
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iron cyclopentadienone complexes derived from C2-symmetric bis-propargylic alcohols; preparation and applications to catalysis†
Roy
Hodgkinson‡
,
Alessandro
Del Grosso‡
,
Guy
Clarkson
and
Martin
Wills
*
Department of Chemistry, Warwick University, Coventry, CV4 7AL, UK. E-mail: m.wills@warwick.ac.uk; Tel: +44 (0)24 7652 3260
First published on 28th January 2016
Abstract
A series of complexes containing the iron-cyclopentadienone structure were prepared by cyclising bis-propargylic alcohols and their derivatives with iron pentacarbonyl. The resulting complexes were characterised and tested in the catalysis of ketone reduction and alcohol oxidation. The complexes are competent catalysts for ketone reduction and alcohol oxidations.
Introduction
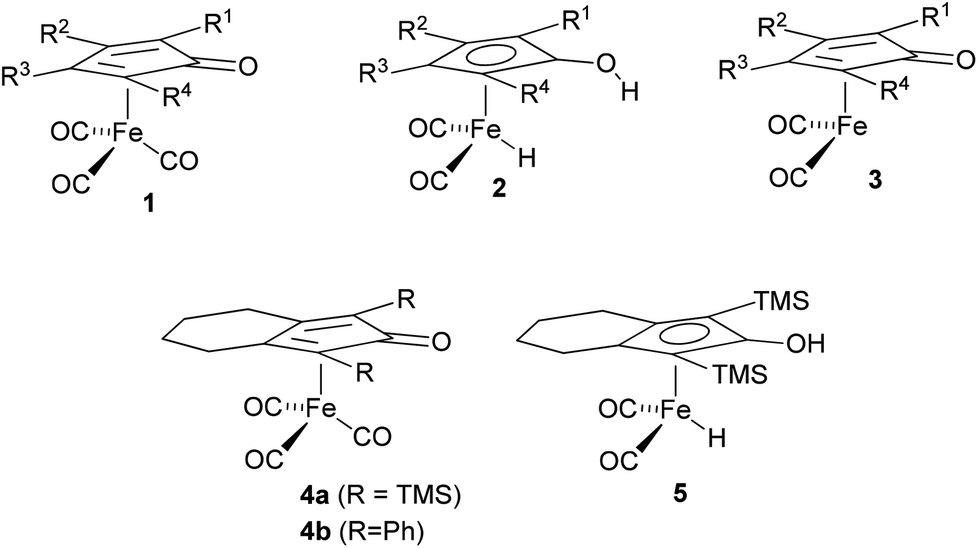
There is a growing interest in the application of iron-based complexes for the catalysis of asymmetric transformations, primarily due to its relative low cost and toxicity relative to more commonly used catalysts based on precious metals.1 Iron cyclopentadienone complexes (general structure 1) have recently emerged as promising reagents for hydrogenation reactions and for hydrogen transfer processes.2–13 Key to their application in this capacity is the formation of the derived hydrides of general structure 2, which can be achieved in situ using a number of activating agents, or through formation and isolation of the hydride prior to use. Complex 2 can transfer two atoms of hydrogen to an acceptor such as a ketone or imine and in doing so is converted to the unsaturated form 3. Complex 2 can be regenerated from 3 using a reducing agent such as formic acid or an alcohol (in the case of asymmetric transfer hydrogenation – ATH) or hydrogen gas (as in the case of asymmetric pressure hydrogenation – APH). The hydrogen transfers are believed to take place through a cyclic transition state as depicted in Fig. 1.13 The complexes have also been used in alkyne and alkene reductions, using a derivative in which the OH bond on the Cp ring is modified.14 | ||
| Fig. 1 Proposed mode of hydride transfer from iron hydride complexes to a ketone. | ||
| Entry | Application | Author, year | Catalyst used | Activator |
|---|---|---|---|---|
| 1 | C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O hydrogenation O hydrogenation |
Casey et al. 2007 and 2009![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4 4 |
5 | n/a |
| 2 | CO hydrogenation |
Berkessel et al. 20115 |
6 | UV light |
| 3 | CO reduction |
Wills et al. 20126a |
7 and derivatives. | TMAO |
| 4 | CO reduction – aldehyde and ketone |
Beller et al. 20127a,b |
4 | K2CO3 |
| 5 | CO reduction – using formaldehyde and H2O |
Wu et al. 201511 |
4 | Na2CO3 |
| 6 | CO reduction |
Gennari et al. 20158 |
8 | TMAO |
| 7 | CHOH oxidn | Funk et al. 20109a |
4 and derivatives | TMAO |
| 8 | CHOH oxidn | Guan et al. 20109b |
5 | n/a |
| 9 | CHOH oxidn | Williams et al. 20099c |
1 (all R = Ph) and 9. | H2O |
| 10 | CHOH oxidn | Wills et al. 20116b |
1 (all R = Ph) and derivatives of 7 | TMAO |
| 11 | Reductive amination | Quintard et al. 2012, 201310b,c,e |
4, and derivatives of 7, 9, 10, 11 | TMAO |
| 12 | CO and CN reduction |
Renaud et al. 201310d |
Cationic derivatives of 11 | TMAO |
| 13 | CN reduction |
Beller et al. 2013, 20117c,d |
5 | Phosphonic acid |
| 14 | Reductive hydroamination of alkynes | Beller et al. 20127e |
5 | Phosphonic acid |
| 15 | ‘Hydrogen borrowing’ | Feringa et al. 201412a |
4, derivative of 4 | TMAO |
| Zhao et al. 201512c |
||||
| 16 | ‘Hydrogen borrowing’ | Wills et al. 201512b |
1 (all R = Ph) | TMAO |
| 17 | Alkene and alkyne reduction | Nakazawa et al. 201414 |
Derivative of 5 | n/a OH of Cp is alkylated |
| 18 | Alkylation of beta-ketoester | Quintard et al. 201310a |
4 | TMAO |
| 19 | Amide to nitrile | Sortais et al. 201512d |
NHC-containing complex | UV light |
Results and discussion
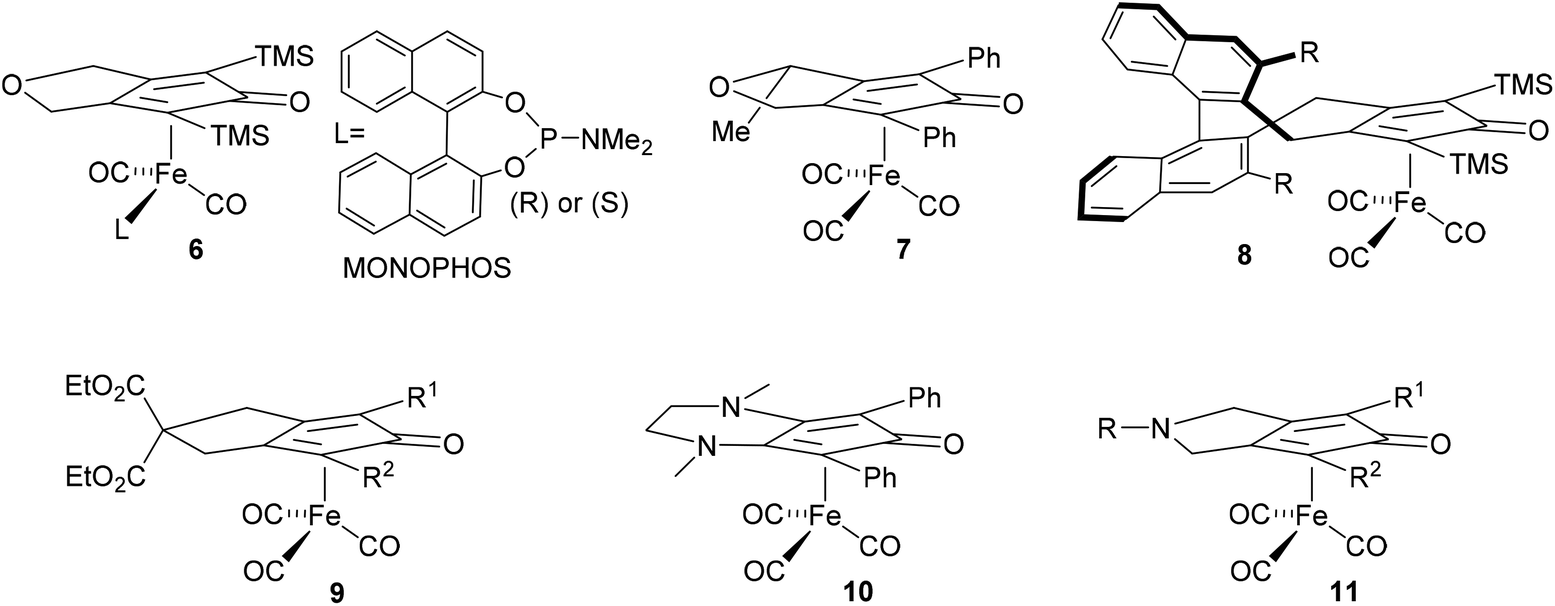
In earlier unrelated work, we demonstrated an efficient route to the synthesis of diol 12 in high enantioselectivity through the asymmetric reduction of the precursor diketone.16 Treatment of 12 with Fe(CO)5 (130 °C, 20 h, sealed tube) resulted in the formation of complex 13 in 76% yield. The breaking of the C2-symmetry of the substrate was clear in the product, with distinct signals in the 1H-NMR spectrum observed for each of the methine protons adjacent to the hydroxyl groups. In addition a racemic/meso mixture of diols was prepared by reduction of the precursor diketone with NaBH4. When this was cyclised in the same manner, the peaks of the racemic product could be observed but in addition the presence of the meso product was confirmed by the observation of extra methine resonances (ESI†). A complex with a hydroxyl group in the fused ring has been reported by Pearson et al.2c In addition, it was possible to prepare each of the O-protected ethers 14–17 and to convert these to the corresponding complexes 18–21 in the same manner (Scheme 1). Each of the catalysts were characterised by NMR, MS and IR analysis, following purification by chromatography on silica gel, to which the complexes are stable. As expected, characteristic peaks were observed in the 1H-NMR spectrum for the diastereoisomeric protecting groups. In addition, taking the OBn and OTBDMS derivatives as substrates, it was possible to substitute one CO for MONOPHOS using the TMAO-activated process described by Berkessel.5 Both diastereomeric combinations of the resulting complexes 22–25 were formed, i.e. derived from both enantiomers of MONOPHOS, in order to examine the possibility of matched and mismatched stereocontrol in subsequent applications. | ||
| Scheme 1 Synthesis of iron(cyclopentadienone) catalysts. Reagents and conditions: (i) NaH, THF, BnBr, tBu4NI, rt, 24 h (for 14); TBDMSCl, Imidazole, DMF, o/n, rt (for 15); TIPSCl, imidazole, DMF; o/n, rt (for 16); TBDPSCl, imidazole, DMF, o/n, rt (for 17). (ii) 3.0 eq. Fe(CO)5, toluene, 130 °C, 20 h. (iii) MONOPHOS (2.0 eq.), TMAO (2.0 eq.), toluene, 60 °C, o/n. | ||
Although complexes 22 and 23 appeared to be single isomers, each diastereoisomer of complexes 24 and 25 bearing OTBDMS groups were found to contain a ca. 10% of the other diastereoisomer. This suggested that either racemisation of the BINOL component during the preparation of MONOPHOS or during its subsequent complexation had taken place. Racemisation of the diether ligand prior to or during complexation seems unlikely as this would have been expected to lead to formation of some of the meso isomer, but this was not observed. The lack of diastereoisomeric impurities in 22 and 23 may be the result of purification from the minor diastereoisomers during the isolation procedure.
The complexes 13, 18–21 were first applied to the asymmetric reduction of the representative substrate acetophenone, using both asymmetric transfer hydrogenation (ATH) and pressure hydrogenation (APH) conditions. The complexes were also used in the oxidation of racemic 1-phenylethanol using acetone as a hydrogen acceptor. In all cases the active catalyst was generated in situ using methods previously reported, and in some cases comparisons with the unfunctionalised catalyst 4b2b were also made.2–12,15 In the ATH reactions (Table 2 shows selected results, further results are given in the ESI, Table S1†), at 60 °C using a 5:2 (molar) formic acid:triethylamine azeotrope (FA/TEA) and 10 mol% catalyst, full conversion was observed in several cases however the asymmetric inductions were extremely low and a significant amount of formate co-product was also formed, presumably through formylation of the initial alcohol product (confirmed to be of the same absolute configuration as the alcohol).6a TMAO was added to ensure full activation of the catalysts efficiently although, as demonstrated using unsubstituted 4b, it could be omitted from the reaction6a at the cost of a slower activation; a reaction complete in 24 h using TMAO reached just 52% conversion in the same time without TMAO (entries 1–3). In this respect, however the diol complex 13 appeared to be less sensitive to the additive (entries 4–7). Although the enantioselectivities were low, the OH and OBn complexes gave products of opposite configurations to those observed with the O-silylated complexes (entries 8–15). The use of lower catalyst loadings (5%, 1%) gave much lower conversions, as did lowering the temperature to 40 °C (ESI†). Another clear trend was the observation of improved ees when using the more hindered silyl-substituted complexes, for both the alcohol and formate products.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Loading/mol% | Activator | Time | Alcohol/% | Formate/% | Alcohol ee/% | Formate ee/% |
| a Reaction conditions; [S] = ca. 1.0 M, TMAO (1 eq. relative to complexes) unless otherwise indicated, % conversions are given and the balance is unreduced ketone. b [S] = 1.45 M. c Not determined. | ||||||||
| 1b | 4b | 10 | None | 24 h | 44 | 8 | — | — |
| 2b | 4b | 10 | None | 5 days | 78 | 22 | — | — |
| 3b | 4b | 10 | TMAO | 24 h | 87 | 13 | — | — |
| 4 | 13 | 10 | TMAO | 5 h | 19 | 5 | 9(S) | 6(S) |
| 5 | 13 | 10 | TMAO | 24 h | 78 | 14 | 8(S) | 8(S) |
| 6b | 13 | 10 | None | 24 h | 69 | 10 | 8(S) | 8(S) |
| 7b | 13 | 10 | TMAO | 24 h | 83 | 14 | 8(S) | 9(S) |
| 8 | 18 | 5 | TMAO | 24 h | 13 | 33 | 2(S) | 3(S) |
| 9 | 18 | 10 | TMAO | 24 h | 65 | 22 | 3(S) | 1(R) |
| 10 | 19 | 5 | TMAO | 24 h | 29 | 54 | 18(R) | 16(R) |
| 11 | 19 | 10 | TMAO | 24 h | 86 | 12 | 17(R) | 16(R) |
| 12 | 20 | 5 | TMAO | 24 h | 24 | 5 | 23(R) | |
| 13 | 20 | 10 | TMAO | 24 h | 76 | 23 | 22(R) | 24(R) |
| 14 | 21 | 5 | TMAO | 24 h | 24 | 26 | 21(R) | 22(R) |
| 15 | 21 | 10 | TMAO | 24 h | 79 | 12 | 24(R) | 18(R) |

A similar pattern emerged for the pressure hydrogenation reactions (APH; Table 3), although some unusual observations were made with respect to the method of activation. Control reactions run under nitrogen indicated that significant background transfer hydrogenation was also operating (entries 4–6, 12–14), however the conversions were lower. The similar ees (for 13) observed in the absence of hydrogen gas indicated that the catalyst was still operating in the reaction. Initially, unsubstituted complex 4b was tested and this gave 100% conversion under 30 bar hydrogen pressure at 80 °C in 24 h in an iPrOH/water solvent mixture, provided that an activator; either K2CO37a,b or TMAO,15 was added (entries 1–3). Given this precedent, K2CO3 was used in all the subsequent tests. Unfortunately, the substituted catalysts were not as active as 4b, giving much lower conversions under the same conditions (entries 8, 17, 24, 30). No advantage was gained from running the reactions at the higher temperature of 100 °C (entries 9, 18, 21, 25, 30); catalyst decomposition was suspected) or for a longer time (72 h) at a lower temperature of 60 °C (entries 16, 20, 23, 28). Whilst most of the other complexes behaved in a similar manner to 4b, requiring some form of activation for best results, for diol-containing complex 13, omission of the K2CO3 (and no other activator) resulted in full acetophenone reduction (entry 7), as did the use of 1 mol% of TMAO (entry 10). The use of 1.5 mol% of TMAO, however, gave a lower conversion, possibly due to partial catalyst decomposition (entry 11). The use of 1 mol% of TMAO also proved to be the most effective way to activate the other catalysts where tested (entries 19, 26, 31). The asymmetric induction was not improved in either case however and remained modest, not exceeding 20% ee in any case, although again the more hindered silylated complexes gave the best enantioselectivities. This may reflect the distant separation of the chiral centres from the likely reduction centre, which requires some further optimisation to extend its influence to the transition state of the reduction; the trend in the results suggests that a further increase in the steric hindrance of the groups on the ‘bridging C-4’ unit could provide a route to such improvements.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Activator | Time/h | T/°C | Conv./% | eeb/% |
| a Reaction conditions; 1 mol% catalyst, [S] = ca.. 1.0 M unless otherwise indicated, 30 bar H2, iPrOH/H2O (0.5 mL/0.2 mL) used as solvent. b [S] = 1.9 M. c Control reaction run under nitrogen (1 atm) with no hydrogen present. | ||||||
| 1b | 4b | None | 18 | 80 | 11 | — |
| 2b | 4b | K2CO3 (5%) | 18 | 80 | 100 | — |
| 3b | 4b | TMAO (1%) | 18 | 80 | 100 | — |
| 4b,c | 4b | None | 18 | 80 | 1 | — |
| 5b,c | 4b | K2CO3 (5%) | 18 | 80 | 51 | — |
| 6b,c | 4b | TMAO (1%) | 18 | 80 | 69 | — |
| 7b | 13 | None | 18 | 80 | 100 | 5(S) |
| 8b | 13 | K2CO3 (5%) | 18 | 80 | 18 | 9(S) |
| 9 | 13 | K2CO3 (5%) | 24 | 100 | 7 | 3(S) |
| 10b | 13 | TMAO (1%) | 18 | 80 | 100 | 8(S) |
| 11b | 13 | TMAO (1.5%) | 18 | 80 | 79 | 6(S) |
| 12b,c | 13 | None | 18 | 80 | 52 | 8(S) |
| 13b,c | 13 | K2CO3 (5%) | 18 | 80 | 6 | 7(S) |
| 14b,c | 13 | TMAO (1%) | 18 | 80 | 53 | 8(S) |
| 15b | 18 | None | 18 | 80 | 55 | 7(S) |
| 16 | 18 | K2CO3 (5%) | 72 | 60 | 29 | 9(S) |
| 17b | 18 | K2CO3 (5%) | 18 | 80 | 93 | 8(S) |
| 18 | 18 | K2CO3 (5%) | 24 | 100 | 18 | 6(S) |
| 19b | 18 | TMAO (1%) | 18 | 80 | 100 | 7(S) |
| 20 | 19 | K2CO3 (5%) | 72 | 60 | 34 | 13(R) |
| 21 | 19 | K2CO3 (5%) | 24 | 100 | 31 | 11(R) |
| 22 | 20 | None | 18 | 80 | 42 | 14(R) |
| 23 | 20 | K2CO3 (5%) | 72 | 60 | 8 | 19(R) |
| 24 | 20 | K2CO3 (5%) | 18 | 80 | 59 | 15(R) |
| 25 | 20 | K2CO3 (5%) | 24 | 100 | 9 | 13(R) |
| 26 | 20 | TMAO (1%) | 18 | 80 | 100 | 13(R) |
| 27 | 21 | None | 18 | 80 | 31 | 20(R) |
| 28 | 21 | K2CO3 (5%) | 72 | 60 | 25 | 19(R) |
| 29 | 21 | K2CO3 (5%) | 18 | 80 | 34 | 18(R) |
| 30 | 21 | K2CO3 (5%) | 24 | 100 | 12 | 6(R) |
| 31 | 21 | TMAO (1%) | 18 | 80 | 72 | 15(R) |

Given the unexpected result obtained when K2CO3 was omitted from the reaction with diol catalyst 13, we questioned whether the addition of water to a dry solvent could reproduce this effect. This proved to be the case; in dry THF, 31% reduction of acetophenone was observed after 18 h at 80 °C, possibly due to trace amounts of adventitious water, however as increasing amounts of water were added, the conversion increased, reaching 99% when 100 mol% (1 eq.) relative to substrate, and 100% when a 5/2 THF/water solvent mixture was used – although no change to the ee was observed (ESI, Table S2†). A 1H-NMR study of the reaction revealed the formation of an iron hydride complex, suggesting that water was initiating the formation of the active species. A similar effect, although to a lesser extent, created by addition of water was also observed when toluene was used as solvent (ESI, Table S3†).
Reductions of acetyl cyclohexane (ATH or APH) and of 3,3-dimethyl-2-butanone (pinacolone, ATH) also worked using 10 mol% catalyst although the products were racemic or of low ee (see ESI, Tables S5 and S6†). In the APH of 3,3-dimethyl-2-butanone, using 1 mol% of catalyst 13 and 18–21, (60 °C, 72 h, 30 bar H2), conversions were generally low (ESI, Table S7†). Due to the low conversions, accurate ees could not be determined, however these were generally in the range of 30–38%.
The complexes proved to be efficient at the catalysis of alcohol oxidation using acetone as hydrogen acceptor, a process which has been reported for a number of iron(cyclopentadienyl) catalysts.6b,9 However both enantiomers of substrate were oxidised with little selectivity (Table 4), although with excellent conversions, particularly when 10 mol% of catalyst was used. The catalyst loading could be reduced to 5 mol% in some cases although at 1 mol% loading, incomplete conversion was observed (ESI, Table S4†). The observed enantiomeric excesses indicated that no significant level of kinetic resolution was taking place in the oxidations.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Loading/mol% | Time/h | Alcohol/% | Ketone/% | Alcohol ee/% |
| a Reaction conditions; 60 °C, [S] = 0.19 M, acetone solvent, TMAO (1 eq. relative to complexes). | ||||||
| 1 | 13 | 10 | 5 | 53 | 47 | 8(R) |
| 2 | 13 | 10 | 24 | 4 | 96 | 28(R) |
| 3 | 18 | 10 | 5 | 15 | 85 | 12(R) |
| 4 | 18 | 10 | 24 | 0 | 100 | n/a |
| 5 | 18 | 5 | 5 | 12 | 88 | 20(R) |
| 8 | 19 | 10 | 5 | 3 | 97 | 31(S) |
| 9 | 19 | 10 | 24 | 0 | 100 | n/a |
| 10 | 19 | 5 | 5 | 50 | 50 | 6(S) |
| 11 | 19 | 5 | 24 | 0 | 100 | n/a |
| 14 | 20 | 10 | 24 | 4 | 96 | 5(R) |
| 15 | 20 | 5 | 24 | 68 | 32 | 5(S) |
| 17 | 21 | 10 | 5 | 8 | 92 | 45(S) |
| 18 | 21 | 10 | 24 | 0 | 100 | n/a |
| 19 | 21 | 5 | 5 | 82 | 18 | 2(S) |
| 20 | 21 | 5 | 24 | 0 | 100 | n/a |

The design of the MONOPHOS-containing catalysts 22–25 was anticipated to be capable of delivering improved results since; (i) MONOPHOS is known to be compatible with complexes of this type and (ii) the additional element of chirality in the ligand could be matched or mismatched to that on the cyclohexyl ring.5 In the event, 22–25 exhibited very low activity in the applications in which they were tested, despite the use of several methods to activate them. Results for the ATH of acetophenone and 3,3-dimethyl-2-butanone with the MONOPHOS complexes are given in the ESI (Table S8†). Using 10 mol% catalyst, for 24 h at 25–60 °C, and activation with TMAO, conversions were below ca. 7%. Performing the reaction using TMAO (in the dark or in the presence of light), or blue or UV light (365 nm) to activate the precatalyst, no improvement was observed. In one case, the use of the OTBDMS 24 catalyst (60 °C, 24 h) in the reduction of acetophenone gave 12.2% alcohol (18.0% ee) and 8.9% formate (12.4% ee), both of R configuration. The same outcome was observed in pressure hydrogenation tests. Using 1 mol% catalyst, 60 °C, 72 h, gave <5% conversion and ees of less than 10% (ESI, Table S9†).
To examine the effect of a phosphine we added increasing amounts of triphenylphosphine to 1 mol% of 13 under APH conditions (ESI, Table S10†). In this case, as the amount of PPh3 increased, the conversion (80 °C, 18 h) decreased and with 2% PPh3 relative to 1 mol% catalyst 13, no reduction was achieved. This may indicate the formation of a less reactive phosphine-containing species similar to 22–25. This study was also carried out using MONOPHOS as an additive and a similar reduction in activity was observed although the enantioselectivity was not significantly changed (ESI, Table S11†). Funk and Moyer have described closely related complexes which demonstrate lower activity towards hydrogen transfer when a CO ligand was replaced by a phosphine, and our results mirror these.9a
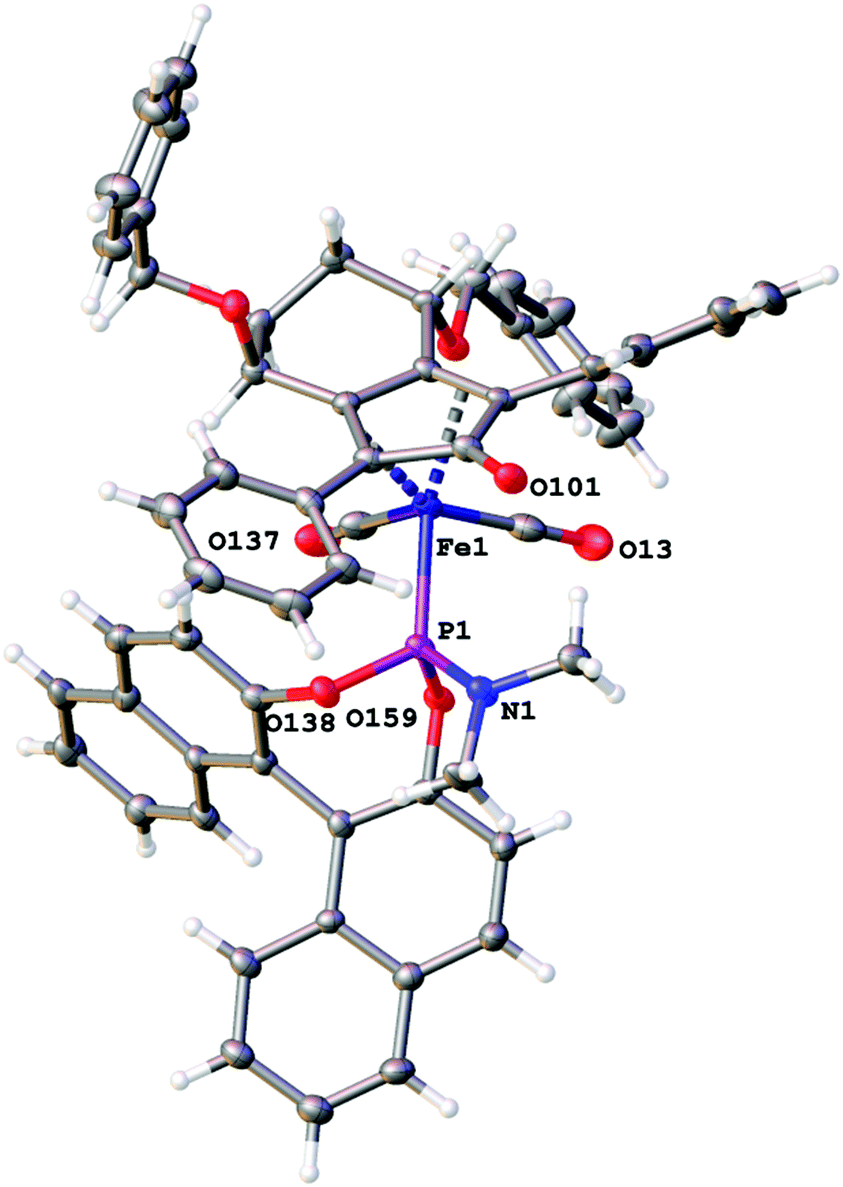
The X-ray crystallographic structures of complexes 4b2b (Fig. 2), 22 (Fig. 3) and 23 (Fig. 4) were obtained (see ESI† for full details). In the case of 23, two independent molecules of similar conformation were observed (see ESI† for full detail of each structure). Both complexes 22 and 23 are quite congested and hindered, particularly in comparison with 4b, which benefits from a more ‘open’ structure. This may account for the observed low reactivity of the derived hydride from the MONOPHOS-containing complexes, not only in comparison with 4b, but also compared to complex 6 reported by Berkessel, which does not contain substituents on the carbon backbone.
 | ||
| Fig. 2 The X-ray crystallographic structure of 4b (CCDC 1431241). | ||
 | ||
| Fig. 3 The X-ray crystallographic structure of complex (SS,S)-22 (CCDC 1431242). | ||
 | ||
| Fig. 4 The X-ray crystallographic structure of complex (SS,R)-23 (CCDC 1431243). | ||
In order to investigate in more detail the reasons for the slow reactivity of 22–25 an 1H-NMR experiment was carried out using catalyst 24 under hydrogenation conditions in a sealed NMR tube (Scheme 2). Upon irradiation over a period of 4 h, a major signal for an iron hydride 26 was observed at −12.11 ppm (J = 88.6 Hz) and a minor doublet at −12.18 ppm (J = 80.1 Hz) in ratio of ca. 12:1 which are tentatively assigned as diastereoisomers at the Fe atom based on analogy with Berkessel's observations.5 There was also a further doublet at −11.40 ppm tentatively assigned to the hydride from complex 25 present in 24. This was subsequently confirmed by independent formation of the hydride from complex 25 (see ESI†).
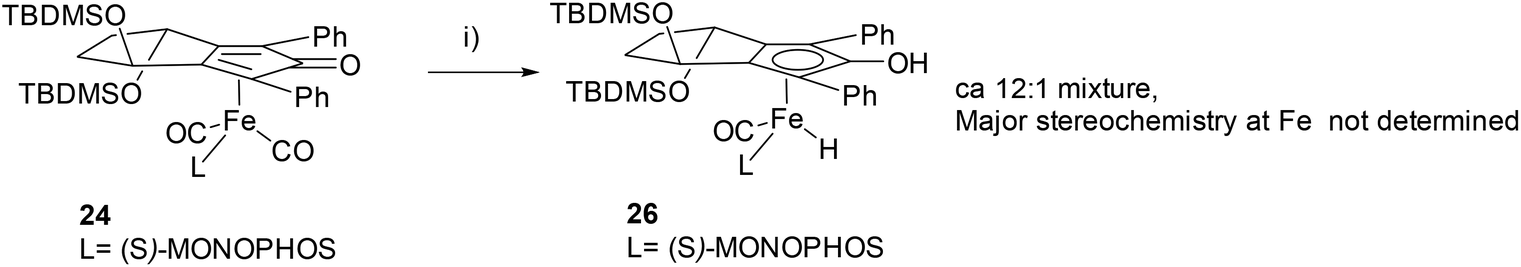 | ||
| Scheme 2 Generation of hydride from iron(cyclopentadienone) catalyst 24. Reagents and conditions: (i) irradiation (365 nm), 4 h, sealed tube, 4 bar H2 gas. | ||
In our case the high ratio of Fe isomers did not translate into a high enantioselectivity in the reduction. However a sample of added acetophenone was not significantly reduced in the reaction after heating the reaction to 80 °C for 3 days, whilst the peak at −12.18 ppm became the major species, together with some additional peaks which corresponded to 24 although this was not reisolated (see ESI†). This strongly indicates that whilst an iron hydride does form in the reaction, the transfer of hydrogen from the complex to the substrate is very slow. Likewise the high level of steric hindrance created in the complex through the introduction of a bulky phosphine may also explain the observed dramatic reduction in reactivity upon addition of triphenylphosphine in the APH tests described above. However, electronic effects could also be important; a computational study on this class of complex13c has revealed that increasing the acidity of the CpOH group can contribute to greater activity. Hence replacement of the electron-withdrawing CO with either PPh3 or MONOPHOS could reduce the acidity of the CpOH and hence reduce catalytic activity. The authors of the computational study indicated that phosphines containing electron-donating groups, of moderate steric size, could potentially increase the CpOH acidity and hence the activity, and this remains the subject of future studies. Similar observations have been made in experimental studies on the ruthenium analogues of the iron catalysts used in this study; in particular the rapid and reversible formation of a bond from the substrate carbonyl to the OH of the CpOH has been shown to be the first step of the catalytic mechanism.17
In conclusion, we have prepared a series of enantiomerically-pure (cyclopentadienone)iron complexes through the cyclisation of a C2-symmetric diol and its derivatives. Replacement of a CO with a phosphorus-donor ligand has also been achieved. The complexes are competent catalysts for the reduction of ketones under a range of conditions and for alcohol oxidation however no significant enantiomeric inductions were achieved in these transformations. An ideal catalyst might benefit from a balance between the size of the phosphorus-donor and any groups on the other part of the complex.
Experimental section
General
Solvents and reagents for the synthesis of complexes and catalytic reactions were degassed prior to use and all reactions were carried out under either a nitrogen or argon atmosphere. All heated experiments were conducted using thermostatically controlled oil baths. Reactions were monitored by TLC using aluminum backed silica gel 60 (F254) plates, visualized using UV 254 nm and phosphomolybdic acid (PMA), potassium permanganate or vanillin dips as appropriate. Flash column chromatography was carried out routinely using 60 micrometer silica gel. Reagents were used as received from commercial sources unless otherwise stated. 1H NMR spectra were recorded on a Bruker DPX (300, 400 or 500 MHz) spectrometer. Chemical shifts are reported in δ units, parts per million relative to the singlet at 7.26 ppm for chloroform and 0.00 ppm for TMS. Coupling constants (J) are measured in Hertz. IR spectra were recorded on a Perkin-Elmer Spectrum One FT-IR Golden Gate. Mass spectra were recorded on a Bruker Esquire2000 or a Bruker MicroTOF mass spectrometer. Melting points were recorded on a Stuart Scientific SMP 1 instrument and are uncorrected. GC analysis was performed using a Hewlett Packard 5890. Dry solvents were purchased and used as received.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) 16.
16.
In a flask 1,8-diphenylocta-1,7-diyne-3,6-dione16 (2.66 g, 9.79 mmol, 1 eq.) was added and dissolved in DCM (9.8 mL) and azeotrope formic acid/triethylamine (5
 :2 mixture; 6.5 mL). To this mixture was added (S,S)-Teth-TsDpen RuCl16 (62 mg, 0.10 mmol, S/C: 100:1) and the mixture was heated to 35 °C and stirred for 20 h. The reaction was cooled to rt and a saturated solution of sodium hydrogen carbonate (50 mL) was added and the reaction was extracted with DCM (3 × 50 mL). The organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The crude material was further purified by column chromatography in 20% to 40% EtOAc/petroleum ether to yield (S,S)-12 as a orange oil, which was recrystallized from heptane to give a off-white solid (2.71 g, 9.33 mmol, 95%, ee: 99%, de: 97.6%). mp: 74–76 °C, (lit: 79–80 °C); IR(neat) 3211, 3054, 2960, 2924, 2069, 2005, 1979, 1662, 1653, 1597, 1559, 1489, 1456, 1442, 1337, 1268, 1177, 1157, 1070, 1041, 1026, 1010 cm−1; δH (500 MHz, CDCl3) 7.42–7.45 (4H, m, ArH), 7.28–7.33 (6H, m, ArH), 4.76 (2H, s, CHOH), 2.35 (1H, br. s, OH), 2.34 (1H, br. s, OH), 2.05–2.15 (4H, m, CH2); δC (125 MHz, CDCl3) 131.7 (4C), 128.5 (2C), 128.3 (4C), 122.5 (2C), 89.5 (2C), 85.3 (2C), 62.5 (2C), 33.4 (2C); m/z (ESI+) 313.1 (M + Na, 100%).
:2 mixture; 6.5 mL). To this mixture was added (S,S)-Teth-TsDpen RuCl16 (62 mg, 0.10 mmol, S/C: 100:1) and the mixture was heated to 35 °C and stirred for 20 h. The reaction was cooled to rt and a saturated solution of sodium hydrogen carbonate (50 mL) was added and the reaction was extracted with DCM (3 × 50 mL). The organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The crude material was further purified by column chromatography in 20% to 40% EtOAc/petroleum ether to yield (S,S)-12 as a orange oil, which was recrystallized from heptane to give a off-white solid (2.71 g, 9.33 mmol, 95%, ee: 99%, de: 97.6%). mp: 74–76 °C, (lit: 79–80 °C); IR(neat) 3211, 3054, 2960, 2924, 2069, 2005, 1979, 1662, 1653, 1597, 1559, 1489, 1456, 1442, 1337, 1268, 1177, 1157, 1070, 1041, 1026, 1010 cm−1; δH (500 MHz, CDCl3) 7.42–7.45 (4H, m, ArH), 7.28–7.33 (6H, m, ArH), 4.76 (2H, s, CHOH), 2.35 (1H, br. s, OH), 2.34 (1H, br. s, OH), 2.05–2.15 (4H, m, CH2); δC (125 MHz, CDCl3) 131.7 (4C), 128.5 (2C), 128.3 (4C), 122.5 (2C), 89.5 (2C), 85.3 (2C), 62.5 (2C), 33.4 (2C); m/z (ESI+) 313.1 (M + Na, 100%).
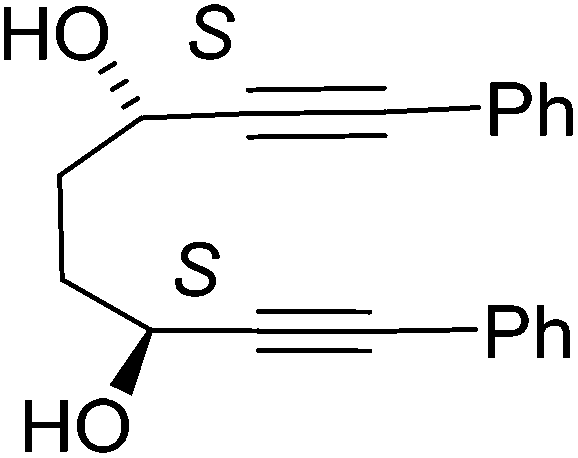
In a pressure tube (3S,6S)-1,8-diphenylocta-1,7-diyne-3,6-diol 12 (500 mg, 1.72 mmol, 1.00 eq.) and iron pentacarbonyl (1.01 g, 0.7 mL, 5.16 mmol, 3.00 eq.) were dissolved in anhydrous toluene (5 mL), the mixture was then degassed (bubbling nitrogen, 10 minutes) and the tube was sealed. The mixture was heated to 130 °C for 20 h. After cooling to rt, the mixture was diluted with EtOAc (10 mL) and passed through a celite plug washing through with EtOAc (100 mL). The solvent was removed and the product was purified by column chromatography in 5% to 10% EtOAc/petroleum ether to yield the product 13 as an orange solid (600 mg, 1.31 mmol, 76%). [α]24D = +244 (c 0.008 in CHCl3); mp: 97–99 °C (dec.); (found (EI): M+ + H, 459.0533. C24H19FeO6 requires M, 459.0526); IR(neat) 3051, 2951, 2064, 1990, 1702, 1657, 1539, 1500, 1441, 1358, 1262, 1217, 1189, 1096, 1069, 1029 cm−1; δH (500 MHz, CDCl3) 7.85–7.87 (2H, m, ArH), 7.78–7.79 (2H, m, ArH), 7.35–7.7.42 (6H, m, ArH), 5.09 (1H, br q, J = 4.7 Hz, CHOH), 4.83 (1H, br q, J = 3.4 Hz, CHOH), 2.37–2.42 (1H, m, CHH), 2.37 (1H, d, J = 3.1 Hz, OH), 2.28–2.33 (1H, m, CHH), 2.28 (1H, d, J = 3.4 Hz, OH), 1.85–1.90 (1H, m, CHH), 1.70–1.75 (1H, m, CHH); δC (125 MHz, CDCl3) 208.0 (3C), 169.7, 131.2, 130.7, 129.9 (2C), 129.3 (2C), 129.0 (2C), 128.8 (2C), 128.5, 128.4, 102.4, 102.2, 82.2, 80.2, 63.0, 61.9, 28.3, 26.8; m/z (ESI+) 481.0 (M + Na, 100%), 459.0 (80%).

A suspension of (3S,6S)-1,8-diphenylocta-1,7-diyne-3,6-diol 12 (300 mg, 1.03 mmol, 1.0 eq.) and sodium hydride (60% dispersion in mineral oil, 86 mg, 2.16 mmol, 2.1 eq.) in THF (10 mL) was stirred at rt for 30 min. Then benzyl bromide (369 mg, 257 μL, 2.16 mmol, 2.1 eq.) and tetrabutylammonium iodide (192 mg, 0.52 mmol, 0.5 eq.) were added and the mixture was stirred at rt for 24 h. The reaction was quenched using a saturated ammonium chloride solution (50 mL) and extracted with EtOAc (3 × 50 mL) and the combined organic extracts were washed with brine (3 × 50 mL). The crude material was purified by column chromatography on silica gel in petroleum ether to 15% EtOAc/petroleum ether to yield the product 14 as a yellow oil (448 mg, 0.95 mmol, 92%). [α]24D: −119 (c 0.54 in CHCl3); HRMS: (found (EI): M+ + Na, 493.2135. C34H30NaO2 requires M, 493.2138); IR(neat) 3062, 3030, 2927, 2856, 1598, 1572, 1489, 1453, 1442, 1389, 1370, 1332, 1280, 1254, 1226, 1203, 1178, 1149, 1086, 1066, 1026 cm−1; δH (500 MHz, CDCl3) 7.45–7.47 (4H, m, ArH), 7.39–7.41 (4H, m, ArH), 7.35–7.37 (3H, m, ArH), 7.30–7.34 (9H, m, ArH), 4.86 (2H, AB, JAB = 11.8 Hz, CHHPh), 4.60 (2H, AB, JAB = 11.8 Hz, CHHPh), 4.38–4.40 (2H, m, CHO), 2.07–2.17 (4H, m, CH2); δC (125 MHz, CDCl3) 138.0, (2C), 131.8 (4C), 128.4 (4C), 128.4 (2C), 128.3 (2C), 128.1 (4C), 127.7 (4C), 122.8 (2C), 88.0 (2C), 86.2 (2C), 70.8 (2C), 68.8 (2C), 31.7 (2C); m/z (ESI+) 493.2 (M+ Na, 100%), 509.2 (M + K − H, 45%).

In a pressure tube ((3S,6S)-3,6-bis(benzyloxy)octa-1,7-diyne-1,8-diyl)dibenzene 14 (450 mg, 0.96 mmol, 1.00 eq.) and iron pentacarbonyl (564 mg, 0.38 μL, 2.88 mmol, 3.00 eq.) were dissolved in anhydrous toluene (5 mL), the mixture was then degassed (bubbling nitrogen, 10 minutes) and the tube was sealed. The mixture was heated to 130 °C for 20 h. After cooling to rt and dilution with EtOAc (10 mL) it was passed through a celite plug washing through EtOAc (100 mL). Solvent was removed under reduced pressure and then the product was purified by column chromatography on silica gel in 5% to 20% EtOAc/petroleum ether to yield the product 18 as an orange solid (420 mg, 0.66 mmol, 69%). [α]24D = +321 (c 0.007 in CHCl3); mp: 66–68 °C (dec.); (found (EI): M+ + H, 639.1472. C38H31FeO6 requires M, 639.1465); IR(neat) 3059, 3030, 2928, 2860, 2063, 2009, 1984, 1635, 1540, 1498, 1439, 1393, 1363, 1319, 1213, 1081, 1051, 1026 cm−1; δH (500 MHz, CDCl3) 7.79–7.82 (2H, m, ArH), 7.71–7.74 (2H, m, ArH), 7.35–7.43 (7H, m, ArH), 7.29–7.32 (3H, m, ArH), 7.24–7.26 (2H, m, ArH), 7.19–7.20 (2H, m, ArH), 6.93–6.95 (2H, m, ArH), 4.69 (1H, br. s, CHO), 4.58 (1H, ABq, JAB = 10.7 Hz, CHH), 4.41 (1H, ABq, JAB = 10.7 Hz, CHH), 4.38 (1H, ABq, JAB = 10.7 Hz, CHH) 4.36 (1H, br. s, CHO), 4.12 (1H, ABq, JAB = 10.6 Hz, CHH), 2.13–2.23 (3H, m, CHHCH2), 2.08–2.11 (1H, m, CHH); δC (125 MHz, CDCl3) 208.2 (3C), 170.2, 137.0, 136.9, 131.4, 131.0, 130.1 (2C), 130.0 (2C), 128.4 (2C), 128.4 (2C), 128.4 (2C), 128.3 (2C), 128.0 (2C), 128.0 (4C), 127.9 (2C), 100.7, 99.9, 83.3, 81.8, 72.3, 71.7, 70.2, 67.8, 21.7, 21.5; m/z (ESI+) 661.1 (M + Na, 100%), 639.2 (43%).

In a flask (3S,6S)-1,8-diphenylocta-1,7-diyne-3,6-diol 12 (500 mg, 1.72 mmol, 1.0 eq.) was dissolved in DMF (20 mL). To the mixture were added imidazole (293 mg, 4.30 mmol, 2.5 eq.) and tert-butyl(chloro)dimethylsilane (648 mg, 4.30 mmol, 2.5eq.) and the mixture was stirred overnight at rt. The reaction was quenched with water (100 mL) and diluted with EtOAc (150 mL) and the organic fraction was washed with water (3 × 50 mL) and brine (3 × 50 mL) then dried using Na2SO4 then concentrated under reduced pressure. Purification by column chromatography on silica gel using 5% to 10% EtOAc/petroleum ether yielded the product 15 as a yellow oil (850 mg, 1.64 mmol, 95%). [α]23D: −34 (c 0.48 in CHCl3); HRMS: (found (EI): M+ + Na, 541.2931. C32H46NaO2Si2 requires M, 541.2929); IR(neat) 2954, 2928, 2885, 2855, 1598, 1471, 1462, 1443, 1407, 1388.9, 1361, 1338, 1281, 1251, 1188, 1083, 1004 cm−1; δH (500 MHz, CDCl3) 7.42–7.44 (4H, m, ArH), 7.29–7.32 (6H, m, ArH), 4.66–4.69 (2H, m, CHOC), 1.94–2.03 (4H, m, CH2), 0.95 (18H, s, tBu), 0.20 (6H, s, CH3), 0.17 (6H, s, CH3); δC (125 MHz, CDCl3) 131.6 (4C), 128.2 (4C), 128.1 (2C), 123.1 (2C), 90.8 (2C), 84.3 (2C), 63.2 (2C), 34.4 (2C), 25.9 (6C) 18.3 (2C), −4.3 (2C), −4.9 (2C); m/z (ESI+) 541.3 (M + Na, 100%).
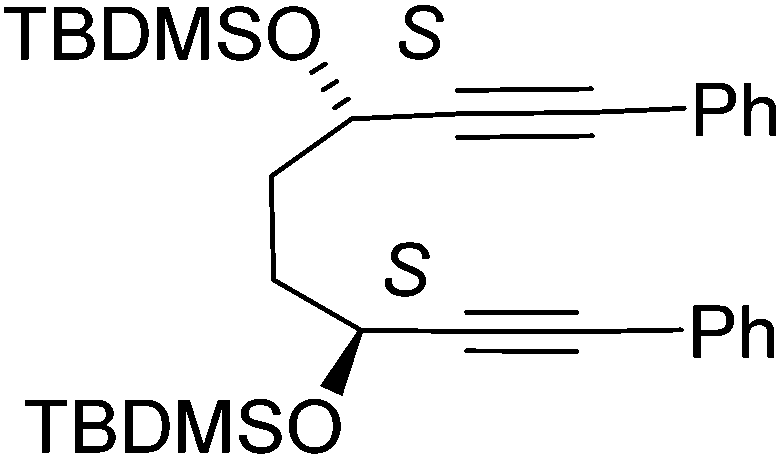
In a pressure tube ((3S,6S)-3,6-bis(tert-butyldimethylsilyloxy)octa-1,7-diyne-1,8-diyl)di benzene 15 (500 mg, 0.96 mmol, 1.0 eq.) and iron pentacarbonyl (564 mg, 0.38 μL, 2.88 mmol, 3.0 eq.) were dissolved in anhydrous toluene (5 mL). The mixture was then degassed (bubbling nitrogen, 10 minutes) and the tube was sealed. The mixture was heated to 130 °C for 20 h. After cooling to rt and dilution with EtOAc (10 mL) and passed through a celite plug washing through with EtOAc (100 mL), solvent was removed under reduced pressure and the product purified by column chromatography on silica gel using 5% to 10% EtOAc/petroleum ether to yield the product 19 as an orange solid (430 mg, 0.63 mmol, 65%). [α]24D = +321 (c 0.007 in CHCl3); mp: 73–75 °C (dec.); (found (EI): M+ + H, 687.2265. C36H47FeO6Si2 requires M, 687.2256); IR(neat) 2953, 2928, 2883, 2855, 2063, 2011, 1984, 1500, 1470, 1439, 1361, 1253, 1198, 1099, 1055, 1005 cm−1; δH (500 MHz, CDCl3) 7.72–7.74 (2H, m, ArH), 7.62–7.64 (2H, m, ArH), 7.40–7.43 (3H, m, ArH), 7.35–7.40 (2H, m, ArH), 7.29–7.34 (1H, m, ArH), 5.01–5.02 (1H, m, CHOTBDMS), 4.88–4.90 (1H, m, CHOTBDMS), 2.28–2.38 (2H, m, CHHCHH), 1.83–1.87 (1H, m, CHH), 1.76–1.80 (1H, m, CHH), 0.86 (9H, s, tBu), 0.71 (9H, s, tBu), 0.09 (3H, s, CH3), −0.006 (3H, s, CH3), −0.34 (3H, s, CH3), −0.58 (3H, s, CH3); δC (125 MHz, CDCl3) 208.6 (3C), 170.6, 131.2, 130.9, 130.7 (2C), 130.0 (2C), 128.5 (2C), 128.4 (2C), 128.1, 128.0, 103.6, 102.2, 83.9, 80.7, 63.4, 61.7, 27.5, 26.1, 26.1 (3C), 25.7 (3C), 18.3, 18.0, −4.4, −4.5, −5.1, −5.8; m/z (ESI+) 709.2 (M + Na, 100%), 678.2 (54%). 1018.

In a flask (3S,6S)-1,8-diphenylocta-1,7-diyne-3,6-diol 12 (500 mg, 1.72 mmol, 1.0 eq.) was dissolved in DMF (20 mL). To this solution was added imidazole (293 mg, 4.30 mmol, 2.5 eq.) and triisopropyl(chloro)silane (829 mg, 920 μL, 4.30 mmol, 2.5 eq.) and the mixture was left to stir overnight at rt. The reaction was quenched with water (50 mL) and diluted with EtOAc (50 mL) and the organic fraction was washed with water (3 × 50 mL) and brine (3 × 50 mL). The organic fraction was dried using Na2SO4 and concentrated under reduced pressure. Purification by column chromatography on silica gel using 2% to 3% EtOAc/petroleum ether yielded the product 16 as a yellow oil (620 mg, 1.03 mmol, 60%). [α]23D: −5 (c 0.46 in CHCl3); HRMS: (found (EI): M+ + Na, 625.3876. C38H58NaO2Si2 requires M, 625.3868); IR(neat) 2941, 2891, 2865, 1598, 1489, 1444, 1384, 1366, 1338, 1253, 1171, 1093, 1059, 1012 cm−1; δH (500 MHz, CDCl3) 7.39–7.41 (4H, m, ArH), 7.28–7.30 (6H, m, ArH), 4.80 (2H, br. s, CHOSi), 2.06–2.07 (4H, m, CH2), 1.15–1.20 (6H, m, CH(CH3)2), 1.12 (18H, d, J = 9.8 Hz, CH(CH3)2), 1.11 (18H, d, J = 9.8 Hz, CH(CH3)2); δC (125 MHz, CDCl3) 131.6 (4C), 128.2 (4C), 128.0 (2C), 123.2 (2C), 91.1 (2C), 84.2 (2C), 63.2 (2C), 34.4 (2C), 18.1 (12C), 12.3 (6C); m/z (ESI+) 625.4 (M + Na, 100%), 595.4 (65%).

In a pressure tube ((3S,6S)-3,6-bis(triisopropylsilyloxy)octa-1,7-diyne-1,8-diyl)dibenzene 16 (500 mg, 0.83 mmol, 1.00 eq.) and iron pentacarbonyl (488 mg, 0.33 μL, 2.49 mmol, 3.00 eq.) and dissolved in anhydrous toluene (5 mL), the tube was then degassed (bubbling nitrogen, 10 minutes) and the tube was sealed. The tube was heated to 130 °C for 20 h, then cooled to rt and diluted with EtOAc (10 mL). The solution was passed through a celite plug washing through EtOAc (100 mL). The solvent was removed under reduced pressure and the product was purified by column chromatography on silica gel in 5% to 30% EtOAc/petroleum ether to yield the product20 as a yellow solid (326 mg, 0.42 mmol, 51%). [α]29D: −136 (c 0.028 in CHCl3); mp: 57–59 °C (dec.); (found (EI): M+ + Na, 793.3019. C42H58FeNaO6Si2 requires M, 793.3015); IR(neat) 2942, 2890, 2865, 2063, 2012, 1982, 1650, 1501, 1462, 1446, 1364, 1136, 1104, 1054, 1014 cm−1; δH (500 MHz, CDCl3) 7.64–7.66 (2H, m, ArH), 7.51–7.53 (2H, m, ArH), 7.29–7.38 (6H, m, ArH), 5.24 (1H, t, J = 4.6 Hz, CHOTIPS), 5.21 (1H, t, J = 3.7 Hz, CHOTIPS), 2.32–2.45 (2H, m, CHHCHH), 1.90–1.96 (1H, m, CHH), 1.81–1.87 (1H, m, CHH), 0.93–0.94 (9H, m, TIPS), 0.86–0.90 (30H, m, TIPS), 0.61 (3H, sept, J = 7.5 Hz, CH(CH3)); δC (125 MHz, CDCl3) 208.5 (3C), 170.8, 131.3 (2C), 131.1, 131.0, 130.1 (2C), 128.4 (2C), 128.2 (2C), 128.0, 127.8, 104.4, 102.8, 85.3, 80.7, 63.8, 63.1, 29.0, 28.4, 18.3 (6C), 18.0 (6C), 13.1 (3C), 12.9 (3C);m/z (ESI+) 793.3 (M + Na, 100%), 771.3 (86%).
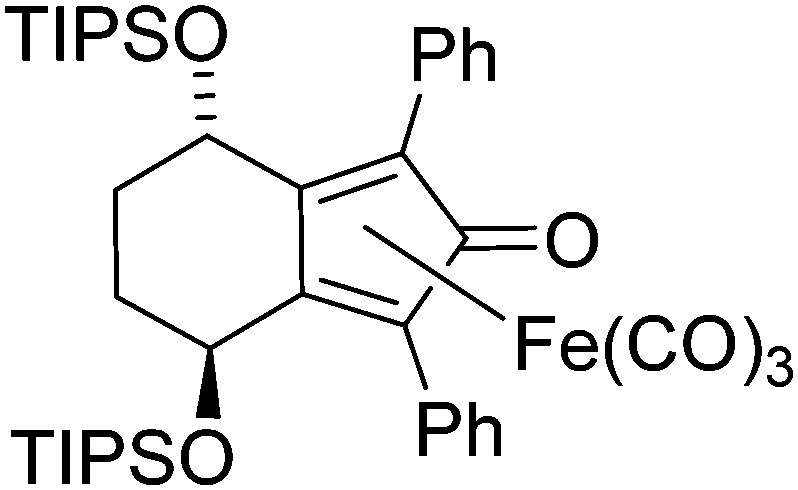
In a flask (3S,6S)-1,8-diphenylocta-1,7-diyne-3,6-diol 12 (500 mg, 1.72 mmol, 1.0 eq.) was added and dissolved in DMF (20 mL). To the solution was added imidazole (293 mg, 4.30 mmol, 2.5 eq.) and tert-butyl(chloro)diphenylsilane (1.18 g, 1.15 mL, 4.30 mmol, 2.5 eq.) and the mixture was left to stir overnight at rt. The reaction was quenched with water (50 mL) and diluted with EtOAc (50 mL) and the organic fraction was washed with water (3 × 50 mL) and brine (3 × 50 mL). The organic extract were dried using Na2SO4 and concentrated under reduced pressure. Purification by column chromatography on silica gel using 5% to 6% EtOAc/petroleum ether yielded the product 17 as a yellow oil (939 mg, 1.22 mmol, 71%). [α]23D: −94 (c 0.7 in CHCl3); (found (EI): M+ + Na, 789.3558. C52H54NaO2Si requires M, 789.3555); IR(neat) 3070, 3048, 2957, 2929, 2889, 2855, 1589, 1489, 1471, 1443, 1427, 1390, 1361, 1341, 1306, 1261, 1189, 1106, 1082, 1028, 1006 cm−1; δH (500 MHz, CDCl3) 7.78 (4H, d, J = 6.7 Hz, ArH), 7.71 (4H, d, J = 6.7 Hz, ArH), 7.37–7.40 (7H, m, ArH), 7.30–7.34 (6H, m, ArH), 7.21–7.27 (5H, m, ArH), 7.17–7.18 (4H, m, ArH), 4.62 (2H, br. s, CHOSi), 2.01–2.03 (4H, m, CH2), 1.10 (18H, s, C(CH3)3); δC (125 MHz, CDCl3) 136.1 (4C), 135.9 (4C), 133.9 (2C), 133.7 (2C), 131.5 (2C), 129.7 (2C), 129.5 (2C), 128.1 (2C), 128.0 (2C), 127.6 (4C), 127.4 (2C), 123.1 (2C), 90.5 (2C), 85.0 (2C), 64.0 (2C), 33.9 (2C), 27.0 (6C), 19.4 (2C); m/z (ESI+) 789.4 (M + Na, 100%), 805.3 (50%).

In a pressure tube ((3S,6S)-3,6-bis(dimethylphenylsilyloxy)octa-1,7-diyne-1,8-diyl)dibenzene 17 (500 mg, 0.65 mmol, 1.00 eq.) and iron pentacarbonyl (382 mg, 0.26 μL, 1.95 mmol, 3.00 eq.) was dissolved in anhydrous toluene (5 mL), the tube was then degassed (bubbling nitrogen, 10 minutes) and the tube was sealed. The tube was heated to 130 °C for 20 h. After cooling to rt and diluted with EtOAc (10 mL) the solution was passed through a celite plug washing through EtOAc (100 mL). The solvent was removed under reduced pressure and the product purified by column chromatography on silica gel in 5% to 20% EtOAc/petroleum ether to yield the product 21 as an orange solid (220 mg, 0.24 mmol, 36%). [α]23D: −39 (c 0.018 in CHCl3); mp: 80–82 °C (dec); (found (EI): M+ + Na, 957.2710. C56H54FeNaO6Si2 requires M, 957.2702); IR(neat) 3050, 2930, 2893, 2856, 2063, 2011, 1985, 1645, 1589, 1500, 1488, 1471, 1444, 1427, 1391, 1362, 1188, 1104, 1042, 1006 cm−1; δH (500 MHz, CDCl3) 7.60 (2H, d, J = 7.0 Hz, ArH), 7.44–7.50 (5H, m, ArH), 7.27–7.41 (17H, m, ArH), 7.21–7.25 (3H, m, ArH), 7.14–7.19 (3H, m, ArH), 5.21 (1H, t, J = 4.2 Hz, CHOSi), 4.93 (1H, t, J = 4.8 Hz, CHOSi), 1.95–2.01 (1H, m, CHH), 1.83–1.89 (1H, m, CHH), 1.50–1.55 (1H, m, CHH), 1.45–1.48 (1H, m, CHH), 0.73 (9H, s, tBu), 0.64 (9H, s, tBu); δC (125 MHz, CDCl3) 208.4 (3C), 170.5, 135.9 (2C), 135.7 (2C), 135.7 (2C), 135.7 (2C), 134.4, 133.6, 133.0, 132.9, 131.2 (2C), 131.0, 131.0, 129.9, 129.8, 129.7 (2C), 129.7, 129.6, 128.6 (2C), 128.2 (2C), 128.2, 127.7, 127.7 (2C), 127.5 (2C), 127.5 (2C), 127.4 (2C), 104.6, 101.7, 84.5, 80.5, 65.0, 64.5, 28.3, 27.5, 27.1 (3C), 26.6 (3C), 19.0, 18.9; m/z (ESI+) 935.3 (M + H, 100%), 957.3 (75%).

An oven dried flask, cooled under N2, fitted with a reflux condenser was charged with (S)-BINOL (2.35 g, 8.2 mmol, 1 eq.) and flushed with N2. To this solid, toluene (50 mL) was added followed by P(NMe2)3 (3.08 g, 3.4 mL, 12.5 mmol, 1.5 eq.). The reaction mixture was refluxed for 9 hours, after cooling to rt the toluene was removed and the product passed through a silica plug using 200 mL of 20% EtOAc/petroleum ether then washing the plug with 200 mL of DCM which contained the product as a white solid (2.84 g, 7.9 mmol, 96%). [α]31D: +552 (c 0.68 in CHCl3); mp: 198–200 °C (lit: 190–191 °C); IR(neat) 3056, 3013, 2976, 2923, 2896, 2840, 2798, 1616, 1588, 1502, 1482, 1458, 1446, 1429, 1407, 1357, 1324, 1294, 1267, 1229, 1203, 1184, 1151, 1142, 1124, 1067 cm−1; δH (500 MHz, CDCl3) 7.95 (1H, d, J = 8.9 Hz, ArH), 7.88–7.91 (3H, m, ArH), 7.50 (1H, d, J = 8.9 Hz, ArH), 7.33–7.42 (5H, m, ArH), 7.22–7.28 (2H, m, ArH), 2.55 (6H, d, JPH = 10.0 Hz, CH3); δC (125 MHz, CDCl3) 150.0 (2C), 149.5, 132.9, 132.6, 131.4, 130.8, 130.3, 128.4, 128.3, 127.0, 126.9, 126.1 (2C), 124.8, 124.6, 124.0, 122.8, 122.1, 36.0; δP (202 MHz, CDCl3) 162.2, m/z (ESI+) 360.0 (M + H, 100%), 382.0 (45%).
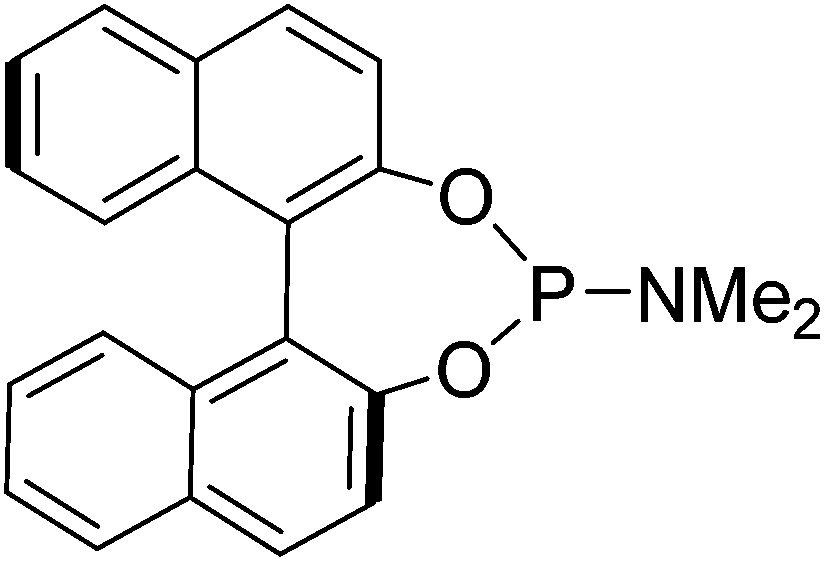
Prepared as described above using (R)-BINOL (2.35 g, 8.2 mmol, 1 eq.) and P(NMe2)3 (3.08 g, 3.4 mL, 12.5 mmol, 1.5 eq.) to give the product as a white solid (2.80 g, 7.8 mmol, 95%). [α]30D = −674 (c 0.23 in CHCl3); mp: 196–198 °C (lit: 190–191 °C); IR(neat) 3056, 3013, 2975, 2923, 2896, 2841, 2806, 1615, 1588, 1503, 1482, 1459, 1446, 1430, 1407, 1357, 1324, 1295, 1267, 1229, 1204, 1184, 1152, 1142, 1067 cm−1; δH (500 MHz, CDCl3) 7.97 (1H, d, J = 8.9 Hz, ArH), 7.89–7.92 (3H, m, ArH), 7.50 (1H, d, J = 8.9 Hz, ArH), 7.33–7.42 (5H, m, ArH), 7.22–7.28 (2H, m, ArH), 2.54 (6H, d, JPH = 10.0 Hz, CH3); δC (125 MHz, CDCl3) 150.0 (2C), 149.5, 132.8, 132.6, 131.4, 130.7, 130.3, 130.0, 128.3, 128.3, 127.0, 126.9, 126.1 (2C), 124.8, 124.6, 122.8, 122.1, 122.0, 36.0; δP (202 MHz, CDCl3) 162.1, m/z (ESI+) 360.0 (M + H, 100%).

To a degassed and foil-covered flask a solution of tricarbonyl (4S,7S)-4,7-bis(benzyloxy)-1,3-diphenyl-4,5,6,7-tetrahydro-2H-inden-2-one iron 18 (620 mg, 0.97 mmol, 1 eq.) and (S)-N,N-dimethyldinaphtho[2,1-d:1′,2′-f][1,3,2]dioxa phosphepin-4-amine (698 mg, 1.94 mmol, 2 eq.) in anhydrous toluene (20 mL) was added trimethylamine-N-oxide (146 mg, 1.94 mmol, 2 eq.) at rt. The reaction was heated to 60 °C overnight, the reaction was cooled to rt and the toluene was removed. The material was passed through a silica plug using initially 10% EtOAc/pentane and flushing the plug with 30% EtOAc/pentane to give 22 as a yellow solid (554 mg, 0.80 mmol, 65%). [α]29D: +120 (c 0.19 in CHCl3); mp: 97–99 °C (dec.); (found (EI): M+, 970.2596. C59H49FeNO7PSi2 requires M, 970.2592); δH (500 MHz, CDCl3) 8.29 (2H, d, J = 7.2 Hz, ArH), 7.90 (1H, d, J = 8.1 Hz, ArH), 7.84 (1H, d, J = 8.2 Hz, ArH), 7.81 (1H, d, J = 8.9 Hz, ArH), 7.70 (1H, d, J = 8.9 Hz, ArH), 7.81 (2H, d, J = 7.3 Hz, ArH), 7.42–7.46 (5H, m, ArH), 7.35–7.40 (5H, m, ArH), 7.17–7.22 (4H, m, ArH), 7.07–7.16 (4H, m, ArH), 6.94 (1H, d, J = 8.9 Hz, ArH), 6.91 (1H, d, J = 7.3Hz, ArH), 6.82 (2H, t, J = 7.6 Hz, ArH), 6.58 (2H, d, J = 6.9 Hz, ArH), 4.87 (1H, br. s, CH), 4.70 (1H, ABq, JAB = 11.3 Hz, CH2Ph), 4.64 (1H, ABq, JAB = 11.3 Hz, CH2Ph), 4.25 (1H, ABq, JAB = 10.1 Hz, CH2Ph), 4.20 (1H, br. s, CH), 3.92 (1H, ABq, JAB = 10.1 Hz, CH2Ph), 2.24–2.30 (1H, m, CHH), 2.14 (6H, d, J = 10.0 Hz, NCH3), 1.95–2.09 (3H, m, CHHCH2); δC (125 MHz, CDCl3) 214.9 (d, JPC = 19.1), 211.0 (d, J = 25.1 Hz), 166.1 (d, J = 4.0 Hz), 149.5 (d, J = 12.1 Hz), 147.9 (d, J = 6.0 Hz), 137.9, 137.4, 134.4, 134.5, 132.8, 132.6, 131.3, 131.1, 130.1, 128.9 (2C), 129.1 (2C), 128.3 (2C), 128.3, 128.1 (2C), 128.1 (2C), 128.0 (2C), 127.6 (2C), 127.3, 127.2, 126.9 (2C), 126.7 (2C), 126.2 (2C), 125.9 (2C), 125.0 (2C), 122.9 (2C), 122.5 (d, JPC = 3.0 Hz), 122.3 (d, JPC = 2.0 Hz), 120.9 (2C), 97.3, 93.6, 81.3, 80.4, 72.6, 71.3, 71.0, 69.0, 37.2, 37.1, 21.1, 20.4; δP (242 MHz, CDCl3) 188.6; m/z (ESI+) 970.2 (M + H, 100%). The compound was assumed to be light sensitive and was stored in the dark.

To a degassed and foil-covered flask a solution of tricarbonyl (4S,7S)-4,7-bis(benzyloxy)-1,3-diphenyl-4,5,6,7-tetrahydro-2H-inden-2-one iron 18 (560 mg, 0.88 mmol, 1 eq.) and (R)-N,N-dimethyldinaphtho[2,1-d:1′,2′-f][1,3,2]dioxa phosphepin-4-amine (633 mg, 1.76 mmol, 2 eq.) in anhydrous toluene (20 mL) was added trimethylamine-N-oxide (132 mg, 1.76 mmol, 2 eq.) at rt. The reaction heated to 60 °C overnight, the reaction was cooled to rt and the toluene was removed. The material was passed through a silica plug using initially using 10% EtOAc/pentane and flushing the plug with 30% EtOAc/pentane to isolated as a yellow solid (572 mg, 0.59 mmol, 67%). [α]29D: +300 (c 0.08 in CHCl3); mp: 117–119 °C (dec.); δH (500 MHz, CDCl3) 8.37 (2H, d, J = 7.3 Hz, ArH), 8.16 (2H, d, J = 8.6 Hz, ArH), 7.84 (3H, dd, J = 6.1 Hz, ArH), 7.59 (1H, d, J = 8.9 Hz, ArH), 7.14–7.49 (21H, m, ArH), 6.91–6.93 (2H, m, ArH), 5.91 (1H, d, J = 8.9 Hz, ArH), 5.12 (1H, br. s, CH), 4.69 (1H, ABq, JAB = 11.0 Hz, CH2Ph), 4.58 (1H, ABq, JAB = 10.4 Hz, CH2Ph), 4.54 (1H, ABq, JAB = 11.0 Hz, CH2Ph), 4.40 (1H, ABq, JAB = 10.4 Hz, CH2Ph), 4.15 (1H, br. s, CH), 2.20–2.32 (2H, m, CH2), 2.13 (3H, s, NCH3), 2.11 (3H, s, NCH3), 2.08 (2H, br. s, CH2); δC (125 MHz, CDCl3) 212.9 (d, JPC = 23.1 Hz), 211.6 (d, JPC = 24.1 Hz), 165.2 (d, JPC = 4.0 Hz), 149.4 (d, JPC = 10.0 Hz), 147.9, 137.7, 137.5, 134.7, 134.3, 133.4, 132.7, 132.5, 131.2 (d, JPC = 15.1 Hz), 131.4, 130.8, 130.4, 130.2, 129.9, 129.8, 129.5, 129.0, 128.8, 128.6, 128.6, 128.5, 128.4, 128.3 (3C), 128.1, 128.0, 127.7, 127.5, 127.1, 127.0, 126.8, 126.3, 125.9, 125.2, 125.0, 124.3, 124.0, 123.1 (d, JPC = 3.0 Hz), 120.9, 117.8, 97.3, 97.2, 93.9, 81.9, 71.9, 71.8, 71.2, 69.4, 37.2 (2C), 20.9, 20.2; δP (242 MHz, CDCl3) 193.6; m/z (ESI+) 970.2 (M + H, 100%). The compound was assumed to be light sensitive and was stored in the dark. The sample contained ca. 5% EtOAc by mass.

To a degassed solution of tricarbonyl((4S,7S)-4,7-bis((tert-butyldimethylsilyl)oxy)-1,3-diphenyl-4,5,6,7-tetrahydro-2H-inden-2-one) iron 19 (600 mg, 0.90 mmol, 1 eq.) and (S)-N,N-dimethyl dinaphtho[2,1-d:1′,2′-f][1,3,2]dioxa phosphepin-4-amine (647 mg, 1.80 mmol, 2 eq.) in anhydrous toluene (50 mL) was added trimethylamine-N-oxide (135 mg, 1.80 mmol, 2 eq.) at rt. This was degassed and heated to 60 °C overnight, then cooled to rt and toluene was removed under reduced pressure. This was further purified by column chromatography using 10% to 30% EtOAc/pentane to yield a orange solid (570 mg, 0.56 mmol, 62%). mp: >150 °C (dec.); [α]30D: +97 (c 0.08 in CHCl3); (found (EI): M+, 1018.3381. C57H65FeNO7PSi2 requires M, 1018.3383); IR(neat) 2952, 2927, 2854, 2008, 1952, 1713, 1591, 1500, 1253, 1227, 1156, 1099, 1050, 1004, 800, 772, 749, 671, 565 cm−1; δH (500 MHz, CDCl3) 8.43 (2H, d, J = 7.4 Hz, ArH), 7.90 (2H, d, J = 8.3 Hz, ArH), 7.82 (1H, d, J = 8.2 Hz, ArH), 7.78 (1H, d, J = 8.9 Hz, ArH), 7.73 (1H, d, J = 9.2 Hz, ArH), 7.52 (2H, d, J = 7.4 Hz, ArH), 7.36–7.48 (6H, m, ArH), 7.14–7.22 (3H, m, ArH), 7.07 1H, d, J = 8.6 Hz, ArH), 6.99 (1H, d, J = 8.9 Hz, ArH), 6.73 (1H, t, J = 7.3 Hz, ArH), 6.64 (2H, t, J = 7.6 Hz, ArH), 5.11 (1H, s, CHOSi), 4.62 (1H, s, CHOSi), 2.36–2.43 (1H, m, CH2), 2.17–2.27 (1H, m, CH2), 2.10 (6H, d, J = 10.0 Hz, N(CH3)2), 1.74–1.79 (2H, m, CH2), 0.98 (9H, s, C(CH3)3), 0.58 (9H, s, C(CH3)3), 0.25 (3H, s, SiCH3), 0.19 (3H, s, SiCH3), −0.13 (3H, s, SiCH3), −0.78 (3H, s, SiCH3); δC (125 MHz, CDCl3) 215.0 (d, JPC = 16.9 Hz), 211.6 (d, JPC = 24.9 Hz), 166.4 (d, JPC = 3.9 Hz), 149.4 (d, JPC = 11.9 Hz), 148.0 (d, JPC = 6.9 Hz), 134.5, 134.1, 132.9, 132.6, 131.3, 131.1, 130.1, 130.0 (2C), 129.8, 129.5 (2C), 128.2, 128.0, 127.9, 127.3, 127.1 (2C), 126.9, 126.7, 126.1, 126.0, 125.8, 125.0 (2C), (d, JPC = 2.5 Hz, ArH), 122.4 (d, JPC = 3.0 Hz, ipso), 122.3, 120.9, 100.3, 95.2, 81.2, 79.5 (d, JPC = 2.0 Hz, CpH), 64.2, 63.4, 37.0 (d, J = 5.9 Hz), 27.2, 26.3 (3C), 25.7 (3C), 25.3, 18.8, 18.0, −3.3, −4.7, −4.9, −5.9; δP (242 MHz, CDCl3) 187.8; m/z (ESI+) 1018.2 (M+, 100%). The compound was found to be light sensitive and decomposed in solution. The 1H-NMR spectrum and subsequent hydride formation experiments indicated the presence of ca. 10% of the diastereomer 25 (see ESI†) and traces of other impurities.

To a degassed solution of tricarbonyl (4S,7S)-4,7-bis(benzyloxy)-1,3-diphenyl-4,5,6,7-tetrahydro-2H-inden-2-one iron 19 (600 mg, 0.90 mmol, 1 eq.) and (R)-N,N-dimethyl dinaphtho[2,1-d:1′,2′-f][1,3,2]dioxa phosphepin-4-amine (647 mg, 1.80 mmol, 2 eq.) in anhydrous toluene (50 mL) was added trimethylamine-N-oxide (135 mg, 1.80 mmol, 2 eq.) at rt. This was degassed and heated to 60 °C overnight, then cooled to rt and toluene was removed under reduced pressure. The product was further purified by column chromatrography using 10% to 30% EtOAc/pentane yielding a orange solid (550 mg, 0.54 mmol, 60%) mp: >150 °C (dec.); [α]30D: +142 (c 0.05 in CHCl3); IR(neat) 2950, 2927, 2854, 2010, 1957, 1608, 1591, 1500, 1463, 1446, 1292, 1253, 1155, 1100, 1052, 980, 850, 826, 720, 562 cm−1; δH (500 MHz, CDCl3) 8.28–8.30 (4H, m, ArH), 7.83–7.86 (3H, m, ArH), 7.62 (1H, d, J = 8.8 Hz, ArH), 7.41–7.46 (3H, m, ArH), 7.36–7.40 (3H, m, ArH), 7.31–7.34 (3H, m, ArH), 7.14–7.24 (4H, m, ArH), 6.09 (1H, d, J = 9.0 Hz, ArH), 5.40 (1H, br. s, CHOSi), 4.60 (1H, br. s, CHOSi), 2.42 (1H, t, J = 13.7 Hz, CH2), 2.23 (1H, t, J = 13.9 Hz, CH2), 2.07 (6H, d, J = 9.7 Hz, N(CH3)2), 1.86 (1H, d, J = 13.7 Hz, CH2), 1.78 (1H, d, J = 8.8 Hz, CH2), 0.94 (9H, s, C(CH3)3), 0.67 (9H, s, C(CH3)3), 0.27 (3H, s, SiCH3), 0.16 (3H, s, SiCH3), 0.07 (3H, s, SiCH3), −0.37 (3H, s, SiCH3); δC (125 MHz, CDCl3) 213.1 (d, J = 20.9 Hz), 212.3 (d, JPC = 23.9 Hz), 165.6 (d, J = 3.9 Hz), 165.6 (d, J = 3.99 Hz), 149.4 (d, J = 9.9 Hz), 148.0 (d, J = 6.9 Hz), 134.9, 134.5, 132.7, 132.5, 131.2, 131.1, 130.0, 129.8, 129.6 (2C), 128.3, 128.2, 128.0, 127.7 (2C), 127.1, 126.9, 126.8, 126.3, 125.9, 125.2, 125.0, 123.1, 122.7 (2C), 121.7, 120.9, 99.4, 96.8, 82.0, 76.9, 64.9, 63.7, 37.1 (2C, d, JPC = 5.0 Hz), 27.4, 26.4 (3C), 25.8 (3C), 25.7, 18.8, 18.1, −3.2, −4.4, −4.9, −5.3; δP (242 MHz, CDCl3) 193.4; m/z (ESI+) 1018.2 (M+, 100%). The compound was found to be light sensitive and decomposed in solution. The 1H-NMR spectrum and subsequent hydride formation experiments indicated the presence of ca. 10% of the diastereomer 24 (see ESI†).

Procedure for reduction of ketones with formic acid/triethylamine (FA/TEA)
Acetophenone (100 mg, 0.83 mmol, 1.0 eq.), Catalyst (0.10 eq./10 mol%) and FA/TEA (5:2 azeotrope, 830 μL) were added to a flask under nitrogen, degassed and stirred for 10 min. TMAO (0.1 eq./10 mol%) was added to the reaction and the mixture was heated at the temperature indicated for the time stated. At the end of this time the reaction was allowed to cool to rt and EtOAc:hexane (1:4, ca. 10 mL) was added to dilute the sample. This solution was passed through celite and then silica gel. The residue was taken up in EtOAc:hexane (1:4, ca. 10 mL × 2) and the solutions filtered passed through celite and then silica gel. This served to remove residues of catalyst to give the product in quantitative conversion as assessed by the mass balance. Removal of solvent gave the product which was analyzed by GC and NMR.6a A sample of the formate derivative of known major configuration was prepared and this is described below. When following conversion over time, a small sample was taken from the reaction and treated as described above. An authentic (commercial) sample of the reduction product of 3,3-dimethyl-2-butanone was used to establish the GC conditions. Chiral GC analysis; (i) for racemic 1-phenylethanol (CP-Chiralsil-Dex-Cβ 25 m × 0.25 mm × 0.25 μm, T = 110 °C, P = 18 psi, He gas) (R) isomer 12.95 min, (S) isomer 14.24 min. The ketone has a RT of 6.03 min (features in the asymmetric sample of 32% ee). 1-Phenylethyl formate (racemic); CP-Chiralsil-Dex-Cβ 25 m × 0.25 mm × 0.25 μm, T = 110 °C, P = 18 psi, He gas) (S) isomer; 7.62 min, (R) isomer; 8.55 min. 3,3-Dimethyl-2-butanol. 70 °C, Racemic CP-Chiralsil-Dex-Cβ 25 m × 0.25 mm × 0.25 μm, T = 110 °C, P = 18 psi, He gas) (R) isomer; 8.41 min, (S) isomer; 8.77 min.
Procedure for reduction of ketones with hydrogen gas
Illustration with acetophenone: acetophenone (100 mg, 0.83 mmol), catalyst (0.01 eq./1 mol%), and iPrOH (0.5 mL) were added to a small test tube containing a stirrer bar. A solution of K2CO3 (5.8 mg, 0.042 mmol) in water (0.2 mL), or TMAO (1 mol%) was added, then the test tube was sealed in a Parr hydrogenator and charged with hydrogen to 30 bar, venting once. The sealed vessel was heated to the temperature indicated and stirred for the time given in the table. At the end of this time, the reaction was allowed to cool to rt, the pressure was carefully released and the sample was worked up and analyzed as previously described.Procedure for oxidation of alcohols with acetone as the hydrogen acceptor
Acetophenone (100 mg, 0.83 mmol, 1.0 eq.), Catalyst (0.10 eq./10 mol%) and acetone (4.3 mL) were added to a pressure tube, degassed and stirred for 10 min. TMAO (0.1 eq./10 mol%) was added, the reaction tube was sealed and the mixture was heated at the temperature indicated for the time stated. At the end of this time the reaction was allowed to cool to rt and the sample was worked up and analyzed as previously described.Synthesis of (S)-1-phenylethyl formate via a Mitsunobu reaction
To an oven dried flask was added acetophenone (250 mg, 243 μL, 2.08 mmol, 1 eq.) and formic acid triethylamine azeotrope (5
 :2) (1.39 mL). To this solution was added 3C-(R,R)-Teth-TsDpenRuCl catalyst16 (2.5 mg, 0.004 mmol, S/C: 500:1) and the mixture was heated to 40 °C overnight. The reaction was cooled to rt and diluted in EtOAc and passed through a silica plug, the solvent was removed to yield a yellow oil (250 mg, 2.05 mmol). To this solution was added formic acid (94 mg, 77 μL, 2.05 mmol, 1 eq.) and triphenylphosphine (538 mg, 2.05 mmol, 1 eq.) as a anhydrous THF (10 mL) solution. The reaction was cooled to 0 °C and DIAD (415 mg, 404 μL, 2.05 mmol, 1 eq.) as a single portion. The reaction was allowed to warm to rt and left overnight. The THF was removed and the water was added and the aqueous solution was extracted with DCM (3 × 50 mL). The organic solvent was dried with Na2SO4 and the solvent was removed under reduced pressure to yield a yellow oil. This contained diisopropyl hydrazine-1,2-dicarboxylate (20% by NMR) no further purification was attempted. On GC this showed peak 1 at 7.72 (75.4%, S) and peak 2 at 8.75 (24.6%, R) (ee = 51%). δH (400MHz, CDCl3) 8.07 (1H, s, C(O)H), 7.27–7.36 (5H, m, ArH), 6.00 (1H, q, J = 6.7 Hz, CH(O)CH3), 1.58 (3H, d, J = 6.5 Hz, CH3). This data was consistent with that previously reported.6b A racemic sample was prepared according to the procedure in this reference.
:2) (1.39 mL). To this solution was added 3C-(R,R)-Teth-TsDpenRuCl catalyst16 (2.5 mg, 0.004 mmol, S/C: 500:1) and the mixture was heated to 40 °C overnight. The reaction was cooled to rt and diluted in EtOAc and passed through a silica plug, the solvent was removed to yield a yellow oil (250 mg, 2.05 mmol). To this solution was added formic acid (94 mg, 77 μL, 2.05 mmol, 1 eq.) and triphenylphosphine (538 mg, 2.05 mmol, 1 eq.) as a anhydrous THF (10 mL) solution. The reaction was cooled to 0 °C and DIAD (415 mg, 404 μL, 2.05 mmol, 1 eq.) as a single portion. The reaction was allowed to warm to rt and left overnight. The THF was removed and the water was added and the aqueous solution was extracted with DCM (3 × 50 mL). The organic solvent was dried with Na2SO4 and the solvent was removed under reduced pressure to yield a yellow oil. This contained diisopropyl hydrazine-1,2-dicarboxylate (20% by NMR) no further purification was attempted. On GC this showed peak 1 at 7.72 (75.4%, S) and peak 2 at 8.75 (24.6%, R) (ee = 51%). δH (400MHz, CDCl3) 8.07 (1H, s, C(O)H), 7.27–7.36 (5H, m, ArH), 6.00 (1H, q, J = 6.7 Hz, CH(O)CH3), 1.58 (3H, d, J = 6.5 Hz, CH3). This data was consistent with that previously reported.6b A racemic sample was prepared according to the procedure in this reference.
Acknowledgements
We thank the Leverhulme Trust (grant no. RPG-374) for financial support to RH, the EPSRC (grant no. EP/M006670/1) for financial support to ADG and Johnson Matthey Catalysis and Chiral Technologies for a gift of Teth-TsDpenRuCl catalyst. The X-ray diffraction instrument was obtained through the Science City Project with support from the AWM and part funded by the ERDF.References
- (a) C. Bolm, J. Legros, J. Le Paih and L. Zani, Chem. Rev., 2004, 104, 6217–6254 CrossRef CAS PubMed; (b) S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317–3321 CrossRef CAS PubMed; (c) S. Gaillard and J.-L. Renaud, ChemSusChem, 2008, 1, 505–509 CrossRef CAS PubMed; (d) G. Bauer and K. A. Kirchner, Angew. Chem., Int. Ed., 2011, 50, 5798–5800 CrossRef CAS PubMed; (e) K. Junge, K. Schroder and M. Beller, Chem. Commun., 2011, 47, 4849–4859 RSC; (f) M. Darwish and M. Wills, Catal. Sci. Technol., 2012, 2, 243–255 RSC; (g) K. Gopalaiah, Chem. Rev., 2013, 113, 3248–3296 CrossRef CAS PubMed; (h) A. Quintard and J. Rodriquez, Angew. Chem., Int. Ed., 2014, 53, 4044–4055 CrossRef CAS PubMed; (i) I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed.
- (a) G. N. Schrauzer, J. Am. Chem. Soc., 1959, 81, 5307–5310 CrossRef CAS; (b) A. J. Pearson, R. J. Shively Jr. and R. A. Dubbert, Organometallics, 1992, 11, 4096–4104 CrossRef CAS; (c) A. J. Pearson and R. J. Shively Jr., Organometallics, 1994, 13, 578–584 CrossRef CAS; (d) A. J. Pearson and J. B. Kim, Org. Lett., 2002, 4, 2837–2840 CrossRef CAS PubMed; (e) E. Weiss, R. G. Merenyl and W. Huber, Chem. Ind., 1960, 407–408 CAS.
- (a) H.-J. Knölker, J. Heber and C. H. Mahler, Synlett, 1992, 1002–1004 CrossRef; (b) H.-J. Knölker, E. Baum, H. Goesman and R. Klauss, Angew. Chem., Int. Ed., 1999, 38, 2064–2066 CrossRef; (c) H.-J. Knölker, E. Baum, H. Goesmann and R. Klauss, Angew. Chem., Int. Ed., 1999, 38, 702–705 CrossRef; (d) T. N. Plank, J. L. Drake, D. K. Kim and T. W. Funk, Adv. Synth. Catal., 2012, 354, 597–601 CrossRef CAS.
- (a) C. P. Casey and H. Guan, J. Am. Chem. Soc., 2007, 129, 5816–5817 CrossRef CAS PubMed; (b) C. P. Casey and H. Guan, J. Am. Chem. Soc., 2009, 131, 2499–2507 CrossRef CAS PubMed.
- (a) A. Berkessel, S. Reichau, A. Von der Höh, N. Leconte and J.-M. Nordörfl, Organometallics, 2011, 30, 3880–3887 CrossRef CAS; (b) A. Berkessel and A. Van der Höh, ChemCatChem, 2011, 3, 861–867 CrossRef.
- (a) J. P. Hopewell, J. E. D. Martins, T. C. Johnson, J. Godfrey and M. Wills, Org. Biomol. Chem., 2012, 10, 134–145 RSC; (b) T. C. Johnson, G. J. Clarkson and M. Wills, Organometallics, 2011, 30, 1859–1868 CrossRef CAS.
- (a) A. Tlili, J. Schranck, H. Neumann and M. Beller, Chem. – Eur. J., 2012, 18, 15935–15939 CrossRef CAS PubMed; (b) S. Fleischer, S. Zhou, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 5120–5124 CrossRef CAS PubMed; (c) S. Fleischer, S. Zhou, S. Werkmeister, K. Junge and M. Beller, Chem. – Eur. J., 2013, 4997–5003 CrossRef CAS PubMed; (d) S. Zhou, S. Fleischer, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 5120–5124 CrossRef CAS PubMed; (e) S. Fleischer, S. Werkmeister, S. Zhau, C. Junge and M. Beller, Chem. – Eur. J., 2012, 18, 9005–9010 CrossRef CAS PubMed.
- P. Gajewski, M. Renom-Carrasco, S. V. Facchini, L. Pignataro, L. Lefort, J. G. de Vries, R. Ferraccioli, A. Forni, U. Piarulli and C. Gennari, Eur. J. Org. Chem., 2015, 1887–1893 CrossRef CAS.
- (a) S. A. Moyer and T. W. Funk, Tetrahedron Lett., 2010, 51, 5430–5433 CrossRef CAS; (b) M. G. Coleman, A. N. Brown, B. A. Bolton and H. Guan, Adv. Synth. Catal., 2010, 352, 967–970 CrossRef CAS; (c) M. K. Thorson, K. L. Klinkel, J. Wang and T. J. Williams, Eur. J. Inorg. Chem., 2009, 295–302 CrossRef CAS.
- (a) A. Quintard, T. Constantieux and J. Rodriquez, Angew. Chem., Int. Ed., 2013, 52, 12883–12887 CrossRef CAS PubMed; (b) S. Moulin, H. Dentel, A. Pagnoux-Ozherelyeva, S. Galliard, A. Poater, L. Cavallo, J.-F. Lohier and J.-L. Renaud, Chem. – Eur. J., 2013, 19, 17881–17890 CrossRef CAS PubMed; (c) A. Pagnoux-Ozherelyeva, N. Pannetier, M. D. Mbaye, S. Gaillard and J.-L. Renaud, Angew. Chem., Int. Ed., 2012, 51, 4976–4980 CrossRef CAS PubMed; (d) D. S. Merel, M. Elie, J.-L. Lohier, S. Gaillard and J.-L. Renaud, ChemCatChem, 2013, 5, 2939–2945 CrossRef CAS; (e) T.-T. Thai, D. S. Mérel, A. Poater, S. Gaillard and J.-L. Renaud, Chem. – Eur. J., 2015, 21, 7066–7070 CrossRef CAS PubMed.
- K. Natte, W. Li, S. Zhou, H. Neumann and X.-F. Wu, Tetrahedron Lett., 2015, 56, 1118–1121 CrossRef CAS.
- (a) T. Yan, B. L. Feringa and K. Barta, Nat. Commun., 2014, 5, 5602 CrossRef CAS PubMed; (b) A. J. Rawlings, L. J. Diorazio and M. Wills, Org. Lett., 2015, 17, 1086–1089 CrossRef CAS PubMed; (c) H.-J. Pan, T. Ng and Y. Zhao, Chem. Commun., 2015, 51, 11907–11910 RSC; (d) S. Elangovan, S. Quintero-Duque, V. Dorcet, T. Roisnel, L. Norel, C. Darcel and J.-B. Sortais, Organometallics, 2015, 34, 4521–4528 CrossRef CAS.
- (a) H. Zhang, D. Chen, Y. Zhang, G. Zhang and J. Liu, Dalton Trans., 2010, 39, 1972–1978 RSC; (b) X. Lu, Y. Zhang, P. Yun, M. Zhang and T. Li, Org. Biomol. Chem., 2013, 11, 5264–5277 RSC; (c) X. Lu, Y. Zhang, N. Turner, M. Ahang and T. Li, Org. Biomol. Chem., 2014, 12, 4361–4371 RSC; (d) X. Lu, R. Cheng, N. Turner, Q. Liu, M. Zhang and X. Sun, J. Org. Chem., 2014, 79, 9355–9364 CrossRef CAS PubMed.
- M. Kamitani, Y. Nishiguchi, R. Tada, M. Itazaki and H. Nakazawa, Organometallics, 2014, 33, 1532–1535 CrossRef CAS.
- (a) T.-Y. Luh, Coord. Chem. Rev., 1984, 60, 255–276 CrossRef CAS; (b) B. Dasgupta and W. A. Donaldson, Tetrahedron Lett., 1998, 39, 343–346 CrossRef CAS; (c) A. J. Pearson and Y. Kwak, Tetrahedron Lett., 2005, 46, 5417–5419 CrossRef CAS; (d) N. A. Bailey, V. S. Jassal, R. Vefghi and C. White, J. Chem. Soc., Dalton Trans., 1987, 2815–2822 RSC; (e) A. J. Pearson and R. J. Shively Jr., Organometallics, 1994, 13, 578–584 CrossRef CAS; (f) H.-J. Knölker, E. Baum and J. Heber, Tetrahedron Lett., 1992, 36, 7647–7650 CrossRef.
- Z. Fang and M. Wills, J. Org. Chem., 2013, 78, 8594–8605 CrossRef CAS PubMed.
- (a) C. P. Casey, N. A. Strotman, S. E. Beetner, J. B. Johnson, D. C. Priebe, T. E. Vos, B. Khodavandi and I. A. Guzei, Organometallics, 2006, 25, 1230–1235 CrossRef CAS; (b) C. P. Casey and H. Guan, Organometallics, 2011, 31, 2631–2638 CrossRef PubMed.
- R. Hulst, N. K. de Vries and B. L. Feringa, Tetrahedron: Asymmetry, 1994, 5, 699–708 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1431241–1431243. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt04610f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |