
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence[RuCl2(η6-p-cymene)] complexes bearing phosphinous acid ligands: preparation, application in C–H bond functionalization and mechanistic investigations†
Lionel V.
Graux
a,
Michel
Giorgi
b,
Gérard
Buono
a and
Hervé
Clavier
*a
aAix Marseille Université, Centrale Marseille, CNRS, iSm2 UMR 7313, 13397, Marseille, France. E-mail: herve.clavier@univ-amu.fr
bAix Marseille Université Spectropole, Fédération des Sciences Chimiques de Marseille, 13397 Marseille, France
First published on 25th February 2016
Abstract
A series of [RuCl2(η6-p-cymene)] complexes bearing phosphinous acid (PA) ligands have been straightforwardly prepared from the dimer [RuCl2(p-cymene)]2 and secondary phosphine oxides (SPOs) and fully characterized. The steric parameter quantification of PAs, other L ligands and η6-p-cymene allowed a better comprehension of the coordination chemistry of these types of complexes and explained the absence of coordination in the case of bulky SPOs such as Ad2P(O)H. These complexes were tested in the C–H activation/functionalization of 2-phenylpyridine and a good activity was obtained at 80 °C for the complex exhibiting the highest steric bulk. A study on halide effects, either on the ruthenium complex or for the aryl halide partner, has also been carried out showing drastic differences. Further investigations on halide effects were performed notably by using a cationic ruthenacycle which was found to be an intermediate for the reaction. In order to rationalize the role played by the phosphinous acid, a mechanism involving a concerted metallation deprotonation favored by a phosphinito species has been proposed.
Introduction
Considered for a long time as a main challenge in organic synthesis, the C–H activation/functionalization has emerged recently as a powerful tool to prepare complex molecules starting from readily available raw materials.1 In this quest, the use of transition metal catalysis is at the origin of groundbreaking discoveries and now various catalytic systems using palladium,2 rhodium,3 ruthenium,4 copper,5 platinum,6 nickel,7 cobalt,8 f-block elements,9etc. are recognized as highly efficient for C–H bond activation. In these systems, major advances were accomplished by clear mechanistic studies which allowed for deeper understanding of the mechanism, notably the role of ligands and/or additives. For instance, Catellani developed a Pd-catalyzed regioselective C–H bond functionalization of arenes using norbornene as a covalent linker.10 Alternatively, it was demonstrated that C–H activation could occur according to a σ-bond metathesis pathway,11 so called concerted metallation deprotonation (CMD). In such a process, the intermolecular abstraction of the proton by acetato ligands favors the metallacycle formation.12Whereas pioneering studies on catalytic C(sp2)–H activation were carried out using Ru-based complexes,13 catalytic systems using Pd or Rh have attracted much attention over the last decade. Nevertheless, interesting Ru-based catalytic systems have been reported in the literature mainly using the dimer [RuCl2(p-cymene)]2 in association with carboxylate additives.4,14 During our research involving secondary phosphine oxides (SPO) and phosphinous acids (PA) as ligands in transition metal catalysis,15 the catalytic system using [RuCl2(p-cymene)]2 and SPO developed by Ackermann in 2005 caught our attention (Scheme 1).16 A Ru(II) complex, prepared in situ from [RuCl2(η6-p-cymene)]2 and a sterically hindered diadamantylphosphine oxide, was found to be very efficient for ortho C–H bond activation of the 2-phenylpyridine 1 and coupling with chlorobenzene to yield quantitatively compound 3 without any traces of monoarylated 2. Moreover, this catalytic system showed a high efficiency for various substrates.17 We wondered about the role played by the SPO or its tautomeric phosphinous acid form in the C–H activation process. Whereas several mechanistic hypothesis have been postulated,4,17c none of them was further investigated.
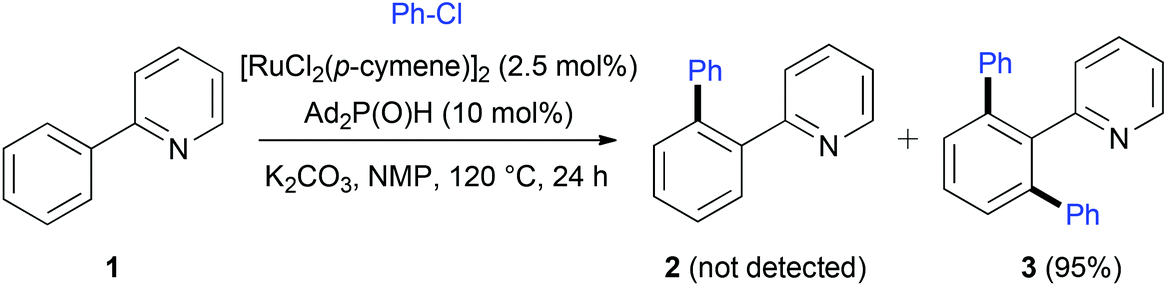 | ||
| Scheme 1 C–H activation/functionalization mediated by [RuCl2(p-cymene)]2 and SPO. | ||
Therefore, we wished to investigate the mechanism of this C–H bond activation/functionalization through the synthesis of well-defined [RuCl2(η6-p-cymene)(PA)] complexes and to study their performances in catalysis. Control experiments have been carried out to corroborate the mechanistic proposal. The results of our study are reported herein.
Results and discussion
We started to study the coordination chemistry of SPOs with [RuCl2(p-cymene)]2. The synthesis of [RuCl2(η6-arene)(PA)] has been previously reported in the literature,18 notably two complexes [Ru(η6-p-cymene)Cl2(PA)] 5a and 5l were already described by the groups of Tyler19 and Leung20 respectively (Fig. 1) and 5a has been successfully applied to nitrile hydratation.19,21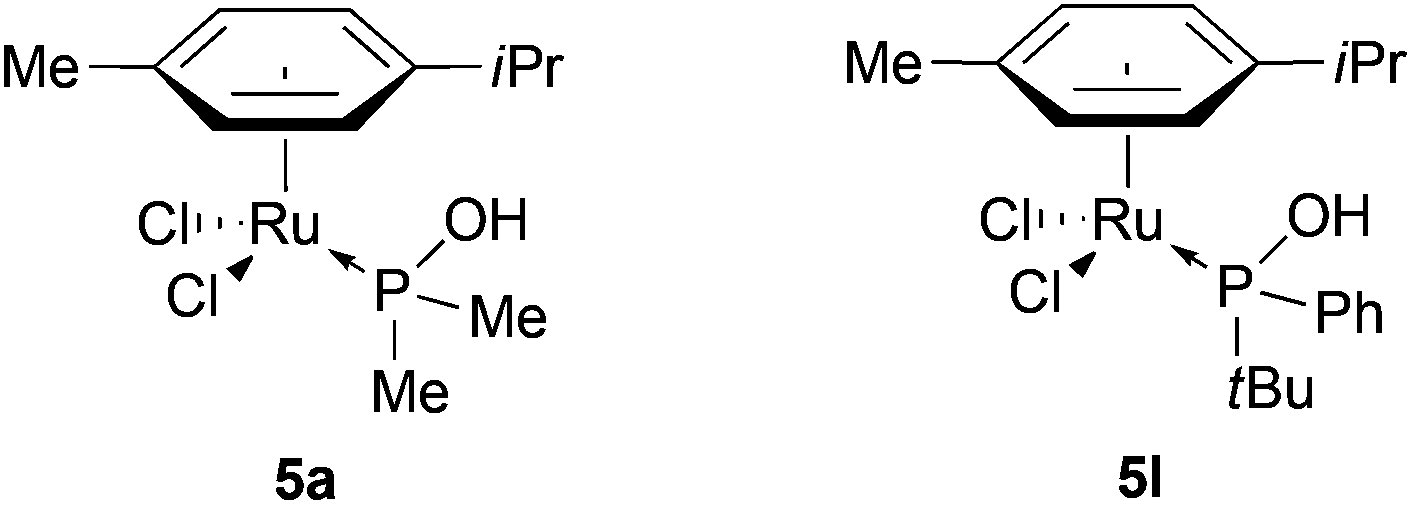 | ||
| Fig. 1 [RuCl2(η6-p-cymene)(PA)] complexes described in the literature. | ||
Following the same procedure, [RuCl2(p-cymene)]2 was treated with various secondary phosphine oxides 4 in THF at 25 °C to afford the expected complexes 5 (Table 1).22 Of note, in dichloromethane or toluene, the reaction was sluggish compared to THF. Excepted for Cy2P(O)H 4b which required 24 h of reaction, other SPOs, either symmetrical ones (entries 2–4) or unsymmetrical ones (entries 7–11), were found very reactive. After only 2–3 h, complexes 5 were isolated in almost quantitative yields. Surprisingly, with bulky SPOs such as Ad2P(O)H 4f or tBu2P(O)H 4g, we were unable to obtain the corresponding complexes 5 (entries 5 and 6). Despite prolonged reaction times and/or heating to 110 °C, their formation could not be detected by NMR spectroscopy.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | t (h) | Complex | Yield (%) |
| a Reaction conditions: [RuCl2(η6-p-cymene)]2, SPO (2.2 equiv.), THF (0.5 M), 25 °C. b Reaction performed in 1,4-dioxane at 110 °C. NR = no reaction. | |||||
| 1 | Cy | Cy | 24 | 5b | 89 |
| 2 | Ph | Ph | 2 | 5c | 96 |
| 3 | p-F-C6H4 | p-F-C6H4 | 6 | 5d | 84 |
| 4 | 3,5-Me2-C6H3 | 3,5-Me2-C6H3 | 3 | 5e | 91 |
| 5b | Ad | Ad | 24 | 5f | NR |
| 6b | tBu | tBu | 24 | 5g | NR |
| 7 | Me | Ph | 3 | 5h | 74 |
| 8 | nBu | Ph | 3 | 5i | 53 |
| 9 | Bn | Ph | 3 | 5j | 88 |
| 10 | Cy | Ph | 3 | 5k | 98 |
| 11 | tBu | Ph | 3 | 5l | 99 |

The well-defined [RuCl2(η6-p-cymene)(PA)] 5 were characterized by 1H,2313C, and 31P NMR spectroscopies, as well as mass spectrometry. The coordination of the phosphinous acid to the metallic center through the phosphorus atom was confirmed by NMR spectroscopy and the absence of 1J(P,H) coupling. Moreover, 31P NMR spectroscopy showed new resonances between 105–130 ppm with a significant shift to a lower field compared to SPOs (Δδ between 68 and 89 ppm, Table 2).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
| Entry | SPO | 31P{1H} (ppm) | Complex 5 | 31P{1H} (ppm) |
|---|---|---|---|---|
| a Measured in CDCl3 at 25 °C. | ||||
| 1 | 4b | 50.0 | 5b | 129.3 |
| 2 | 4c | 21.4 | 5c | 107.1 |
| 3 | 4d | 18.2 | 5d | 105.6 |
| 4 | 4e | 22.8 | 5e | 107.0 |
| 5 | 4h | 20.2 | 5h | 109.5 |
| 6 | 4i | 28.0 | 5i | 113.1 |
| 7 | 4j | 29.6 | 5j | 109.4 |
| 8 | 4k | 36.7 | 5k | 114.2 |
| 9 | 4l | 47.4 | 5l | 115.5 |
To establish unambiguously the structure of [RuCl2(η6-p-cymene)(PA)] complexes 5, suitable crystals for single-crystal X-ray diffraction studies were obtained for most of the compounds. As depicted in Fig. 2, all complexes adopt a distorted octahedral structure with the expected three-legged pseudotetrahedral “piano-stool” geometry around the Ru atom and the η6-coordination of the p-cymene. Two chlorine atoms and the phosphorus occupy the other three positions. Bond lengths are similar to those reported for analogous complexes (Table 3); the Ru–Cl bond distances range from 2.3943(10) to 2.4395(7) Å, the Ru–P bond lengths from 2.3009(8) to 2.3680(7) Å and the longest distances have been observed with cyclohexyl and tert-butyl P-substituents (entries 1, 6 and 7). This seems to be the result of a higher steric congestion of PA ligands. The P–O bond lengths (all close to 1.60 Å) are consistent with a P–O single bond. The O⋯Cl distances ranging from 2.961(2) to 3.070(2) Å – except for 5h, 3.175(2) Å, entry 4 – indicate a hydrogen bonding with chlorines. Of note, with 5h, the hydrogen bonding takes place intermolecularly.
 | ||
| Fig. 2 Molecular structures of complexes 5b–d, 5h–i and 5k–l represented at 50% ellipsoid probability. Most of the H atoms have been omitted for clarity. | ||
| Entry | Complex | Ru(1)–Cl(1) (Å) | Ru(1)–Cl(2) (Å) | Ru(1)–P(1) (Å) | P(1)–O(1) (Å) | O⋯Cl (Å) | Ru(1)–Cavg (Å) |
|---|---|---|---|---|---|---|---|
| 1 | 5b | 2.4323(10) | 2.3943(10) | 2.3376(9) | 1.600(3) | 2.988(3) | 2.203(4) |
| 2.4320(10) | 2.4198(9) | 2.3379(9) | 1.602(3) | 3.005(3) | 2.214(4) | ||
| 2.4296(10) | 2.4072(9) | 2.3445(10) | 1.612(3) | 3.027(3) | 2.215(4) | ||
| 2 | 5c | 2.4171(8) | 2.4156(8) | 2.3120(8) | 1.602(2) | 3.070(2) | 2.216(3) |
| 3 | 5d | 2.4147(9) | 2.3970(8) | 2.3009(8) | 1.605(2) | 3.011(3) | 2.212(3) |
| 4 | 5h | 2.4024(6) | 2.4197(6) | 2.3130(6) | 1.6034(19) | 3.175(2) | 2.210(3) |
| 5 | 5i | 2.4295(11) | 2.4032(11) | 2.3017(10) | 1.604(3) | 2.988(3) | 2.209(4) |
| 6 | 5k | 2.3966(8) | 2.4389(9) | 2.3342(8) | 1.601(2) | 3.009(2) | 2.212(3) |
| 7 | 5l | 2.4395(7) | 2.4124(7) | 2.3680(7) | 1.6098(19) | 2.961(2) | 2.214(3) |
In order to gain more insights into the coordination chemistry of [RuCl2(p-cymene)(L)] and particularly to address the issue of the absence of reaction with bulky SPOs Ad2P(O)H 4f or tBu2P(O)H 4g, we quantified the steric parameter of phosphinous acids and other ligands L (phosphine, phosphite and N-heterocyclic carbene (NHC)) through the percentage buried volume (%Vbur).24,25 Selected values have been gathered in Table 4.23 The %Vbur for PA ligands span from 21.9 to 24.6%. These values are significantly lower than those calculated on AuCl(PA) complexes.15g As expected Ph2POH is more sterically demanding than Ph2PH (entries 3 and 9) but less than Ph2PCH2OH, Ph2PnBu, PPh3 or P(OPh)3 (entries 9–13). The highest %Vbur calculated in [RuCl2(p-cymene)(L)] complexes is for the NHC IMes (1,3-dimesityl-imidazol-2-ylidene) (27.9%) which is in the lower range for this ligand (entry 14).24 These low %Vbur values and their narrow range attest probably that the other ligands in the coordination sphere of the Ru are sterically demanding and the L ligand has therefore to minimize its size. The %Vbur of the η6-p-cymene has also been quantified in 24 X-ray structures and the size of this ligand was found to be relatively invariant with an average value of 47.5% and a standard deviation of 0.35%.23 As intuitively expected, the η6-p-cymene occupies almost half of the Ru coordination sphere and does not show any structural flexibilities. Therefore, the coordination of sterically hindered ligands, such as Ad2POH or tBu2POH, seems unlikely. Indeed, calculations performed on other complexes shown that %Vbur of tBu2POH and Ad2POH are around 30.5 and 31.5, respectively.23
| Entry | L | d(Ru–L) (Å) | %Vbur (L) |
|---|---|---|---|
| a Parameters used for SambVca calculations: 3.50 Å for the sphere radius, exact distances between the ligand and the metal were considered, hydrogen atoms were omitted and bond radii scaled by 1.17. | |||
| 1 | Me2POH | 2.3078(10) | 21.9 |
| 2 | Cy2POH | 2.3376(9) | 24.4 |
| 3 | Ph2POH | 2.3120(8) | 24.6 |
| 4 | (p-F-C6H4)2POH | 2.3009(8) | 24.6 |
| 5 | MePhPOH | 2.3130(6) | 22.7 |
| 6 | nBuPhPOH | 2.3017(10) | 22.7 |
| 7 | CyPhPOH | 2.3342(8) | 24.1 |
| 8 | tBuPhPOH | 2.3680(7) | 24.4 |
| 9 | Ph2PH | 2.313(2) | 23.2 |
| 10 | Ph2PCH2OH | 2.3516(8) | 25.1 |
| 11 | Ph2PnBu | 2.352(2) | 25.6 |
| 12 | PPh3 | 2.3438(6) | 26.8 |
| 13 | P(OPh)3 | 2.2642(8) | 24.7 |
| 14 | IMes | 2.142(4) | 27.9 |
Having prepared a series of well-defined [RuCl2(η6-p-cymene)(PA)], we then tested their catalytic performances in C–H activation using 2-phenylpyridine 1 as the benchmark substrate using Ackermann's conditions (Table 5). A control experiment showed that at 120 °C, [RuCl2(p-cymene)]2 without addition of SPO was competent for C–H functionalization since 88% of diarylated product 3 was isolated (entry 1). At 100 °C, only minute amounts of products 2 and 3 were obtained with [RuCl2(p-cymene)]2 (entry 2) whereas adding either 5 or 10 mol% of Ad2POH allowed to isolate around 80% of diarylated product 3 (entries 3 and 4). However, lowering the reaction temperature to 80 °C led to the loss of activity of the dimer [RuCl2(p-cymene)]2 (entry 5). On one hand, at this temperature, [RuCl2(p-cymene)]2 in association with Ad2P(O)H 4f gave only small amounts of 2 and 3 (entry 6). On the other hand, well-defined [RuCl2(η6-cymene)(PA)] 5a–e and 5i–k were found much more competent and significant quantities of C–H functionalized products were isolated (entries 7–14). Since the reactions did not reach completion, ca. 1:1 mixtures of mono- and diarylated products were obtained. The well-defined complexes performed slightly better than in situ-generated complexes (entries 14 and 15) and addition of an extra quantity of SPO reduced the catalytic activity.23 To our delight, with RuCl2(η6-p-cymene)(tBuPhPOH)] 5l, only compound 3 was formed with an almost quantitative yield (entry 16). Of note, after only 2 h of the reaction, a 1:1 mixture of mono- and diarylated products was observed with 5l (entry 17). The electronic properties of phosphinous acids did not influence the catalytic activities of the resulting complexes. For example Ph2POH-containing 5c performed better than 5b bearing the more electron rich Cy2POH rather than 5d bearing electron withdrawing ligand (p-F-C6H4)2POH (entries 8–10). In contrast, higher activities have been obtained for more sterically demanding PA ligands, i.e. tBuPhPOH and (3,5-Me2-C6H3)2POH (entries 16 and 11, respectively). Importantly, complexes bearing phosphine or phosphite exhibited interesting activities upon C–H activation (entries 18–20). In particular, [RuCl2(η6-p-cymene)(PPh3)] was found to be more selective for mono-functionalization of 2-phenylpyridine 1 (entry 18).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Additive | Yield (%) | |
| 2 | 3 | |||
| a Reaction conditions: 2-phenylpyridine 1 (142 μL, 1 mmol), chlorobenzene (220 μL, 2.2 mmol, 2.2 equiv.), K2CO3 (415 mg, 3 mmol, 3 equiv.), 5 mol% of Ru complex (2.5 mol% for [RuCl2(p-cymene)]2), NMP (2 mL), 80 °C, and 24 h. b Reaction was performed at 120 °C. c Reaction was performed at 100 °C. d Reaction time: 2 h. | ||||
| 1b | [RuCl2(p-cymene)]2 | 0 | 88 | |
| 2c | RuCl2(p-cymene)]2 | 8 | 2 | |
| 3c | RuCl2(p-cymene)]2 | Ad2P(O)H 4f (5 mol%) | 0 | 80 |
| 4c | RuCl2(p-cymene)]2 | Ad2P(O)H 4f (10 mol%) | 0 | 83 |
| 5 | [RuCl2(p-cymene)]2 | 0 | 0 | |
| 6 | [RuCl2(p-cymene)]2 | Ad2P(O)H 4f (10 mol%) | 7 | 3 |
| 7 | [RuCl2(η6-p-cymene)(Me2POH)] 5a | 14 | 15 | |
| 8 | [RuCl2(η6-p-cymene)(Cy2POH)] 5b | 12 | 11 | |
| 9 | [RuCl2(η6-p-cymene)(Ph2POH)] 5c | 27 | 22 | |
| 10 | [RuCl2(η6-p-cymene)((p-F-C6H4)2POH)] 5d | 19 | 11 | |
| 11 | [RuCl2(η6-p-cymene)((3,5-Me2-C6H3)2POH)] 5e | 32 | 32 | |
| 12 | [RuCl2(η6-p-cymene)(nBuPhPOH)] 5i | 19 | 19 | |
| 13 | [RuCl2(η6-p-cymene)(BnPhPOH)] 5j | 19 | 11 | |
| 14 | [RuCl2(η6-p-cymene)(CyPhPOH)] 5k | 22 | 54 | |
| 15 | [RuCl2(p-cymene)]2 | CyPhP(O)H 4k (5 mol%) | 21 | 19 |
| 16 | [RuCl2(η6-p-cymene)(tBuPhPOH)] 5l | 0 | 89 | |
| 17d | [RuCl2(η6-p-cymene)(tBuPhPOH)] 5l | 14 | 13 | |
| 18 | [RuCl2(η6-p-cymene)(PPh3)] | 58 | 6 | |
| 19 | [RuCl2(η6-p-cymene)(PCy3)] | 18 | 5 | |
| 20 | [RuCl2(η6-p-cymene)(P(OPh)3)] | 19 | 15 | |
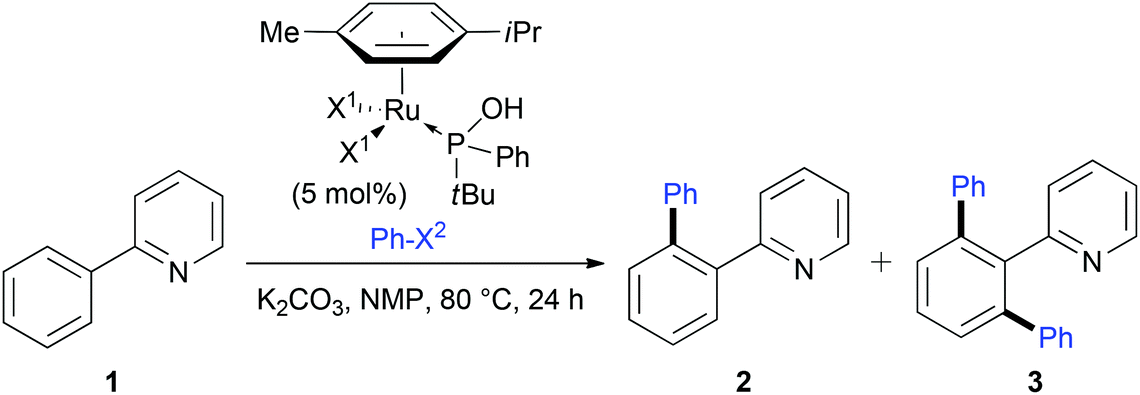
In Ru-mediated C–H activation, the anionic ligands played an important role, for example carboxylate-containing complexes are extremely efficient due to chelation-assisted C–H activations.1k,4a Surprisingly, to the best of our knowledge, complexes bearing bromide and iodide ligands have not been investigated so far. Bromide and iodide counterparts bearing tBuPhPOH phosphinous acid were prepared with good yield following the same protocol than for chloride analogues (6l and 7l respectively, Scheme 2). The activity of these catalysts was compared for C–H functionalization of 2-phenylpyridine 1 and as a function of the aryl halide partner (Table 6). Unexpectedly, drastic differences were observed as only low yields of C–H functionalization and with an almost equimolar mixture of mono- and diarylation were obtained (entries 2 and 3). Moreover, whereas aryl bromides are regularly used as coupling partners,26 at 80 °C they exhibited a significantly lower activity than their chlorine counterpart (entries 1, 2 and 4).27 We assume that this is due to an exchange of halogens between the aryl halide to the metal center occurring through the oxidative addition. Halide effects in transition metal catalysis are often difficult to rationalize,28 however, we believe that the iodide atoms increase the congestion around the metal center and decrease its reactivity.29
 | ||
| Scheme 2 Synthesis of bromide- and iodide-containing [RuX2(η6-p-cymene)(PA)] complexes. | ||
|
|
||||
|---|---|---|---|---|
| Entry | X1 (complex) | Ph-X2 | Yield (%) | |
| 2 | 3 | |||
| a Reaction conditions: 2-phenylpyridine 1 (142 μL, 1 mmol), aryl halide (2.2 mmol, 2.2 equiv.), K2CO3 (415 mg, 3 mmol, 3 equiv.), complex (5 mol%), NMP (2 mL), and 24 h. | ||||
| 1 | Cl (5l) | Ph-Cl | 0 | 89 |
| 2 | Br (6l) | Ph-Br | 23 | 18 |
| 3 | I (7l) | Ph-I | 14 | 8 |
| 4 | Cl (5l) | Ph-Br | 6 | 21 |
| 5 | Cl (5l) | Ph-I | 3 | 5 |
| 6 | Br (6l) | Ph-I | 3 | 7 |
From these results, a halide inhibition was suspected; the chlorine dependence was therefore investigated through the use of additives (Table 7). The addition of one equivalent of LiCl in the reaction mixture quenched indeed the C–H activation process (entries 1 and 2). A significant loss of activity was also observed with 1 equiv. of tetrabutylammonium chloride (entry 3). Nevertheless, addition of a stoichiometric amount of silver salt did not boost the catalytic performances of complexes exhibiting a moderate activity such as complexes 5a and 5b (entries 5 and 8). Higher quantities of silver salt led to the inhibition of the C–H functionalization (entries 6 and 9). These results suggest that chlorine atoms play a key role in the C–H activation process.
|
|
||||
|---|---|---|---|---|
| Entry | Complex 5 | Additives | Yield (%) | |
| 2 | 3 | |||
| a Reaction conditions: 2-phenylpyridine 1 (142 μL, 1 mmol), chlorobenzene (220 μL, 2.2 mmol, 2.2 equiv.), K2CO3 (415 mg, 3 mmol, 3 equiv.), complex (5 mol%), NMP (2 mL), and 24 h. | ||||
| 1 | 5l (R1, R2 = Ph, tBu) | None | 0 | 89 |
| 2 | 5l (R1, R2 = Ph, tBu) | LiCl (1 equiv.) | 0 | 0 |
| 3 | 5l (R1, R2 = Ph, tBu) | nBu4NCl (1 equiv.) | 9 | 6 |
| 4 | 5a (R1, R2 = Me, Me) | None | 14 | 15 |
| 5 | 5a (R1, R2 = Me, Me) | AgBF4 (5 mol%) | 14 | 16 |
| 6 | 5a (R1, R2 = Me, Me) | AgBF4 (1 equiv.) | 0 | 0 |
| 7 | 5b (R1, R2 = Cy, Cy) | None | 12 | 11 |
| 8 | 5b (R1, R2 = Cy, Cy) | AgBF4 (5 mol%) | 9 | 8 |
| 9 | 5b (R1, R2 = Cy, Cy) | AgBF4 (1 equiv.) | 2 | 2 |

In order to gain insight into the mechanism, in particular the role of the PA ligand, and as PAs are considered as labile ligands due to a weak M–PA bond,15d we attempted to determine if the PA remains in the coordination sphere of the ruthenium all along the catalytic cycle. For this purpose, the ruthenium cyclometallated complex 8 was prepared30 and tested in catalysis (Table 8). As anticipated, at 120 °C, this catalyst performed well with 91% of diarylated compound 3 isolated (entry 1). However, at 80 °C, no reaction occurred, even when the ligand tBuPhP(O)H 4l was added (entries 2 and 3). On one hand, the addition of a substoichiometric amount of silver salts such as AgBF4 and AgSbF6 allowed the formation small quantities of products 2 and 3 (entries 4 and 5). On the other hand, the combination of the silver salt with OPS tBuPhP(O)H 4l led to restore almost completely the activity of the catalytic system (entries 6 and 7).
a
|
|
||||
|---|---|---|---|---|
| Entry | Additives | T (°C) | Yield (%) | |
| 2 | 3 | |||
| a Reaction conditions: 2-phenylpyridine 1 (142 μL, 1 mmol), chlorobenzene (220 μL, 2.2 mmol, 2.2 equiv.), K2CO3 (415 mg, 3 mmol, 3 equiv.), complex 8 (21.3 mg, 5 mol%), NMP (2 mL), and 24 h. | ||||
| 1 | None | 120 | 0 | 91 |
| 2 | None | 80 | 0 | 0 |
| 3 | tBuPhP(O)H 4l (5 mol%) | 80 | 0 | 0 |
| 4 | AgBF4 (6 mol%) | 80 | 5 | 4 |
| 5 | AgSbF6 (6 mol%) | 80 | 7 | 8 |
| 6 | AgBF4 (6 mol%), tBuPhP(O)H 4l (5 mol%) | 80 | 26 | 52 |
| 7 | AgSbF6 (6 mol%), tBuPhP(O)H 4l (5 mol%) | 80 | 26 | 56 |

We were also able to prepare quantitatively the cationic ruthenacycle 9c bearing a phosphinous acid ligand by the treatment of complex 8 with OPS 4c in the presence of silver tetrafluoroborate in a stoichiometric amount (Scheme 3). Unfortunately 9c was found to exhibit no activity under our optimal conditions (80 °C). However, when this reaction was performed in the presence of 5 mol% of tetrabutylammonium chloride, 9c displayed moderate activity with the formation of 11% of mono arylated product 2 and 14% of 3. This represents only a slight decrease of the performance compared to catalyst 5c (27% of 2 and 22% of 3, Table 5, entry 9). To further investigate this chlorine effect, complex 9c was treated with 5 equiv. of LiCl in CDCl3 at room temperature, and rapidly 31P NMR spectroscopy showed a new resonance at 108.4 ppm with complete disappearance of the signal at 114.7 ppm after only one hour. Unfortunately, this new compound was found to be unstable and could not be isolated and fully characterized.
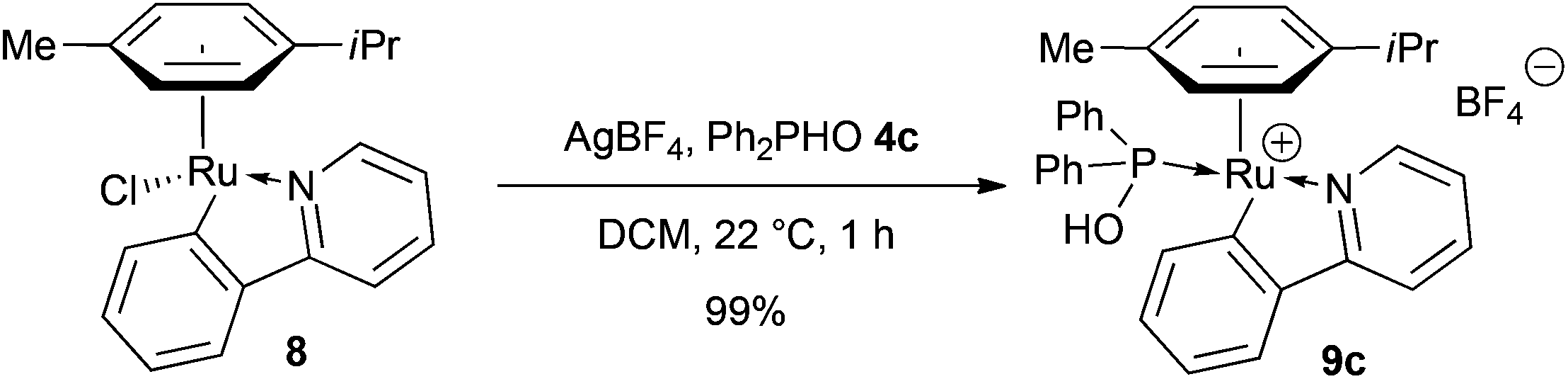 | ||
| Scheme 3 Preparation of the cationic ruthenacycle 9c. | ||
With these results in mind, we propose the following mechanism for C–H activation/functionalization catalyzed by [RuCl2(η6-p-cymene)(PA)] complexes 5 (Scheme 4).31 The mechanism initiates by a base-promoted loss of HCl to give the 18e ruthenium κ2-PO-phosphinito species A. A transition to a κ1 coordination frees a coordination site for the pyridine-containing substrate. At this stage, the CDM mechanism may be triggered by the phosphinito which intercepts the ortho-proton with a concomitant release of the chlorine atom to lead to the cationic ruthenacycle C. Of note, C corresponds to the isolated complex 9c. According to our observations dealing with the reactivity of 9c with LiCl, we believe that the chlorine counterattacks the metallic center and prompts to pyridine-moiety decoordination to afford species D. This step might be at the origin of the observed halide effect; the counterattack is probably less favored with more bulky halides such as bromide and iodide. Finally, the rate determining oxidative addition4d,32 occurring possibly through a single electron transfer process31 and the reductive elimination give the coupling product 2 and release complex 5.
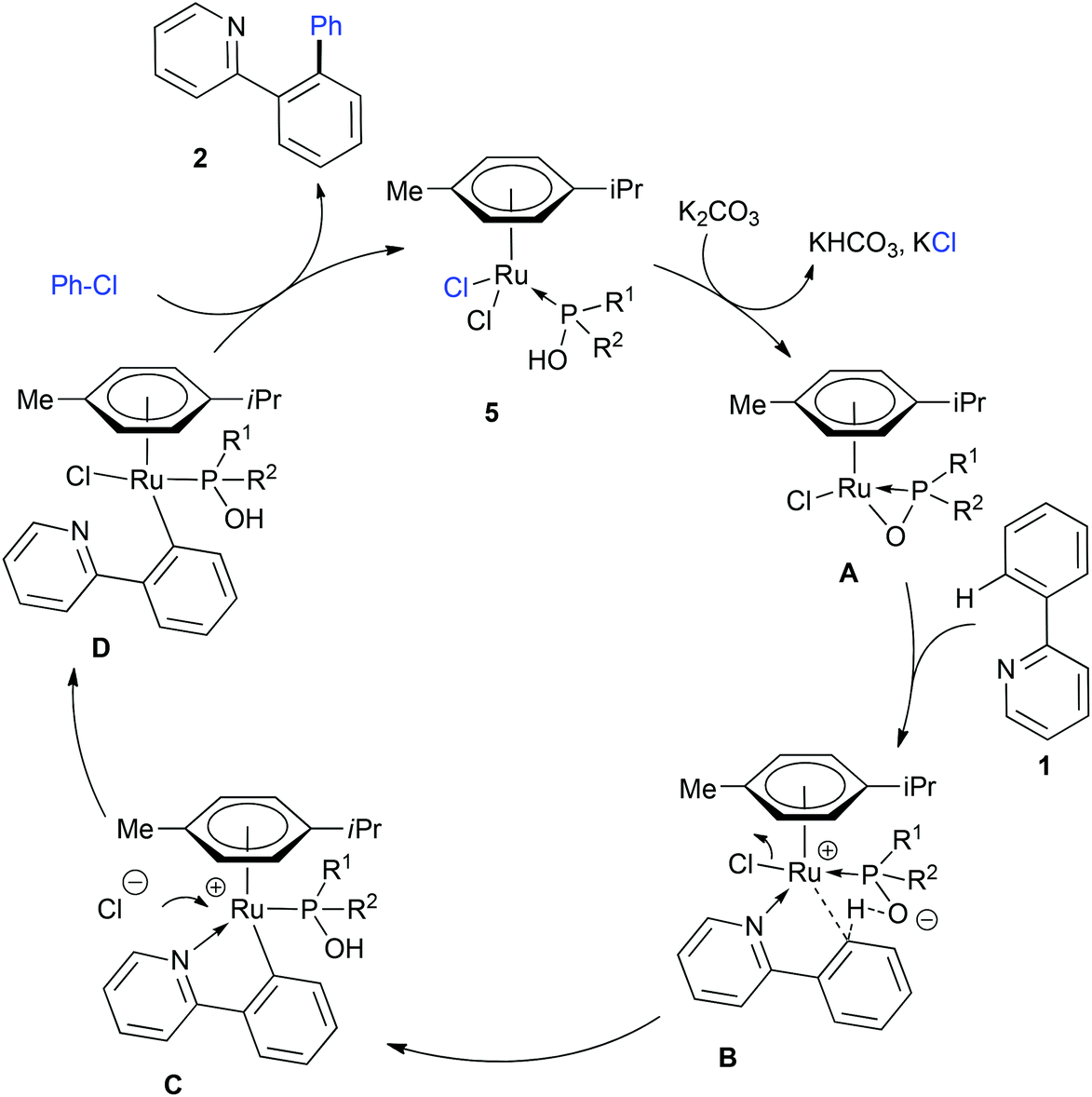 | ||
| Scheme 4 Proposed mechanism for the C–H activation mediated by [RuCl2(η6-p-cymene)(PA)] complexes. | ||
Conclusions
In summary, a series of [RuCl2(η6-p-cymene)] complexes bearing phosphinous acid (PA) ligands has been straightforwardly prepared starting from the dimer [RuCl2(p-cymene)]2 and secondary phosphine oxides. These complexes were fully characterized, notably by single-crystal X-ray diffraction which allowed us to calculate the percentage buried volumes of PAs and other phosphorus-based ligands. This quantification of the steric parameter led to a better comprehension of the coordination chemistry for these types of complexes and explained the absence of coordination in the case of bulky SPOs such as Ad2P(O)H. The performances of these complexes were evaluated in C–H activation/functionalization of 2-phenylpyridine. At 80 °C, an efficient complex bearing the bulkier PA was identified. A thorough comparison of aryl halides as coupling partners revealed a sharp halide effect. Further investigations allowed us to establish a halide dependence: large quantities of chlorine inhibited the C–H activation/functionalization but stoichiometric amounts were also necessary. On the basis of these results and the isolation of a reaction intermediate, a mechanism involving successively the formation of κ2-PO-phosphinito species, a concerted metallation deprotonation favored by the phosphinito, a chlorine counterattack and single electron transfer oxidative addition were proposed. Since the hindered Ad2P(O)H does not coordinate to the metal center even though it improves the C–H activation, we believe that it participates in the CDM as an outer-sphere base. An enantioselective version of C–H activation/functionalization taking advantage of the P-stereogenic center of PA ligands is currently under investigation in our laboratory.Experimental
All reagents were obtained from commercial sources and used as received. Secondary phosphine oxides 4c and 4e–g were obtained from chemical suppliers. Other SPOs were prepared according to literature procedures: 4b,334d,334h,344i,344j,354k15a and 4l.34 THF was purified and dried over the Braun solvent purification system (MB-SPS-800). Analytical Thin Layer Chromatography (TLC) was carried out on a Merck silica gel 60 F254. The products were revealed by ultraviolet light (254 or 366 nm) and stained with dyeing reagents solutions. Flash chromatography was performed on a Combiflash® Companion or with Merck silica gel 60 (230–400 mesh). 1H, 13C, 31P and 19F NMR spectra were recorded in CDCl3 at ambient temperature on Bruker Avance III 300 or 400 spectrometers operating at 300 and 400 MHz respectively for 1H. 13C, 31P and 19F nuclei were observed with 1H decoupling. Solvent residual signals were used as internal standards.36 Chemical shifts (δ) and coupling constants (J) are given in ppm and Hz respectively. HRMS were recorded on a SYNAPT G2 HDMS (Waters) or on a QStar Elite (Applied Biosystems SGIEX) equipped with an Atmospheric Pressure Ionization (API) source. Mass spectra were obtained using a Time Of Flight (TOF) analyser. X-ray diffraction: intensity data were collected on a Bruker-Nonius KappaCCD diffractometer using MoKα radiation (0.71073 Å) at 293(2) K. Data reduction was performed using the HKL-2000 software package. The structure was resolved using the software SIR9237 by direct methods and refined using SHELXL-97.38 For compound 5d, intensity data were collected on an Agilent SuperNova AtlasS2 diffractometer using MoKα radiation (0.71073 Å) at 293(2) K. Data reduction was performed using the CrysAlisPro software package (version 1.171.37.31). The structure was resolved using the software SHELXS-97 by direct methods and refined using SHELXL-2013-4. The CIF files of compounds 5b–d, 5h, 5i, 5k and 5l have been deposited with CCDC numbers 1434226–1434232, respectively.General procedure for the synthesis of complexes [RuCl2(η6-arene)(PA)] 5
In a Schlenk flask, a solution of ruthenium dimer [RuCl2(η6-p-cymene)]2 (61.2 mg, 0.1 mmol, 2 equiv. of ruthenium) and secondary phosphine oxide (0.22 mmol, 2.2 equiv.) in THF (2 mL) was stirred at room temperature for 3 h. The reaction mixture was half-concentrated and n-hexane (10 mL) was added to initiate precipitation. The red precipitate was filtered off and washed with n-hexane. Recrystallisation from DCM/n-hexane gave crystals of the desired product.Acknowledgements
This work was supported by the Ministère de l'enseignement supérieur et de la recherche (L. G. Ph.D. grant), the CNRS, AMU, and Centrale Marseille. Dr Valérie Monnier and Christophe Chendo (Spectropole, Fédération des Sciences Chimiques de Marseille) are gratefully acknowledged for MS analyses. We thank Matthieu Ortega for the preparation of SPOs, Dr Alphonse Tenaglia for fruitful discussions and Dr Julie Broggi for useful comments on the manuscript. We are indebted to both referees for relevant comments and suggestions.Notes and references
- For selected recent reviews on C–H activation, see: (a) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107–1295 RSC; (b) G. Qiu and J. Wu, Org. Chem. Front., 2015, 2, 169–178 RSC; (c) L. Yang and H. Huang, Chem. Rev., 2015, 115, 3468–3417 CrossRef CAS PubMed; (d) B. Liu, F. Hu and B.-F. Shi, Dalton Trans., 2015, 5, 1883–1881 Search PubMed; (e) B. Li and P. H. Dixneuf, Chem. Soc. Rev., 2013, 42, 5744–5767 RSC; (f) J. J. Mousseau and A. B. Charette, Acc. Chem. Res., 2013, 46, 412–424 CrossRef CAS PubMed; (g) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236–10254 CrossRef CAS PubMed; (h) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS PubMed; (i) K. Hirano and M. Miura, Synlett, 2011, 294–307 CAS; (j) J. Wencel-Delord, T. Droge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740–4761 RSC; (k) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed; (l) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885–1898 RSC; (m) R. Jazzar, J. Hitce, A. Renaudat, J. Sofack-Kreutzer and O. Baudoin, Chem. – Eur. J., 2010, 16, 2654–2672 CrossRef CAS PubMed.
- (a) N. Dastbaravardeh, M. Christakakou, M. Haider and M. Schnuerch, Synthesis, 2014, 1421–1439 Search PubMed; (b) R. Giri, S. Thapa and A. Kafle, Adv. Synth. Catal., 2014, 356, 1395–1411 CrossRef CAS; (c) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed; (d) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Commun., 2010, 46, 677–685 RSC; (e) X. Chen, K. M. Engle, D. H. Wang and J. Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed.
- (a) F. Hu, Y. Xia, C. Ma, Y. Zhang and J. Wang, Chem. Commun., 2015, 51, 7986–7995 RSC; (b) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651–3678 RSC; (c) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed.
- (a) L. Ackermann, Acc. Chem. Res., 2014, 47, 281–295 CrossRef CAS PubMed; (b) V. S. Thirunavukkarasu, S. I. Kozhushkov and L. Ackermann, Chem. Commun., 2014, 50, 29–39 RSC; (c) S. I. Kozhushkov and L. Ackermann, Chem. Sci., 2013, 4, 886–896 RSC; (d) P. B. Arockiam, C. Bruneau, P. H. Dixneuf and H. Pierre, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed.
- (a) X.-H. Cai and B. Xie, Synthesis, 2015, 737–759 CrossRef CAS; (b) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074–1086 CrossRef CAS PubMed.
- U. Fekl and K. I. Goldberg, Adv. Inorg. Chem., 2003, 54, 259–320 CrossRef CAS.
- (a) S. A. Johnson, Dalton Trans., 2015, 44, 10905–10913 RSC and references cited therein; (b) N. Yoshikai, Chem. Rec., 2011, 11, 242–251 CrossRef PubMed.
- K. Gao and N. Yoshikai, Acc. Chem. Res., 2014, 47, 1208–1219 CrossRef CAS PubMed.
- P. L. Arnold, M. W. McMullon, J. Rieb and F. E. Kühn, Angew. Chem., Int. Ed., 2015, 54, 82–100 CrossRef CAS PubMed.
- For a recent review, see: (a) M. Catellani, E. Motti and N. Della, Acc. Chem. Res., 2008, 41, 1512–1522 CrossRef CAS PubMed. For early discovery and significant development of Catellani reaction: (b) M. Catellani and G. P. Chiusoli, J. Organomet. Chem., 1982, 239, C35–C37 CrossRef CAS; (c) M. Catellani and M. C. Fagnola, Angew. Chem., Int. Ed. Engl., 1994, 33, 2421–2422 CrossRef; (d) M. Catellani, F. Frignani and A. Rangnoni, Angew. Chem., Int. Ed. Engl., 1997, 36, 119–122 CrossRef CAS; (e) M. Lautens and S. Piguel, Angew. Chem., Int. Ed., 2000, 39, 1045–1046 CrossRef CAS; (f) F. Faccini, E. Motti and M. Catellani, J. Am. Chem. Soc., 2004, 126, 78–79 CrossRef CAS PubMed; (g) C. Bressy, D. Alberico and M. Lautens, J. Am. Chem. Soc., 2005, 127, 13148–13145 CrossRef CAS PubMed; (h) B. Mariampillai, J. Alliot, M. Li and M. Lautens, J. Am. Chem. Soc., 2007, 129, 15372–15379 CrossRef CAS PubMed; (i) A. Martins, B. Mariampillai and M. Lautens, Top. Curr. Chem., 2009, 292, 1–33 CrossRef; (j) N. Della, C. G. Maestri and M. Catellani, Chem. – Eur. J., 2009, 15, 7850–7853 CrossRef PubMed; (k) P.-X. Shen, X.-C. Wang, P. Wang, R.-Y. Zhu and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 11574–11577 CrossRef CAS PubMed.
- (a) V. I. Sokolov, L. L. Troitskaya and O. A. Reutov, J. Organomet. Chem., 1979, 182, 537–546 CrossRef CAS; (b) D. L. Davies, S. M. A. Donald and S. A. Macgregor, J. Am. Chem. Soc., 2005, 127, 13754–13755 CrossRef CAS PubMed; (c) W. J. Tenn III, K. J. H. Young, G. Bhalla, J. Oxgaard, W. A. Goddard III and R. A. Periana, J. Am. Chem. Soc., 2005, 127, 14172–14189 CrossRef PubMed; (d) Y. Feng, M. Lail, K. A. Barakat, T. R. Cundari, T. B. Gunnoe and J. L. Petersen, J. Am. Chem. Soc., 2005, 127, 14174–14175 CrossRef CAS PubMed.
- (a) D. Garcia-Cuadrado, A. A. C. Braga, F. Maseras and A. M. Echavarren, J. Am. Chem. Soc., 2006, 128, 1066–1067 CrossRef CAS PubMed; (b) D. García-Cuadrado, P. de Mendoza, A. A. C. Braga, F. Maseras and A. M. Echavarren, J. Am. Chem. Soc., 2007, 129, 6880–6886 CrossRef PubMed; (c) M. Lafrance and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 16496–16497 CrossRef CAS PubMed; (d) S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 10848–10849 CrossRef CAS PubMed; (e) K. M. Engle, P. S. Thuy-Boun, M. Dang and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 18183–18193 CrossRef CAS PubMed.
- (a) L. N. Lewis and J. F. Smith, J. Am. Chem. Soc., 1986, 108, 2728–2735 CrossRef CAS; (b) E. J. Moore, W. R. Pretzer, T. J. O'Connell, J. Harris, L. LaBounty, L. Chou and S. S. Grimmer, J. Am. Chem. Soc., 1992, 114, 5888–5890 CrossRef CAS; (c) S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529–531 CrossRef CAS; (d) F. Kakiuchi, S. Kan, K. Igi, N. Chatani and S. Murai, J. Am. Chem. Soc., 2003, 125, 1698–1699 CrossRef CAS PubMed.
- For selected references, see: (a) S. Oi, Y. Fukita, N. Watanuli, S. Miyano and Y. Inoue, Org. Lett., 2001, 3, 2579–2581 CrossRef CAS PubMed; (b) I. Özdemir, S. Demir, B. Çetinkaya, C. Gourlauoen, F. Maseras, C. Bruneau and P. H. Dixneuf, J. Am. Chem. Soc., 2008, 130, 1156–1157 CrossRef PubMed; (c) P. B. Arochiam, C. Fischmeister, C. Bruneau and P. H. Dixneuf, Angew. Chem., Int. Ed., 2010, 49, 6629–6632 CrossRef PubMed; (d) E. Ferrer Flegeau, C. Bruneau, P. H. Dixneuf and A. Jutand, J. Am. Chem. Soc., 2011, 133, 10161–10170 CrossRef PubMed; (e) S. R. Chipudi, I. Khan and H. W. Lam, Angew. Chem., Int. Ed., 2012, 51, 12115–12119 CrossRef PubMed; (f) J. Zhang and T.-P. Loh, Chem. Commun., 2012, 48, 11232–11234 RSC; (g) M. C. Reddy and M. Jeganmohan, Chem. Commun., 2013, 49, 481–483 RSC; (h) R. K. Chinnagolla, S. Pimparkar and M. Jeganmohan, Chem. Commun., 2013, 49, 3703–3705 RSC; (i) G. Rouquet and N. Chatani, Chem. Sci., 2013, 4, 2201–2208 RSC; (j) P. B. Arochiam, C. Fischmeister, C. Bruneau and P. H. Dixneuf, Green Chem., 2013, 15, 67–71 RSC; (k) D. G. Johnson, J. M. Lynam, N. S. Mistry, J. M. Slattery, R. J. Thatcher and A. C. Whitwood, J. Am. Chem. Soc., 2013, 135, 2222–2234 CrossRef CAS PubMed; (l) N. Hofmann and L. Ackermann, J. Am. Chem. Soc., 2013, 135, 5877–5884 CrossRef CAS PubMed; (m) M. Shang, S.-H. Zeng, S.-Z. Sun, H.-X. Dai and J.-Q. Yu, Org. Lett., 2013, 15, 5286–5289 CrossRef CAS PubMed; (n) P. M. Liu and C. G. Frost, Org. Lett., 2013, 15, 5862–5865 CrossRef CAS PubMed; (o) Z. Zhang, H. Jiang and Y. Huang, Org. Lett., 2014, 16, 5976–5979 CrossRef CAS PubMed; (p) S. Nakanowatari and L. Ackermann, Chem. – Eur. J., 2014, 20, 5409–5413 CrossRef CAS PubMed.
- (a) J. Bigeault, L. Giordano and G. Buono, Angew. Chem., Int. Ed., 2005, 44, 4753–4757 CrossRef CAS PubMed; (b) J. Bigeault, L. Giordano, I. De Riggi, Y. Gimbert and G. Buono, Org. Lett., 2007, 9, 3567–3570 CrossRef CAS PubMed; (c) T. Achard, L. Giordano, A. Tenaglia, Y. Gimbert and G. Buono, Organometallics, 2010, 29, 3936–3950 CrossRef CAS; (d) D. Martin, D. Moraleda, T. Achard, L. Giordano and G. Buono, Chem. – Eur. J., 2011, 17, 12729–12740 CrossRef CAS PubMed; (e) T. Achard, A. Lepronier, Y. Gimbert, H. Clavier, L. Giordano, A. Tenaglia and G. Buono, Angew. Chem., Int. Ed., 2011, 50, 3352–3356 CrossRef PubMed; (f) H. Clavier, A. Lepronier, N. Bengobesse-Mintsa, D. Gatineau, H. Pellissier, L. Giordano, A. Tenaglia and G. Buono, Adv. Synth. Catal., 2013, 355, 403–408 CAS; (g) F. Schröder, C. Tugny, E. Salanouve, H. Clavier, L. Giordano, D. Moraleda, Y. Gimbert, V. Mouriès-Mansuy, J.-P. Goddard and L. Fensterbank, Organometallics, 2014, 33, 4051–4056 CrossRef; (h) L. V. Graux, M. Giorgi, G. Buono and H. Clavier, Organometallics, 2015, 34, 1864–1871 CrossRef CAS; (i) P. Nava, H. Clavier, Y. Gimbert, L. Giordano, G. Buono and S. Humbel, ChemCatChem, 2015, 7, 3848–3854 CrossRef CAS.
- L. Ackermann, Org. Lett., 2005, 7, 3123–3125 CrossRef CAS PubMed.
- (a) L. Ackermann, A. Althammer and R. Born, Angew. Chem., Int. Ed., 2006, 45, 2619–2622 CrossRef CAS PubMed; (b) L. Ackermann, R. Born and P. Álvarez-Bercedo, Angew. Chem., Int. Ed., 2007, 46, 6364–6367 CrossRef CAS PubMed; (c) L. Ackermann, R. Vincente and A. Althammer, Org. Lett., 2008, 10, 3123–3125 Search PubMed; (d) L. Ackermann and M. Mulzer, Org. Lett., 2008, 10, 5043–5045 CrossRef CAS PubMed; (e) L. Ackermann and A. V. Lygin, Org. Lett., 2011, 13, 3332–3335 CrossRef CAS PubMed.
- R. Krafczyk, H. Thönnessen, P. G. Jones and R. Schmutzler, J. Fluorine Chem., 1997, 83, 159–166 CrossRef CAS.
- S. M. M. Knapp, T. J. Sherbow, R. B. Yelle, J. J. Juliette and D. R. Tyler, Organometallics, 2013, 32, 3744–3752 CrossRef CAS.
- E. Y. Y. Chan, Q.-F. Zhang, Y.-K. Sau, S. M. F. Lo, H. H. Y. Sung, I. D. Williams, R. K. Haynes and W.-H. Leung, Inorg. Chem., 2004, 43, 4921–4926 CrossRef CAS PubMed.
- E. Tomás-Mendivil, L. Menéndez-Rodriguez, J. Francos, P. Crochet and V. Cadierno, RSC Adv., 2014, 4, 63466–63474 RSC.
- During the redaction of the manuscript, the synthesis of other [RuCl2(η6−-arene)(PA)] complexes, including 5c was reported, see: E. Tomás-Mendivil, V. Cadierno, M. I. Menéndez and R. López, Chem. – Eur. J., 2015, 21, 16874–16886 CrossRef PubMed.
- For details, see ESI.†.
- H. Clavier and S. P. Nolan, Chem. Commun., 2010, 49, 841–861 RSC.
- (a) A. C. Hillier, W. J. Sommer, B. S. Yong, J. L. Petersen, L. Cavallo and S. P. Nolan, Organometallics, 2003, 22, 4322–4326 CrossRef CAS; (b) A. Poater, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, Eur. J. Inorg. Chem., 2009, 1759–1766 CrossRef CAS; (c) https://www.molnac.unisa.it/OMtools/sambvca.php .
- For selected examples, see: (a) N. Y. P. Kumar, R. Jeyachandran and L. Ackermann, J. Org. Chem., 2013, 78, 4145–4152 CrossRef PubMed; (b) M. Seki, RSC Adv., 2014, 4, 29131–29133 RSC.
- For a rare example of thorough comparison between aryl halides, see ref. 17b.
- K. Fagnou and M. Lautens, Angew. Chem., Int. Ed., 2002, 41, 26–47 CrossRef CAS.
- The ionic radii of Cl and I are 167 pm and 207 pm, and their covalent radii are 99 pm and 133 pm, respectively; J. E. Huheey, E. A. Keiter and R. L. Keiter, in Inorganic Chemistry: Principles of Structure and Reactivity, Harper Collins, New York, 1993, ch. 13 Search PubMed.
- B. Li, T. Roisnel, C. Darcel and P. H. Dixneuf, Dalton Trans., 2012, 41, 10934–10937 RSC.
- During the reviewing process of this manuscript, a similar mechanism was proposed by Ackermann, see: D. Zell, S. Warrataz, D. Gelman, S. J. Garden and L. Ackermann, Chem. – Eur. J., 2016, 22, 1248–1252 CrossRef CAS PubMed.
- L. Ackermann, R. Vicente, H. K. Potukuchi and V. Pirovano, Org. Lett., 2010, 12, 5032–5035 CrossRef CAS PubMed.
- C. A. Busacca, J. C. Lorenz, N. Grinberg, N. Haddad, M. Hrapchak, B. Latli, H. Lee, P. Sabila, A. Saha, M. Sarvestani, S. Shen, R. Varsolona, X. Wei and C. H. Senannayake, Org. Lett., 2005, 7, 4277–4280 CrossRef CAS PubMed.
- A. Leyris, J. Bigeault, D. Nuel, L. Giordano and G. Buono, Tetrahedron Lett., 2007, 48, 5247–5250 CrossRef CAS.
- T. L. Emmick and R. L. Letsinger, J. Am. Chem. Soc., 1968, 90, 3459–3465 CrossRef CAS.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343–350 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and spectral data. CCDC 1434226–1434232. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt04683a |
| This journal is © The Royal Society of Chemistry 2016 |