
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectronic communication in phosphine substituted bridged dirhenium complexes – clarifying ambiguities raised by the redox non-innocence of the C4H2- and C4-bridges†
Yan
Li
a,
Olivier
Blacque
a,
Thomas
Fox
a,
Sandra
Luber
a,
Walther
Polit
b,
Rainer F.
Winter
b,
Koushik
Venkatesan
a and
Heinz
Berke
*a
aDepartment of Chemistry, University of Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland. E-mail: hberke@chem.uzh.ch
bFachbereich Chemie der Universität Konstanz, Universitätstrasse 10, D-78457 Konstanz, Germany
First published on 3rd March 2016
Abstract
The mononuclear rhenium carbyne complex trans-[Re(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CSiMe3)(C–Me)(PMe3)4][PF6] (2) was prepared in 90% yield by heating a mixture of the dinitrogen complex trans-[ReCl(N2)(PMe3)4] (1), TlPF6, and an excess of HCCSiMe3. 2 could be deprotonated with KOtBu to the vinylidene complex trans-[Re(CCSiMe3)(
CSiMe3)(C–Me)(PMe3)4][PF6] (2) was prepared in 90% yield by heating a mixture of the dinitrogen complex trans-[ReCl(N2)(PMe3)4] (1), TlPF6, and an excess of HCCSiMe3. 2 could be deprotonated with KOtBu to the vinylidene complex trans-[Re(CCSiMe3)(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CCH2)(PMe3)4] (3) in 98% yield. Oxidation of 3 with 1.2 equiv. of [Cp2Fe][PF6] at −78 °C gave the Cβ–C′β coupled dinuclear rhenium biscarbyne complex trans-[(Me3SiCC)(PMe3)4ReC–CH2–CH2–CRe(PMe3)4(CCSiMe3)][PF6]2 (5) in 92% yield. Deprotonation of 5 with an excess of KOtBu in THF produced the diamagnetic trans-[(Me3SiCC)(PMe3)4ReCCH–CHCRe(PMe3)4(CCSiMe3)] complex (E-6(S)) in 87% yield with an E-butadienediylidene bridge. Density functional theory (DFT) calculations of E-6(S) confirmed its singlet ground state. The Z-form of 6 (Z-6(S)) could not be observed, which is in accord with its DFT calculated 17.8 kJ mol−1 higher energy. Oxidation of E-6 with 2 equiv. of [Cp2Fe][PF6] resulted in the stable diamagnetic dicationic trans-[(Me3SiCC)(PMe3)4ReC–CHCH–CRe(PMe3)4(CCSiMe3)][PF6]2 complex (E-6[PF6]2) with an ethylenylidene dicarbyne structure of the bridge. The paramagnetic mixed-valence (MV) complex E-6[PF6] was obtained by comproportionation of E-6(S) and E-6[PF6]2 or by oxidation of E-6(S) with 1 equiv. of [Cp2Fe][PF6]. The dicationic trans-[(Me3SiCC)(PMe3)4ReC–CC–CRe(PMe3)4(CCSiMe3)][PF6]2 (7[PF6]2) complex, attributed a butynedi(triyl) bridge structure, was obtained by deprotonation of E-6[PF6]2 with KOtBu followed by oxidation with 2 equiv. of [Cp2Fe][PF6]. The neutral complex 7 could be accessed best by reduction of 7[PF6]2 with KH in the presence of 18-crown-6. According to DFT calculations 7 possesses two equilibrating electronic states: diamagnetic 7(S) and triplet 7(F) with ferromagnetically coupled spins. The latter is calculated to be 5.2 kcal mol−1 lower in energy than 7(S). There is experimental evidence that 7(S) prevails in solution. 7 could not be isolated in the crystalline state and is unstable transforming mainly by H-abstraction to give E-6(S). UV-Vis-NIR spectroscopy for the dinuclear rhenium complexes E-6(S), E-6[PF6] and E-6[PF6]2, as well as EPR spectroscopic and variable-temperature magnetization measurements for the MV complex E-6[PF6] were also conducted. Spectro-electrochemical reduction studies on 7[PF6]2 allowed the characterization of the mono- and direduced forms of 7+ and 7 by means of IR- and UV-Vis-NIR-spectroscopy and revealed the chemical fate of the higher reduced form.
CCH2)(PMe3)4] (3) in 98% yield. Oxidation of 3 with 1.2 equiv. of [Cp2Fe][PF6] at −78 °C gave the Cβ–C′β coupled dinuclear rhenium biscarbyne complex trans-[(Me3SiCC)(PMe3)4ReC–CH2–CH2–CRe(PMe3)4(CCSiMe3)][PF6]2 (5) in 92% yield. Deprotonation of 5 with an excess of KOtBu in THF produced the diamagnetic trans-[(Me3SiCC)(PMe3)4ReCCH–CHCRe(PMe3)4(CCSiMe3)] complex (E-6(S)) in 87% yield with an E-butadienediylidene bridge. Density functional theory (DFT) calculations of E-6(S) confirmed its singlet ground state. The Z-form of 6 (Z-6(S)) could not be observed, which is in accord with its DFT calculated 17.8 kJ mol−1 higher energy. Oxidation of E-6 with 2 equiv. of [Cp2Fe][PF6] resulted in the stable diamagnetic dicationic trans-[(Me3SiCC)(PMe3)4ReC–CHCH–CRe(PMe3)4(CCSiMe3)][PF6]2 complex (E-6[PF6]2) with an ethylenylidene dicarbyne structure of the bridge. The paramagnetic mixed-valence (MV) complex E-6[PF6] was obtained by comproportionation of E-6(S) and E-6[PF6]2 or by oxidation of E-6(S) with 1 equiv. of [Cp2Fe][PF6]. The dicationic trans-[(Me3SiCC)(PMe3)4ReC–CC–CRe(PMe3)4(CCSiMe3)][PF6]2 (7[PF6]2) complex, attributed a butynedi(triyl) bridge structure, was obtained by deprotonation of E-6[PF6]2 with KOtBu followed by oxidation with 2 equiv. of [Cp2Fe][PF6]. The neutral complex 7 could be accessed best by reduction of 7[PF6]2 with KH in the presence of 18-crown-6. According to DFT calculations 7 possesses two equilibrating electronic states: diamagnetic 7(S) and triplet 7(F) with ferromagnetically coupled spins. The latter is calculated to be 5.2 kcal mol−1 lower in energy than 7(S). There is experimental evidence that 7(S) prevails in solution. 7 could not be isolated in the crystalline state and is unstable transforming mainly by H-abstraction to give E-6(S). UV-Vis-NIR spectroscopy for the dinuclear rhenium complexes E-6(S), E-6[PF6] and E-6[PF6]2, as well as EPR spectroscopic and variable-temperature magnetization measurements for the MV complex E-6[PF6] were also conducted. Spectro-electrochemical reduction studies on 7[PF6]2 allowed the characterization of the mono- and direduced forms of 7+ and 7 by means of IR- and UV-Vis-NIR-spectroscopy and revealed the chemical fate of the higher reduced form.
I. Introduction
Organometallic rigid-rod dinuclear complexes consisting of a rigid π-conjugated organic carbyl or hydrocarbyl bridge CxHy with redox-active metal end groups of the type [LnMCxHyMLn] (M = metal; L = ligand) have recently received considerable attention due to their potential function in molecular electronic devices.1 In this paper we will address the tetracarbyl (C4) and butadiene-1,4-di(ylidene) (C4H2) units as rigid π-conjugated bridging moieties of rhenium based complexes. Rigid molecules of these kinds are denoted as molecular wires potentially providing molecular conductance between the remote ends.1,2 For instance, Gladysz and co-workers reported a series of dinuclear rhenium complexes with μ-Cx carbyl chains (x ≤ 20) composed of up to ten alkynyl units.3 Voltammetric studies revealed that with increasing chain lengths, the potential difference ΔE1/2 between the two mainly metal derived redox processes decreases and the oxidation becomes increasingly irreversible. In other words, the longer the chain length, the smaller is the electronic interaction between the remote redox sites and the higher is the reactivity of the (hydro)carbyl chain. The HOMO and LUMO of bridged polyacetylenic systems ([LnMCxMLn], x = even number) are in most cases π-type molecular orbitals with only limited π-delocalization, large HOMO–LUMO gaps, and quite low polarizabilities of the bridges also preventing strong metal–bridge–metal interactions. Furthermore, for molecules serving as junctions in single-molecule conductivity devices the energetic alignment of the HOMO or the LUMO with the Fermi level EF of the metal electrodes favors electron transfer via a resonant conductance mechanism.4 This conductance mechanism can be addressed by cyclic voltammetry (CV) and by UV-Vis-NIR spectroscopy. Here the interaction of the metal centers can be quantified on the basis of the characteristic parameters (energy![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) , extinction coefficient ε and band-width at half height Δ1/2) of the intervalence charge transfer (IVCT) absorptions of dinuclear mixed-valence complexes.23,24 According to their interaction strengths mixed-valence systems have been grouped into three classes by Robin & Day5 and an extension of this view has been given with regard to charged species by Kaupp et al.6
, extinction coefficient ε and band-width at half height Δ1/2) of the intervalence charge transfer (IVCT) absorptions of dinuclear mixed-valence complexes.23,24 According to their interaction strengths mixed-valence systems have been grouped into three classes by Robin & Day5 and an extension of this view has been given with regard to charged species by Kaupp et al.6
The through-bridge electronic interaction in dinuclear complexes has a significant impact on their chemical and physical properties, which greatly depends on the type of metal centers, the ancillary ligands and the bridge.2t,3,7 Based on the ease of synthetic access, stability, and favorable electronic properties, the relatively short butadiene-1,4-di(ylidene) C4H2 and butadiynediyl C4 chains were anticipated to be appropriate bridges for strong electronic interactions between the metal centers.3 Therefore intensive investigations have been carried out on complexes of the type [LnMC4MLn] with different transition metal centers, such as Mn,8 Fe,3a,9 Re,10 Ru,11 Pt,12 W, and Mo.13 However, only complexes with metal centers, which could take over the role of bi- or trifunctionalized linking units possess the potential for the build-up of oligo- or polynuclear complexes. Mono-functionalized bridged metal centers are functioning as end groups and are in connectivity terms stopper units. In addition bi- or trifunctionalized metal centers would allow the introduction of special types of functionalities for, for instance, their hook up to electrodes, which is the key to their function as junctions in molecular conductance devices.
Therefore we sought to construct trans-bifunctional dinuclear rhenium complexes equipped with C4H2 and C4 bridges and a trimethylsilyl acetylide functional group, the latter eventually enabling a stable and electronically strong coupling to gold electrodes as previously demonstrated in the case of organic oligoynes14,15 and of appropriate dinuclear complexes by pre-measurement removal of the silyl groups16 or by subsequent conversion of the silyl into stannyl groups, which allow spontaneous removal of the tin groups upon contact with the gold surface.2b,c,16b
The C4H2 and C4 bridges belong to the class of redox non-innocent ligands17 and are by this property expected to enable the terminal binding of metal centers by various canonical forms (Scheme 1). These bridges are prone to electronic flexibility in their σ- and π-bonding adjusting electronically via varying electron counts at the bridges’ termini. It would be natural to assume that in the case of metal attachment the C2H4 or C4 bridging systems adopt to the electron demands of electron-precise metal centers. The bridges’ π electrons are variably distributed between CC π bonding electrons and electrons π donated to the metal centers. The π electrons donated to a metal center account for varying strengths of the metal–carbon bonds and the π electrons of the bridge account for different π stabilizations within the bridges (Scheme 1).
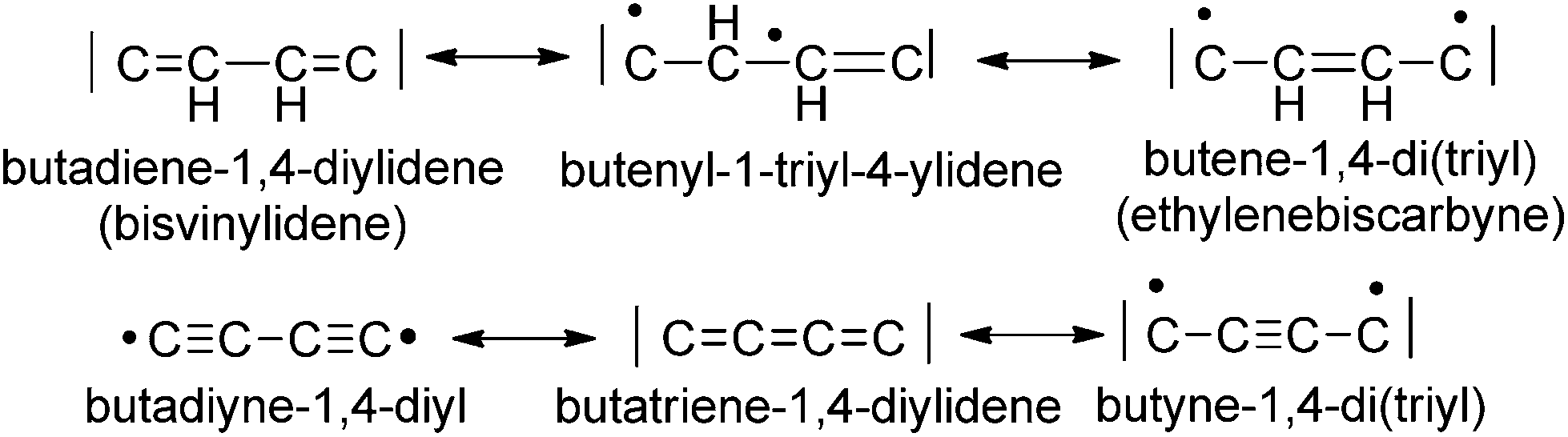 | ||
| Scheme 1 Different canonical forms of the C4H2 and the C4 ligand bridges. | ||
For instance, the butadiyne-1,4-diyl bridging unit is expected to have the lowest π-delocalization between the metal centers13a,18 of all possible forms of the C4 bridge, since the number of π electrons donated to the metal centers is zero and the π-delocalization of diynes is quite low.19,20
In special cases, when the M–C bond is strongly covalent, the bridging ligands will sometimes not adjust to the electron demand of the metal centers. This in turn might lead to ligand-dominated redox-processes and the so-called “non-innocent” behaviour.17,21 It maybe interesting to note that the isomeric forms of the C4H2 and the C4 bridge are expected to be related by bond-stretch isomerism revealing different carbon–carbon bond lengths as free molecules and as ligands in complexes.22
For the development of dinuclear complexes with conjugated C4H2 and C4 bridges we selected the 16e− rhenium fragment [(RCC)(PMe3)4Re], which is bifunctional in the sense that it allows for hooking up to electrodes via the RCC group and can act at the same time as one terminus of bridging systems. The targeted dinuclear rhenium complexes were anticipated to be analogous to manganese-based ones reported by our group earlier and to be related to systems with [X(diphosphine)2W] end groups possessing one electron less per metal site. Both types of systems should therefore differ in the preferred canonical form of the bridge and in their electronic properties.8,13c,23 Rhenium as a heavier transition element was expected to render structurally stable complexes and to form strong ligand bonds concomitant with higher barriers for ligand exchange. To avoid difficulties in the synthetic access to [LnReC4H2ReLn] and [LnReC4ReLn] complexes, we tried to circumvent the direct introduction of the C4H2 and C4 bridges by ligand exchange by the alternative construction of μ-C4Hn (n = 0, 2 or 4) dinuclear complexes by oxidative C–C coupling processes, i.e. by ‘dimerizing’ mononuclear C2Hn (n = 0, 1 or 2) complex units. Our work on manganese coupling chemistry8a,23a,b,e,f,24 and related work on niobium,25 tungsten,13a molybdenum,13a,26 manganese,18a,27 rhenium,28 iron,9,29,34 and ruthenium30 complexes demonstrated the synthetic utility of the oxidative coupling of metal alkynyls or of oxidative dehydro-dimerizations of metal vinylidenes.18b,31
A key point of our rhenium based endeavors to access dinuclear rhenium complexes of the type [(Me3SiCC)(PMe3)4ReC4HnRe(PMe3)4(CCSiMe3)] with n = 0, 2 or 4 was thus to synthetically access the mononuclear rhenium vinylidene species trans-[Re(CCSiMe3)(CCH2)(PMe3)4] and couple that complex to the non-conjugated C4H4 bridged system. From there we sought to access the C4H2 and C4 bridges by successive oxidative dehydrogenations.
II. Results and discussion
IIa. Synthesis and characterization of mononuclear rhenium complexes
The synthesis of the dinuclear rhenium complex trans-[(Me3SiCC)(PMe3)4ReC–CH2–CH2–CRe(PMe3)4(CCSiMe3)][PF6]2 started from the dinitrogen complex trans-[ReCl(N2)(PMe3)4] (1). From this precursor the mononuclear rhenium carbyne complex trans-[Re(CCSiMe3)(CMe)(PMe3)4][PF6] (2) was formed in 90% yield by heating in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture with TlPF6 and by the addition of excess HCCSiMe3 in N,N-diisopropylethylamine (DIPEA)/THF at 95 °C (Scheme 2).
:1 mixture with TlPF6 and by the addition of excess HCCSiMe3 in N,N-diisopropylethylamine (DIPEA)/THF at 95 °C (Scheme 2).
 | ||
| Scheme 2 | ||
The mononuclear rhenium carbyne complex 2 readily underwent deprotonation using excess KOtBu in THF to form the corresponding mononuclear rhenium vinylidene complex trans-[Re(CCSiMe3)(CCH2)(PMe3)4] (3) in 98% yield18b,31c (Scheme 2). Re-protonation of 3 could be accomplished using HCl to recover the carbyne complex trans-[Re(CCSiMe3)(C–Me)(PMe3)4]Cl (4) as a chloride salt, which in turn could be deprotonated with KOtBu to regenerate 3.
The mononuclear rhenium complexes 2, 3 and 4 were characterized by NMR, IR, elemental analyses and mass spectroscopy. The 1H NMR spectrum of trans-[Re(CCSiMe3)(CCH2)(PMe3)4] (3) displayed a characteristic quintet for the vinylidene protons at 1.35 ppm (4JPH = 3.5 Hz). In the 13C{1H} NMR spectrum two resonances were found at 301.6 ppm (2JPC = 11.9 Hz) and 87.8 ppm that could be assigned to the Cα and Cβ atoms of the vinylidene group. The other two resonances at 150.1 ppm and 67.8 ppm were attributed to the Cα and Cβ atoms of the acetylide moiety8a,18,23a,b,f,27,28 presumably reflecting an extraordinary electron-richness. The IR spectrum of 3 showed strong ν(CC) and ν(CC) bands at 1558 and 1982 cm−1, respectively.
The carbyne complexes trans-[Re(CCSiMe3)(C–Me)(PMe3)4][PF6] (2) and trans-[Re(CCSiMe3)(C–Me)(PMe3)4]Cl (4) exhibited in the 1H NMR spectra a characteristic quintet for the Mecarbyne protons at 1.27 ppm (4JPH = 4.0 Hz) and 1.31 ppm (4JPH = 4.0 Hz), respectively, while the 13C{1H} NMR spectra of 2 and 4 displayed respective resonances for the Cα atoms at 284.5 or 284.9 ppm. Their intensity was, however, too low to allow extraction of the apparently small JPC values. Two additional 13C{1H} NMR resonances at 134.7 and 125.7 ppm or at 135.0 ppm and 125.8 ppm for 2 and 4 confirmed the presence of the acetylide groups. In the IR spectra ν(CC) bands for 2 and 4 were observed at 2029 cm−1 or 2025 cm−1, respectively. A singlet resonance in the 31P{1H} NMR spectra complied with the trans-arrangements of the alkynyl and carbyne ligands in 2, 3 and 4.
IIb. Synthesis and characterization of C4Hn bridged dirhenium complexes
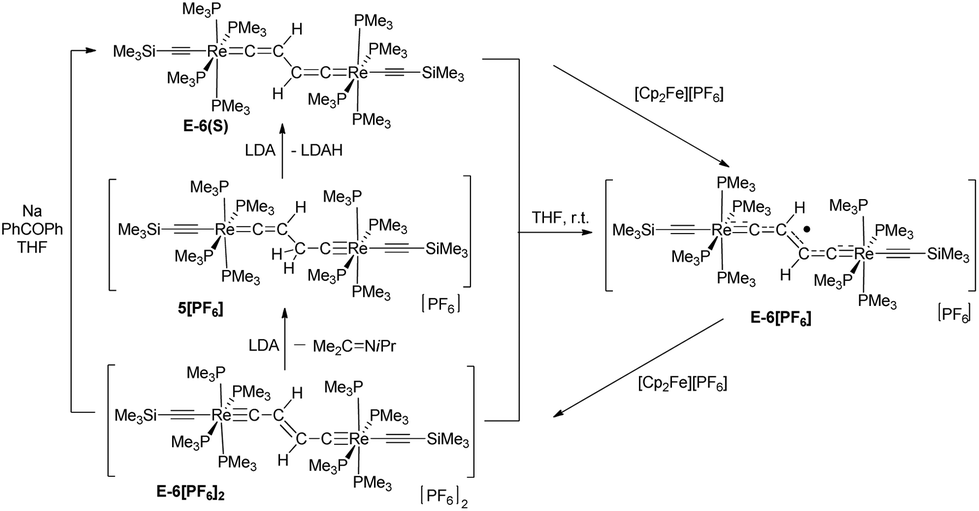
Oxidative C–C coupling was demonstrated to be an effective method for the build-up of the bridges of dinuclear μ-C4H2 and μ-C4 complexes.18a,23a,b,f,27,28,30 To adjust this method to dinuclear rhenium complexes with C4H2 bridges, we initially tried a variation of the oxidative coupling of 3 with electron and proton removal in the presence of a base. However, the reaction of 3 with [Cp2Fe][PF6] in the presence of quinuclidine, DBU, or KOtBu inevitably resulted in a mixture of complexes: the mononuclear rhenium carbyne complex trans-[Re(CCSiMe3)(C–Me)(PMe3)4][PF6] (2), the dinuclear rhenium biscarbyne complex trans-[(Me3SiCC)(PMe3)4ReC–CH2–CH2–CRe(PMe3)4(CCSiMe3)][PF6]2 (5), and the dicationic bisvinylidene complex trans-[(Me3SiCC)(PMe3)4ReCCH–CHCRe(PMe3)4(CCSiMe3)][PF6]2 (6[PF6]2). This complex reaction behavior was partly attributed to the highly basic nature of 3, which competed with the base added to deprotonate the oxidatively formed radical cation [3]+ giving 2 and the acetylide complex trans-[(Me3SiCC)Re(CC–Me)(PMe3)4] and, by subsequent oxidation, the trans-[(Me3SiCC)Re(CCMe)(PMe3)4]+ cation, which dimerized by C–C coupling to give 6[PF6]2. We anticipated that a better control of the resulting products could be achieved by performing the reaction in the absence of a base at low temperatures27 to accumulate [3]+ at higher concentrations, thus promoting the second order recombination reaction to 5. Oxidation of 3 was carried out at −78 °C by adding a THF solution of 3 dropwise to an excess of [Cp2Fe][PF6] in CH3CN/THF (1:3 ratio) leading to a 92% yield of the coupled dicationic dirhenium biscarbyne complex trans-[(Me3SiCC)(PMe3)4ReC–CH2–CH2–CRe(PMe3)4(CCSiMe3)][PF6]2 (5) as the sole product (Scheme 3).
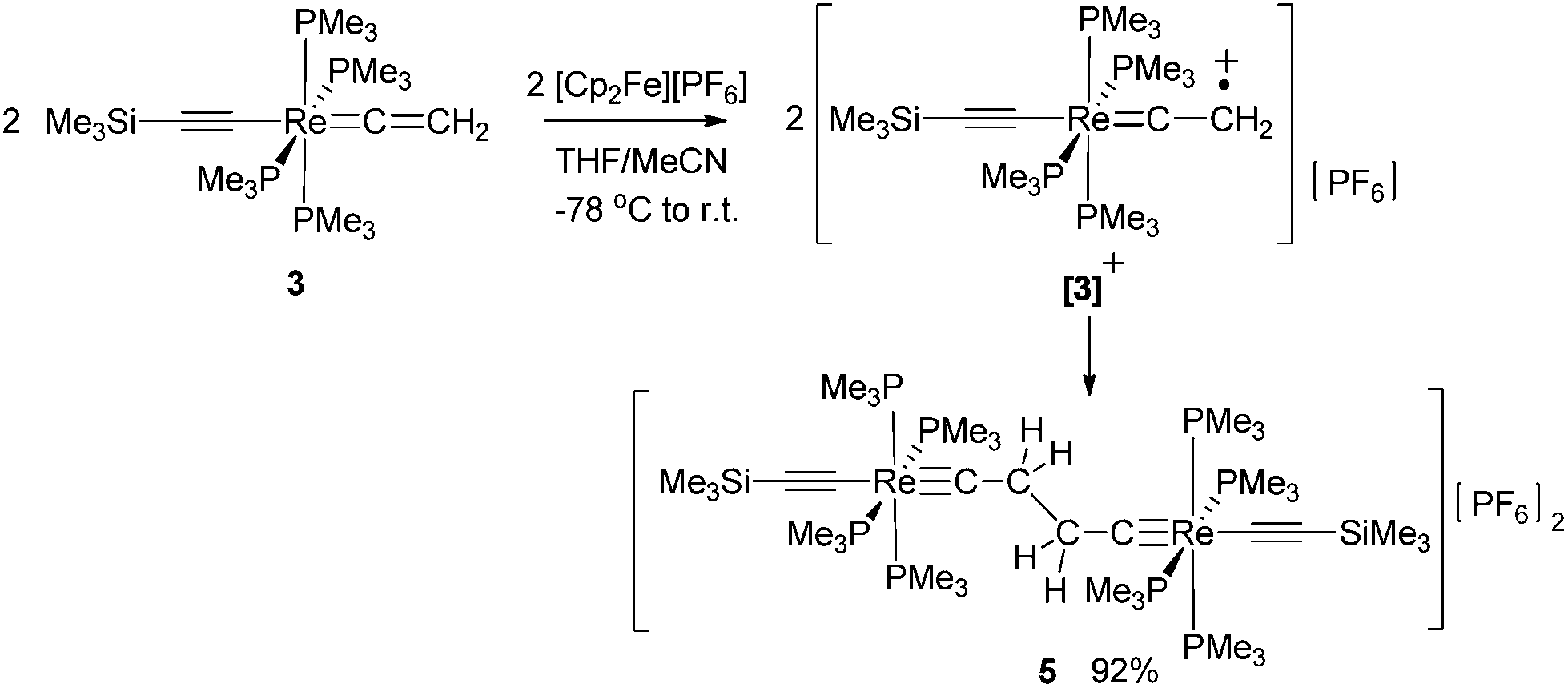 | ||
| Scheme 3 | ||
The 1H NMR spectrum of 5 displayed three signals. A resonance at 1.56 ppm was ascribed to the CH2 protons of the bridge and two other signals at 1.75 ppm and 0.05 ppm were attributed to the protons of the PMe3 and the SiMe3 groups, respectively. In the 13C{1H} NMR spectrum resonances appeared for the Cα and Cβ atoms of the μ-biscarbyne ligand at 279.9 ppm (2JPC = 13.8 Hz) and 46.4 ppm. Two additional resonances of the 13C{1H} NMR spectrum at 134.0 ppm (2JPC = 19.4 Hz) and 126.6 ppm were attributed to the terminal acetylide groups; additionally a ν(CC) band at 2027 cm−1 in the IR spectrum was also diagnostic for that moiety. In the 31P NMR spectrum the resonance for the PMe3 ligands appeared at −42.2 ppm, while the [PF6]− anion gave rise to a septet at −146.6 ppm. The composition of 5 was confirmed by a correct elemental analysis.
5 could be deprotonated by applying an excess of KOtBu yielding the brownish-green bisvinylidene complex E-[(Me3SiCC)(PMe3)4ReCCH–CHCRe(PMe3)4(CCSiMe3)] (E-6(S)) in 87% yield (Scheme 4). The use of lithium diisopropylamide (LDA) as a base at room temperature in THF furnished a somewhat lower yield (83%), but the work-up procedure to E-6(S) was found to be more facile in this case. It is quite remarkable that the Z-isomer Z-6(S) could not be traced at any stage of the reaction to 5. Z-6(S) might indeed form in these reactions. Kohn–Sham density functional theory (DFT) calculations indicate that Z-6(S) is at a high energy of 17.8 kcal mol−1 above E-6(S) and transforms quickly into E-6(S) (see section III). An alternative deactivation pathway of Z-6(S) could be its dehydrogenation (Z-6(S) → 7(F) + H2) assuming a low activation barrier for this process (Scheme 6). The latter process was calculated by DFT to be energetically downhill by −7.8 kcal mol−1. In this context it should be mentioned that the dehydrogenation reaction of E-6(S) (E-6(S) → 7(F) + H2) was calculated to be energetically uphill by 10.0 kcal mol−1 and is therefore anticipated not to occur. The Z-6(S) dehydrogenation process to 7(F) could make up for the 13 or 17% of the missing yield of E-6(S) in its formation process along Scheme 4. It should be mentioned at this point that the open-shell structure E-6(F) with ferromagnetically coupled electrons is an excited state of E-6(S) at a too high electronic energy to be reached by thermal activation and is therefore expected to be non-existent at room temperature in solution.
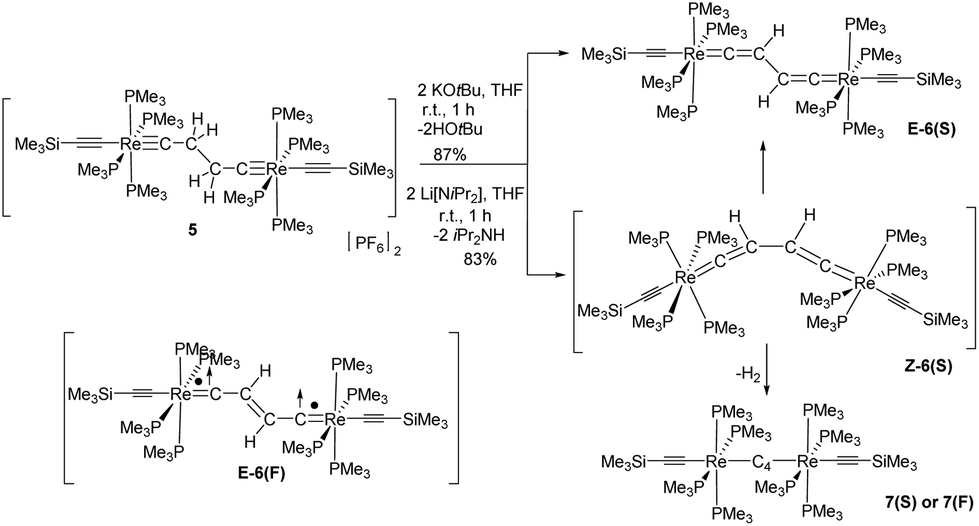 | ||
| Scheme 4 Synthesis of E-6(S)via deprotonation of 5. Generation, isomerization and dehydrogenation of the potential intermediate complex Z-6(S). Sketch of E-6(F) as an activated state of E-6. | ||
The 1H NMR spectrum of E-6(S) in THF-d8 at room temperature revealed a ‘normal’ Hvinylidene chemical shift of 2.50 ppm; but its doublet structure (J = 4 Hz) was unexpected and indicated a peculiar phenomenon. The solid state structure of E-6(S) (Fig. 1a) possesses an approximate planarity of the [Re]CCH–CHC[Re] moiety. On this basis, the mentioned coupling can be interpreted as 4JPH coupling originating from the trans-coplanar arrangement of the closest Hvinylidene atom (plane (Hvinylidene, C2, C1)) to the strongly ‘bent-back’ P3 atom (plane (P3, Re, C1) of Fig. 1a and Scheme 5). The coupling effect would be strongest, if the ReP4 fragment is subject to hindered rotation, which prevents averaging on the NMR time scale with the other three non-coupled P atoms (4JPH = 0).
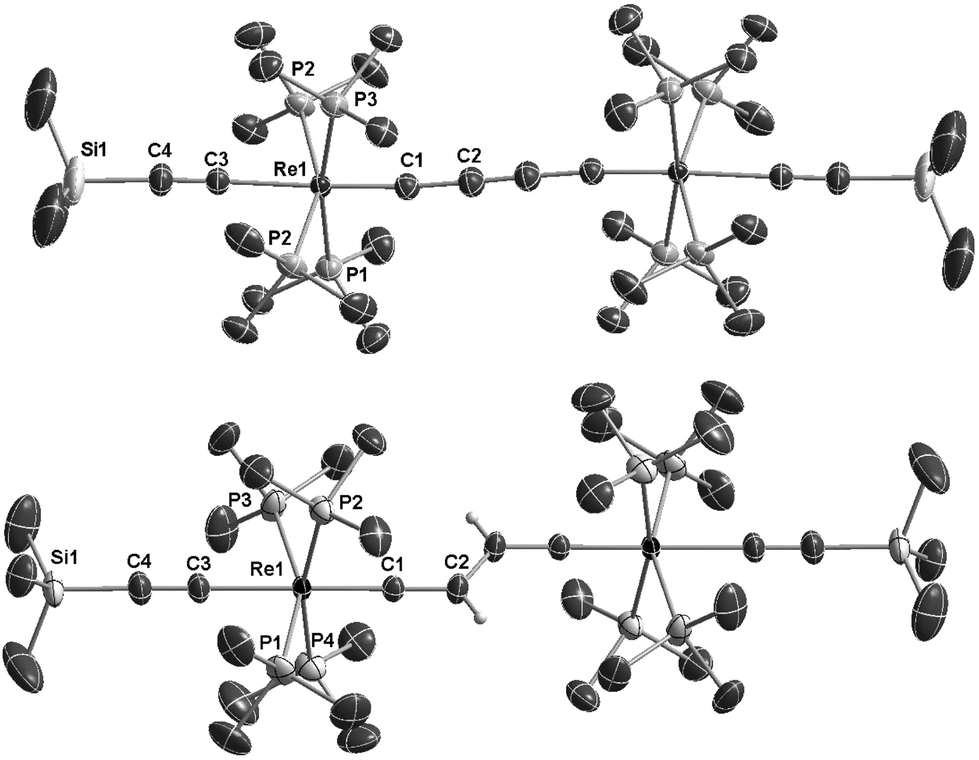 | ||
| Fig. 1 ORTEP like drawing of (a) of the neutral dinuclear rhenium butadienediylidene complex trans-[(Me3SiCC)(PMe3)4ReC4H2Re(PMe3)4(CCSiMe3)] (E-6(S)) (bottom); (b) of the dicationic dinuclear rhenium complex trans-[(Me3SiCC)(PMe3)4ReC–CC–CRe(PMe3)4(CCSiMe3)][PF6]27[PF6]2 (top) (50% probability level of thermal ellipsoids; solvate molecules, the [PF6]− counterions and selected hydrogen atoms are omitted for clarity). | ||
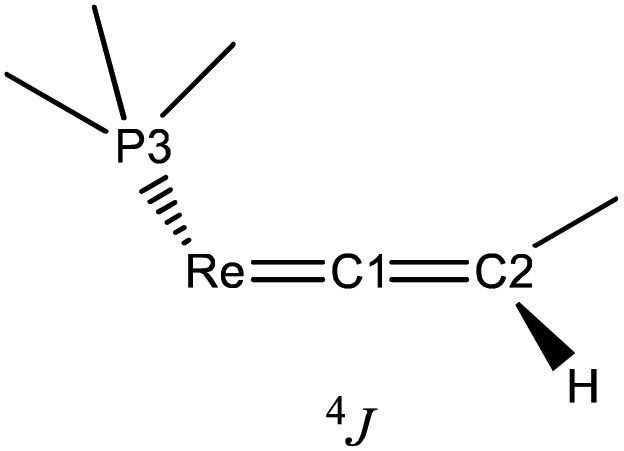 | ||
| Scheme 5 Sketch of the trans-coplanar arrangement of P3 with Hvinylidene of E-6(S). | ||
The 13C, 31P and 29Si NMR signals of E-6(S) are in agreement with the presence of a diamagnetic compound. In the 13C{1H} NMR spectrum of E-6(S) in THF-d8 at 10 °C two characteristic signals appeared, which were assigned to the Cα and Cβ vinylidene atoms at 309.4 ppm (quint, 2JPC = 12.8 Hz) and 96.7 ppm. Two additional resonances at 152.8 ppm (2JPC = 16.0 Hz) and 127.8 ppm were attributed to the Cα and Cβ atoms of the acetylide groups. The 31P and the 29Si NMR spectra showed singlet resonances of E-6(S) at δ = −40.5 ppm (31P NMR) and at δ = −31.8 ppm (29Si NMR).
The solid state IR and Raman spectra displayed a strong ν(CC) band at 1975 cm−1 (IR) or 1973 cm−1 (Raman) for the terminal acetylide moieties. Bands at 1543 cm−1 (IR) and 1581 cm−1 (Raman) were attributed to the νas(CCHCHC) vibration of the bridge (IR) and to the corresponding νs(CCHCHC) vibration (Raman). The molecular structure of E-6(S) obtained by single crystal X-ray diffraction (vide infra) also demonstrated the E-configuration for this molecule and, moreover, the bond distances in the bridge reflected the singlet state of the butadiene-1,4-diylidene(bisvinylidene) canonical form (Scheme 1).
As shown in Scheme 6E-6(S) could be oxidized by two equiv. of [Cp2Fe][PF6] to yield the diamagnetic dicationic complex E-[(Me3SiCC)(PMe3)4ReC–CHCH–CRe(PMe3)4(CCSiMe3)][PF6]2 (E-6[PF6]2) preserving the E-configuration, but adopting an ethylenylidene dicarbyne canonical structure of the bridge (see also X-ray diffraction study of E-6[PF6]2 summarized in Table 1 and displayed in the ESI†) The 1H NMR spectrum of E-6[PF6]2 showed a unique resonance for both vinylidene protons at 5.82 ppm in the typical chemical shift range of olefinic protons, but in a shift range distinct from the corresponding resonances of E-6(S) indicating a significantly different electronic structure. E-6[PF6]2 revealed resonances for the Cα and Cβ nuclei at 265.3 ppm and 145.8 ppm, again appearing in a chemical shift range distinct from that of E-6(S). Due to their low intensities we could not extract the JPC values. Additional 13C NMR resonances at 135.5 ppm and 130.9 ppm were attributed to the acetylide groups. In the 31P NMR spectrum a resonance at −43.8 ppm was ascribed to the P nuclei of the PMe3 ligands. The characteristic signal for the [PF6]− anion appeared as a septet at −143.9 ppm. Although reduction of E-6[PF6]2 to E-6(S) could be carried out with an excess of Na/benzophenone, this reaction was achieved with better control using lithium diisopropylamide (LDA) acting initially as a hydride transfer agent leading to dimethylisopropyl imine32 and the hydride added putative intermediate species 5[PF6], which becomes subsequently deprotonated by LDA to form E-6(S) (Scheme 6).
 | ||
| Scheme 6 Redox reactions of E-6(S), E-6[PF6] and E-6[PF6]2 and comproportionation of E-6(S) and E-6[PF6]2 to yield E-6[PF6]. | ||
| Bond | 5 | E-6(S) | E-6(S) DFT** | E-6[PF6] | E-6[PF6]2 | 7(S) DFT** | 7(F) DFT** | 7[PF6]2 |
|---|---|---|---|---|---|---|---|---|
| Re1–C1 | 1.7834 (19) | 1.904 (2) | 1.899 (1.912) | 1.851 (6) | 1.805 (5) | 1.943 (1.965) | 2.038 (1.972) | 1.814 (4) |
| C1–C2 | 1.491 (3) | 1.337 (3) | 1.322 (1.333) | 1.375 (8) | 1.414 (7) | 1.270 (1.276) | 1.234 (1.274) | 1.363 (5) |
| C2–C2′ | 1.513 (4) | 1.469 (4) | 1.462 (1.465) | 1.388 (10) | 1.331 (10) | 1.295 (1.305) | 1.345 (1.301) | 1.197 (8) |
| P1–Re1–P3* | 156.20 (2) | 155.45 (2) | 155.24 (155.14) | 156.49 (6) | 170.3 (4) | 164.27 (163.86) | 164.16 (163.32) | 169.20 (3) |
| P5–Re2–P7* | — | — | 156.46 (6) | — | — | |||
| P2–Re–P4* | 171.22 (2) | 165.36 (2) | 163.89 (163.61) | 166.62 (6) | 155.7 (4) | 155.20 (155.33) | 157.76 (160.20) | 155.13 (4) |
| P6–Re–P8* | — | — | 166.71 (6) | — | — | |||
| Re⋯Re | 7.323 | 7.416 | 7.348 (7.400) | 7.371 | 7.282 | 7.718 (7.786) | 7.915 (7.797) | 7.536 |
The comproportionation reaction of E-6(S) with E-6[PF6]2 or the oxidation of E-6(S) with one equiv. of [Cp2Fe][PF6] produced the stable mixed valence complex E-6[PF6] (Scheme 5). Only broad signals were observed in the 1H NMR spectra for E-6[PF6] indicating paramagnetic behavior. For E-6[PF6] and the dicationic species E-6[PF6]2, the IR spectra showed weak acetylenic bands at 1987 cm−1 and 2022 cm−1, respectively. In the Raman spectra the corresponding bands were assigned at 2001 cm−1 and 2017 cm−1. ν(C4) bands of the C4H2 bridge could not be observed for E-6[PF6] and E-6[PF6]2 in the IR. In the case of E-6[PF6] this can be viewed as an indication of strong electron delocalization on the IR time scale (10−13 s).13c The diamagnetic dicationic ethylenylidene biscarbyne complex E-6[PF6]2 showed good solubility and stability in polar solvents, such as CH2Cl2 and MeCN. In contrast, neutral E-6(S) complexes and monocationic E-6[PF6] decomposed quickly in CH2Cl2, and over longer period of times also in MeCN, and they underwent facile oxidation in the solid state and in solution. E-6(S) is stable for several months under an inert atmosphere, but should be stored as a solid at −30 °C.
IIc. Characterization of C4 bridged dirhenium complexes
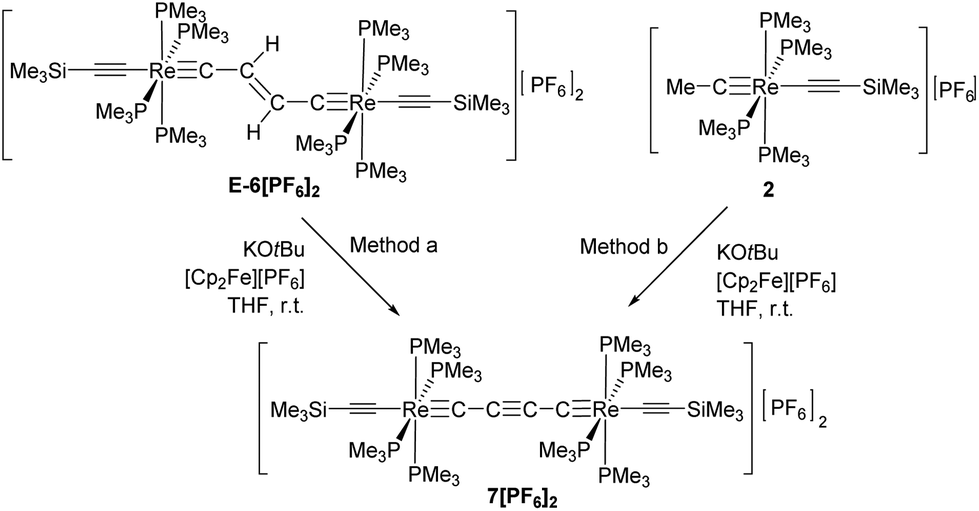
The dicationic trans-[(Me3SiCC)(PMe3)4ReC4Re(PMe3)4(CCSiMe3)][PF6]2 complex (7[PF6]2) could be obtained by repetitive cycles of deprotonations and oxidations starting from E-6[PF6]2 or from 2 following method a or method b as depicted in Scheme 7.
 | ||
| Scheme 7 | ||
In the 1H NMR spectrum the dicationic complex 7[PF6]2 showed singlet resonances for the PMe3 ligands at 1.82 ppm and for the trimethylsilyl groups at 0.26 ppm. The 13C{1H} NMR spectrum revealed characteristic resonances for Cα and Cβ at 235.0 and 95.8 ppm, but the intensities of these signals were too low to extract JPC couplings. The resonances of the acetylide groups appeared at 135.5 ppm and 128.4 ppm. In the 31P NMR spectrum a singlet resonance at −43.6 ppm was ascribed to the PMe3 ligands. The characteristic signal for the [PF6]− counterion appeared as a septet at −143.9 ppm. The IR spectrum of 7[PF6]2 revealed a weak band at 2013 cm−1 attributed to a vibrational band of the acetylenic substituents. Vibrational bands for the C4 moiety could not be found, which supports the notion of a highly symmetric bridging unit of 7[PF6]2.
The reduction of the dicationic species 7[PF6]2 was attempted by treatment with Na/Hg or Na/benzophenone, but these reactions resulted in a mixture of many components. Using KH in the presence of 18-crown-6 in THF the neutral trans-[(Me3SiCC)(PMe3)4ReC4Re(PMe3)4(CCSiMe3)] complex 7 was formed. As depicted in Scheme 7 and according to DFT calculations (section III), two different electronic structures have to be considered for 7: diradical 7(F) with a but-2-ynediylidene bridge and with ferromagnetically coupled electrons and diamagnetic 7(S) with a butatrienediylidene bridge. The unpaired spins of 7(F) would be delocalized over the entire ReC4Re moiety and occupy two degenerate perpendicular π orbitals, which according to Hund's rule should lead to a ferromagnetic electronic state. 7(S) possesses a cumulenic bridge (Scheme 1) providing 2-electrons to each electron-precise Re(I) center, thus establishing a diamagnetic molecule. The MO description of the cumulenic structure is particularly complex, since it is based on different occupancies of the two π planes of the bridge. The inequality of these π planes becomes further amplified in the complex 7(S) by interaction with the unequal π planes of the rhenium centers strongly distinguished in their binding capacity via dislocation of the PMe3 ligands from planarity (section IId). These electronic states of 7 are expected to co-exist at room temperature in solution. However, experimentally it was not possible to distinguish these isomers or to determine the ratio at which these isomers equilibrate.
It should be mentioned at this point that another conceivable isomeric form of 7 is based on a diacetylenic canonical form of the bridge (Schemes 1 and 8). The diacetylenic bridge would need to formally accept one σ electron from each rhenium center to form a diacetylide ligand bridging two low-spin d5 Re(II) fragments with δ type singly occupied d-orbitals.33 The δ type d-orbitals would be arranged perpendicular to the main axis of the molecule incapable of interacting with π-type orbitals of the bridge and excluding conjugation. These remote radical centers could therefore coexist in this isomer of 7 as ferromagnetically coupled SOMOs (Scheme 8). However, according to the DFT calculations in section III the δ orbitals of such rhenium based complexes are located within the “t2g” set at relatively low energies well below the HOMO/SOMO levels. An isomer of 7 with a diacetylenic bridge is therefore very unlikely and is thus omitted from the subsequent discussion.
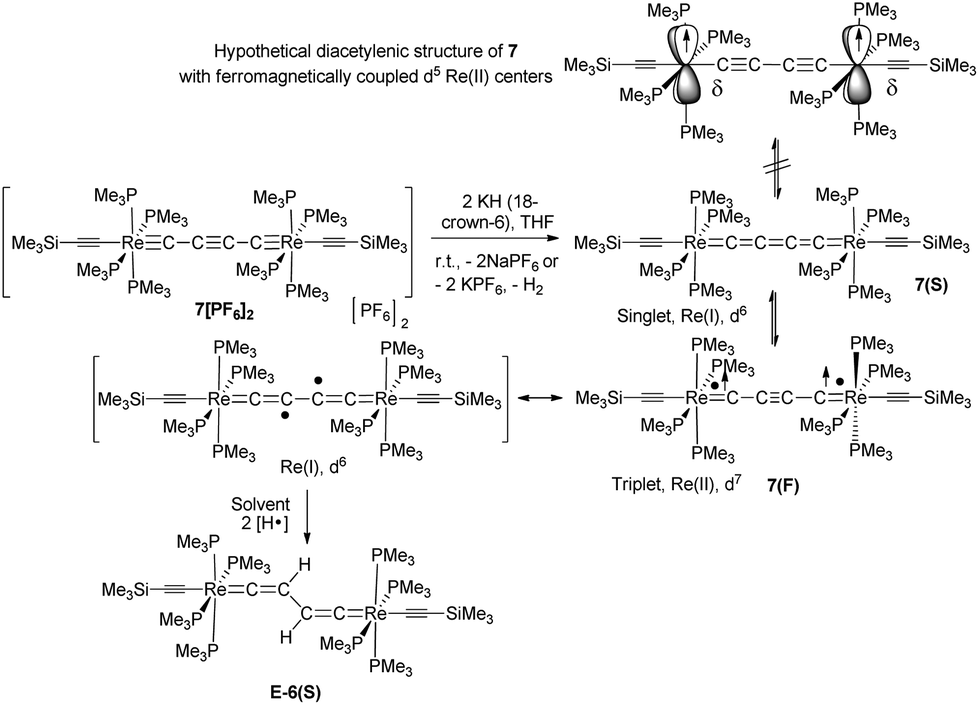 | ||
| Scheme 8 Synthesis, possible electronic structures, and hydrogen abstraction of the isomers of 7 (7(F) and 7(S)). | ||
7 is indeed quite unstable and very reactive reflecting either the high nucleophilicity and basicity of 7(S) or the open-shell diradical character of 7(F). Various attempts to isolate 7 failed. At room temperature in solution the main decomposition product was found to be 6(S), which is likely formed through H-abstraction from the solvent by any form of 7 (Scheme 7). Formally this reaction could be envisaged to occur preferably with 7(F) in a resonance structure with radical centers at Cβ and C′β. H-abstraction of 7 to form E-6(S) could further be substantiated by spectro-electrochemical studies (vide infra) identifying E-6(S) as a subsequent product of 7 by IR and UV-Vis spectroscopy (section IVc). Nuclei in close vicinity to radical centers are excluded from NMR identification due to paramagnetic broadening and shifting of the signals. Therefore, in 7(F) only NMR signals of atoms distant to the radical centers may be observable as broadened or somewhat broadened resonances. In solutions of 7 slightly broadened 1H NMR resonances could be detected at 1.10 and 0.53 ppm, which were assigned to the protons of the MeP and MeSi groups, respectively, but the 29Si spectrum showed only a very broad signal and 13C NMR and 31P NMR signals could not be observed at all. The 1H NMR signal broadening of the methyl resonances of 7 was, however, rather low. From these observations we conclude that, in solution, 7(S) is the prevalent species, coexisting with 7(F), rapidly equilibrating at room temperature on the NMR time scale. Additional evidence for the prevalence of 7(S) as the main solution component of 7 was derived from the spectro-electrochemical studies (vide infra) in THF. For instance, the appearance and position of a cumulenic type ν(C4) IR band at 1758 cm−1 speaks for the existence of 7(S) in solutions of 7. We note here that for the related complex [(η5-MeC5H4)(dmpe)MnCCMn(dmpe)(η5-MeC5H4)] diamagnetic and paramagnetic states have also been postulated to equilibrate in solution.23c
It should be mentioned at this point that in particular the spectro-electrochemical studies (section IVc) provided clear evidence that the mono-oxidized species 7[PF6] is existent in solution.
IId. Structural features of the [P4Re]C4H2[ReP4] and [P4Re]C4[ReP4] units of 5, E-6(S), E-6[PF6], E-6[PF6]2 and 7[PF6]2
The dinuclear rhenium compounds 5, E-6(S), E-6[PF6], E-6[PF6]2 and 7[PF6]2 could be structurally characterized. The structures of 5, E-6[PF6] and E-6[PF6]2 are described in the ESI.† ORTEP like drawings of E-6(S) and 7[PF6]2 are displayed in Fig. 1 and selected bond lengths and angles of 5, E-6(S), E-6[PF6], E-6[PF6]2 and 7[PF6]2 are summarized in Table 1.The Re1–C1 bond of trans-[(Me3SiCC)(PMe3)4ReC–CH2–CH2–CRe(PMe3)4(CCSiMe3)][PF6]2 (5) (see the ESI†) showed a ReC distance of 1.7834(19) Å, slightly longer than the expected range (1.75–1.72 Å) of the sum of covalent radii of the ReC(sp) unit. The C1–C2 and C2–C2′ bond lengths of 1.491(3) Å and 1.513(4) Å fall into the range of C(sp)–C(sp3) and C(sp3)–C(sp3) single bonds.
The structures of E-6(S), E-6[PF6], and E-6[PF6]2 all revealed E-configurations of the bridging units. The C4H2 bridges of E-6(S), E-6[PF6], and E-6[PF6]2 show features of delocalized systems, for instance the Re1–C1 bond distance of E-6 (1.904(2) Å) is significantly shorter than the ReC distance of 2.046(8) Å of the vinylidene complex trans-[ReCl(CCHPh)(dppe)2].34 Upon stepwise oxidation to E-6[PF6] and E-6[PF6]2 the Re–C1 bonds shorten to 1.851(6) Å and to 1.805(5) Å, respectively, indicating gradual adoption of a triple bond character. The C1–C2 bond length 1.337(3) Å of E-6(S) lies within the CC double bond range 1.33–1.38 Å, while the same bonds of E-6[PF6] and E-6[PF6]2 with 1.414(7) Å and 1.375(8) Å, respectively, elongate and move closer in distance toward C(sp)–C(sp2) single bonds averaging at 1.43 Å.35 The C2–C2′ distance of 1.469(4) Å of E-6(S) is in agreement with a single bond between two sp2 hybridized carbon atoms,35 while the same bonds in E-6[PF6] and E-6[PF6]2 are 1.388(10) Å and 1.331(10) Å, respectively, within the range of CC double bonds 1.33–1.38 Å. In conclusion, according to Table 1, upon oxidation of the neutral complex E-6(S) to the dicationic complex E-6[PF6]2, the Re1–C1 distances gradually shorten coinciding with a change in the bond order from ReC to ReC. The C1–C2 distances gradually lengthen from a CC double to a C–C single bond, while the C2–C2′ distances shorten from a C–C single bond to a CC double bond. The Re⋯Re distances gradually decrease from the neutral complex E-6(S) to the corresponding dicationic complex E-6[PF6]2. All these observations clearly indicate that the oxidation of E-6(S) causes the sp2 C4H2 linkage to transform from a bisvinylidene to an ethylenylidene biscarbyne structure (Scheme 1). These data coincide with the results of the NMR experiments that upon oxidation of E-6(S) to E-6[PF6]2, the resonances of Cα showed an up-field shift, while those of Cβ and H moved downfield.
The Re1–C1 bond distance of 7[PF6]2 of 1.814(4) Å is close to the corresponding bond length of E-6[PF6]2 of 1.805(4) Å. Taken together with the short C2–C2′ bond of 1.197(8) Å, this provides clear evidence for an alkynediyl biscarbyne canonical form of the bridge in 7[PF6]2.
The phosphine ligands of the ReP4 subunits of all complexes 5, E-6(S), E-6[PF6], E-6[PF6]2, and 7[PF6]2 may not just play the role of ancillary ligands being ‘innocent’ bystanders, but, in most cases, seem to actively contribute to stabilizing the respective valence structure of the C4Hy bridge (see section III). We first note that the ReP4 ‘equatorial’ arrangements as subunits of pseudo-octahedral complexes are not planar, but are significantly distorted towards C2v symmetry with one trans P–Re–P angle significantly smaller than 180° (about 156°) and bent back toward the acetylide ligands and with the other trans P–Re–P angle being between 166° and 170° and bent towards the C4H2 or C4 bridge as schematically sketched in Scheme 9.
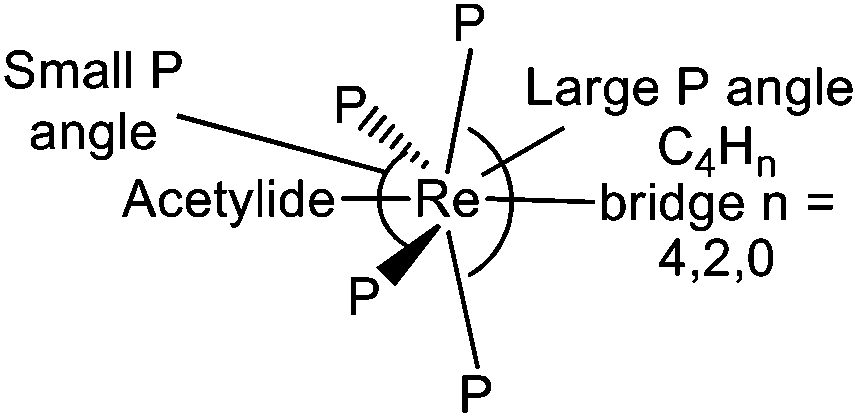 | ||
| Scheme 9 Sketch of trans phosphine angle distortions of the ReP4 ‘equatorial’ plane toward a C2v local symmetry for complexes 5, E-6(S), E-6[PF6], E-6[PF6]2, and 7[PF6]2. | ||
As was already discussed, the structural and compositional changes of the bridges of 5, E-6(S), E-6[PF6], E-6[PF6]2, and 7[PF6]2 are also reflected in the 13C{1H} NMR spectra. This lets us conclude that the carbon framework in the alkyne biscarbyne complex 7[PF6]2 is more electron-rich than that in the ethylenylidene biscarbyne complex 6[PF6]2. The C1–C2 bond length in 7[PF6]2 of 1.363(5) Å is slightly shorter than a typical C(sp)–C(sp) single bond (1.377 Å), but close to the corresponding C1–C2 bond distance in the tungsten complex [I(dppe)2WC–CC–CW(dppe)2I] with a bond length of 1.34(1) Å.13c The C2–C2′ distance of 1.197(8) Å is within the CC triple bond range (1.18–1.20 Å). Therefore the structure of 7[PF6]2 consists of two symmetrically arranged [trans-X(PMe3)4Re] fragments linked by a C4 system resembling a canonical alkynediyl biscarbynic structure. Owing to the practically linear structure of the Re–C4Hn–Re entity, the Re⋯Re distance in 7[PF6]2 of 7.536 Å is longer than the corresponding distance in E-6[PF6]2. The alkynediyl biscarbynic structure of the C4 chain in 7[PF6]2 differs from the all-cumulenic butatrienediylidene one in the dicationic dinuclear rhenium complex [(Cp*)(NO)(PPh3)ReC4Re(PPh3)(NO)(Cp*)][PF6]2 reported by Gladysz and coworkers. The results clearly reflect the influence of the different electron counts and redox properties of the terminal metal entities and of the ancillary ligand frameworks eliciting different valence structures of the [Re]–C4–[Re] linkages.
III. DFT calculations on the electronic states of E-6 and 7 revealing redox non-innocent structures
A closer insight into the electronic structures of the complexes E-6 and 7 was expected from a thorough DFT analysis based on an appropriate methodological approach to enable the description and proper evaluation of open and closed shell electronic configurations (for details, see the ESI†).IIIa. Qualitative orbital representation of the pseudo-octahedral [(acetylide)(PMe3)4Re(bridge)] complex unit
The ligand field picture of the pseudo-octahedral [(acetylide)(PMe3)4Re(bridge)] complex is expected to reveal three non-bonding d-orbitals at the rhenium center. According to their parent octahedral coordination geometries (Oh symmetry) these occupied orbitals are denoted as “t2g”. Due to the observed apparently sterically induced lowering of the symmetry of the ReP4 units to C2v, splitting of the “t2g” orbitals into two π-type d/p hybridized donor orbitals and one δ-type orbital occurs (Scheme 10).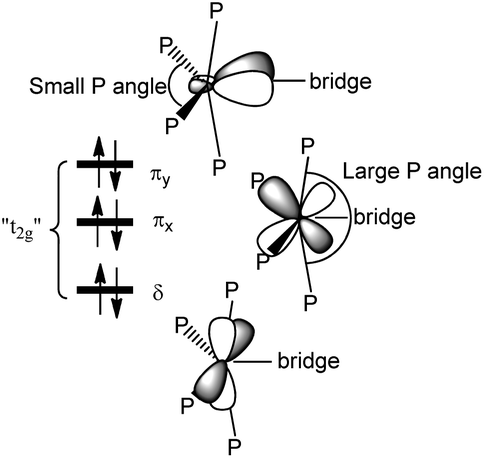 | ||
| Scheme 10 Qualitative representation of the filled non-bonding d-orbitals (“t2g”) of an [(acetylide)(PMe3)4Re(bridge)] unit with two π type orbitals (πx and πy) and one δ type orbital. | ||
In the small P angle plane (πy) the extent of the d/p hybridization is larger and the larger side of the hybrid lobes is directed toward the bridge. The extent of d/p hybridization in the large P angle plane (πx) is smaller and the larger side of the hybrid lobes is directed away from the bridge. The C2v distortion thus makes the π plane of the smaller P angle plane (πy) more electron donating to the side of the bridge and the π plane of the larger P angle plane (πx) less electron donating toward the bridge. The π interactions are thus anisotropic to the bridge side with the consequence that the strongest π orbital interaction results in π anisotropic orbitals of a bridge possessing for instance a πy acceptor orbital and a πx donor orbital.33 This mechanism of an optimum electronic fit seems to be in operation for the complexes E-6(S), E-6[PF6], E-6[PF6]2 and 7(S) supporting the binding in certain ReC4H2Re and ReC4Re moieties. For the carbyne complex 5 and for 7[PF6]2 involving a conical ReC carbyne interaction the C2v distortion of the ReP4 fragment is anticipated to be primarily of steric origin without additional support from the rhenium–bridge interaction.
IIIb. DFT calculations on trans-[(Me3SiC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) C)(PMe3)4ReC4H2Re(PMe3)4(CCSiMe3)] (E-6(S)) and trans-[(Me3SiCC)(PMe3)4ReC4Re(PMe3)4(CCSiMe3)] (7)
C)(PMe3)4ReC4H2Re(PMe3)4(CCSiMe3)] (E-6(S)) and trans-[(Me3SiCC)(PMe3)4ReC4Re(PMe3)4(CCSiMe3)] (7)
We carried out structure optimizations for trans-[(Me3SiCC)(PMe3)4ReC4H2Re(PMe3)4(CCSiMe3)] (E-6(S) and trans-[(Me3SiCC)(PMe3)4ReC4Re(PMe3)4(CCSiMe3)] (7) by means of Kohn–Sham DFT (for details, see Computational methodology). If not mentioned otherwise, the BHLYP hybrid functional with 50% Hartree–Fock exchange was employed.36 As shown for organic mixed-valence compounds,37 it is important to include a proper amount of the exact exchange, since standard non-hybrid and hybrid exchange–correlation functionals give a too delocalized description.38
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) C)(PMe3)4ReC4H2Re(PMe3)4(CCSiMe3)] (E-6).
The possible isomers of the neutral molecule E-6 are the diamagnetic geometric isomers E-6(S) and Z-6(S) and the open-shell, diradical alternatives E-6(F) and E-6(A) (Scheme 1) possessing SOMOs with either anti- or ferromagnetically coupled electrons. Isomers Z-6(F) and Z-6(A) were not considered due to the expected very high electronic energies for these molecules and the fact that none of them was experimentally observed.
C)(PMe3)4ReC4H2Re(PMe3)4(CCSiMe3)] (E-6).
The possible isomers of the neutral molecule E-6 are the diamagnetic geometric isomers E-6(S) and Z-6(S) and the open-shell, diradical alternatives E-6(F) and E-6(A) (Scheme 1) possessing SOMOs with either anti- or ferromagnetically coupled electrons. Isomers Z-6(F) and Z-6(A) were not considered due to the expected very high electronic energies for these molecules and the fact that none of them was experimentally observed.
E-6(S) was calculated to have the lowest electronic energy constituting the ground state of E-6 (Scheme 10). The calculations for the singlet diradical structure E-6(A) did not converge (the same was found, if the PBE039 or B3LYP density functionals36a,b,40 were used) indicating that this state is perhaps also not of practical relevance. The E-6(F) state could be optimized with different density functionals (BHLYP/B3LYP/BP86)36a,41 leading to slightly different electronic energies, but all of these energies were at levels much higher (34.1/35.3/28.4 kcal mol−1) than that of E-6(S) (Scheme 11). Given the large energy difference E-6(F) can be considered an electronic excited state of E-6(S).
 | ||
| Scheme 11 Scheme of calculated electronic energies for two isomers of E-6 and 7 and their isomerization and dehydrogenation reactions. Energies in kcal mol−1 obtained from DFT geometry optimizations applying different exchange–correlation density functionals indicated underneath the energy values. | ||
The potential energy surface of E-6(S) was theoretically explored further by a thermodynamic evaluation calculating first the geometric Z isomer Z-6(S), which could not be detected in solution. By structural optimizations Z-6(S) turned out to be 17.8 kcal mol−1 higher energy than E-6(S). This makes its existence in solution at room temperature very unlikely. One can safely assume that, if formed, it will spontaneously convert to E-6(S) with a very low barrier. Another possible deactivation pathway is dehydrogenation to 7. For the thermally stable E-6(S) the dehydrogenation process is uphill in energy (Scheme 12).
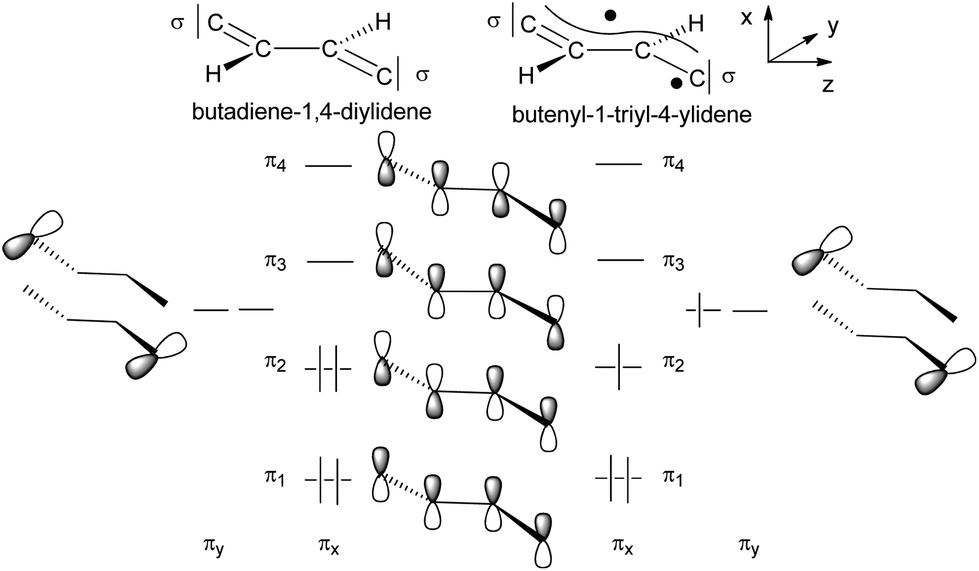 | ||
| Scheme 12 Schematic MO picture of two electronic states of the C4H2 bridge. | ||
In the following section the electronic structures of E-6(S) and E-6(F) are discussed, which will be accomplished by a fragmental view of the molecules dividing the molecules up into the [(acetylide)(PMe3)4Re] fragment with the π type large and small P angle plane dRe orbitals combined with π orbitals of the different bridge states.
The qualitative MO picture of the C4H2 bridge (Scheme 12) shows two perpendicular π systems denoted as πx and πy, which have different electron occupancies for the closed shell or open shell electronic configurations of butadiene-1,4-diylidene and butenyl-1-triyl-4-ylidene. Scheme 11 will be used to identify the bridge contributions of prominent MOs of E-6(S) and E-6(F) as they are obtained from the DFT calculations.
The butadiene-1,4-diylidene bridge of E-6(S) possesses two main components for π-type orbital interactions with the rhenium fragments. First there is a net weak π donor interaction in the πx plane originating from the interaction with the filled π2 orbital, which is complemented by the π acceptor interaction with the empty π3 orbital. Second in the πy plane the bridge behaves as a strong π acceptor due to the interactions with the empty carbene p orbitals and the dπ orbitals at each rhenium center. The main destabilizing component of the π donor interaction is found in the HOMO of E-6(S) and can be viewed as the antibonding component dRe(πx)−π2, where π2 is out-of-phase with the dRe(πx) orbitals (Fig. 2, bottom left). The antibonding dRe(πx)−π2 orbital has as a bonding counterpart dRe(πx)+π2 lying at a quite low energy. Of further significance for the overall orbital description of E-6(S) is the HOMO−1 (Fig. 2, bottom right), which is composed of the bonding combination of π3 with the in-phase-combination of dRe(πx) orbitals in the small P angle plane dRe(πx)+π3. The HOMO−1 thus has the effect of a π acceptor interaction counteracting and attenuating partly the effect of π electron donation. The overlay of the electron densities of these π interactions with the mentioned main contributions of the [Re]CC(H)C(H)C[Re] fragment describes well the bridge's C1–C2 and C2–C2′ full and partial double bond characters. The minor influence of HOMO−1 can be recognized by (a slight) elongation of the C1–C2 and C1′–C2′ bonds of E-6(S) and a greater influence on the shortening of the C2–C2′ toward a double bond (cf. Table 1). Selected calculated structural parameters of E-6(S) are compiled in Table 1 in comparison with the data obtained from the X-ray diffraction study demonstrating very good agreement in distances and angles.
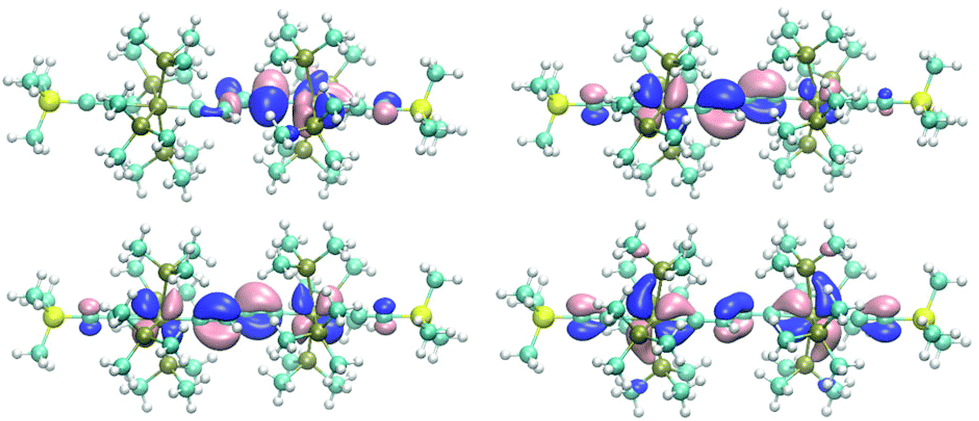 | ||
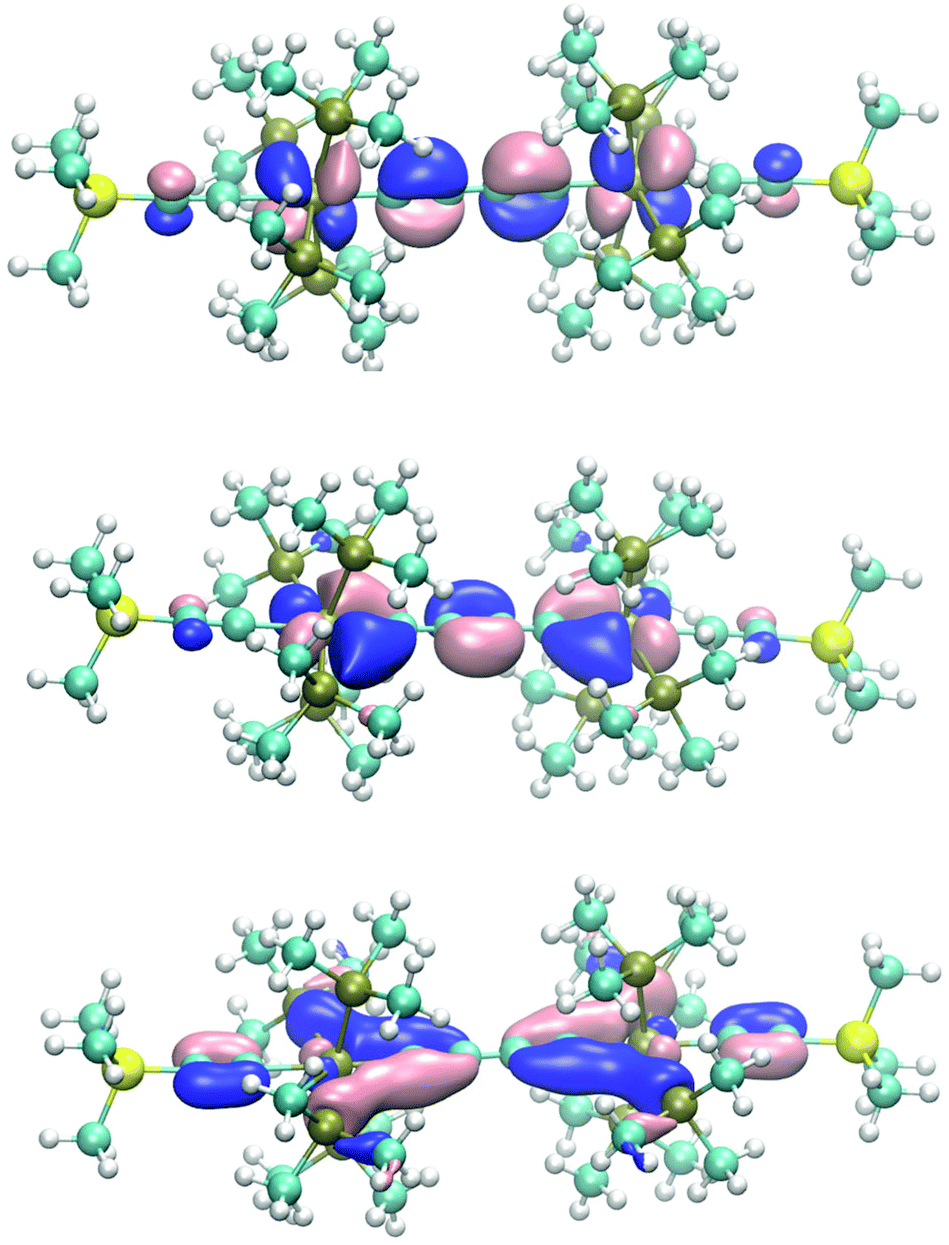
| Fig. 2 Top: views of the two SOMOs of E-6(F) demonstrating for both orbitals relative strong localization on the carbenic (rhenium–carbon)bridge bond (dRe−πy) (left, HOMO) and the (±dRe(πx))−π2 orbital (right, HOMO−1). Bottom: orbital plots of the HOMO (left) and HOMO−1 (right) of E-6(S). | ||
C–CHCH–C[Re]).
C)(PMe3)4ReC4Re(PMe3)4(CCSiMe3)] (7).
In contrast to complex E-6, where the diradical structure E-6(F) is energetically too high to be considered as a ground state or close to it, DFT calculations on 7 suggest that the diradical structure of 7(F) is slightly lower in energy by 5.3/0.9 kcal mol−1 than the singlet state 7(S) using the BHYLP/B3LYP density functional (Scheme 11). Given the relatively high confidence intervals for the electronic energies of the hybrid type DFT calculations, we have to consider both configurations energetically close so that they may co-exist at room temperature in solution. It is also possible that 7(S) is even of a lower energy than 7(F). The calculated main structural parameters of 7(S) and 7(F) are compiled in Table 1 and according to these both have very similar structures. The slightly longer Re–C bond lengths of 7(F) may reflect a somewhat higher degree of the antibonding character of the metal–carbon bonds. Despite these small structural differences, these molecules may constitute examples of bond length isomerism.21e,42
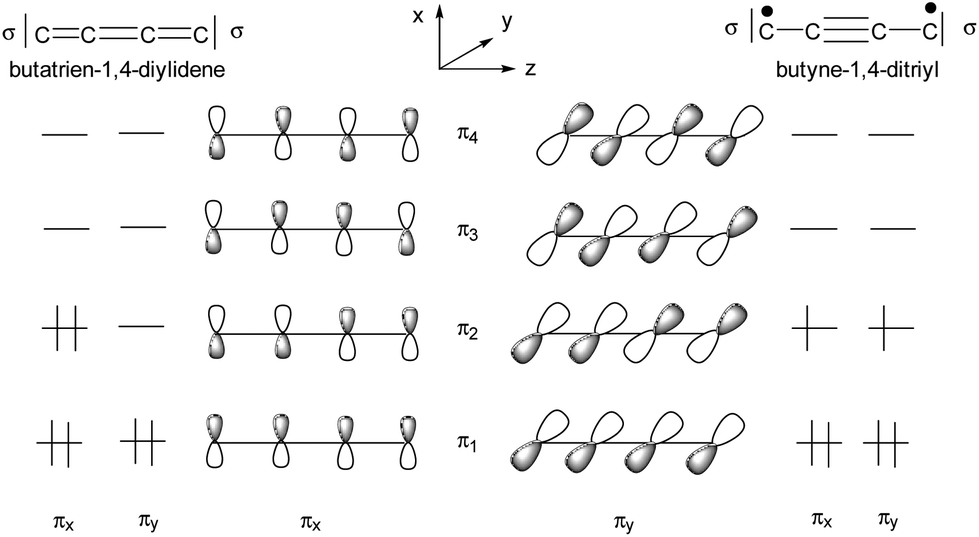 | ||
| Scheme 13 Schematic MO picture of two electronic states of the C4 bridge. | ||
Given these basic orbital descriptions of the bridge we continue to first analyze the compositions of the 7(F) orbitals by attachment of (acetylide)Re(PMe3)4 fragments on both sides of the butyne-1,4-di(triyl) bridge. The two π2 SOMO orbitals interact with the filled πx and πy orbitals of the rhenium centers and get transformed into SOMOs of 7(F) forming two half-filled π donor interactions antibonding with respect to the rhenium–carbon interactions of the dRe(πx,πy)−π2 orbital character. Fig. 3 (top) shows the orbital drawing of one of the – on the DFT basis – nearly degenerate SOMOs. The bonding orbital counterparts of the SOMOs are the dRe(πx,πy)+π2 orbitals, occupied by two electrons, which are located at quite low electronic energies. These two 3-electron π donor interactions amount to a total binding balance of the rhenium–carbon π bond order of ½ each. In each π plane (πx and πy) there is however an additional π acceptor interaction involving π3 of the πx and πy (dRe(πx,πy)+π3), which could in the best case reach a maximum bond order of 1 at each rhenium center. Counting in addition the two σ electron pairs of the bridge contributing a bond order of 1 to the (Re–carbon)bridge bond a total bond order of 2½ would result. Since the SOMOs of 7(F) bear the bonding character of π2 of the bridge, these lie at lower electronic energies than the SOMOs of E-6(F), which have more antibonding character. The relatively low energy and the extensive delocalization of the SOMOs of 7(F) is also the cause for the competitive total electronic energies of isomeric 7(F) and 7(S) (vide infra).
 | ||
| Fig. 3 Top: orbital representation of one of the perpendicular SOMO π orbitals of 7(F). Bottom: plot of the calculated spin density of 7(F) (B3LYP/def2-TZVP/COSMO; grey: isosurface at 0.002 e Bohr−3, blue; isosurface at −0.002 e Bohr−3). | ||
In line with the overlaid shapes of the SOMOs of 7(F), the spin density (spin-down (β) electronic densities) in Fig. 3 is conical along the main axis of the molecule and to a large extent positive on the rhenium centers, the carbon atoms of the bridge, and the acetylenic β atoms. A significant excess of the β spin density is also obtained on the acetylenic α atoms presumably via a spin polarization mechanism.43
As shown in Scheme 12 the butatriene-1,4-di(ylidene) electron occupancy of six π electrons of a C4 bridge would be unequal with respect to the πx and πy planes, which in the case of 7(S), causes anisotropy in the π interactions with the rhenium centers. Since the πx and πy planes of the rhenium centers are different in p,d hybridization and energy, they are anisotropic in character (Schemes 8 and 9). This leads to cooperativity in the interactions with the likewise anisotropic π orbitals of the bridge (the stronger π donor plane of the rhenium center interacts with the stronger π acceptor plane of the bridge and vice versa). The HOMO (πx plane), HOMO−1 (πy plane) and the second π acceptor interaction (πy plane) of 7(S) (Fig. 4, bottom) indeed reflect the highly anisotropic character of the ReC4Re π interactions consisting thus mainly of one π donor and two π acceptor interactions with the rhenium center.
 | ||
| Fig. 4 Three orbital plots selected from the orbitals of 7(S) in qualitative order of their energies. Top: HOMO, middle: first π acceptor interaction of 7(S) (HOMO−1, πy plane) bottom: plot of the second πy acceptor orbital at low electronic energy. | ||
The HOMO represents an antibonding interaction between the dRe(πx) orbital of the large P angle plane (Scheme 9) with π2 of the bridge. Due to the small bending toward the bridge orbital overlap is reduced on the bridge side, thus decreasing the repulsive character of the 4-electron interaction of dRe(πx) with π2 of the bridge (HOMO, Fig. 4, top).33 The πy MOs of the bridge – coplanar with the stronger πy donor orbitals of the rhenium fragment – are prone to very strong π acceptor interactions engaging even two dRe(πy)+(π2,π3) π-type interactions. The dRe(πy) orbitals at each rhenium center of 7(S) form an in-phase and an out-of-phase orbital combination, which at one time each interacts in a bonding fashion with π3 (HOMO−1, middle of Fig. 4) or with π2 of the bridge to establish the second π acceptor orbital lying at low electronic energies (Fig. 4, bottom). In a localized bonding picture these two π bonding orbitals would account for one π bond at each rhenium center and together with the σ bonds the ReC double bond character results.
IV. Electrochemical and spectroscopic studies of E-6(S) and 7
IVa. Cyclic voltammetry (CV) studies of the dinuclear rhenium complexes E-6(S), E-6[PF6]2 and 7[PF6]2
Voltammograms of E-6[PF6]2 (Fig. 2a) recorded in THF displayed two reversible waves at E1/2 = −1.292 V and E1/2 = −1.749 V. The electrochemical and structural data in concert support a stepwise reduction of E-6[PF6]2. This is accompanied by an electronic re-adjustment of the C4H2 linkage from an ethylene biscarbyne to a bis(vinylidene) structure revealing that the redox processes are to a great extent bridge-centered as is also mandated by the HOMO of E-6[PF6]2 as shown in Fig. 2. The potential difference ΔE1/2 between the two redox waves of 0.457 V results in a Kc value of 7.1 × 107. Cyclic voltammetry measurements of E-6(S) gave identical results apart from the fact that the forward peaks were now anodic instead of cathodic. In comparison with the manganese analog [(MeC5H4)(dmpe)MnC–CHCH–CMn(dmpe)(MeC5H4)][PF6]2, the values of the two redox couples of E-6[PF6]2 are more negative and the Kc value is smaller (Table 2). In accordance with the CV study, neutral E-6 is very easy to oxidize (Fig. 5).
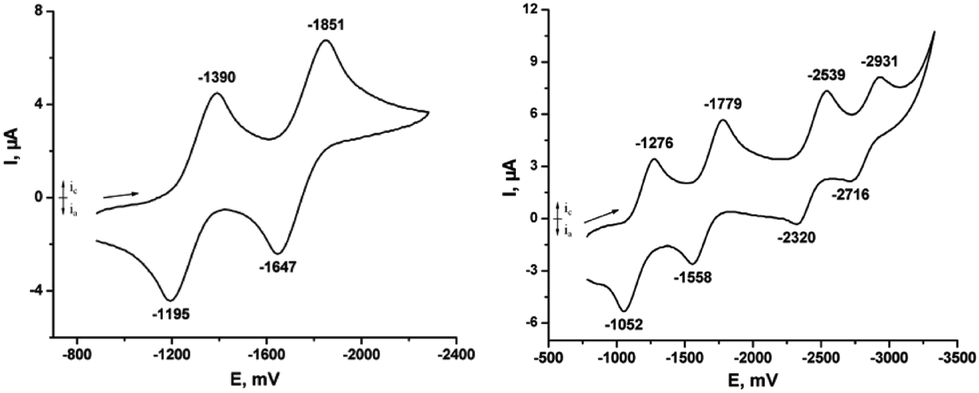 | ||
| Fig. 5 Cyclic voltammogram of E-6[PF6]2 in 0.1 M THF solution of [n-Bu4N][PF6], Au electrode; E vs. Fc0/+; scan rate = 100 mV s−1; 20 °C. CV of 7[PF6]2 in 0.1 M THF solution of [nBu4N][PF6], Au electrode; E vs. Fc0/+; scan rate = 100 mV s−1; 20 °C. | ||
| Complexes | Couple 1 E1/2 (V) | Couple 2 E1/2 (V) | ΔE (V) | K c | Ref. |
|---|---|---|---|---|---|
| [(Me3SiCC)(PMe3)4ReC–CHCH–CRe(PMe3)4(Me3SiCC)][PF6]2 (E-6[PF6]2) |
−1.292 | −1.749 | 0.457 | 7.1 × 107 | Present work |
| [(MeC5H4)(depe)MnC–CHCH–CMn(depe)(MeC5H4)][PF6]2 |
−0.820 | −1.386 | 0.576 | 6.6 × 109 | 23d |
| [(MeC5H4)(dmpe)Mn–CC–Mn(dmpe)(MeC5H4)][PF6]2 |
−0.847 | −1.835 | 0.988 | 8.6 × 1016 | 23d |
| [(Me3SiCC)(PMe3)4ReC–CC–CRe(PMe3)4(Me3SiCC)]2[PF6]2 (7[PF6]2) |
−1.164 | −1.668 | 0.504 | 4.7 × 108 | Present work |
| [(Cp*)(NO)(PPh3)Re–CC–CC–Re(PPh3)(NO)(Cp*)] |
0.06 | 0.59 | 0.53 | 1.1 × 109 | 10a |
The CV of trans-[(Me3SiCC)(PMe3)4ReC4Re(PMe3)4(CCSiMe3)][PF6]2 (7[PF6]2) in THF showed four separate waves with high degrees of chemical reversibility corresponding to four consecutive reductions from [7]2+ → 7+ → [7] → [7]− → [7]2−. The potential difference ΔE1/2 between the first redox waves of 7[PF6]2 is 0.504 V resulting in a Kc value of 4.7 × 108 with 7[PF6] as an in principle isolable intermediate redox state as also revealed in the spectro-electrochemical studies. However, attempts to prepare 7[PF6] were unsuccessful. The third and fourth redox processes have very negative potentials, which make the corresponding reduced species [7]− and [7]2− difficult to prepare by chemical methods. For [7]− and [7]2− we have to assume bridge-centered reduction steps. The ΔE1/2 values of the complexes 6[PF6]2 (0.475 V) and 7[PF6]2 (0.504 V) are very close. The only difference in these complexes are the bridges, therefore one might conclude that the strengths of electronic communication through the C4H2 and the C4 bridges are very similar.
IVb. NIR evidence for through-bridge electronic interaction in E-6(S), E-6[PF6] and E-6[PF6]2
To further assess the extent of the electronic interaction between the two redox-active rhenium centers of E-6(S), E-6[PF6] and E-6[PF6]2, the UV-vis spectra of E-6(S), E-6[PF6] and E-6[PF6]2 were examined (Fig. 6). Due to limited solubility and stability in other solvents, the neutral complexes could be studied in THF only and the examination of a solvent effect of the MV complexes could not be performed. The UV-vis spectral data and IVCT absorption data for the MV complexes are listed in Tables 2 and S3.†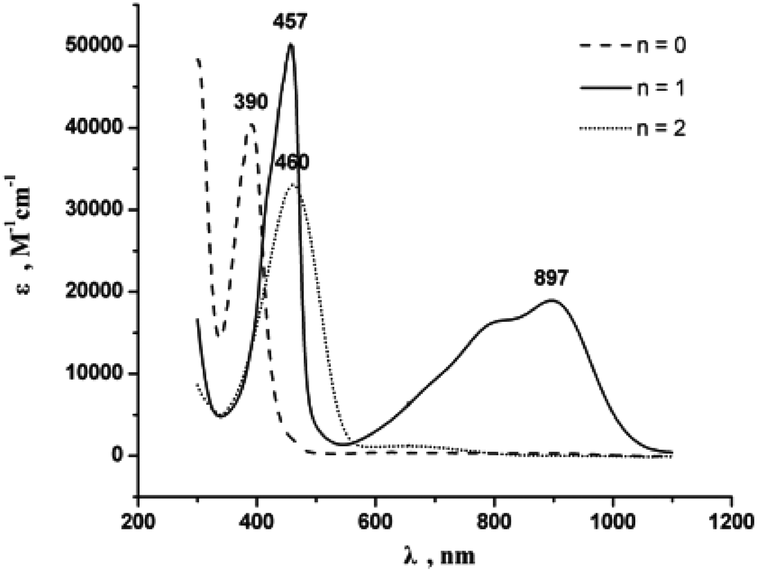 | ||
| Fig. 6 UV-vis spectra of [E-6]n+ (n = 0 in THF; n = 1 and n = 2 in CH3CN, ambient temperature, 5 × 10−5 M). | ||
The UV-vis spectra of all complexes E-6(S), E-6[PF6], and E-6[PF6]2 show intense absorption bands in the visible and ultraviolet region, which can be attributed to a metal-to-ligand charge transfer (MLCT) transition (Fig. 6 and Table S3 in the ESI†). In the visible and near-infrared region an additional absorption band is observed at approximately 897 nm (ε is about 1.90 × 104 M−1 cm−1) with a notable shoulder at ∼800 nm for the MV complex E-6[PF6]. There are no counterparts in the spectra of the corresponding neutral complex E-6(S) and the dicationic complex E-6[PF6]2, so that they may be identified as intervalence charge transfer (IVCT) bands. Observations of multiple IVCT bands have been reported previously for the MV complexes {[(MeC5H4)(dmpe)Mn]2(C–CPhCPh–C)}+,24b {[(η5-C5Me5)(NO)(PPh3)Re]2(μ-CC–CC)}+,10a {[(C5Me5)(dppm)Ru]2(μ-CC–CC)}+30 and {[(C5Me5)(dppm)Fe]2(μ-CC–X–CC)}+ (X = 2,5-C4H2S, –C4–).44 One explanation considered is spin–orbit coupling, which is of particular relevance in third-row transition metals.45 An alternative assignment is that one absorption is a LMCT transition, and the other a MLCT transition.10a,24b,30,44,46 Considering the strongly metal–ligand delocalized nature of the frontier MOs, the underlying transitions may be best viewed as π → π* type transitions within a conjugated open-shell metal–organic π-system with only a limited amount of charge transfer between the individual constituents.30,47 On the premise that the observed absorptions can be viewed as IVCT transitions, a Gaussian analysis of the IVCT absorption band of the MV complex was performed in order to calculate the electronic coupling energy Hab.44,46 The spectrum of the mixed valence complex E-6[PF6] can be deconvoluted into three Gaussian bands A, B, and C (ESI†), where band B is the major component, and with a tail of the MLCT band observed in the visible and ultraviolet regions. Spectral data extracted from the IVCT band shape analyses are summarized in Table S3 (ESI).† The observed bandwidths of the three bands at half height (Δ1/2) are narrower than those predicted from the equation Δν1/2(cm−1) = (2310·νmax)1/2,7a,48 which is in agreement with a class III character for the MV complex. In addition, the delocalization parameters Γ as calculated from the equation Γ = 1 − Δν1/2/(2310νmax)1/2,7a,49 are larger than (A, B) or close to (C) 0.5 for all bands. Based on the CV and NIR spectroscopic data, the MV complex E-6[PF6] can be described as a class III MV compound with the odd electron fully delocalized over the Re2C4 core. Based on the values of Table 3 electronic coupling energies HabIII for band A, band B, and band C of E-6[PF6] are 5.43 × 103 cm−1, 6.10 × 103 cm−1, and 7.24 × 103 cm−1, respectively.
| Complex | λ max (nm) | ν max (cm−1) | ε max (M−1 cm−1) |
|---|---|---|---|
| E-6(S) | 390 | 2.56 × 104 | 4.04 × 104 |
| E-6[PF6] | 457 | 2.19 × 104 | 5.02 × 104 |
| 897 | 1.14 × 104 | 1.90 × 104 | |
| E-6[PF6]2 | 460 | 2.17 × 104 | 3.30 × 104 |
C–)Pd(PEt3)2Cl]+ and [{Cp*Re(NO)(PPh3)(μ-CC–)}2-Pd(PEt3)2]+ with g = 2.110–2.121, A185/187Re = 190 G) and [{Cp*Re(NO)(PPh3)}2(μ-CC–CC)]+ (g = 2.018, A185/187Re = 98 G).51 The latter compound was reported to have about equal metal/C4 ligand contributions to the SOMO (i.e. ca. 25% of the total spin density on each Re,atom).52 Even in the case of the heterobimetallic [{Cp*Re(NO)(PPh3)}(μ-CC–CC){Fe(dppe)Cp*}]+, where the rhenium contribution to the overall spin density is reduced to ca. 17%, Re hyperfine interactions of 48 G were readily discerned.11b This all argues for an even lower metal/higher bridge contribution to the SOMO of E-6[PF6]. On the other hand, the non-observability of Re hyperfine interactions does not allow us to draw any conclusion on the extent of spin delocalization within the [Re–C4H2–Re]+ array.
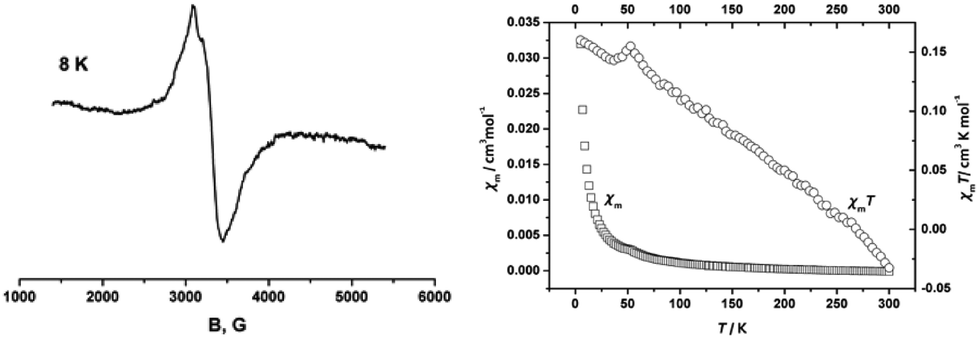 | ||
| Fig. 7 (a) EPR spectrum of E-6[PF6] at 8 K in CH3CN glass; (b) plots of χm and χmT vs. T for E-6[PF6]. | ||
Variable-temperature magnetic susceptibility measurements were carried out in the temperature range from 5 K to 300 K. The magnetic properties are presented in the form of χm and χmT vs. T plots in Fig. 6b. The MV complex E-6[PF6] showed typical paramagnetic behavior with the magnetic susceptibility χm dropping abruptly from 5 K to 50 K then gradually and the χmT value decreasing from 5 K to 300 K. This is a manifestation of the paramagnetism expected from the odd electron in this molecule.23c
IVc. Spectro-electrochemical studies on 7[PF6]2
The high chemical reactivity of the reduced forms of complex 7[PF6]2 prevented their isolation as pure compounds. In order to characterize them by IR- and UV-Vis-NIR-spectroscopy we resorted to their generation in situ by means of spectro-electrochemistry. Inside an OTTLE (optically transparent thin layer electrolysis) cell three consecutive reductions of 7[PF6]2 could be followed. Just like in the solid state, 7[PF6]2 only showed a weak ν(CC) stretching vibration at 2018 cm−1 in the THF/NBu4PF6 electrolyte. Upon reduction to 7+ species the original ν(CC) band was replaced by a much stronger absorption at 1992 cm−1 and a much weaker one at 1612 cm−1. During the second reduction to the neutral complex 7 the former ν(CC) band gave way to an even stronger absorption at 1957 cm−1, while the band at 1612 cm−1 was replaced by a stronger one at 1738 cm−1 (Fig. 8). Both these processes were fully reversible as ascertained by the presence of clean isosbestic points and full recovery of the original spectrum of 7[PF6]2 upon exhaustive back electrolysis at a potential sufficiently positive of the 7+/[7] and 7+/[7]2+ redox waves. Identical results were obtained using the 1,2-C2H4Cl2/NBu4PF6 electrolyte ([7]2+: ν(CC) = 2016 cm−1; 7+: ν(CC) = 1991 cm−1, ν(C4) = 1615 cm−1; 7: ν(CC) = 1958 cm−1, ν(C4) = 1738 cm−1). Low energies of ν(CC) stretching bands are not without precedence in C4-bridged dimetal complexes and are indicative of the cumulenic character of the all-carbon bridge as in 7(S) (Scheme 6).11a,23b,f,53 Of note is the rather strong shift of the ν(CC) bands assigned to the terminal alkynyl ligands upon stepwise reduction. It is much stronger than in comparable C4-bridged dimanganese complexes (2026 → 2016 → 2010 cm−1).23b,f,24a This points to significant contributions of the terminal alkynyl ligands to the HOMO of [7]2+ and the SOMO of 7+ and hence an enhanced delocalization of the charge onto the terminal alkynyl ligands for the heavier congener.
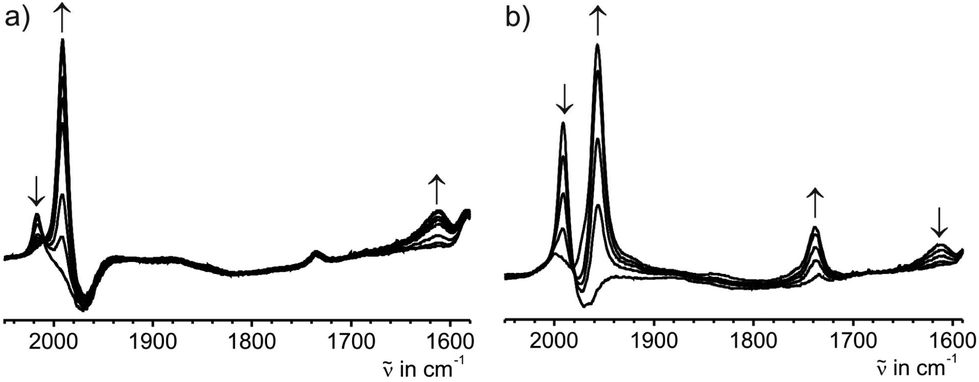 | ||
| Fig. 8 IR spectroscopic changes during (a) the first and (b) the second reduction of complex 7[PF6]2 in THF/NBu4PF6 at r.t. | ||
Attempts to further reduce 7 to [7]− resulted in a sequence of two consecutive electron transfers and subsequent protonation steps to ultimately yield complex E-6. This is clearly seen from the growth of ν(CC) and ν(CC) bands at 1972 and 1545 cm−1 during this process. These bands are virtually identical to those of pristine solid E-6. Furthermore, stepwise re-oxidation of the exhaustively reduced solution first gave a ν(CC) band at 1999 cm−1 and, after the final re-oxidation step, a band at 2022 cm−1 in agreement with the ATR data of complexes E-6[PF]6 and E-6[PF6]2. When solutions of 7[PF6] were on partially electrolyzed at potentials negative of the 7/[7]− wave the spectroscopic features of species 6 and 7 were detected without any detectable intermediate [7]− (Fig. 9a). Stepwise reoxidation of such incompletely reduced solutions first produced mixtures of complexes 7[PF6] and E-6[PF6] and then of E-6[PF6]2 and 7[PF6]2 (Fig. 9b and c).
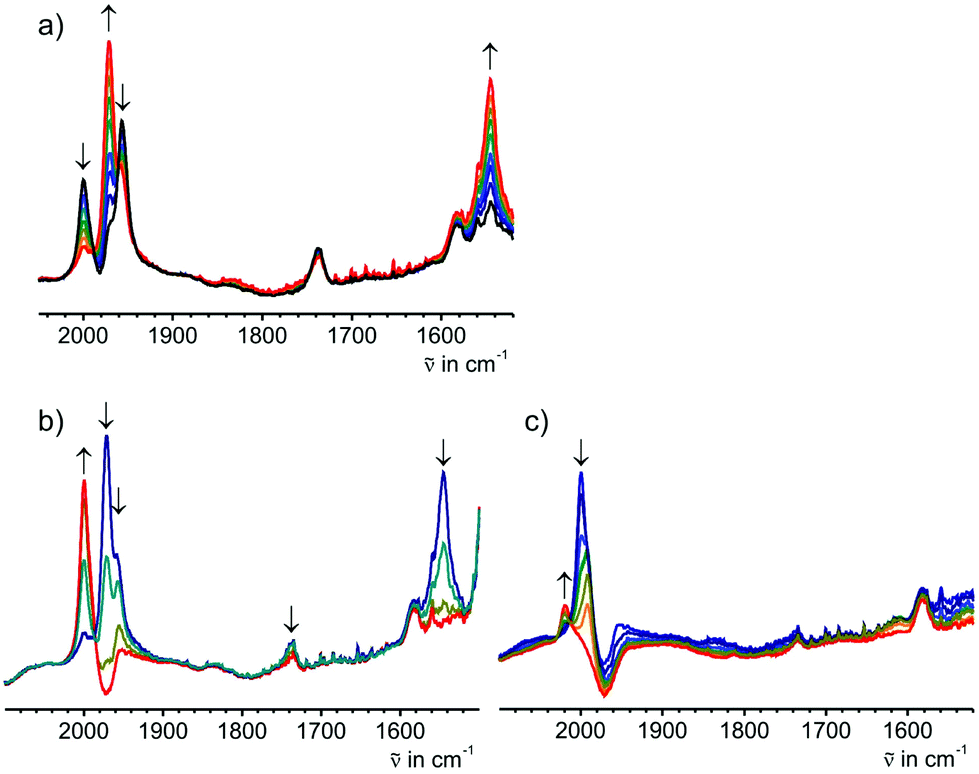 | ||
| Fig. 9 (a) IR spectroscopic changes upon reduction of complex 7[PF6] at negative potentials for the 7/[7]− wave showing the intermediate growth of the CC band of 7 at 1955 cm−1 and the accompanying growth of the ν(CC) and the ν(CC) bands of E-6 at 1972 and 1544 cm−1. (b, c) stepwise re-oxidation of these mixtures at (b) positive potentials of the 7/7+ and E-6/[E-6]+ waves and (c) at positive potentials of the 7+/[7]2+ and [E-6]+/[E-6]2+ waves. | ||
In situ reduction of 7[PF6]2 under UV/Vis/NIR monitoring also occurred in two well-resolved steps to first produce the radical cation 7+ and then neutral 7, again with clean isosbestic points for each individual step (Fig. 10). During the first reduction, the prominent near UV band of [7]2+ peaking at 376 nm sharpened and red-shifted into the visible range to produce an intense peak at 409 nm, while a plateau-like absorption of [7]2+ with peak positions at 510 and 570 nm develops into a considerably more richly structured absorption with individual peaks at 498, 533, 588, and 696 nm. In the NIR a much weaker and likewise structured absorption is seen in the 900 to 1500 nm range (see the inset of Fig. 10a). This band is also seen in IR-spectroelectrochemistry as a rising baseline near the high-energy limit of the detector. None of these bands readily qualifies for an IVCT transition of a mixed-valent system. On the other hand structured absorptions of like appearance are routinely seen when redox processes of complexes with carbon-rich ligands produce ligand centered radicals. Alkynyl or vinyl complexes are typical examples.2o,47a,54 Further reduction to 7 caused an intensity decrease and a splitting of the high-energy band into separate peaks at 358, 390, and 404 nm, as well as the growth of red-shifted bands at 590, 718, and ca. 910 nm. Both these steps were largely reversible with ca. 90 or 85% optical yields of recovered [7]2+ upon back-electrolysis. Further reduction of 7 produced a solution with the spectroscopic features of complex E-6, i.e. a strong band near 380 nm and an even stronger absorption peaking at 302 nm. Taken together, the emerging picture from our spectro-electrochemical studies is that of ligand dominated reductions of [7]2+ with extensive delocalization of charge and spin over the entire RCC–Re–C4–Re–CCR chain and no evidence for a mixed-valent behavior of 7+. This latter finding is in contrast to other C4-bridged complexes with more metal centered redox orbitals and also to [E-6]+.
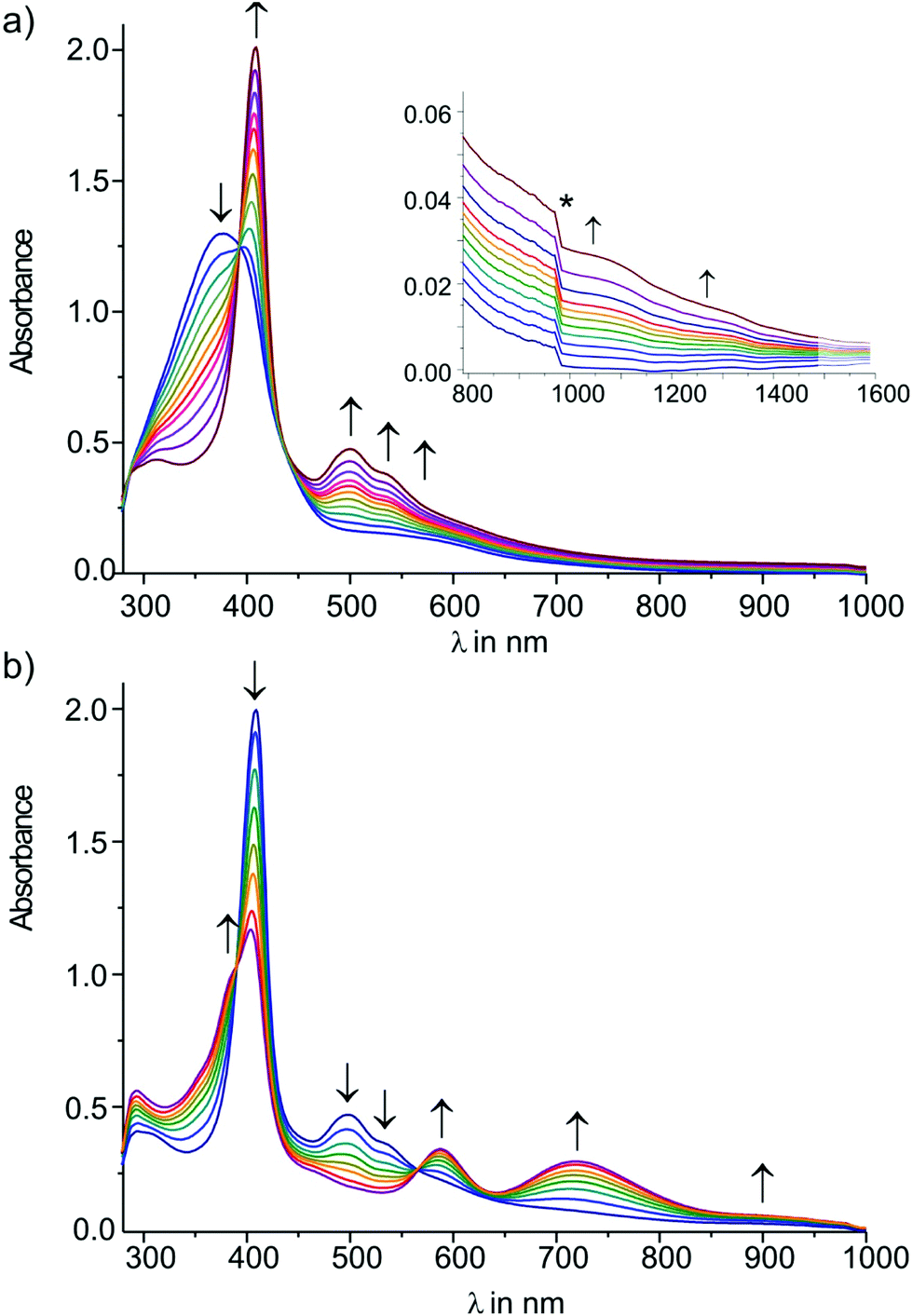 | ||
| Fig. 10 UV/Vis spectroscopic changes during (a) the first and (b) the second reduction of complex 7[PF6]2 in THF/NBu4PF6 at r.t. | ||
Conclusions
Starting from the mononuclear rhenium vinylidene complex trans-[Re(CCSiMe3)(CCH2)(PMe3)4] (3) a series of oxidatively coupled dinuclear complexes trans-[(Me3SiCC)(PMe3)4ReC4HnRe(PMe3)4(CCSiMe3)]m+ (n = 2, 0; m = 0, 1, 2) were prepared. For n = 2 and for m = 0 the respective singlet and triplet states E-6(S) and 7(S) as well as E-6(S) and 7(S) were evaluated. For any of these species it was possible to change the redox state indicated by the charges m = 1,2 of the molecules, which brought about changes in the preferred canonical forms of the bridges. This indicates strong bridge contributions to the respective frontier MOs, i.e. a non-innocent character of the C4Hx-bridges. Oxidation of E-6(S) to E-6[PF6]2 went along with the transformation from a butadienediylidene ([Re]CCH–CHC[Re]) into an ethylenylidene(ditriyl) ([Re]C–CHCH–C[Re]) valence structure. The MV complex of this series of compounds has a large Kc of 7.1 × 107, high electronic coupling energy Hab and a high delocalization parameter Γ and can be safely described as a class III MV compound with an intrinsically delocalized {[M]–π-bridge–[M]}+ entity. The diradical and the cumulenic closed-shell isomers ([Re](·)C–CC–C(·)[Re]) 7(F) ([Re]CCCC[Re]) 7(S) are calculated to be close to isoenergetic. Due to the instability of these species, they could not be fully characterized so no firm conclusion on the prevalent ground state structure can be drawn. By hydrogen abstraction 7 transforms into E-6(S). Cyclic voltammetry (CV) studies and IR- and UV-Vis-NIR-spectro-electrochemistry studies for the reduction of 7[PF6]2 revealed a large Kc of 4.7 × 108 and hence a high thermodynamic stability of the radical monocation 7[PF6]. Structured bands in the visible and NIR of 7[PF6] are indicative of a strongly ligand-centered rather than a purely rhenium centered radical, thus defying its classification as a classical mixed-valent species. Further studies are ongoing in our group for functionalization of the terminal group of E-6 and 7 with the appropriate end groups sticking to gold electrodes suitable for single molecule conductance measurements.
Acknowledgements
Funding from the University of Zurich is gratefully acknowledged. We thank Dr Thomas Fox for his help with the NMR studies.Notes and references
- A. Burgun, F. Gendron, C. J. Sumby, T. Roisnel, O. Cador, K. Costuas, J. F. Halet, M. I. Bruce and C. Lapinte, Organometallics, 2014, 33, 2613–2627 CrossRef CAS.
- (a) M. I. Bruce, M. L. Cole, K. Costuas, B. G. Ellis, K. A. Kramarczuk, C. Lapinte, B. K. Nicholson, G. J. Perkins, B. W. Skelton, A. H. White and N. N. Zaitseva, Z. Anorg. Allg. Chem., 2013, 639, 2216–2223 CrossRef CAS; (b) F. Schwarz, G. Kastlunger, F. Lissel, H. Riel, K. Venkatesan, H. Berke, R. Stadler and E. Lortscher, Nano Lett., 2014, 14, 5932–5940 CrossRef CAS PubMed; (c) F. Lissel, F. Schwarz, O. Blacque, H. Riel, E. Lortscher, K. Venkatesan and H. Berke, J. Am. Chem. Soc., 2014, 136, 14560–14569 CrossRef CAS PubMed; (d) T. Tanaka and A. Osuka, Chem. Soc. Rev., 2015, 44, 943–969 RSC; (e) E. Leary, A. La Rosa, M. T. Gonzalez, G. Rubio-Bollinger, N. Agrait and N. Martin, Chem. Soc. Rev., 2015, 44, 920–942 RSC; (f) D. M. Guldi, H. Nishihara and L. Venkataraman, Chem. Soc. Rev., 2015, 44, 842–844 RSC; (g) S. Rigaut, Dalton Trans., 2013, 42, 15859–15863 RSC; (h) J. R. Heath, Annu. Rev. Mater. Res., 2009, 39, 1–23 CrossRef CAS; (i) R. L. Carroll and C. B. Gorman, Angew. Chem., Int. Ed., 2002, 41, 4379–4400 CrossRef; (j) N. J. Tao, Nat. Nanotechnol., 2006, 1, 173–181 CrossRef CAS PubMed; (k) F. Chen, J. Hihath, Z. F. Huang, X. L. Li and N. J. Tao, Annu. Rev. Phys. Chem., 2007, 58, 535–564 CrossRef CAS PubMed; (l) V. V. Zhirnov and R. K. Cavin, Nat. Mater., 2006, 5, 11–12 CrossRef CAS PubMed; (m) T. Kurita, Y. Nishimori, F. Toshimitsu, S. Muratsugu, S. Kume and H. Nishihara, J. Am. Chem. Soc., 2010, 132, 4524–4525 CrossRef CAS PubMed; (n) N. Tuccitto, V. Ferri, M. Cavazzini, S. Quici, G. Zhavnerko, A. Licciardello and M. A. Rampi, Nat. Mater., 2009, 8, 41–46 CrossRef CAS PubMed; (o) N. Szesni, M. Drexler, J. Maurer, R. F. Winter, F. de Montigny, C. Lapinte, S. Steffens, J. Heck, B. Weibert and H. Fischer, Organometallics, 2006, 25, 5774–5787 CrossRef CAS; (p) W. Kaim and G. K. Lahiri, Angew. Chem., Int. Ed., 2007, 46, 1778–1796 CrossRef CAS PubMed; (q) K. Kowalski, M. Linseis, R. F. Winter, M. Zabel, S. Záliš, H. Kelm, H. J. Krüger, B. Sarkar and W. Kaim, Organometallics, 2009, 28, 4196–4209 CrossRef CAS; (r) F. Paul and C. Lapinte, Coord. Chem. Rev., 1998, 178, 431–509 CrossRef; (s) P. Belser, S. Bernhard, C. Blum, A. Beyeler, L. De Cola and V. Balzani, Coord. Chem. Rev., 1999, 192, 155–169 CrossRef; (t) V. Balzani, A. Juris, M. Venturi, S. Campagna and S. Serroni, Chem. Rev., 1996, 96, 759–833 CrossRef CAS PubMed; (u) P. F. H. Schwab, M. D. Levin and J. Michl, Chem. Rev., 1999, 99, 1863–1933 CrossRef CAS PubMed.
- (a) H. J. Jiao, K. Costuas, J. A. Gladysz, J. F. Halet, M. Guillemot, L. Toupet, F. Paul and C. Lapinte, J. Am. Chem. Soc., 2003, 125, 9511–9522 CrossRef CAS PubMed; (b) R. Dembinski, T. Bartik, B. Bartik, M. Jaeger and J. A. Gladysz, J. Am. Chem. Soc., 2000, 122, 810–822 CrossRef CAS.
- H. Song, Y. Kim, Y. H. Jang, H. Jeong, M. A. Reed and T. Lee, Nature, 2009, 462, 1039–1043 CrossRef CAS PubMed.
- M. B. Robin and P. Day, Prog. Inorg. Chem., 1967, 10, 247 CAS.
- M. Parthey and M. Kaupp, Chem. Soc. Rev., 2014, 43, 5067–5088 RSC.
- (a) P. Aguirre-Etcheverry and D. O'Hare, Chem. Rev., 2010, 110, 4839–4864 CrossRef CAS PubMed; (b) A. Ceccon, S. Santi, L. Orian and A. Bisello, Coord. Chem. Rev., 2004, 248, 683–724 CrossRef CAS; (c) H. Adams, P. J. Costa, M. Newell, S. J. Vickers, M. D. Ward, V. Felix and J. A. Thomas, Inorg. Chem., 2008, 47, 11633–11643 CrossRef CAS PubMed.
- (a) K. Venkatesan, T. Fox, H. W. Schmalle and H. Berke, Organometallics, 2005, 24, 2834–2847 CrossRef CAS; (b) K. Venkatesan, F. J. Fernandez, O. Blacque, T. Fox, M. Alfonso, H. W. Schmalle and H. Berke, Chem. Commun., 2003, 2006–2008 RSC; (c) S. Kheradmandan, K. Heinze, H. W. Schmalle and H. Berke, Angew. Chem., Int. Ed., 1999, 38, 2270–2273 CrossRef CAS.
- N. LeNarvor, L. Toupet and C. Lapinte, J. Am. Chem. Soc., 1995, 117, 7129–7138 CrossRef CAS.
- (a) M. Brady, W. Q. Weng, Y. L. Zhou, J. W. Seyler, A. J. Amoroso, A. M. Arif, M. Böhme, G. Frenking and J. A. Gladysz, J. Am. Chem. Soc., 1997, 119, 775–788 CrossRef CAS; (b) Y. L. Zhou, J. W. Seyler, W. Q. Weng, A. M. Arif and J. A. Gladysz, J. Am. Chem. Soc., 1993, 115, 8509–8510 CrossRef CAS; (c) V. W. W. Yam, V. C. Y. Lau and K. K. Cheung, Organometallics, 1996, 15, 1740–1744 CrossRef CAS.
- (a) M. I. Bruce, P. J. Low, K. Costuas, J. F. Halet, S. P. Best and G. A. Heath, J. Am. Chem. Soc., 2000, 122, 1949–1962 CrossRef CAS; (b) F. Paul, W. E. Meyer, L. Toupet, H. J. Jiao, J. A. Gladysz and C. Lapinte, J. Am. Chem. Soc., 2000, 122, 9405–9414 CrossRef CAS.
- K. Onitsuka, N. Ose, F. Ozawa and S. Takahashi, J. Organomet. Chem., 1999, 578, 169–177 CrossRef CAS.
- (a) B. E. Woodworth, P. S. White and J. L. Templeton, J. Am. Chem. Soc., 1997, 119, 828–829 CrossRef CAS; (b) R. L. Roberts, H. Puschmann, J. A. K. Howard, J. H. Yamamoto, A. J. Carty and P. J. Low, Dalton Trans., 2003, 1099–1105 RSC; (c) S. N. Semenov, O. Blacque, T. Fox, K. Venkatesan and H. Berke, J. Am. Chem. Soc., 2010, 132, 3115–3127 CrossRef CAS PubMed.
- M. Gulcur, P. Moreno-Garcia, X. T. Zhao, M. Baghernejad, A. S. Batsanov, W. J. Hong, M. R. Bryce and T. Wandlowski, Chem. – Eur. J., 2014, 20, 4653–4660 CrossRef CAS PubMed.
- X. T. Zhao, C. C. Huang, M. Gulcur, A. S. Batsanov, M. Baghernejad, W. J. Hong, M. R. Bryce and T. Wandlowski, Chem. Mater., 2013, 25, 4340–4347 CrossRef CAS.
- (a) A. Kuzume, U. Zhumaev, J. F. Li, Y. C. Fu, M. Fueg, M. Estevez, Z. Borjas, T. Wandlowski and A. Esteve-Nunez, Phys. Chem. Chem. Phys., 2014, 16, 22229–22236 RSC; (b) W. J. Hong, H. Li, S. X. Liu, Y. C. Fu, J. F. Li, V. Kaliginedi, S. Decurtins and T. Wandlowski, J. Am. Chem. Soc., 2012, 134, 19425–19431 CrossRef CAS PubMed.
- K. Costuas and S. Rigaut, Dalton Trans., 2011, 40, 5643–5658 RSC.
- (a) L. N. Novikova, M. G. Peterleitner, K. A. Sevumyan, O. V. Semeikin, D. A. Valyaev and N. A. Ustynyuk, Appl. Organomet. Chem., 2002, 16, 530–536 CrossRef CAS; (b) N. A. Ustynyuk, O. V. Gusev, L. N. Novikova, M. G. Peterleitner, L. I. Denisovich, T. A. Peganova, O. V. Semeikin and D. A. Valyaev, J. Solid State Electrochem., 2007, 11, 1621–1634 CrossRef CAS.
- (a) R. P. Orenha, R. Vessecchi and S. E. Galembeck, Struct. Chem., 2015, 26, 365–373 CrossRef CAS; (b) A. A. Zavitsas, N. Matsunaga and D. W. Rogers, J. Phys. Chem. A, 2008, 112, 5734–5741 CrossRef CAS PubMed.
- T. Flitcroft, H. A. Skinner and M. C. Whiting, Trans. Faraday Soc., 1957, 53, 784–790 RSC.
- (a) W. Kaim, Inorg. Chem., 2011, 50, 9752–9765 CrossRef CAS PubMed; (b) W. Kaim, Eur. J. Inorg. Chem., 2012, 343–348 CrossRef CAS; (c) S. Záliš, R. F. Winter and W. Kaim, Coord. Chem. Rev., 2010, 254, 1383–1396 CrossRef; (d) M. D. Ward and J. A. McCleverty, J. Chem. Soc., Dalton Trans., 2002, 275–288 RSC; (e) P. Comba, A. Hauser, M. Kerscher and H. Pritzkow, Angew. Chem., Int. Ed., 2003, 42, 4536–4540 CrossRef CAS PubMed.
- (a) G. Parkin, Acc. Chem. Res., 1992, 25, 455–460 CrossRef CAS; (b) Y. Jean, A. Lledos, J. K. Burdett and R. Hoffmann, J. Am. Chem. Soc., 1988, 110, 4506–4516 CrossRef CAS; (c) J. A. Labinger, C. R. Chim., 2002, 5, 235–244 CrossRef CAS.
- (a) F. J. Fernandez, M. Alfonso, H. W. Schmalle and H. Berke, Organometallics, 2001, 20, 3122–3131 CrossRef CAS; (b) F. J. Fernandez, K. Venkatesan, O. Blacque, M. Alfonso, H. W. Schmalle and H. Berke, Chem. – Eur. J., 2003, 9, 6192–6206 CrossRef CAS PubMed; (c) S. Kheradmandan, K. Venkatesan, O. Blacque, H. W. Schmalle and H. Berke, Chem. – Eur. J., 2004, 10, 4872–4885 CrossRef CAS PubMed; (d) K. Venkatesan, O. Blacque and H. Berke, Dalton Trans., 2007, 1091–1100 RSC; (e) K. Venkatesan, O. Blacque, T. Fox, M. Alfonso, H. W. Schmalle and H. Berke, Organometallics, 2004, 23, 1183–1186 CrossRef CAS; (f) K. Venkatesan, O. Blacque, T. Fox, M. Alfonso, H. W. Schmalle, S. Kheradmandan and H. Berke, Organometallics, 2005, 24, 920–932 CrossRef CAS.
- (a) K. Venkatesan, O. Blacque and H. Berke, Organometallics, 2006, 25, 5190–5200 CrossRef CAS; (b) D. Unseld, V. V. Krivykh, K. Heinze, F. Wild, G. Artus, H. Schmalle and H. Berke, Organometallics, 1999, 18, 1525–1541 CrossRef CAS.
- A. Antiñolo, A. Otero, M. Fajardo, C. García-Yebra, C. López-Mardomingo, A. Martín and P. Gómez-Sal, Organometallics, 1997, 16, 2601–2611 CrossRef.
- R. L. Beddoes, C. Bitcon, R. W. Grime, A. Ricalton and M. W. Whiteley, J. Chem. Soc., Dalton Trans., 1995, 2873–2883 RSC.
- L. N. Novikova, M. G. Peterleitner, K. A. Sevumyan, O. V. Semeikin, D. A. Valyaev, N. A. Ustynyuk, V. N. Khrustalev, L. N. Kuleshova and M. Y. Antipin, J. Organomet. Chem., 2001, 631, 47–53 CrossRef CAS.
- D. A. Valyaev, O. V. Semelkin, M. G. Peterleitner, Y. A. Borisov, V. N. Khrustalev, A. M. Mazhuga, E. V. Kremer and N. A. Ustynyuk, J. Organomet. Chem., 2004, 689, 3837–3846 CrossRef CAS.
- R. S. Iyer and J. P. Selegue, J. Am. Chem. Soc., 1987, 109, 910–911 CrossRef CAS.
- M. I. Bruce, B. G. Ellis, P. J. Low, B. W. Skelton and A. H. White, Organometallics, 2003, 22, 3184–3198 CrossRef CAS.
- (a) M. I. Bruce, Chem. Rev., 1991, 91, 197–257 CrossRef CAS; (b) D. A. Valyaev, O. V. Semeikin and N. A. Ustynyuk, Coord. Chem. Rev., 2004, 248, 1679–1692 CrossRef CAS; (c) C. Bruneau and P. Dixneuf, Metal Vinylidenes and Allenylidenes in Catalysis: From Reactivity to Application in Synthesis, Wiley-VCH Verlag GmbH & Co. KgaA, Weinheim, 2008 Search PubMed; (d) H. Werner, Coord. Chem. Rev., 2004, 248, 1693–1702 CrossRef CAS.
- (a) M. Newcomb and M. T. Burchill, J. Am. Chem. Soc., 1984, 106, 8276–8282 CrossRef CAS; (b) M. Newcomb and M. T. Burchill, J. Am. Chem. Soc., 1984, 106, 2450–2451 CrossRef CAS; (c) M. Jasinski, G. Mloston, A. Gebert and H. Heimgartner, Phosphorus, Sulfur Silicon Relat. Elem., 2015, 190, 1281–1284 CrossRef CAS.
- T. A. Albright, J. Burdettt and M. Whangbo, Orbital Interactions in Chemistry, John Wiley & Sons, Inc., Hoboken, New Jersey, USA, 2013 Search PubMed.
- A. J. L. Pombeiro, A. Hills, D. L. Hughes and R. L. Richards, J. Organomet. Chem., 1988, 352, C5–C7 CrossRef CAS.
- D. R. Lide and H. P. Frederikse, CRC Handbook of Chemistry and Physics, CRC press, Boca Raton, 1995 Search PubMed.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed; (b) C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS; (c) A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- (a) M. Renz and M. Kaupp, J. Phys. Chem. A, 2012, 116, 10629–10637 CrossRef CAS PubMed; (b) M. Renz, K. Theilacker, C. Lambert and M. Kaupp, J. Am. Chem. Soc., 2009, 131, 16292–16302 CrossRef CAS PubMed; (c) M. Renz, M. Kess, M. Diedenhofen, A. Klamt and M. Kaupp, J. Chem. Theory Comput., 2012, 8, 4189–4203 CrossRef CAS PubMed.
- (a) V. Coropceanu, C. Lambert, G. Nöll and J. L. Brédas, Chem. Phys. Lett., 2003, 373, 153–160 CrossRef CAS; (b) V. Coropceanu, M. Malagoli, J. M. Andre and J. L. Brédas, J. Am. Chem. Soc., 2002, 124, 10519–10530 CrossRef CAS PubMed.
- (a) J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter, 1992, 45, 13244–13249 CrossRef; (b) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed; (c) J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986, 33, 8822–8824 CrossRef.
- P. Gütlich, H. A. Goodwin and D. N. Hendrickson, Angew. Chem., Int. Ed., 1994, 33, 425–427 CrossRef.
- E. Ruiz, J. Cirera and S. Alvarez, Coord. Chem. Rev., 2005, 249, 2649–2660 CrossRef CAS.
- S. Le Stang, F. Paul and C. Lapinte, Organometallics, 2000, 19, 1035–1043 CrossRef CAS.
- (a) D. M. D'Alessandro and F. R. Keene, Chem. Soc. Rev., 2006, 35, 424–440 Search PubMed; (b) K. D. Demadis, C. M. Hartshorn and T. J. Meyer, Chem. Rev., 2001, 101, 2655–2685 CrossRef CAS PubMed.
- J. R. Reimers and N. S. Hush, Inorg. Chem., 1990, 29, 4510–4513 CrossRef CAS.
- (a) W. Y. Man, J. L. Xia, N. J. Brown, J. D. Farmer, D. S. Yufit, J. A. K. Howard, S. H. Liu and P. J. Low, Organometallics, 2011, 30, 1852–1858 CrossRef CAS; (b) F. Lissel, T. Fox, O. Blacque, W. Polit, R. F. Winter, K. Venkatesan and H. Berke, J. Am. Chem. Soc., 2013, 135, 4051–4060 CrossRef CAS PubMed.
- (a) N. S. Hush, Coord. Chem. Rev., 1985, 64, 135–157 CrossRef CAS; (b) N. S. Hush, Prog. Inorg. Chem., 1967, 391 CrossRef CAS.
- B. S. Brunschwig, C. Creutz and N. Sutin, Chem. Soc. Rev., 2002, 31, 168–184 RSC.
- J. A. Weil and J. R. Bolton, Electron Paramagnetic Resonance: Elementary Theory and Practical Applications, Wiley, New York, 1994 Search PubMed.
- (a) W. Q. Weng, T. Bartik, M. Brady, B. Bartik, J. A. Ramsden, A. M. Arif and J. A. Gladysz, J. Am. Chem. Soc., 1995, 117, 11922–11931 CrossRef CAS; (b) J. W. Seyler, W. Q. Weng, Y. L. Zhou and J. A. Gladysz, Organometallics, 1993, 12, 3802–3804 CrossRef CAS.
- C. Herrmann, J. Neugebauer, J. A. Gladysz and M. Reiher, Inorg. Chem., 2005, 44, 6174–6182 CrossRef CAS PubMed.
- (a) M. I. Bruce, K. Costuas, T. Davin, B. G. Ellis, J. F. Halet, C. Lapinte, P. J. Low, M. E. Smith, B. W. Skelton, L. Toupet and A. H. White, Organometallics, 2005, 24, 3864–3881 CrossRef CAS; (b) M. I. Bruce, K. Costuas, T. Davin, J. F. Halet, K. A. Kramarczuk, P. J. Low, B. K. Nicholson, G. J. Perkins, R. L. Roberts, B. W. Skelton, M. E. Smith and A. H. White, Dalton Trans., 2007, 5387–5399 RSC.
- (a) M. A. Fox, R. L. Roberts, W. M. Khairul, F. Hartl and P. J. Low, J. Organomet. Chem., 2007, 692, 3277–3290 CrossRef CAS; (b) M. A. Fox, J. D. Farmer, R. L. Roberts, M. G. Humphrey and P. J. Low, Organometallics, 2009, 28, 5266–5269 CrossRef CAS; (c) N. Gauthier, N. Tchouar, F. Justaud, G. Argouarch, M. P. Cifuentes, L. Toupet, D. Touchard, J. F. Halet, S. Rigaut, M. G. Humphrey, K. Costuas and F. Paul, Organometallics, 2009, 28, 2253–2266 CrossRef CAS; (d) J. Maurer, R. F. Winter, B. Sarkar, J. Fiedler and S. Zalis, Chem. Commun., 2004, 1900–1901 RSC; (e) J. Maurer, M. Linseis, B. Sarkar, B. Schwederski, M. Niemeyer, W. Kaim, S. Zalis, C. Anson, M. Zabel and R. F. Winter, J. Am. Chem. Soc., 2008, 130, 259–268 CrossRef CAS PubMed; (f) J. Maurer, B. Sarkar, B. Schwederski, W. Kaim, R. F. Winter and S. Záliš, Organometallics, 2006, 25, 3701–3712 CrossRef CAS; (g) F. Pevny, E. Di Piazza, L. Norel, M. Drescher, R. F. Winter and S. Rigaut, Organometallics, 2010, 29, 5912–5918 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details describing the syntheses as well as giving details of the NMR spectroscopy, refinement details and crystallographic data for the X-ray diffraction studies, deconvoluted UV-Vis-NIR spectra and computational results. CCDC 859304–859309. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt04768d |
| This journal is © The Royal Society of Chemistry 2016 |