
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interaction of europium and curium with alpha-amylase†
Astrid
Barkleit
*,
Anne
Heller‡
,
Atsushi
Ikeda-Ohno
and
Gert
Bernhard
Helmholtz-Zentrum Dresden-Rossendorf, Institute of Resource Ecology, P.O. Box 510119, 01314 Dresden, Germany. E-mail: a.barkleit@hzdr.de
First published on 27th January 2016
Abstract
The complexation of Eu(III) and Cm(III) with the protein α-amylase (Amy), a major enzyme in saliva and pancreatic juice, was investigated over wide ranges of pH and concentration at both ambient and physiological temperatures. Macroscopic sorption experiments demonstrated a strong and fast binding of Eu(III) to Amy between pH 5 and 8. The protein provides three independent, non-cooperative binding sites for Eu(III). The overall association constant of these three binding sites on the protein was calculated to be log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K = 6.4 ± 0.1 at ambient temperature. With potentiometric titration, the averaged deprotonation constant of the carboxyl groups (the aspartic and glutamic acid residues) of Amy was determined to be pKa = 5.23 ± 0.14 at 25 °C and 5.11 ± 0.24 at 37 °C. Time-resolved laser-induced fluorescence spectroscopy (TRLFS) revealed two different species for both Eu(III) and Cm(III) with Amy. In the case of the Eu(III) species, the stability constants were determined to be logβ11 = 4.7 ± 0.2 and logβ13 = 12.0 ± 0.4 for Eu:Amy = 1:1 and 1:3 complexes, respectively, whereas the values for the respective Cm(III) species were logβ11 = 4.8 ± 0.1 and logβ13 = 12.1 ± 0.1. Furthermore, the obtained stability constants were extrapolated to infinite dilution to make our data compatible with the existing thermodynamic database.
K = 6.4 ± 0.1 at ambient temperature. With potentiometric titration, the averaged deprotonation constant of the carboxyl groups (the aspartic and glutamic acid residues) of Amy was determined to be pKa = 5.23 ± 0.14 at 25 °C and 5.11 ± 0.24 at 37 °C. Time-resolved laser-induced fluorescence spectroscopy (TRLFS) revealed two different species for both Eu(III) and Cm(III) with Amy. In the case of the Eu(III) species, the stability constants were determined to be logβ11 = 4.7 ± 0.2 and logβ13 = 12.0 ± 0.4 for Eu:Amy = 1:1 and 1:3 complexes, respectively, whereas the values for the respective Cm(III) species were logβ11 = 4.8 ± 0.1 and logβ13 = 12.1 ± 0.1. Furthermore, the obtained stability constants were extrapolated to infinite dilution to make our data compatible with the existing thermodynamic database.
Introduction
Radioactive heavy metals represent a serious health risk to humans due to their chemo and/or radiotoxicity. Trivalent actinides (An(III)), such as Am(III) or Cm(III), are man-made radioactive elements that are exclusively generated in nuclear reactors, whereas lanthanides (Ln(III)), which are commonly used as their non-radioactive analogs, are naturally occurring elements having wide technological and medical applications.1–3Via different pathways, all these heavy metals can potentially be taken up into the body, posing a serious health threat to humans.4–6 Because An(III) and Ln(III) are considered to have no essential function in the human body, little is known about their biochemical behavior from their uptake, in vivo transport and “metabolism” to their final accumulation or excretion. Our recent investigations on the in vitro speciation of U(VI), Cm(III), and Eu(III) in various body fluids (saliva, urine and sweat) proved experimentally that organic bio-molecules strongly interact with the metal ions and dominate their speciation under certain conditions.7–9 These studies supposed that in addition to small organic molecules (e.g. lactate or citrate), bio-macromolecules, such as proteins or enzymes, are also potential binding partners for these elements under bio-relevant conditions.For the last few decades, the speciation of An(III) and Ln(III) has been studied extensively in blood media with the aim to understand their complexation behavior with blood proteins (e.g. albumin and transferrin).4,10–18 However, little is known about their speciation in the gastrointestinal tract and their complexation behavior with digestive proteins.4,16–18 There have been several attempts to simulate the speciation of An(III) and Ln(III) in the gastrointestinal tract based on thermodynamic data.19,20 However, the accuracy of such calculations strongly depends on the model applied and the database used.21 In the case wherein the chemical species existing in the real system are not described correctly (or in the worst case, missing) in the thermodynamic database, the resultant speciation could be misleading and/or contradict the experimental results. As a matter of fact, such inconsistency between thermodynamically modeled and experimentally determined speciation has been recently reported for the Eu(III), Cm(III) and U(VI) speciation in several biological media.7,8,22
To improve the reliability of the thermodynamic database for bio-macromolecules, this study aims to experimentally investigate the complexation behavior of Eu(III) and Cm(III) with α-amylase (Amy), one of the most important digestive proteins. The enzyme Amy (α-1,4-glucan-4-glucanhydrolase; EC 3.2.1.1.) is one of the major enzymes in salivary and pancreatic secretions of mammals, and catalyzes the hydrolysis of the α-1,4-glycosidic linkages of polysaccharides such as starch or glycogen.23,24 Human salivary and pancreatic Amys, as well as porcine pancreatic Amys, show considerable similarities in sequence and three-dimensional structure.25–27 They consist of 496 amino acid residues and have a molecular mass of ∼55 kDa.28–31 The protein binds one calcium and one chloride ion per molecule and provides 50 carboxyl groups from both aspartic acid (Asp) and glutamic acid (Glu) residues,30,31 which can act as major binding sites for metal ions. The Amy monomers are composed of three domains: domain A (residues 1–99 and 169–404) contains a central α/β-barrel of eight parallel strands, domain B (residues 100–168) consists of several helices and β-strands forming an open loop structure, and domain C is composed of eight β-strands, which form a compact Greek-key β-barrel. The active site, comprising the catalytic residues Asp197, Glu233 and Asp300, is found in domain A. The chloride ion is placed near the active site and is coordinated by the side chains of arginine (Arg195 and Arg337) and asparagine (Asn298) residues, which could increase the enzyme activity. Calcium is located between domains A and B and is coordinated by a histidine residue (His201) from domain A and Asn100, Arg158 and Asp167 from domain B. The presence of calcium is necessary for the activity and the structural stability of the protein.25–27,32–35
In this study, we report the complexation of Eu(III) and Cm(III) with Amy over a wide pH range at ambient and physiological temperatures. The deprotonation constants of the protein's functional groups were determined by potentiometric titrations, whereas the stability constants of the metal–protein complexes were determined by macroscopic sorption experiments combined with time-resolved laser-induced fluorescence spectroscopy (TRLFS). TRLFS is a very sensitive method to study the complexation behavior of luminescent ions at trace concentrations relevant for in vivo conditions.36,37 In this study, the luminescent metal ions Eu(III) and Cm(III) have been employed as representatives for Ln(III) and An(III), respectively.
Materials and methods
Sorption experiments
Batch experiments were performed to study the sorption behavior of Eu(III) on Amy (porcine pancreas Amy from Sigma) both in the absence and presence of calcium. The experiments were performed at room temperature as functions of pH, metal and enzyme concentrations, and sorption time. The pH was varied between 3.0 and 8.0, whereas the Eu(III) and Amy concentrations were varied between 10−6 and 10−4 M and between 0.2 and 3.0 g L−1 (3.6 × 10−6 to 5.5 × 10−5 M), respectively. Samples with 1 g L−1 Amy (1.8 × 10−5 M) and 1 × 10−5 to 5 × 10−5 M Eu(III) were additionally spiked with 1 × 10−5 to 5 × 10−5 M Ca. The solubility of Amy is ∼0.1 g L−1, meaning that the Amy used in the batch experiments was in the form of a suspension. The ionic strength was maintained constant at 0.1 M with NaCl for all the experiments.A Eu(III) stock solution was prepared from EuCl3·6H2O (Sigma). Aliquots were diluted with a 0.1 M NaCl solution to obtain the desired Eu concentrations, and the pH was adjusted with HCl and NaOH. Amy was then added and the pH was adjusted again, if necessary. In the case of the experiments in the presence of calcium, aliquots of a CaCl2 stock solution were also added to the solution. For pH- and concentration-dependent experiments, the mixture was shaken for 24 hours at ambient temperature and the pH was adjusted as necessary. Subsequently, the solution was centrifuged for 20 min at 4000 rpm and filtered with 150 μm membrane filters. The pH of the filtrate was measured and the Eu and Ca concentrations were determined by inductively coupled plasma mass spectrometry (ICP-MS ELAN 9000, Perkin-Elmer). Time-dependent experiments were performed at pH = 6.0. Aliquots of the sample solution were acquired at 2 min intervals for the first 10 min, and then at 5 min intervals until 60 min. The collected aliquots were filtered swiftly with 150 μm membrane filters, and the Eu concentration in the filtrate was determined with ICP-MS.
The Ca content of pure Amy was detected randomly with ICP-MS, resulting in 1 mol Ca per mol Amy.
Potentiometric titration
Potentiometric titration experiments were performed to determine the deprotonation constants (pKa) of Amy and the stability constants of Eu(III)–Amy complexes (logβ). All sample preparation and measurements were carried out under N2 atmosphere. For the determination of pKa, 3 mg Amy was dissolved in 30 mL deionized carbonate-free H2O. The resulting solution had a protein concentration of 0.1 g L−1 (1.8 × 10−6 M), which is equivalent to 9 × 10−5 M carboxyl groups (from Asp and Glu). The solution was then adjusted to I = 0.1 M with NaCl, and to pH = 3 with HCl. For the determination of logβ, the Eu(III) stock solution was added to the protein solution resulting in a final Eu concentration of 10−4 M. The solutions were automatically titrated in a thermostatic vessel at both 25.0 ± 0.1 °C and 37.0 ± 0.1 °C with 736 GP Titrino/TiNet 2.50 (Metrohm) using 0.1 M NaOH (carbonate-free, Titrisol, Merck). Dynamic titration was performed using a BlueLine 11 electrode (Schott), with a minimum drift of 0.5 mV min−1 and a delay time of at least 60 s at each pH measurement. Prior to each titration experiment, the electrode was calibrated with standard buffers of pH = 4.008, 6.865 and 9.180 (Schott). Each experiment was performed in triplicate. The experiments for the pure Amy system were carried out in the pH range between 3 and 11, whereas those with Eu(III) were stopped at pH = 7 to avoid the precipitation of Eu(III) hydroxides.
Time-resolved laser-induced fluorescence spectroscopy
TRLFS is another method used to determine the logβ values of metal–Amy complexes. For spectrophotometric titration experiments, Eu(III) samples were prepared from the same Eu(III) stock solution described above, whereas Cm(III) samples were prepared from a stock solution of 248Cm (supplied by the Oak Ridge National Laboratory, U.S. Department of Energy Office of Basic Energy Sciences) in 1 M HClO4. The sample solutions were prepared with metal concentrations of 1 × 10−5 M and 3 × 10−7 M for Eu(III) and Cm(III), respectively. The pH of the solutions was adjusted to 5.5, and the sample solutions were titrated stepwise with 1, 2, or 10 g L−1 Amy in 0.1 M NaCl until the Amy concentration in the sample reached approximately 1 g L−1 (1.8 × 10−5 M protein, which is equivalent to 9 × 10−4 M carboxyl groups). The ionic strength was maintained constant at 0.1 M with NaCl. Another series of pH titration experiments was carried out under the same solution conditions, but by increasing the pH to 8 with NaOH. At each titration step (0.2–0.3 pH values), the sample solution was equilibrated for at least 10 min and TRLFS measurements were performed. Two Eu(III) samples with 1 g L−1 Amy at pH 5.5 and 7.0 were back titrated with 1 × 10−3 M Ca.
TRLFS spectra were obtained at both 25.0 ± 1 °C and 37.0 ± 1 °C using a pulsed flash lamp pumped Nd:YAG-OPO laser system (Powerlite Precision II 9020 laser equipped with a Green PANTHER EX OPO, Continuum, USA). The temperature was maintained using a temperature-controlled cuvette holder (Flash 300™, Quantum Northwest, USA). The laser pulse energy, which was in the range of 1–2 mJ, was monitored with a photodiode. The fluorescence emission spectra were obtained on an optical multi-channel analyzer-system, consisting of an Oriel MS 257 monochromator, a spectrograph with a 300 or 1200 lines per mm grating, and an Andor iStar ICCD camera (Lot-Oriel, Germany). The emission spectra were obtained in the ranges of 440–780 nm (300 lines per mm grating) and 570–650 nm (1200 lines per mm grating) for time-resolved and single measurements, respectively. A constant time window of 1 ms was applied. The excitation wavelengths were 394 nm and 398 nm for Eu(III) and Cm(III), respectively. For time-resolved measurements, 40 to 60 spectra were acquired at delay time intervals between 10 and 50 μs.
Data analysis
The data from the batch sorption experiments at a constant pH were fitted with OriginPro9.0 (OriginLab, USA) using the Hill equation (eqn (1))38 to calculate the number of binding sites (g), the association equilibrium constant (KH) and the Hill coefficient (r) from the experimentally determined saturation function, v, with the unit of “bound M3+ (mol)/Amy (mol)”, and the free metal concentration. | (1) |
The calculations of pKa and logβML were carried out by applying the equations as follows:
| R–LH ⇄ R–L− + H+ Ka = [R–L−] [H+]/[R–LH], | (2) |
| M3+ + xR–L− ⇄ M(R–L)x(3−x)+ β1x = [M(R–L)x(3−x)+]/[M3+] [R–L−]x, | (3) |
The data from potentiometric titration were treated using the program HYPERQUAD2008 (Protonic Software)39 to obtain the pKa and logβ values.
The TRLFS data were analyzed with OriginPro9.0. The Eu(III) spectra were normalized to the peak area of the 5D0 → 7F1 transition peak (585–600 nm), whereas the Cm(III) spectra were normalized to the whole peak area.
The lifetimes of luminescent species were determined according to the equation as follows:
 | (4) |
Using the lifetimes τ (in ms), the number of water molecules in the first coordination shell of the heavy metal ions was estimated using the empirical equations as follows:40,41
| n(H2O) ± 0.5 = (1.07/τ) − 0.62 for Eu(III), | (5) |
| n(H2O) ± 0.5 = (0.65/τ) − 0.88 for Cm(III). | (6) |
The luminescence lifetimes of pure aquo species in H2O are 110 ± 4 μs and 65 ± 2 μs for Eu(III) and Cm(III), respectively.42 These values each correspond to nine water molecules in the first coordination sphere.
The data from spectrophotometric titration were further analyzed using the program SPECFIT43 to calculate the logβ values and the spectra of individual species.
The obtained pKa and logβ values were extrapolated to infinite dilution by applying SIT (Specific Interaction Theory) using the IUPAC software for Ionic Strength Corrections.44
Results and discussion
Sorption of europium onto α-amylase
Batch sorption experiments were performed to get a macroscopic overview of the binding behavior of Eu(III) towards Amy. The sorption behavior of Eu(III) with Amy as a function of pH, at three different Eu(III) concentrations is shown on the left in Fig. 1. Independent of the Eu(III) concentration, the sorption of Eu(III) began at around pH = 4, attained ∼100% of sorption at pH = 5–5.5, and then reached a plateau. Based on these results, the pH value for the subsequent time-dependent sorption experiments was set to 6. At this pH, not only the sorption of Eu(III) reaches ~100% but also the precipitation of Eu(III) hydroxide can be avoided and Amy retains a high enzyme activity. In fact, it has been reported that Amy shows the highest enzyme activity between pH 5.5 and 8.0 with the optimum at pH = 7,29 but at this pH, starting Eu(III) precipitation may interfere with the sorption process. | ||
| Fig. 1 Sorption of Eu(III) on Amy as a function of pH at different constant metal concentrations (left: I = 0.1 M with NaCl, T = 25 °C) and as a function of the sorption time (right: 1 × 10−4 M Eu3+, pH 6.0, I = 0.1 M with NaCl, T = 25 °C). | ||
On the right in Fig. 1, the sorption of Eu(III) at two different Amy concentrations as a function of contact time is shown. The metal ion was sorbed within a very short time. More specifically, the sorption of Eu(III) started within the first few minutes, and reached a plateau after 5 minutes. With regard to the potential ingestion of Eu(III), this time span might be relevant for the contact time with saliva in the mouth, wherein the transit time of the ingested substances can last from seconds up to few minutes.45,46 However, as Amy can also be found in pancreatic juice, one of the digestive fluids,24 this time frame would be particularly relevant to the gastrointestinal tract wherein the retention time of substances is much longer (up to several hours).45,46
When the equimolar amount of Eu(III) was sorbed on Amy, one molar Amy was found to release one molar Ca that was initially retained on Amy. This indicates that Eu(III) replaces the Ca on Amy selectively, which is a well-known process for proteins and other bio-macromolecules.1,47 This replacement happens regardless of the amount of excess Ca, up to the 5-fold excess, compared to the Eu concentration, suggesting a stronger interaction of Eu(III) on Amy than that of Ca.
Fig. 2 shows the binding isotherm of Amy as a function of Eu(III) concentration. By fitting the experimental data with the Hill equation (eqn (1)),38 which is a modification of the Langmuir sorption isotherm, we obtained information about the macroscopic interaction between the metal ions and the enzyme. In eqn (1), r = 1 stands for non-cooperative systems (i.e., identical or non-identical but independent binding sites), r > 1 stands for positively cooperative systems and r < 1 stands for negatively cooperative systems (i.e., interacting binding sites in both cases). Based on the data for Eu(III) in Fig. 2, the r value was calculated to be 1.1 ± 0.2 with g = 3.1 ± 0.1, suggesting that Amy provides three binding sites for Eu(III) with very weak cooperativity, which may be considered as non-cooperative according to the definition of Saboury and Karbassi.48,49
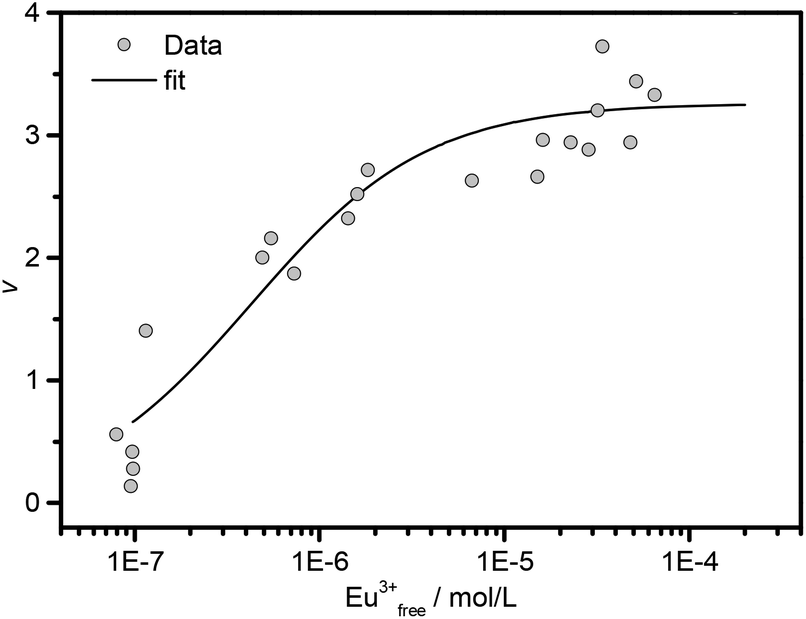 | ||
| Fig. 2 Binding isotherm of Amy (0.2–3.0 g L−1) as a function of Eu(III) concentration, at pH = 6.0, I = 0.1 M with NaCl and T = 25 °C. The fitting curve was obtained according to eqn (1). | ||
The association constant was calculated to be logKH = 6.4 ± 0.1 and is an averaged overall value for the three binding sites of Amy. It has been reported that the Amy from Bacillus subtilis provides two binding sites to Gd(III) with logKH = 4.6.50 Another study on the crystalline Amy from Aspergillus oryzae demonstrated six binding sites for Gd(III).51 Although the number of binding sites estimated for the present Eu(III) system is within these reported values, the association constants differ considerably. These variations could be caused by the structural differences of the different Amys. Depending on the origin, Amys vary in number, composition and sequence of the amino acids.31 For instance, the Amys from Aspergillus oryzae and Bacillus subtilis retain 2 and 3 calcium ions at different binding sites,52,53 and provide more Asp/Glu residues (54 and 66, respectively), compared to the Amy from porcine pancreas (1 bound calcium with 50 Asp/Glu residues). This can consequently cause varying numbers on potential Ln(III) binding sites with varying binding strengths.
Potentiometric titration
| Species | T (°C) | pKa0.1 | pKa0 |
|---|---|---|---|
| Amy–COOH | 25 | 5.23 ± 0.14 | 5.43 |
| 37 | 5.11 ± 0.24 | 5.31 | |
| Amy–NH3+/Amy–OH | 25 | 10.22 ± 0.10 | 10.42 |
| 37 | 10.14 ± 0.21 | 10.34 |
According to the amino acid composition of Amy by the ProtParam Tool on ExPASy,30,31 pKa1 can be assigned to the deprotonation of the carboxylic groups of Glu and Asp. The pKa values of pure Glu and Asp are reported to be 4.15 (ref. 54) and 3.71,54 respectively, being slightly lower than the pKa1 values obtained for Amy. However, due to the concentration of charged functional groups on the protein molecules, a significant electrostatic effect influencing the deprotonation of functional groups on Amy can occur.55 This could explain the higher pKa values of Amy in comparison to those of pure Glu and Asp. In fact, such an enhancement of deprotonation constants was observed for oligopeptides containing Glu and Asp residues.56 Furthermore, the average pKa value of Asp and Glu residues in the protein trypsin is 5.2,57 which is in line with our results.
The second deprotonation constant obtained for Amy can be associated with the deprotonation of amino groups from arginine, asparagine, glutamine, and lysine, and/or the hydroxyl groups from serine, threonine, and tyrosine residues. In fact, the obtained pKa2 values are in very good agreement with the pKa values of amino and hydroxyl groups in bio-macromolecules58,59 and oligopeptides.60 However, based on the obtained pKa values, these functional groups are expected to be protonated under in vivo conditions, suggesting that they have none or little contribution to the complexation of Amy with Eu(III) and Cm(III). Furthermore, it was shown that Ln(III) preferentially binds to carboxylate groups of Asp and Glu residues rather than to amino or hydroxyl groups.61
:1 complex in which Eu(III) is bound via one carboxyl group of a Glu or Asp residue of Amy. Within the range of error, no significant temperature effect was observed, suggesting that the complexation behavior is unaltered in this temperature range. The averaged value is logβ11 = 4.56 ± 0.13. A similar stability constant of logβ11 = 4.18 ± 0.05 was reported for the complexation of Tb(III) with the protein chymotrypsin,62 supporting the validity of our results.
β0 values were obtained by extrapolating the logβ0.1 values to infinite dilution, applying SIT44
| Species | T (°C) | logβ0.1 |
logβ0 |
Method |
|---|---|---|---|---|
| Eu(Amy–COO)2+ | 25 | 4.54 ± 0.13 | 5.20 | Potentiometry |
| 37 | 4.57 ± 0.12 | 5.24 | Potentiometry | |
| 25 | 4.83 ± 0.43 | 5.49 | TRLFS | |
| 37 | 4.51 ± 0.15 | 5.18 | TRLFS | |
| Eu(Amy–COO)3 | 25 | 12.04 ± 0.36 | 13.31 | TRLFS |
| 37 | 12.13 ± 0.14 | 13.43 | TRLFS | |
| Cm(Amy–COO)2+ | 25 | 4.76 ± 0.11 | 5.42 | TRLFS |
| Cm(Amy–COO)3 | 25 | 12.13 ± 0.12 | 13.40 | TRLFS |
Time-resolved laser-induced fluorescence spectroscopy
 | ||
| Fig. 3 Spectrophotometric titration of 1 × 10−5 M Eu(III) (I = 0.1 M with NaCl, T = 25 °C); left: as a function of Amy concentration at pH = 5.5, right: as a function of pH with 1.0 g L−1 Amy. | ||
The results from time-resolved measurements revealed a bi-exponential decay, suggesting that at least two luminescent species are formed in both systems (see Tables S1 and S2 in the ESI†). The luminescence lifetimes were prolonged with increasing protein concentration or pH. This indicates a continuous replacement of water molecules by the coordinative functional groups of the protein, in the first coordination sphere of Eu(III). The direct excitation of the 7F0 → 5D0 transition, which is a non-degenerate transition and consequently yields a single emission line for every non-equivalent Eu(III) species, also supports the formation of two Eu(III)–Amy species (Fig. S1 and experimental details see ESI†).
The obtained TRLFS data were further fitted by applying eqn (2) and (3) to calculate the conditional stability constants for Eu(III)–Amy complexes. That way, two different Eu(III)–Amy complex species could be identified. The obtained values are summarized in Table 2. The stability constant for the first Eu(III)–Amy species was calculated to be logβ11 = 4.8 ± 0.4 at 25 °C, suggesting the presence of a “Eu(III):L = 1:1” complex, where L is the binding carboxylate group of Amy. This stability constant agrees well with that previously determined with potentiometric titration. The stability constant for the second Eu(III)–Amy species was calculated to be logβ13 = 12.0 ± 0.4, indicating the formation of a “1:3” complex. Attempts to fit the data by assuming the “1:2” complex failed. The spectra obtained for the individual Eu(III) species are shown in Fig. S2 in ESI.† In fact, the formation of 1:1 and 1:3 complexes was also observed in the crystal structure of the Aspergillus oryzae-originated Amy complexing Gd(III),51 which supports our results. Furthermore, in the reported Gd(III)–Amy complex, some Gd(III) ions act as a linker to connect different protein molecules, resulting in the oligomerization of the proteins.51 This would suggest that the carboxylate groups of the 1:3 complex identified for our Eu(III)–Amy system may originate from different protein molecules, not from a single molecule.
Based on the complex stability constants obtained from TRLFS measurements, the distribution of Eu(III)–Amy species was calculated as a function of both Amy concentration and pH. The results are shown in Fig. 4. According to this speciation, the measured luminescence lifetimes can be assigned either to one predominant species or to a mixture of several different species. For instance, on the right of Fig. 4, the Eu(III):Amy = 1:1 complex (Eu(Amy-COO)2+) becomes dominant between pH 4 and 5. Therefore, the longer major lifetime observed in this pH range can be assigned to this complex, whereas the shorter minor one of ∼110 μs corresponds to the free aquo ion (see Table S1 in ESI†). When the pH is increased above 5, the second Eu(III)–Amy complex (Eu(Amy-COO)3) becomes dominant with more than a 90% fraction. Consequently, the longer major lifetimes obtained in this pH range can be assigned to this species. However, the minor lifetimes observed at pH > 5 are longer than that of the free Eu3+ aquo ion, but shorter than that of Eu(Amy-COO)2+. This suggests that these shorter minor lifetimes probably originate from mixtures of these two species and/or other minor species (see Table S1 in ESI†). The same approach can be applied to the concentration-dependent series at pH 5.5, wherein the Eu3+ aquo ion and the 1:1 complex dominate the species distribution for Amy concentrations below 0.1 g L−1, but the 1:3 complex becomes dominant for Amy concentrations exceeding 0.4 g L−1 (left figure in Fig. 4). Consequently, considering both TRLFS series, the luminescence lifetimes of the Eu(III)–Amy species can be estimated to be 380 ± 40 μs for Eu(Amy-COO)2+ and 630 ± 50 μs for Eu(Amy-COO)3 (Tables S1 and S2 in ESI†). These luminescence lifetimes correspond to the replacement of 7 and 8 water molecules from the first coordination sphere of Eu(III) within species 1 and 2. Assuming that the carboxylate groups of Amy interact with Eu(III) in a bidentate coordination mode, the complexation by Amy should only exclude 2 and 6 water molecules from the Eu(III) coordination sphere for species 1 and 2, respectively. This suggests that in addition to the coordination of the carboxylate groups of Amy, Eu(III) must be surrounded by other ligands (e.g., Cl−) and/or other functional groups of Amy, which further exclude water molecules from the first coordination sphere of Eu(III). Another possibility is a decrease in total coordination number of Eu(III) to a value smaller than 9 in Amy complexes, due to steric hindrance caused by the protein structure.
 | ||
| Fig. 4 Species distribution of Eu(III)–Amy complexes at 1 × 10−5 M Eu(III) (I = 0.1 M and T = 25 °C); left: as a function of Amy concentration at pH = 5.5, right: as a function of pH with 1.0 g L−1 Amy. | ||
Sorption experiments indicated that Eu(III) selectively replaces the binding sites of Ca on Amy. On Amy molecules, the calcium ion is coordinated by the carboxyl oxygens of Asp167 in a bidentate manner, the carbonyl oxygens of Asn100, His201 and Arg158 and additional two or three water molecules.26,32,33 This could explain the replacement of 7 water molecules from the first coordination sphere of species 1 (i.e., Eu(Amy-COO)2+), when Eu(III) is assumed to be sorbed on the Ca binding site on Amy. On the other hand, the Eu(III) in species 2 is surrounded by three carboxyl groups. This cannot be explained by the simple replacement of Ca binding sites, but could be due to the rearrangement and/or aggregation of protein molecules.
The back titration of the Eu-saturated Amy solution with calcium showed no significant changes in the luminescence spectra, indicating that the binding of Eu(III) in the protein is stronger than that of Ca(II).
In general, similar trends were observed in TRLFS for the series at physiological temperature of 37 °C. The obtained spectra are given in Fig. S3 in ESI.† Within the range of error, the conditional stability constants obtained at this temperature are in good agreement with those at room temperature (Table 2). This further supports and fits the results from the previous potentiometric titration experiments.
β values similar to those obtained for the Eu(III)–Amy complexes (Table 2). The spectra calculated for the individual Cm(III) species are shown in Fig. S4 in ESI.†
 | ||
| Fig. 5 Spectrophotometric titration of 3 × 10−7 M Cm(III) (I = 0.1 M with NaCl, T = 25 °C); left: as a function of Amy concentration at pH = 5.5, right: as a function of the pH with 1.0 g L−1 Amy. | ||
Based on these logβ values, the distribution of Cm(III)–Amy species was calculated as a function of both Amy concentration and pH. The results are given in Fig. 6. According to the Eu(III) system, the measured luminescence lifetimes can be interpreted as either one dominating species or a mixture of several species, based on the speciation information in Fig. 6. The results are summarized in Tables S3 and S4 in ESI.† The luminescence lifetimes for species 1 (Cm(Amy-COO)2+) and species 2 (Cm(Amy-COO)3) are calculated to be 120 ± 10 and 240 ± 40 μs, respectively. This corresponds to the replacement of 5 and 7 water molecules, respectively, in the Cm first coordination sphere of species 1 and 2. Interestingly, this differs from the previous Eu(III) system, indicating that the interaction of Cm(III) with Amy could be unequal to that of Eu(III). However, we also have to consider the difference in metal concentrations (1 × 10−5 M for the Eu(III) system and 3 × 10−7 M for the Cm(III) system), which also affect the interaction of the metals with Amy and may be the cause for the differences observed.
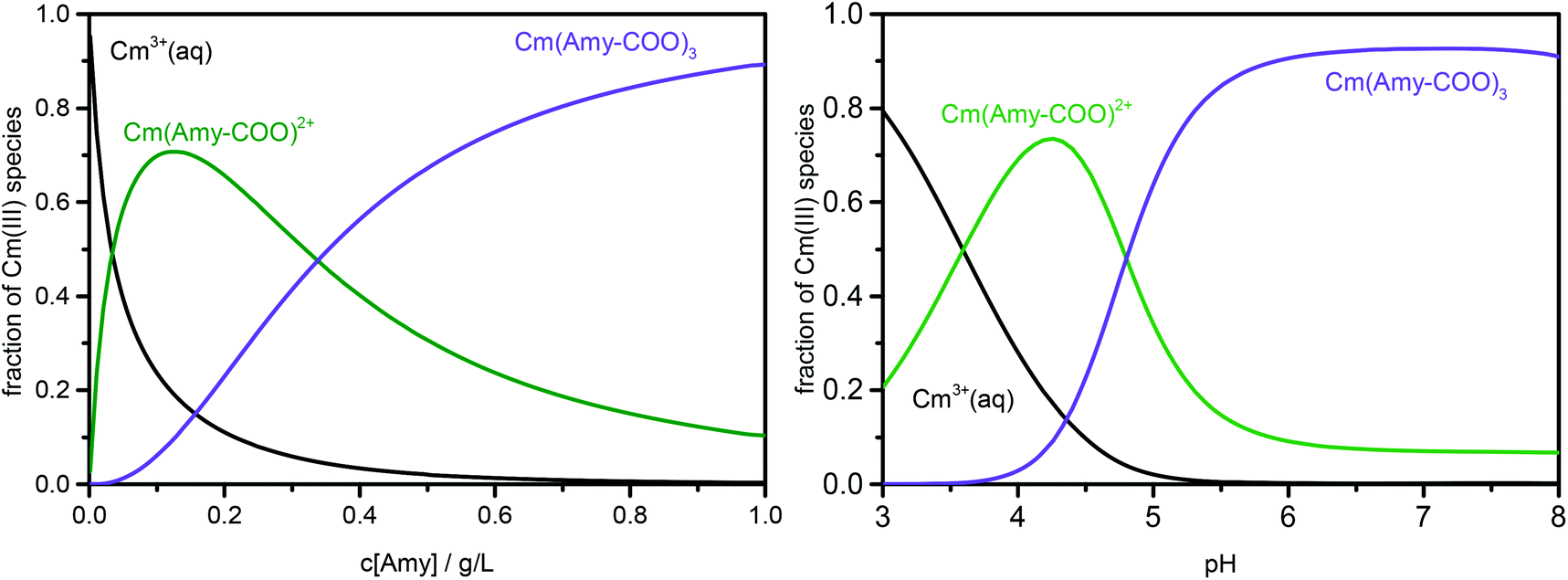 | ||
| Fig. 6 Species distribution of Cm(III)–Amy complexes at 3 × 10−7 M Cm(III) (I = 0.1 M and T = 25 °C); left: as a function of Amy concentration at pH = 5.5, right: as a function of the pH with 1.0 g L−1 Amy. | ||
Conclusions
The interaction of Eu(III) and Cm(III) with the protein α-amylase, an important digestive enzyme in saliva and pancreatic juice, has been studied over wide pH and concentration ranges. Batch sorption experiments showed a strong and fast interaction of Eu(III) with Amy over a wide pH-range, covering also the in vivo conditions in the human gastrointestinal tract. This points to the potential influence of Amy on the biochemical behavior of these trivalent metal cations within the gastrointestinal tract. Potentiometric titrations and TRLFS investigations were performed to successfully calculate the stability constants of M(III)–Amy complexes. These constants are essential for the reliable modelling of chemical speciation and consequently, for the improvement of the transport modelling of Ln(III) and An(III) in the digestive system. This may also allow us to perform more reliable risk assessment in the event of accidental oral incorporation of radioactive heavy metals, and eventually to propose the effective decorporation procedures. The similarity of the stability constants for Eu(III) and Cm(III) confirms the chemical analogy of trivalent lanthanides and actinides, due to similar ionic radii. Nevertheless, while Eu(III) offers the benefit of being non-radioactive, Cm(III) provides the possibility to investigate the complexation behavior at much lower concentrations, which is consequently closer to environmental conditions.Based on the results obtained in this study, the chemical speciation of Eu(III) and Cm(III) in human body fluids associated with the digestive system (e.g., saliva, gastric, pancreatic and bile juice) is currently under investigation. Preliminary results from these on-going studies demonstrate a significant contribution of Amy to the chemical speciation of Eu(III) and Cm(III) in the digestive body fluids such as saliva. Nevertheless, to obtain a comprehensive overview of the potential metabolic pathways of An(III) and Ln(III) in the human body after oral ingestion, we also have to consider other essential proteins and bio-macromolecules in the gastrointestinal tract such as mucin in future investigations.
Acknowledgements
This study was funded by the German Research Council (Deutsche Forschungsgemeinschaft, DFG) under the contract number BE 2234/10-1/2 and the Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung, BMBF) under the contract number 02NUK030F.The authors are indebted to the U.S. Department of Energy, Office of Basic Energy Sciences, for the acquisition of 248Cm via Oak Ridge National Laboratory. The acquisition of 248Cm was materialized through the collaboration between Helmholtz-Zentrum Dresden-Rossendorf (HZDR) and the Lawrence Berkeley National Laboratory (LBNL). We thank Mrs Schaefer for ICP-MS measurements, and Mrs Jähnigen and Mr Zegke for technical assistance.
References
- J. C. G. Bünzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef PubMed.
- J. C. G. Bünzli and S. V. Eliseeva, J. Rare Earths, 2010, 28, 824–842 CrossRef.
- J. C. G. Bünzli, Coord. Chem. Rev., 2015, 293–294, 19–47 CrossRef.
- R. Leggett, E. Ansoborlo, M. Bailey, D. Gregoratto, F. Paquet and D. Taylor, Int. J. Radiat. Biol., 2014, 90, 996–1010 CrossRef CAS PubMed.
- E. Ansoborlo, B. Amekraz, C. Moulin, V. Moulin, F. Taran, T. Bailly, R. Burgada, M. H. Henge-Napoli, A. Jeanson, C. Den Auwer, L. Bonin and P. Moisy, C. R. Chim., 2007, 10, 1010–1019 CrossRef CAS.
- F. Menetrier, D. M. Taylor and A. Comte, Appl. Radiat. Isot., 2008, 66, 632–647 CrossRef CAS PubMed.
- A. Heller, A. Barkleit and G. Bernhard, Chem. Res. Toxicol., 2011, 24, 193–203 CrossRef CAS PubMed.
- A. A. A. Osman, G. Geipel, A. Barkleit and G. Bernhard, Chem. Res. Toxicol., 2015, 28, 238–247 CAS.
- A. Heller, A. Barkleit, H. Foerstendorf, S. Tsushima, K. Heim and G. Bernhard, Dalton Trans., 2012, 41, 13969–13983 RSC.
- D. M. Taylor, J. Alloys Compd., 1998, 271–273, 6–10 CrossRef CAS.
- M. Sturzbecher-Hoehne, C. Goujon, G. J. P. Deblonde, A. B. Mason and R. J. Abergel, J. Am. Chem. Soc., 2013, 135, 2676–2683 CrossRef CAS PubMed.
- G. J. Deblonde, M. Sturzbecher-Hoehne, A. B. Mason and R. J. Abergel, Metallomics, 2013, 5, 619–626 RSC.
- N. Bauer, D. R. Fröhlich and P. J. Panak, Dalton Trans., 2014, 43, 6689–6700 RSC.
- N. Bauer and P. J. Panak, New J. Chem., 2015, 39, 1375–1381 RSC.
- N. Bauer, V. C. Smith, R. T. A. MacGillivray and P. J. Panak, Dalton Trans., 2015, 44, 1850–1857 RSC.
- E. Ansoborlo, O. Prat, P. Moisy, C. Den Auwer, P. Guilbaud, M. Carriere, B. Gouget, J. Duffield, D. Doizi, T. Vercouter, C. Moulin and V. Moulin, Biochimie, 2006, 88, 1605–1618 CrossRef CAS PubMed.
- E. Ansoborlo, L. Bion, D. Doizi, C. Moulin, V. Lourenco, C. Madic, G. Cote, J. Van der Lee and V. Moulin, Radiat. Prot. Dosim., 2007, 127, 97–102 CrossRef CAS PubMed.
- E. Blanchardon, E. Davesne, F. Paquet and M. Bailey, Int. J. Radiat. Biol., 2014, 90, 959–965 CrossRef CAS PubMed.
- L. M. Webb, D. M. Taylor and D. R. Williams, Radiat. Prot. Dosim., 1998, 79, 219–222 CrossRef CAS.
- L. M. Webb, D. M. Taylor and D. R. Williams, J. Alloys Compd., 1998, 271, 112–115 CrossRef.
- L. Bion, E. Ansoborlo, V. Moulin, P. Reiller, R. Collins, R. Gilbin, L. Fevrier, T. Perrier, F. Denison and G. Cote, Radiochim. Acta, 2005, 93, 715–718 CrossRef CAS.
- S. Sachs, A. Heller, S. Weiss, F. Bok and G. Bernhard, Toxicol. in Vitro, 2015, 29, 1555–1568 CrossRef CAS PubMed.
- C. Dawes, A. M. L. Pedersen, A. Villa, J. Ekstrom, G. B. Proctor, A. Vissink, D. Aframian, R. McGowan, A. Aliko, N. Narayana, Y. W. Sia, R. K. Joshi, S. B. Jensen, A. R. Kerr and A. Wolff, Arch. Oral Biol., 2015, 60, 863–874 CrossRef CAS PubMed.
- D. C. Whitcomb and M. E. Lowe, Dig. Dis. Sci., 2007, 52, 1–17 CrossRef CAS PubMed.
- S. Darnis, N. Juge, X. J. Guo, G. Marchis-Mouren, A. Puigserver and J. C. Chaix, Biochim. Biophys. Acta, 1999, 1430, 281–289 CrossRef CAS.
- M. X. Qian, E. Ajandouz, F. Payan and V. Nahoum, Biochemistry, 2005, 44, 3194–3201 CrossRef CAS PubMed.
- N. Ramasubbu, V. Paloth, Y. G. Luo, G. D. Brayer and M. J. Levine, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1996, 52, 435–446 CrossRef CAS PubMed.
- R. P. Agarwal and R. I. Henkin, J. Biol. Chem., 1987, 262, 2568–2575 CAS.
- B. A. Gopal and G. Muralikrishna, Int. J. Food Prop., 2009, 12, 571–586 CrossRef CAS.
- E. Gasteiger, C. Hoogland, A. Gattiker, S. Duvaud, M. R. Wilkins, R. D. Appel and A. Bairoch, Protein Identification and Analysis Tools on the ExPASy Server, in The Proteomics Protocols Handbook, ed. J. M. Walker, Humana Press, 2005, pp. 571–607 Search PubMed.
- P. Artimo, M. Jonnalagedda, K. Arnold, D. Baratin, G. Csardi, E. de Castro, S. Duvaud, V. Flegel, A. Fortier, E. Gasteiger, A. Grosdidier, C. Hernandez, V. Ioannidis, D. Kuznetsov, R. Liechti, S. Moretti, K. Mostaguir, N. Redaschi, G. Rossier, I. Xenarios and H. Stockinger, Nucleic Acids Res., 2012, 40, W597–W603 CrossRef CAS PubMed.
- S. B. Larson, A. Greenwood, D. Cascio, J. Day and A. McPherson, J. Mol. Biol., 1994, 235, 1560–1584 CrossRef CAS PubMed.
- M. Machius, L. Vertesy, R. Huber and G. Wiegand, J. Mol. Biol., 1996, 260, 409–421 CrossRef CAS PubMed.
- S. Z. Fisher, L. Govindasamy, C. Tu, M. Agbandje-McKenna, D. N. Silverman, H. J. Rajaniemi and R. McKenna, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2006, 62, 88–93 CAS.
- E. H. Rydberg, G. Sidhu, H. C. Vo, J. Hewitt, H. C. F. Cote, Y. L. Wang, S. Numao, R. T. A. MacGillivray, C. M. Overall, G. D. Brayer and S. G. Withers, Protein Sci., 1999, 8, 635–643 CrossRef CAS PubMed.
- N. M. Edelstein, R. Klenze, T. Fanghänel and S. Hubert, Coord. Chem. Rev., 2006, 250, 948–973 CrossRef CAS.
- G. Geipel, Coord. Chem. Rev., 2006, 250, 844–854 CrossRef CAS.
- A. V. Hill, J. Physiol., 1910, 40(suppl), iv–vii Search PubMed.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739–1753 CrossRef CAS PubMed.
- W. D. Horrocks and D. R. Sudnick, J. Am. Chem. Soc., 1979, 101, 334–340 CrossRef CAS.
- T. Kimura and G. R. Choppin, J. Alloys Compd., 1994, 213–214, 313–317 CrossRef.
- J. I. Kim, R. Klenze, H. Wimmer, W. Runde and W. Hauser, J. Alloys Compd., 1994, 213/214, 333–340 CrossRef CAS.
- R. A. Binstead, A. D. Zuberbühler and B. Jung, SPECFIT Global Analysis System, version 3.0.37, Spectrum Software Associates, Marlborough, MA, USA, 2005 Search PubMed.
- L. D. Pettit, I. Puigdomenech and H. Wanner, Ionic Strength Corrections for Stability Constants using Specific Interaction Theory (SIT), York, UK, 2004 Search PubMed.
- F. N. Christensen, S. S. Davis, J. G. Hardy, M. J. Taylor, D. R. Whalley and C. G. Wilson, J. Pharm. Pharmacol., 1985, 37, 91–95 CrossRef CAS PubMed.
- A. G. Oomen, C. J. M. Rompelberg, M. A. Bruil, C. J. G. Dobbe, D. P. K. H. Pereboom and A. J. A. M. Sips, Arch. Environ. Contam. Toxicol., 2003, 44, 281–287 CrossRef CAS PubMed.
- K. Djinovic-Carugo and O. Carugo, J. Inorg. Biochem., 2015, 143, 69–76 CrossRef CAS PubMed.
- A. A. Saboury, Biologia, 2002, 57, 221–228 CAS.
- A. A. Saboury and F. Karbassi, Thermochim. Acta, 2000, 362, 121–129 CrossRef CAS.
- A. Levitzki and J. Reuben, Biochemistry, 1973, 12, 41–44 CrossRef CAS PubMed.
- M. Sugahara, M. Takehira and K. Yutani, PLoS One, 2013, 8, e57432 CAS.
- A. M. Brzozowski and G. J. Davies, Biochemistry, 1997, 36, 10837–10845 CrossRef CAS PubMed.
- Z. Fujimoto, K. Takase, N. Doui, M. Momma, T. Matsumoto and H. Mizuno, J. Mol. Biol., 1998, 277, 393–407 CrossRef CAS PubMed.
- A. E. Martell and R. M. Smith, NIST - Critically selected stability constants of metal complexes database, version 7.0, 2003 Search PubMed.
- A. vanderWal, W. Norde, A. J. B. Zehnder and J. Lyklema, Colloids Surf., B, 1997, 9, 81–100 CrossRef.
- C. Kallay, K. Varnagy, G. Micera, D. Sanna and I. Sovago, J. Inorg. Biochem., 2005, 99, 1514–1525 CrossRef CAS PubMed.
- M. Epstein, A. Levitzki and J. Reuben, Biochemistry, 1974, 13, 1777–1782 CrossRef CAS PubMed.
- A. Barkleit, H. Moll and G. Bernhard, Dalton Trans., 2008, 2879–2886 RSC.
- A. Barkleit, H. Moll and G. Bernhard, Dalton Trans., 2009, 5379–5385 RSC.
- I. Sovago, C. Kallay and K. Varnagy, Coord. Chem. Rev., 2012, 256, 2225–2233 CrossRef CAS.
- M. Sugahara, Y. Asada, H. Shimada, H. Taka and N. Kunishima, J. Appl. Crystallogr., 2009, 42, 540–544 CrossRef CAS.
- M. Angeletti, G. Lupidi, A. M. Eleuteri, R. Tacconi, E. Fioretti and M. Coletta, J. Biol. Inorg. Chem., 1997, 2, 320–326 CrossRef CAS.
- H. Moll and G. Bernhard, J. Coord. Chem., 2007, 60, 1795–1807 CrossRef CAS.
- H. Moll, L. Lütke, A. Barkleit and G. Bernhard, Geomicrobiol. J., 2013, 30, 337–346 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5dt04790k |
| ‡ Present address: Technische Universität Dresden, Department of Biology, Institute of Zoology, Professorship of Molecular Cell Physiology and Endocrinology, 01062 Dresden, Germany. |
| This journal is © The Royal Society of Chemistry 2016 |