = 2.60 ppm, 2.69 ppm, 3.04 ppm and 3.07 ppm) were observed in addition to partially overlapping aromatic signals. For the dimethylamides, two singlet resonances were detected at δ = 2.65 ppm and 3.30 ppm, indicative of a trigonal bipyramidal complex geometry with the two dimethylamides either in cis- and trans-position to the phosphine (fac-isomer) or in a syn- and anti-alignment with respect to the phosphine-bound phenyl substituent (mer-isomer).
 | ||
| Scheme 3 Synthesis of C1-symmetric complexes 4-M and 5-M (M = Zr, Hf). Arabic numbers printed in red are used in the experimental part for the NMR signal assignment of bn[NPN]H2, 4-M and 5-M. | ||
Although these isomers cannot be distinguished easily by NMR spectroscopy in solution, it was clearly shown that complexes 4-M are C1-symmetric in solution at room temperature, while averaged CS-symmetric solution structures were observed for complexes 1-M, 2-M and 3-Zr. Even at +80 °C, no signs of a beginning coalescence of the ligand resonances were detected, indicating that complexes 4-M are configurationally stable within this temperature range. Single crystals suitable for X-ray diffraction were obtained by cooling saturated solutions of complexes 4-M (M = Zr, Hf) in Et2O to −40 °C (4-Zr: see Fig. 2, 4-Hf: see Fig. S23 in the ESI,† for selected metrical parameters see Table 2).
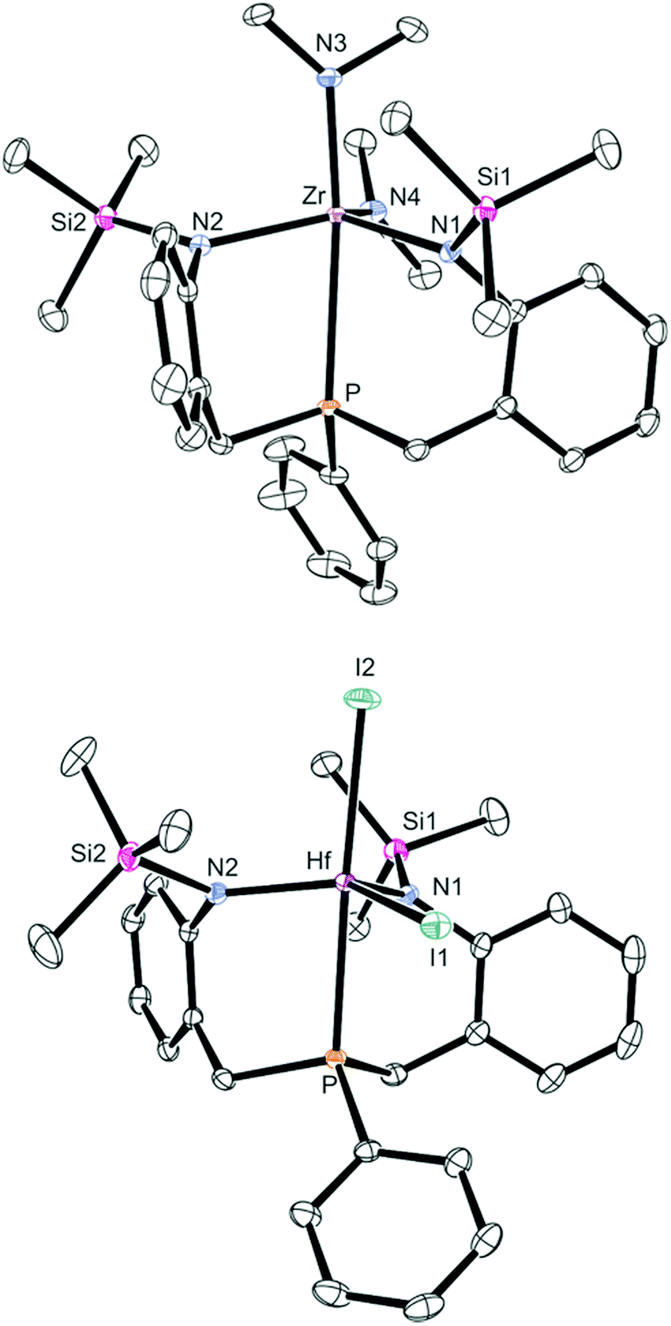 | ||
| Fig. 2 ORTEP diagrams of 4-Zr (top) and 5-Hf (bottom) (hydrogen atoms omitted for clarity, thermal ellipsoids set at 50% probability). Selected bond lengths are summarised in Table 2. | ||
| 4-Zr | 4-Hf | 5-Hf | |
|---|---|---|---|
| M–P (Å) | 2.8922(13) | 2.8725(16) | 2.7560(9) |
| M–N (Å) | 2.1223(13) | 2.1004(18) | 2.0064(15) |
| 2.1343(12) | 2.1176(16) | 2.0363(15) | |
| M–X (Å) | 2.0561(13) | 2.0437(18) | 2.8264(8) |
| 2.0574(14) | 2.0430(18) | 2.8043(8) | |
| P–M–X (°) | 169.95(3) | 170.52(4) | 176.376(9) |
| 92.00(3) | 91.67(5) | 83.33(2) | |
| N–M–N (°) | 121.75(5) | 121.85(7) | 109.57(6) |
| δ(31P) (ppm) | 8.2 | 15.6 | 13.4 |
Inspection of the refined molecular structure of 4-Zr revealed that the central zirconium ion is situated in a trigonal–bipyramidal coordination polyhedron with N1, N2 and N4 occupying the equatorial positions. The phosphine donor atom and N3 were found in axial positions and only a diminutive deviation from an ideal geometry (fac-isomer) was noticed. Accordingly, an averaged Neq–Zr–Neq angle of 118° and a practically linear principal axis (P–Zr–N3 = 170°) was found in the solid state structure.
The important insight gained from this molecular structure is that both ligand sidearms are inequivalent with the trimethylsilyl substituents pointing in different directions, which agrees well with the structure of 4-Zr in solution. This is also the case for the isostructural hafnium derivative 4-Hf.
Attempts to convert complexes 4-M (M = Zr, Hf) to the corresponding halides or triflates via treatment with Me3SiCl or Et3SiOTf led to unidentified mixtures of products. The reactions of 4-M (M = Zr, Hf) with Me3SiI, however, resulted in clean conversions to the corresponding iodides bn[NPN]MI2 (5-M, M = Zr, Hf), which were isolated in acceptable yields (5-Zr: 47% with δ(31P) = 8.5 ppm; 5-Hf: 59% with δ(31P) = 13.4 ppm; see Scheme 3). Particularly noteworthy is the fact that these reactions need to be carried out at low temperatures as the products were found to be thermally sensitive. Interestingly, decomposition of 5-Zr (purified sample) set in within a few hours at room temperature (in C6D6 solution), while purified samples of 5-Hf could be kept at room temperature for approximately two days (in C6D6 solution). Solid samples of both complexes were found to be stable at −40 °C for more than one month. In the 1H NMR spectra of 5-M two different singlet resonances for the trimethylsilyl units were found for each complex and two signals detected in the corresponding 13C{1H} NMR spectra as well. As in the case of 4-M, overall C1-symmetries were deduced from these NMR data and no cross-peaks were observed in the EXSY NMR spectra at room temperature. A crystallographic analysis revealed that complex 5-Hf is C1-symmetric in the solid state as well (see Fig. 2, cf.Table 2 for selected metrical parameters).
With the iodides 5-M available, the focus was once again set on the isolation of alkyl derivatives. Unfortunately, no clean conversions to the desired dialkyl species were observed in the reactions of 5-M with LiCH2SiMe3, Bn2Mg(THF)2 or MeMgBr, not even in the presence of excess of the alky transfer reagent.
In view of these findings, the first step of our ligand screening and optimization procedure was considered complete and three particularly noteworthy observations were made:
(i) Group 4 complexes of both ligands (pr[NPN] and bn[NPN]) were found to adopt C1-symmetric structures in the solid state. This observation was ascribed to the highly flexible phosphine-tethered ligand backbones. Note that this interpretation applies to the solid state structures only, while higher ligand flexibilities might facilitate dynamic processes in solution and thus lead to the observation of higher averaged symmetries in solution.
(ii) In solution, averaged CS-symmetric structures, reminiscent to the CS-symmetric structures of systems A–F (cf.Chart 1), were observed only for the complexes of the N,N′-diphenyl-substituted pr[NPN] ligand and ascribed to a dynamic process that renders both ligand sidearms equivalent on the NMR timescale. Therefore, the hypothesis that the sterically demanding N-trimethylsilyl groups in bn[NPN]MX2 effectively prevent a dynamic equilibration was considered reasonable.
(iii) The low solubilities of complexes 2-M and 3-Zr (bearing the N,N′-diphenyl-substituted pr[NPN] ligand) and the thermal sensitivity of 5-Zr (bearing the N,N′-bis-(trimethylsilyl)-substituted bn[NPN] ligand) were identified as major drawbacks of the individual systems.
Based on these findings we concluded that a control over the overall flexibility of these systems was required to allow for a more predictable fine-tuning. One possibility to achieve this goal without disrupting the six-membered N–C3–P chelates, was to restrain the exceedingly flexible central 10-membered N–C3PC3–N chelate within these systems by crosslinking the sidearms. Out of the several theoretical possibilities to interconnect both sidearms, an implementation of a central six-membered N–C3–N-chelate was considered practical and a system with overall three six-membered chelates was envisioned (see Chart 3). To enhance solubilities, N-tert-butyl and N-SiMe3 substituents were considered and the former chosen in order to exclude Si–N bond cleavage reactions, which might be related to the thermal sensitivities of complexes 5-M (M = Zr, Hf). Thus, the geometrically rigid CS-symmetric “molecular claw”61 shown in Chart 3 was identified as target molecule for the second step of our study.
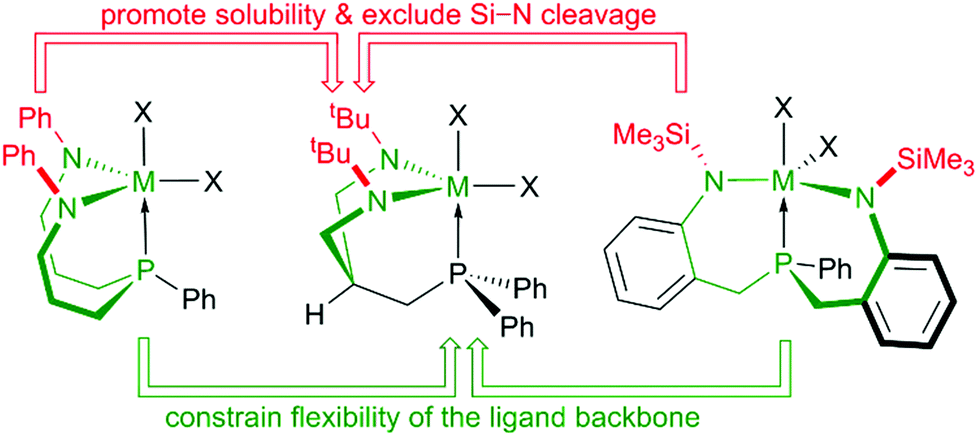 | ||
| Chart 3 Design of the tripodal diamidophosphine [PN2]H2. | ||
Step 2: [PN2]H2 and its group 4 complexes
For the synthesis of the desired [PN2]H2 ligand scaffold, a radical hydrophosphination pathway starting from diphenyl phosphine and N,N′-bis-(tert-butyl)-substituted 2-(aminomethyl)-allylamine62 was pursued. In order to achieve accept-able reaction rates, the latter amine was used as a solvent and the radical initiator (AIBN) only added in small portions over the period of 7 days (see Scheme 4). Using this methodology, the desired protioligand [PN2]H2 was obtained in >95% purity on a 5 g scale. Slow addition of AIBN was found to be of particular importance in order to effectively suppress homocoupling of Ph2PH and avoid a contamination of the product with Ph4P2.63,64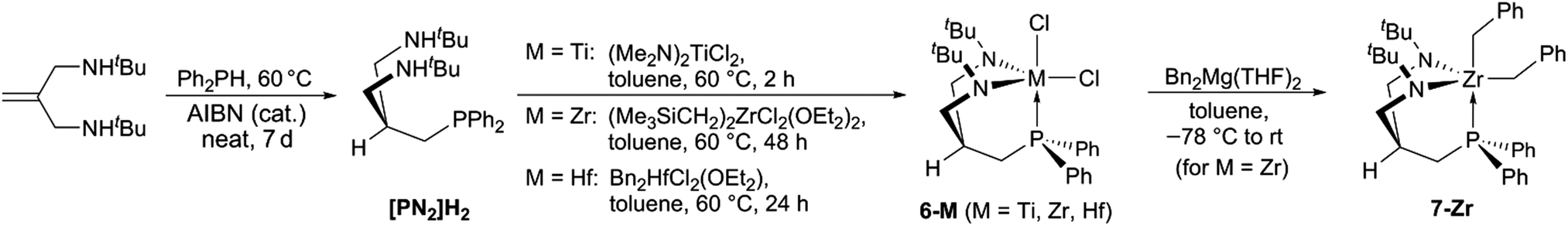 | ||
| Scheme 4 Synthesis of the tripodal protioligand [PN2]H2 and its group 4 complexes 6-M (M = Ti, Zr, Hf) and 7-Zr. | ||
With [PN2]H2 available in adequate quantities, the synthesis of the group 4 dichloro complexes 6-M (M = Ti, Zr, Hf) was targeted (see Scheme 4). For M = Zr, (Me3SiCH2)2ZrCl2(OEt2)265 was identified as the most convenient precursor, while the use of Bn2ZrCl2(OEt2)66 resulted in lower yields of the target complex (6-Zr). Both precursors (Me3SiCH2)2HfCl2(OEt2)265 and Bn2HfCl2(OEt2)66 were suitable for the preparation of 6-Hf, although the use of the former starting material is not recommended due to the difficulties associated with its isolation.67 Complexes (Me2N)2MCl2(L)2 (M = Zr, Hf, L = THF, DME)68–70 were found to be unsuitable for the preparation of 6-Zr and 6-Hf. Interestingly, the titanium homologue [PN2]TiCl2 (6-Ti) can be prepared starting from (Me2N)2TiCl2,71 albeit in modest yields of only 27%. In the proton NMR spectra of each complex 6-M, only one sharp singlet for the tert-butyl groups was found in addition to two geminally coupled signals for the NCH2 units. In the aliphatic region, a phosphorus-coupled signal for the CH2P unit and a multiplet for the central methine-bridgehead was detected for each complex.
In agreement with their CS-symmetric structures in solution, one set of signals was found for the symmetry-related Ph2P-groups in each complex. For titanium and hafnium, single crystals suitable for X-ray diffraction were grown by slowly cooling a boiling toluene solution of 6-Ti or 6-Hf to room temperature (6-Ti: see Fig. 3, 6-Hf: see Fig. S24 in the ESI,†cf.Table 3 for selected metrical parameters). Inspection of the refined molecular structures of 6-Ti and 6-Hf confirmed that the complexes adopt the expected CS-symmetric geometries, although a crystallographic plane of symmetry is only found in the case of titanium. The central metals in both structures were found in trigonal bipyramidal coordination polyhedra with the ligand binding in a facial coordination mode. In both cases, the axial positions of the trigonal bipyramids are occupied by the phosphine and one chloride, while both amides and the second chloride were found in the equatorial positions. The unexceptional bond distances and angles (see Table 3) indicated that both virtually isostructural assemblies are free of strains.
 | ||
| Fig. 3 ORTEP diagrams of 6-Ti (top) and 7-Zr (bottom) (hydrogen atoms omitted for clarity, thermal ellipsoids set at 50% probability). Selected bond lengths and angles are summarised in Table 3. | ||
| 6-Ti | 6-Hf | 7-Zr | |
|---|---|---|---|
| a Recorded in dichloromethane-d2 at room temperature. b Recorded in C6D6 at room temperature. | |||
| M–P (Å) | 2.597(3) | 2.7233(15) | 2.8472(4) |
| M–N (Å) | 1.867(5) | 2.000(3) | 2.0250(12) |
| 1.867(5) | 2.003(3) | 2.0489(12) | |
| M–X (Å) | 2.394(2) | 2.4687(13) | 2.3336(15) |
| 2.327(3) | 2.4367(13) | 2.3292(15) | |
| P–M–X (°) | 179.09(9) | 175.78(3) | 173.89(4) |
| 89.95(10) | 90.94(4) | 87.81(4) | |
| N–M–N (°) | 106.7(3) | 101.99(14) | 103.80(5) |
| δ 31P (ppm) | −8.4a | −1.8a | −17.7b |
The reaction of Bn2Mg(THF)2 with complexes 6-M proceeded cleanly in the case of zirconium and led to the isolation of the corresponding dibenzyl complex 7-Zr as a benzene-soluble pale yellow powder. Thus, our approach to employ tert-butyl groups to promote solubilities was considered successful.
In the proton NMR spectrum of 7-Zr, the signals corresponding to the ligand were easily assigned via comparison to the proton NMR spectrum of 6-Zr and the remaining two sets of signals assigned to two inequivalent benzyl ligands. Based on the different 3JH,P coupling constants of the benzylic methylene protons (0.4 Hz vs. 2.0 Hz),48 these sets of signals were assigned to a trans-P and cis-P-positioned benzyl substituent, which is in line with an overall CS-symmetric structure in solution. In the solid state molecular structure of 7-Zr, a trigonal bipyramidal geometry around zirconium was found with the phosphine and one of the benzyl ligands occupying the axial positions (see Fig. 3).
A comparison of the molecular structures of 6-Ti and 7-Zr (cf.Table 3 and Fig. 3) revealed that a certain flexibility of the ligand is retained allowing for variable metal amide distances (6-Ti: ∼1.85 Å vs.7-Zr: ∼2.03 Å) and different metal phosphine bond lengths (6-Ti: ∼2.60 Å vs.7-Zr: ∼2.85 Å). Therefore, this ligand might be of use not only for the complexation of group 4 metals, but also for coordinating other transition metals.
In contrast to trianionic C3-symmetric “molecular claws”,61,73,74 only a few trimethylene-methane-based mono- and dianionic C1- or CS-symmetric phosphorus-containing tripods are known,75 which effectively support a tridentate coordination to a single metal ion. Interestingly, aggregation or incomplete cage-closure has been observed more frequently.75–78 In view of the “development gap” between [PNP] and [NPN] ligands discussed in the introduction, the new [PN2] system reported herein, certainly adds to fill this void. Nevertheless, the development of amidodiphosphines still seems to be ahead, as a monoanionic silyl-tethered tripodal [NP2] scaffold has already been prepared by Peters and co-workers, while similar silyl-tethered diamidophosphines are so far unknown.79 In view of the trimethylene-methane backbone of the new [PN2] tripod, however, Rüther's neutral bis-(N-methylimidazole-2-yl)-derived phosphine [P(Im)2]80 and Gade's dianionic diamidopyridine [pyN2]81 seem to represent the most closely related systems (see Chart 4). Employing [pyN2]-coordinated zirconium hydrazinediido complexes,82,83 Gade and co-workers have applied their system not only for the stabilization of unusual polyazametallacylces84 and the assembly of a (NSN)2− unit within the coordination sphere of zirconium,85 but also for the catalytic formation of indoles from 1,1-disubstituted hydrazines and alkynes.86–89
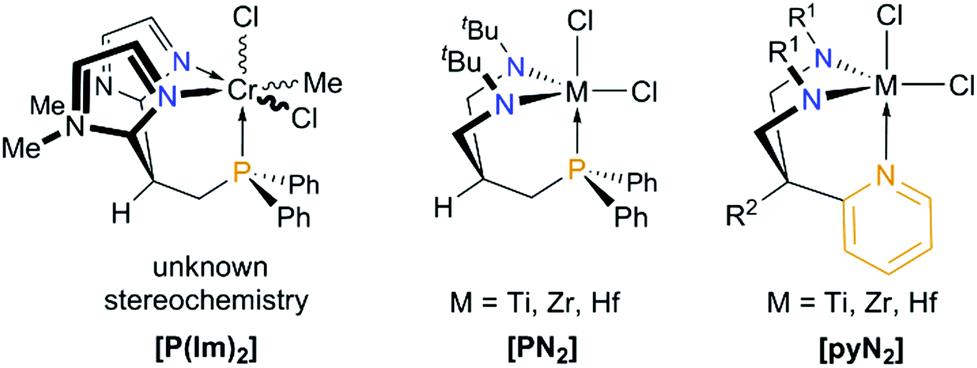 | ||
| Chart 4 Monomeric complexes of the carbon-tethered CS-symmetric “molecular claws” [P(Im)2], [PN2] and [pyN2] (R1 = tBuMe2Si, Me3Si, 3,5-Me2C6H3, C6F5 for R2 = Me and R1 = Me3Si for R2 = H). Note that [pyN2]-coordinated imido complexes of Ti, Zr, Nb, Ta, Mo and W are known as well.72 | ||
Closely related titanium imido derivatives bearing this ligand and their reactivity towards unsaturated substrates have been studied recently by Mountford,90,91 while Schrock made use of this ligand backbone to prepare cationic hafnium alkyl complexes as initiators for the living polymerization of 1-hexene.92,93
Inspired by this work, our current efforts are directed towards the application of the new [PN2]-coordinated group 4 complexes in catalysis and towards the synthesis of related [PN2]-coordinated hydrazinediido complexes.
Experimental
Materials and methods
All manipulations were performed under an atmosphere of dry and oxygen-free argon by means of standard Schlenk or Glovebox techniques. Toluene, THF, pentanes, hexanes and diethylether were purified by passing the solvents through activated alumina columns (MBraun Solvent Purification System). Toluene-d8, THF-d8 and benzene-d6 were refluxed over sodium and purified by distillation. DMSO-d6, CD2Cl2, Me3SiOSiMe3 and dichloromethane were dried over CaH2 and distilled prior to use. Diphenyl phosphine and Me3SiX (X = Cl, OTf, I) were dried over molecular sieves and purified by distillation. NMR spectra were recorded on a Bruker Avance II 400 MHz or a Bruker Avance III 600 MHz spectrometer at room temperature unless noted otherwise. 1H and 13C{1H} NMR spectra were referenced to residual proton signals of the lock solvent.9431P{1H} NMR spectra were referenced to external P(OMe)3 (141.0 ppm with respect to 85%H3PO4 at 0.0 ppm). Microanalyses (C, H, N) were performed at the Department of Chemistry at the University of Heidelberg. Compounds (Me2N)2TiCl271 and (Me2N)2MCl2(DME)2 (M = Zr, Hf)68–70 were synthesised according to literature. AIBN and M(NMe2)4 (M = Ti, Zr, Hf) were purchased from commercial suppliers and used as received. Commercially available solution of Me3SiCH2Li (1.0 M in pentane) and BnMgCl (1.0 M in Et2O) were used for the preparation of (Me3SiCH2)2MCl2(OEt2)2 (M = Zr, Hf),65,67 Bn2MCl2(Et2O) (M = Zr, Hf)66 and Bn2Mg(THF)2.60 Experimental procedures for the synthesis of pr[NPN]H2, bn[NPN]H2 and N,N′-bis-(tert-butyl)-2-(aminomethyl)-allylamine are provided in the ESI.†
Conclusions
In summary, the phosphine-tethered diamidophosphines pr[NPN]H2 and bn[NPN]H2 were prepared and the structures of their group 4 complexes analysed in solution and in the solid state. In contrast to the known [NPN] ligands with five-membered chelates, these systems incorporate six-membered chelates and form complexes that adopt C1-symmetric solid state structures. In solution, an averaged CS-symmetry was found for the pr[NPN]-coordinated species (1-M, 2-M, 3-Zr), but not for the bn[NPN]-coordinated derivatives (4-M, 5-M). Based on these findings, the ligand backbones in pr[NPN]H2 and bn[NPN]H2 were modified and the new facially coordinating diamidophosphine [PN2]H2 was designed. This trimethylene-methane-tethered tripod was then prepared and its coordination chemistry examined employing group 4 metals. In the case of 6-M and 7-Zr, CS-symmetric structures were detected not only in solution, but also in the solid state. The dibenzyl derivative 7-Zr was found to be well-soluble in (commonly) inert solvents (e.g. in C6H6) and was therefore identified as the most promising candidate for future reactivity studies. In a wider context, the three new diamidophosphines reported herein certainly contribute to diminish the “development gap” between [PNP] and [NPN] ligands, although late transition metal complexes of the new ligands have not yet been explored. Nevertheless, the arrangement of the three donor atoms within the new [PN2] system allowed for an adaption to different metal radii (Ti vs. Zr), which provides a basis for further studies directed towards the complexation of other metals. These efforts are currently ongoing in our laboratory in addition to an extensive screening for applications of the new group 4 complexes in catalysis.Acknowledgements
The authors thank the Fonds der Chemischen Industrie (FCI, Li-187/02) and the Deutsche Forschungsgemeinschaft (DFG, BA 4859/1-1) for funding of this work. We thank Prof. Dr L. H. Gade for generous support, fruitful discussions and continued interest in our work.Notes and references
- L.-C. Liang, Coord. Chem. Rev., 2006, 250, 1152–1177 CrossRef CAS.
- M. D. Fryzuk, T. S. Haddad and D. J. Berg, Coord. Chem. Rev., 1990, 99, 137–212 CrossRef CAS.
- P. Orioli and L. Sacconi, Chem. Commun., 1968, 1310–1311 RSC.
- D. Breuer and H. J. Haupt, React. Kinet. Catal. Lett., 1988, 37, 13–18 CrossRef CAS.
- G. T. Venkanna, T. V. M. Ramos, H. D. Arman and Z. J. Tonzetich, Inorg. Chem., 2012, 51, 12789–12795 CrossRef CAS PubMed.
- J. Henning, H. Schubert, K. Eichele, F. Winter, R. Pöttgen, H. A. Mayer and L. Wesemann, Inorg. Chem., 2012, 51, 5787–5794 CrossRef CAS PubMed.
- N. Grueger, H. Wadepohl and L. H. Gade, Dalton Trans., 2012, 41, 14028–14030 RSC.
- L.-C. Liang, P.-S. Chien, Y.-C. Hsiao, C.-W. Li and C.-H. Chang, J. Organomet. Chem., 2011, 696, 3961–3965 CrossRef CAS.
- B. Askevold, M. M. Khusniyarov, E. Herdtweck, K. Meyer and S. Schneider, Angew. Chem., Int. Ed., 2010, 49, 7566–7569 CrossRef CAS PubMed.
- M. T. Whited and R. H. Grubbs, Organometallics, 2008, 27, 5737–5740 CrossRef CAS.
- U. J. Kilgore, C. A. Sengelaub, M. Pink, A. R. Fout and D. J. Mindiola, Angew. Chem., Int. Ed., 2008, 47, 3769–3772 CrossRef CAS PubMed.
- C. M. Fafard, D. Adhikari, B. M. Foxman, D. J. Mindiola and O. V. Ozerov, J. Am. Chem. Soc., 2007, 129, 10318–10319 CrossRef CAS PubMed.
- U. J. Kilgore, X. Yang, J. Tomaszewski, J. C. Huffman and D. J. Mindiola, Inorg. Chem., 2006, 45, 10712–10721 CrossRef CAS PubMed.
- L. Fan, B. M. Foxman and O. V. Ozerov, Organometallics, 2004, 23, 326–328 CrossRef CAS.
- M. Kreye, M. Freytag, P. G. Jones, P. G. Williard, W. H. Bernskoetter and M. D. Walter, Chem. Commun., 2015, 51, 2946–2949 RSC.
- N. Grüger, L.-I. Rodriguez, H. Wadepohl and L. H. Gade, Inorg. Chem., 2013, 52, 2050–2059 CrossRef PubMed.
- C. Cheng, B. G. Kim, D. Guironnet, M. Brookhart, C. Guan, D. Y. Wang, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2014, 136, 6672–6683 CrossRef CAS PubMed.
- Y. Xu, C. A. Rettenmeier, G. T. Plundrich, H. Wadepohl, M. Enders and L. H. Gade, Organometallics, 2015, 34, 5113–5118 CrossRef CAS.
- For related non-innocent aminodiphosphines that are commonly employed in a neutral form, but well-known to support a monoanionic form upon metal complexation, see: (a) D. Milstein, Philos. Trans. R. Soc. London, Ser. A, 2015, 373 CrossRef CAS PubMed, 20140189; (b) M. Feller, E. Ben-Ari, Y. Diskin-Posner, R. Carmieli, L. Weiner and D. Milstein, J. Am. Chem. Soc., 2015, 137, 4634–4637 CrossRef CAS PubMed; (c) Y.-H. Chang, K. Takeuchi, M. Wakioka and F. Ozawa, Organometallics, 2015, 34, 1957–1962 CrossRef CAS; (d) J. I. van der Vlugt and J. N. H. Reek, Angew. Chem., Int. Ed., 2009, 48, 8832–8846 CrossRef CAS PubMed; (e) D. Benito-Garagorri and K. Kirchner, Acc. Chem. Res., 2008, 41, 201–213 CrossRef CAS PubMed.
- M. G. Scheibel, B. Askevold, F. W. Heinemann, E. J. Reijerse, B. de Bruin and S. Schneider, Nat. Chem., 2012, 4, 552–558 CrossRef CAS PubMed.
- B. Askevold, J. T. Nieto, S. Tussupbayev, M. Diefenbach, E. Herdtweck, M. C. Holthausen and S. Schneider, Nat. Chem., 2011, 3, 532–537 CrossRef CAS PubMed.
- M. G. Scheibel, Y. Wu, A. C. Stückl, L. Krause, E. Carl, D. Stalke, B. de Bruin and S. Schneider, J. Am. Chem. Soc., 2013, 135, 17719–17722 CrossRef CAS PubMed.
- A. Glüer, M. Förster, V. R. Celinski, J. Schmedt auf der Günne, M. C. Holthausen and S. Schneider, ACS Catal., 2015, 7214–7217 CrossRef.
- D. A. Smith, D. E. Herbert, J. R. Walensky and O. V. Ozerov, Organometallics, 2013, 32, 2050–2058 CrossRef CAS.
- Y. Zhu, D. A. Smith, D. E. Herbert, S. Gatard and O. V. Ozerov, Chem. Commun., 2012, 48, 218–220 RSC.
- D. Bacciu, C.-H. Chen, P. Surawatanawong, B. M. Foxman and O. V. Ozerov, Inorg. Chem., 2010, 49, 5328–5334 CrossRef CAS PubMed.
- C. Camp, L. C. E. Naested, K. Severin and J. Arnold, Polyhedron, 2016, 103, 157–163 CrossRef CAS.
- S. S. Rozenel, R. Padilla, C. Camp and J. Arnold, Chem. Commun., 2014, 50, 2612–2614 RSC.
- S. S. Rozenel, J. B. Kerr and J. Arnold, Dalton Trans., 2011, 40, 10397–10405 RSC.
- M. Kamitani, K. Searles, C.-H. Chen, P. J. Carroll and D. J. Mindiola, Organometallics, 2015, 34, 2558–2566 CrossRef CAS.
- M. Kamitani, B. Pinter, K. Searles, M. G. Crestani, A. Hickey, B. C. Manor, P. J. Carroll and D. J. Mindiola, J. Am. Chem. Soc., 2015, 137, 11872–11875 CrossRef CAS PubMed.
- M. Kamitani, B. Pinter, C.-H. Chen, M. Pink and D. J. Mindiola, Angew. Chem., Int. Ed., 2014, 53, 10913–10915 CrossRef CAS PubMed.
- M. G. Crestani, A. K. Hickey, B. Pinter, X. Gao and D. J. Mindiola, Organometallics, 2014, 33, 1157–1173 CrossRef CAS.
- M. G. Crestani, A. K. Hickey, X. Gao, B. Pinter, V. N. Cavaliere, J.-I. Ito, C.-H. Chen and D. J. Mindiola, J. Am. Chem. Soc., 2013, 135, 14754–14767 CrossRef CAS PubMed.
- D. S. Levine, T. D. Tilley and R. A. Andersen, Organometallics, 2015, 34, 4647–4655 CrossRef CAS.
- For a rare example of a monoanionic [NPN]-coordinated ruthenium complex, see: M. D. Fryzuk, M. J. Petrella and B. O. Patrick, Organometallics, 2005, 24, 5440–5454 CrossRef CAS.
- L. Morello, P. Yu, C. D. Carmichael, B. O. Patrick and M. D. Fryzuk, J. Am. Chem. Soc., 2005, 127, 12796–12797 CrossRef CAS PubMed.
- M. D. Fryzuk, S. A. Johnson, B. O. Patrick, A. Albinati, S. A. Mason and T. F. Koetzle, J. Am. Chem. Soc., 2001, 123, 3960–3973 CrossRef CAS PubMed.
- J. Ballmann, F. Pick, L. Castro, M. D. Fryzuk and L. Maron, Inorg. Chem., 2013, 52, 1685–1687 CrossRef CAS PubMed.
- J. Ballmann, F. Pick, L. Castro, M. D. Fryzuk and L. Maron, Organometallics, 2012, 31, 8516–8524 CrossRef CAS.
- J. Ballmann, A. Yeo, B. O. Patrick and M. D. Fryzuk, Angew. Chem., Int. Ed., 2011, 50, 507–510 CrossRef CAS PubMed.
- K. D. J. Parker, D. Nied and M. D. Fryzuk, Organometallics, 2015, 34, 3546–3558 CrossRef CAS.
- K. D. J. Parker and M. D. Fryzuk, Organometallics, 2015, 34, 2037–2047 CrossRef CAS.
- Y. Ohki and M. D. Fryzuk, Angew. Chem., Int. Ed., 2007, 46, 3180–3183 CrossRef CAS PubMed.
- T. Zhu, T. C. Wambach and M. D. Fryzuk, Inorg. Chem., 2011, 50, 11212–11221 CrossRef CAS PubMed.
- E. A. MacLachlan, F. M. Hess, B. O. Patrick and M. D. Fryzuk, J. Am. Chem. Soc., 2007, 129, 10895–10905 CrossRef CAS PubMed.
- E. A. MacLachlan and M. D. Fryzuk, Organometallics, 2006, 25, 1530–1543 CrossRef CAS.
- R. R. Schrock, S. W. Seidel, Y. Schrodi and W. M. Davis, Organometallics, 1999, 18, 428–437 CrossRef CAS.
- M. D. Fryzuk, S. A. Johnson and S. J. Rettig, J. Am. Chem. Soc., 1998, 120, 11024–11025 CrossRef CAS.
- M. D. Fryzuk, A. Carter and A. Westerhaus, Inorg. Chem., 1985, 24, 642–648 CrossRef CAS.
- M. D. Fryzuk and P. A. MacNeil, J. Am. Chem. Soc., 1981, 103, 3592–3593 CrossRef CAS.
- M. D. Fryzuk, M. P. Shaver and B. O. Patrick, Inorg. Chim. Acta, 2003, 350, 293–298 CrossRef CAS.
- B. A. MacKay, S. A. Johnson, B. O. Patrick and M. D. Fryzuk, Can. J. Chem., 2005, 83, 315–323 CrossRef CAS.
- T. Zhu, T. C. Wambach and M. D. Fryzuk, Inorg. Chem., 2013, 50, 11212–11221 CrossRef PubMed.
- G. Ménard, H. Jong and M. D. Fryzuk, Organometallics, 2009, 28, 5253–5260 CrossRef.
- E. A. MacLachlan and M. D. Fryzuk, Organometallics, 2005, 24, 1112–1118 CrossRef CAS.
- C. S. Lee, J. H. Park, E. Y. Hwang, G. H. Park, M. J. Go, J. Lee and B. Y. Lee, J. Organomet. Chem., 2014, 772–773, 172–181 CrossRef CAS.
- M. Sietzen, S. Batke, L. Merz, H. Wadepohl and J. Ballmann, Organometallics, 2015, 34, 1118–1128 CrossRef CAS.
- S. Batke, M. Sietzen, H. Wadepohl and J. Ballmann, Inorg. Chem., 2014, 53, 4144–4153 CrossRef CAS PubMed.
- P. J. Bailey, R. A. Coxall, C. M. Dick, S. Fabre, L. C. Henderson, C. Herber, S. T. Liddle, D. Lorono-Gonzalez, A. Parkin and S. Parsons, Chem. – Eur. J., 2003, 9, 4820–4828 CrossRef CAS PubMed.
- L. H. Gade, Acc. Chem. Res., 2002, 35, 575–582 CrossRef CAS PubMed.
- K. Schulze, G. Winkler, W. Dietrich and M. Mühlstaedt, J. Prakt. Chem., 1977, 319, 463–474 CrossRef CAS.
- U. Schmidt, F. Geiger, A. Müller and K. Markau, Angew. Chem., Int. Ed. Engl., 1963, 75, 640–641 CAS.
- H. Fritzsche, U. Hasserodt and F. Korte, Angew. Chem., Int. Ed. Engl., 1963, 75, 1205 CAS.
- H. Brand, J. A. Capriotti and J. Arnold, Organometallics, 1994, 13, 4469–4473 CrossRef CAS.
- T. Boussie, O. Bruemmer, G. M. Diamond, A. M. Lapointe, M. K. Leclerc, C. Micklatcher, P. Sun and X. Bei, US20060025548A1, Symyx Technologies, Inc., USA, 2006 Search PubMed.
- T. H. Warren, R. R. Schrock and W. M. Davis, Organometallics, 1998, 17, 308–321 CrossRef CAS.
- T. H. Warren, G. Erker, R. Fröhlich and B. Wibbeling, Organometallics, 2000, 19, 127–134 CrossRef CAS.
- S. Brenner, R. Kempe and P. Arndt, Z. Anorg. Allg. Chem., 1995, 621, 2121–2124 CrossRef CAS.
- T. Senda, H. Hanaoka, S. Nakahara, Y. Oda, H. Tsurugi and K. Mashima, Macromolecules, 2010, 43, 2299–2306 CrossRef CAS.
- E. Benzing and W. Kornicker, Chem. Ber., 1961, 94, 2263–2267 CrossRef CAS.
- L. H. Gade and P. Mountford, Coord. Chem. Rev., 2001, 216–217, 65–97 CrossRef CAS.
- F. Akagi, Y. Ishida, T. Matsuo and H. Kawaguchi, Dalton Trans., 2011, 40, 2375–2382 RSC.
- F. Akagi, T. Matsuo and H. Kawaguchi, Angew. Chem., Int. Ed., 2007, 46, 8778–8781 CrossRef CAS PubMed.
- G. Huttner, J. Strittmatter and S. Sandhöfner, Comprehensive Coordination Chemistry II, Vol 1, Chapter 1.13: Phosphorus Tripodal Ligands, Elsevier Ltd., 2004 Search PubMed.
- R. Faissner, G. Huttner, E. Kaifer and P. Rutsch, Eur. J. Inorg. Chem., 2003, 1681–1693 CrossRef CAS.
- R. Faissner, G. Huttner, E. Kaifer, P. Kircher, P. Rutsch and L. Zsolnai, Eur. J. Inorg. Chem., 2003, 2219–2238 CrossRef CAS.
- A. J. McAlees, R. McCrindle and A. R. Woon-Fat, Inorg. Chem., 1976, 15, 1065–1074 CrossRef CAS.
- M. T. Whited, E. Rivard and J. C. Peters, Chem. Commun., 2006, 1613–1615 RSC.
- T. Rüther, N. Braussaud and K. J. Cavell, Organometallics, 2001, 20, 1247–1250 CrossRef.
- S. Friedrich, M. Schubart, L. H. Gade, I. J. Scowen, A. J. Edwards and M. McPartlin, Chem. Ber., 1997, 130, 1751–1759 CrossRef CAS.
- H. Herrmann, T. Gehrmann, H. Wadepohl and L. H. Gade, Dalton Trans., 2008, 6231–6241 RSC.
- H. Herrmann, J. L. Fillol, T. Gehrmann, M. Enders, H. Wadepohl and L. H. Gade, Chem. – Eur. J., 2008, 14, 8131–8146 CrossRef CAS PubMed.
- T. Gehrmann, J. L. Fillol, H. Wadepohl and L. H. Gade, Angew. Chem., Int. Ed., 2009, 48, 2152–2156 CrossRef CAS PubMed.
- H. Herrmann, J. L. Fillol, H. Wadepohl and L. H. Gade, Angew. Chem., Int. Ed., 2007, 46, 8426–8430 CrossRef CAS PubMed.
- T. Gehrmann, J. L. Fillol, S. A. Scholl, H. Wadepohl and L. H. Gade, Angew. Chem., Int. Ed., 2011, 50, 5757–5761 CrossRef CAS PubMed.
- S. A. Scholl, H. Wadepohl and L. H. Gade, Organometallics, 2013, 32, 937–940 CrossRef CAS.
- S. A. Scholl, G. T. Plundrich, H. Wadepohl and L. H. Gade, Inorg. Chem., 2013, 52, 10158–10166 CrossRef CAS PubMed.
- T. Gehrmann, G. T. Plundrich, H. Wadepohl and L. H. Gade, Organometallics, 2012, 31, 3346–3354 CrossRef CAS.
- A. D. Schofield, A. Nova, J. D. Selby, A. D. Schwarz, E. Clot and P. Mountford, Chem. – Eur. J., 2011, 17, 265–285 CrossRef CAS PubMed.
- A. D. Schofield, A. Nova, J. D. Selby, C. D. Manley, A. D. Schwarz, E. Clot and P. Mountford, J. Am. Chem. Soc., 2010, 132, 10484–10497 CrossRef CAS PubMed.
- P. Mehrkhodavandi, R. R. Schrock and P. J. Bonitatebus Jr., Organometallics, 2002, 21, 5785–5798 CrossRef CAS.
- P. Mehrkhodavandi and R. R. Schrock, J. Am. Chem. Soc., 2001, 123, 10746–10747 CrossRef CAS PubMed.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional experimental details and compound characterisation data, selected NMR spectra; crystallographic data and details of the structure determinations. CCDC 1440950–1440958. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt04911c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |