
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Access to novel fluorovinylidene ligands via exploitation of outer-sphere electrophilic fluorination: new insights into C–F bond formation and activation†
Lucy M.
Milner
,
Lewis M.
Hall
,
Natalie E.
Pridmore
,
Matthew K.
Skeats
,
Adrian C.
Whitwood
,
Jason M.
Lynam
* and
John M.
Slattery
*
Department of Chemistry, University of York, Heslington, York, YO10 5DD, UK. E-mail: jason.lynam@york.ac.uk; john.slattery@york.ac.uk; Fax: +44 (0)1904322516; Tel: +44 (0)1904322534, +44 (0)1904322610
First published on 24th December 2015
Abstract
Metal vinylidene complexes are widely encountered, or postulated, as intermediates in a range of important metal-mediated transformations of alkynes. However, fluorovinylidene complexes have rarely been described and their reactivity is largely unexplored. By making use of the novel outer-sphere electrophilic fluorination (OSEF) strategy we have developed a rapid, robust and convenient method for the preparation of fluorovinylidene and trifluoromethylvinylidene ruthenium complexes from non-fluorinated alkynes. Spectroscopic investigations (NMR and UV/Vis), coupled with TD-DFT studies, show that fluorine incorporation results in significant changes to the electronic structure of the vinylidene ligand. The reactivity of fluorovinylidene complexes shows many similarities to non-fluorinated analogues, but also some interesting differences, including a propensity to undergo unexpected C–F bond cleavage reactions. Heating fluorovinylidene complex [Ru(η5-C5H5)(PPh3)2(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C{F}R)][BF4] led to C–H activation of a PPh3 ligand to form an orthometallated fluorovinylphosphonium ligand. Reaction with pyridine led to nucleophilic attack at the metal-bound carbon atom of the vinylidene to form a vinyl pyridinium species, which undergoes both C–H and C–F activation to give a novel pyridylidene complex. Addition of water, in the presence of chloride, leads to anti-Markovnikov hydration of a fluorovinylidene complex to form an α-fluoroaldehyde, which slowly rearranges to its acyl fluoride isomer. Therefore, fluorovinylidenes ligands may be viewed as synthetic equivalents of 1-fluoroalkynes providing access to reactivity not possible by other routes.
C{F}R)][BF4] led to C–H activation of a PPh3 ligand to form an orthometallated fluorovinylphosphonium ligand. Reaction with pyridine led to nucleophilic attack at the metal-bound carbon atom of the vinylidene to form a vinyl pyridinium species, which undergoes both C–H and C–F activation to give a novel pyridylidene complex. Addition of water, in the presence of chloride, leads to anti-Markovnikov hydration of a fluorovinylidene complex to form an α-fluoroaldehyde, which slowly rearranges to its acyl fluoride isomer. Therefore, fluorovinylidenes ligands may be viewed as synthetic equivalents of 1-fluoroalkynes providing access to reactivity not possible by other routes.
Introduction
The unique properties of the carbon–fluorine bond can enhance many fundamental properties of biologically relevant molecules including bioavailability, metabolic stability and lipophilicity, with approximately 30% of agrochemicals and 20% of all pharmaceuticals now containing fluorine.1–9 The use of 18F-labelled imaging agents in positron emission tomography (PET) is also a highly active area of medical research.10,11 As such, new facile and selective synthetic methods for the introduction of fluorine into organic frameworks are highly desirable. Of particular interest are examples which offer high selectivity, controlled regiochemistry that is complementary to other methodologies and/or the ability to synthesise C–F bonds in less common environments.The development of novel catalytic procedures for C–F bond formation is highly desirable and can offer significant advantages over traditional stoichiometric processes. Recent years have seen many advances using enzymatic-,12 organo- or photo-catalytic13 methods for the synthesis of fluoro-organic compounds. Alongside these, transition-metal-promoted reactions have played an increasingly prominent role,14 particularly in the formation of aryl fluorides.15 Complementary studies on C–F bond cleavage4,16–20 and hydrodefluorination by metal complexes provide insight into the functionalization of readily available fluorine-rich substrates.13,21–23
Metal-mediated C–F bond forming methodologies using both nucleophilic and electrophilic sources of fluorine have been developed.5,6,15,24 Our interest has focused on reactions involving electrophilic fluorinating reagents, a number of which are commercially available.25,26 They have been utilized in metal-catalyzed C–F bond formation reactions, which are believed to proceed either by oxidative fluorination of palladium27–29 or silver30 to form a high-oxidation-state metal-fluoride complex,31 followed by C–F reductive elimination, or through single-electron processes with either copper32 or palladium catalysts.2,33–35 In addition, a wealth of Lewis-acidic-metal-mediated electrophilic fluorination reactions have been developed, which do not involve metal-based redox chemistry.36–39
We have recently described a new mechanism for the formation of C–F bonds within the coordination sphere of a redox-active metal complex using electrophilic fluorinating reagents (Scheme 1a).40,41 Reaction of pyridylidene complex [I] with a latent source of “F+” results in selective C–F bond formation to give [II]+. Reaction of [II]+ with a further equivalent of 1-fluoro-2,4,6-trimethylpyridinium (FTMP) salt (in the presence of a catalytic amount of a base), or [I] with two equivalents of FTMP, gives the difluorinated analogue [III]+. Experimental and computational studies indicate that this process involves direct fluorination of the ligand, without Ru–F bond formation, and is hence referred to as an outer-sphere electrophilic fluorination (OSEF) reaction.
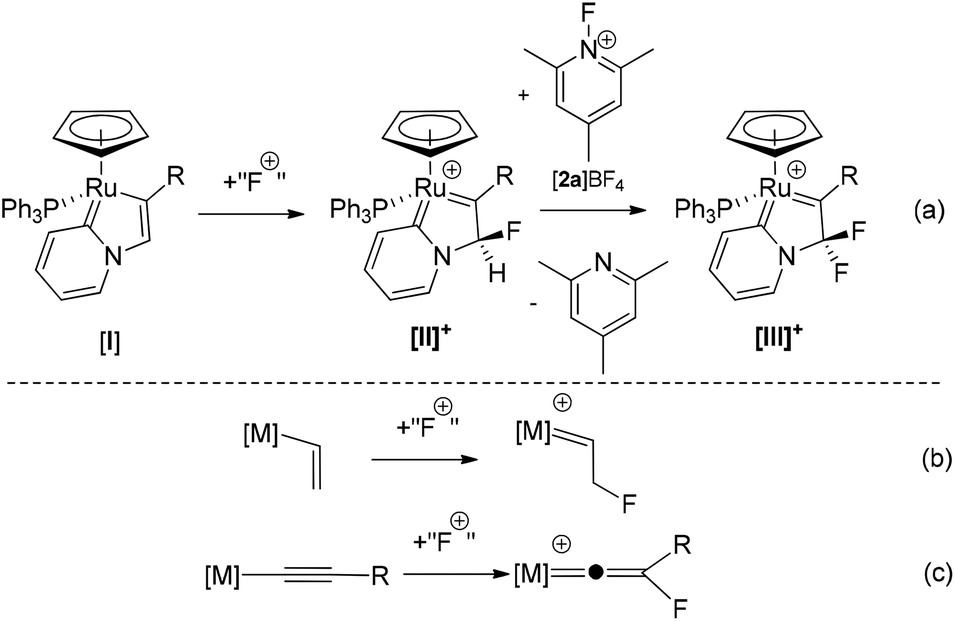 | ||
| Scheme 1 Outer sphere electrophilic fluorination of an organo-ruthenium complex. “F+” = [FTMP]BF4, Selectfluor or N-fluorobenzenesulfonimide (NFSI). | ||
We believe that the OSEF reaction in Scheme 1a is a general process and recent reports that an outer-sphere fluorination step may be involved in palladium-catalysed C–F bond formation42 and the fluorination of alkyne ligands in the coordination sphere of platinum43 support this idea. It is therefore important to gain a deeper understanding of the scope of OSEF reactions and the reactivities of the novel fluorinated ligand systems that are formed.
The conversion of [I] to [II]+ may be viewed as the transformation of an alkenyl ligand to a fluorinated carbene (Scheme 1b) and by analogy we postulated that the corresponding reaction of an alkynyl ligand via OSEF might provide facile and selective formation of very unusual fluorovinylidene ligands (Scheme 1c). Fluorovinylidene complexes have rarely been described – limited examples of dimeric iridium44–47 and iron48 complexes have been reported via activation of fluorinated organic precursors such as H2CCHF (iridium) and CF2(COCl)2 (iron). Since the availability of these precursors with a range of substitution patterns is restricted, this approach does not represent a general method for the synthesis and exploitation of fluorovinylidenes complexes, and hence their reactivity is largely unexplored. However, the potential importance of these ligands as intermediates in catalytic processes is highlighted by the large number of catalytic reactions that proceed via non-fluorinated vinylidene intermediates.49–52
We now demonstrate the successful preparation of fluorovinylidene complexes by OSEF from readily available, non-fluorinated alkynes. Studies of their reactivity clearly demonstrate that these complexes may indeed provide important routes to fluorinated compounds. The fluorovinylidenes ligands may act as fluoroalkyne equivalents therefore offering access to reactivity patterns of compounds which are otherwise difficult to prepare and handle.
Results and discussion
Synthesis and characterization of fluorine- and trifluoromethyl-containing vinylidene complexes
By analogy with related electrophilic additions (H+,53 Cl2, Br2, I2,54 Au(PPh3)+ (ref. 55)), the electrophilic fluorination of the alkynyl ligand in [Ru(η5-C5H5)(–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CR)(PPh3)2] to give fluorinated vinylidenes was targeted. Complexes based on the [Ru(η5-C5H5)(–CCPh)(PPh3)2] were selected as metal-based precursors as they are readily available from the reaction between [Ru(η5-C5H5)Cl(PPh3)2] and a wide range of terminal alkynes.56 Treatment of a CH2Cl2 solution of [Ru(η5-C5H5)(–CCPh)(PPh3)2], [1a], with [FTMP]BF4, [2a]BF4, resulted in an instantaneous colour change from a yellow to a dark green solution from which the fluorinated vinylidene complex [Ru(η5-C5H5)(CC{F}Ph)(PPh3)2]BF4, [3a]BF4, could be isolated in excellent yield (Scheme 2a). No intermediates were observed by NMR spectroscopy between 195 K and 295 K, supporting the hypothesis that [3a]BF4 is being formed by direct fluorination of the alkynyl ligand. Complex [3a]BF4 could also be prepared in a one-pot procedure by treatment of the vinylidene complex [Ru(η5-C5H5)(CC{H}Ph)(PPh3)2]+, [4a]+, with base to generate [1a] in situ followed by reaction with [2a]BF4. This reaction also proceeds in the absence of added base, but the reaction is slightly slower, requiring ca. 30 min to reach completion.
CR)(PPh3)2] to give fluorinated vinylidenes was targeted. Complexes based on the [Ru(η5-C5H5)(–CCPh)(PPh3)2] were selected as metal-based precursors as they are readily available from the reaction between [Ru(η5-C5H5)Cl(PPh3)2] and a wide range of terminal alkynes.56 Treatment of a CH2Cl2 solution of [Ru(η5-C5H5)(–CCPh)(PPh3)2], [1a], with [FTMP]BF4, [2a]BF4, resulted in an instantaneous colour change from a yellow to a dark green solution from which the fluorinated vinylidene complex [Ru(η5-C5H5)(CC{F}Ph)(PPh3)2]BF4, [3a]BF4, could be isolated in excellent yield (Scheme 2a). No intermediates were observed by NMR spectroscopy between 195 K and 295 K, supporting the hypothesis that [3a]BF4 is being formed by direct fluorination of the alkynyl ligand. Complex [3a]BF4 could also be prepared in a one-pot procedure by treatment of the vinylidene complex [Ru(η5-C5H5)(CC{H}Ph)(PPh3)2]+, [4a]+, with base to generate [1a] in situ followed by reaction with [2a]BF4. This reaction also proceeds in the absence of added base, but the reaction is slightly slower, requiring ca. 30 min to reach completion.
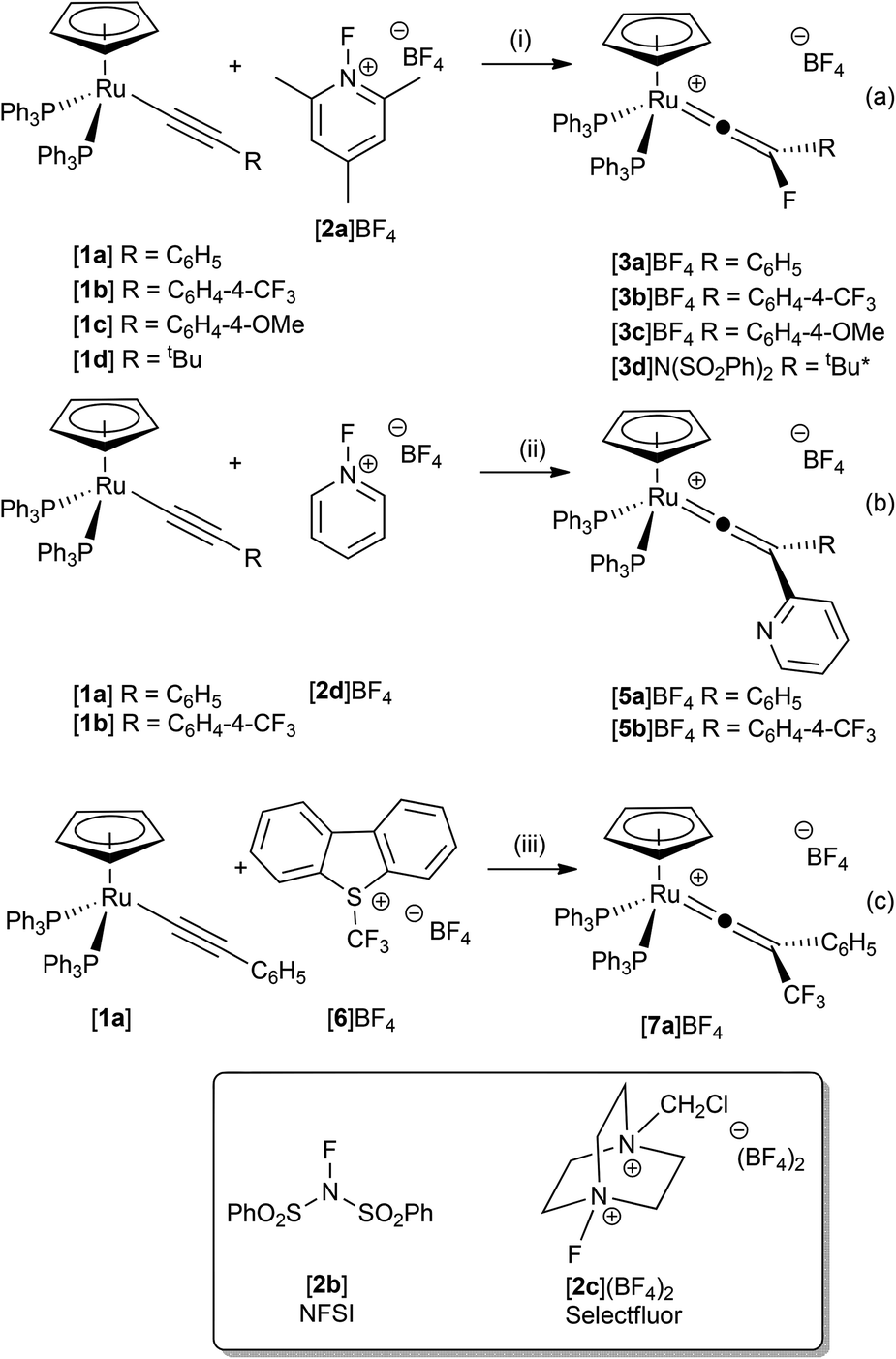 | ||
| Scheme 2 (i) CH2Cl2 solution, -2,4,6-trimethylpyridine. *NFSI was used as the fluorinating agent. (ii) CH2Cl2 solution, –“HF” (iii) CH2Cl2 solution, 4 h, r.t., -dibenzothiophene. | ||
Analogous reactions of ruthenium alkynyl complexes containing both electron-releasing and electron-withdrawing substituents ([1b]–[1d], Scheme 2a) and with a range of electrophilic fluorinating reagents (NFSI, [2b]BF4 and Selectfluor, [2c](BF4)2) proceeded with no notable difference in reaction outcome, demonstrating the generality of the approach. However, reaction of [1a] or [1b] with 1-fluoropyridinium tetrafluoroborate, [2d]BF4, resulted in the formation of the disubstituted vinylidene complexes [5a]BF4 and [5b]BF4 respectively (Scheme 2b), the structure of the latter was confirmed by X-ray crystallography (ESI†). Complex [5a]BF4 appears to have been formed by formal nucleophilic aromatic substitution of [2d]BF4 by the alkynyl ligand in [1a] with concomitant loss of HF.
In a similar manner to electrophilic fluorination, the electrophilic incorporation of –CF3 groups into the framework of organic compounds is also desirable.10,39,57,58 There are only limited examples of CF3-substituted vinylidene complexes, all of which are dimeric species formed from the activation of H2CCHCF3.46 The reaction between [1a] and Umemoto's reagent, [6]BF4, (a latent source of “CF3+”) resulted in the selective formation of [Ru(η5-C5H5)(CC{CF3}Ph)(PPh3)2]BF4, [7a]BF4 (Scheme 2c). The structure of [7a]BF4 was confirmed by a single crystal X-ray diffraction study (ESI†). To the best of our knowledge complexes [3]BF4 are the first examples of fluorovinylidene ligands in mononuclear metal species, and access to these complexes has allowed an evaluation of the effects of fluorine incorporation into vinylidene ligands. For example, the 19F NMR spectrum of [3a]+ displayed a singlet resonance at δ −208.7 for the fluorine atom of the vinylidene ligand and the 13C{1H} NMR spectrum exhibits a doublet of triplets at δ 389.0 (2JCF = 39.3 Hz, 2JCP = 16.2 Hz) for the metal-bound carbon atom and a doublet at δ 196.7 (1JCF = 222.1 Hz) for the fluorine-bearing carbon atom. These resonances are shifted ca. 30 ppm and 70 ppm downfield, respectively, when compared to the hydrogen-substituted analogue [4a]PF6. Indeed the metal-bound carbon is observed at one of the lowest reported chemical shifts for a vinylidene α-carbon.59,60 In CD2Cl2 solution the 1H, 31P and 19F NMR spectra of [3a]BF4 showed no change on cooling to 195 K, indicating that the vinylidene was undergoing rapid rotation on the NMR timescale.
The deep green colour of the fluorovinylidene complexes is due to an absorption band at ca. 690 nm, which showed no significant solvatochromism (ESI†). With the aid of TD-DFT studies this transition was shown to be dominated by HOMO → LUMO character. In the HOMO (Fig. 1) a fluorine lone pair has an anti-bonding interaction with the π-system of the vinylidene ligand (which is itself anti-bonding with respect to a metal-centred orbital). The LUMO has a contribution from a p-orbital on the fluorine and an orbital based on the metal-bound carbon.
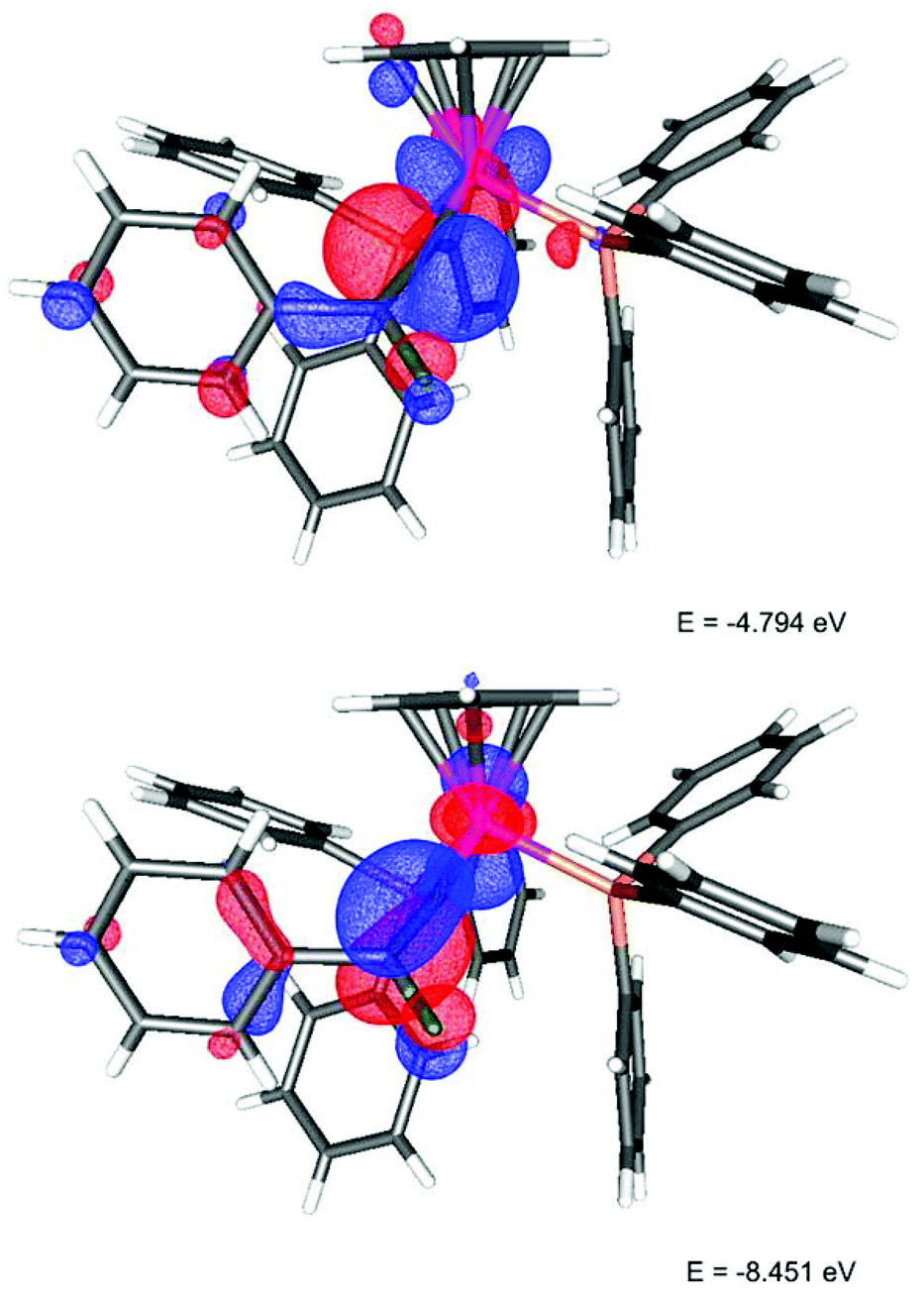 | ||
| Fig. 1 LUMO (top) and HOMO (bottom) of [3a]+. | ||
Additional spectroscopic data of the other halogen-substituted vinylidene complexes [Ru(η5-C5H5)(CC{E}C6H4-4-R)(PPh3)2]X54 (E = Cl,‡ Br and I) and the CF3-containing complex, [7a]BF4, support an interplay between σ- and π-effects on the energy of the HOMO-LUMO transition (Table 1). Good π-donors (e.g. F, Cl) raise the energy of the HOMO, thereby lowering the energy of the HOMO-LUMO transition. In contrast, the strongly (−I) electron-withdrawing groups, such as CF3 in [7a]BF4 display a higher energy transition, presumably due to an opposite effect. This effect is supported by the bathochromic shift observed in methoxy-substituted [3c]BF4 and the hypsochromic shift in [3b]BF4. The interplay between σ- and π-effects appears to also be manifested in the chemical shift of the metal bound carbon atom, as the CF3-subsituted vinylidene ligand has a chemical shift similar to [4a]PF6, but the halogen-substituted variants have a chemical shift range of 66 ppm.
| Complex | 13C δ M = C/ppm | λ max/nm | λ max/nm (calc.) |
|---|---|---|---|
| [3a]BF4 | 389.0 | 691 | 730 |
| [3b]BF4 | 383.5 | 684 | 720 |
| [3c]BF4 | 389.9 | 698 | 764 |
| [3d]N(SO2Ph)2 | 392.4 | 644 | 655 |
| [4a]PF6 | 355.5 | 524 | 537 |
| [7a]BF4 | 340.0 | 488 | 452 |
| [8aCl]BF4 | 353.5 | 615 | 702 |
| [8aBr]BF4 | 340.3 | 627 | 647 |
| [8Br]Br3 | 339.3 | 626 | — |
| [8aI]I3 | 323.6 | 619 | 668 |
|
|||
Complex [3a]PF6 crystallises as two different polymorphs, which have significantly different photophysical properties in the solid state (Fig. 2). One is orange (λmax = 742 nm) and one green (λmax = 692 nm) and they differ primarily in the orientation of the fluorine atom and phenyl group of the vinylidene ligand and the geometry around the β-carbon (ESI†). Dissolution of both crystals in CH2Cl2 gave essentially identical UV-vis and NMR spectra showing that the two crystalline forms convert to the same species in solution. We propose that the observed species is a time-averaged signal for the different rotameric forms. Different agostic isomers of the same organometallic complex have been shown to produce crystals with markedly different electronic spectra,61 however, in the case of [3a]PF6 subtle changes to the structure in the solid state has a pronounced effect on the photophysical properties of the system by altering the energy of the HOMO-LUMO transition.
 | ||
| Fig. 2 Solid-state structures of the [3a]+ cation found in (a) green crystals and (b) orange crystals. Hydrogen atoms removed for clarity and thermal ellipsoids (where shown) are at the 50% probability level. | ||
Reactions of fluorine-containing vinylidene complexes
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) CPh.
The facile syntheses of complexes [3]BF4 has allowed an exploration of the reactivity of these species to evaluate the effects of fluorine incorporation into vinylidene ligands. In the case of half-sandwich ruthenium complexes with either mono-62,63 or di-substituted vinylidene ligands64 reaction with NCMe has been shown to reverse the alkyne-vinylidene tautomerisation and generate [Ru(η5-L)(NCMe)(PPh3)2]+, (L = C5H5, [9]+) liberating the corresponding alkynes HCCR and PhCCR respectively. These studies have permitted detailed kinetic insight into the nature of the alkyne-vinylidene interconversion.65 However, heating a sample of [3a]+ in NCMe-d3 did not result in an analogous reaction to form [9]+ and FCCPh. Instead, the major product from the reaction was [10a]BF4, in which a coordinated alkene ligand has been formed from a combination of vinylidene and phosphine ligands, the latter having undergone an orthometallation process (Scheme 3). The 31P NMR spectrum of [10a]BF4 exhibits two resonances at δ 46.6 (dd, 3JPF = 48.6 Hz; 3JPP = 4.4 Hz) and δ 42.3 (dd, 3JPF = 16.2 Hz; 3JPP = 4.4 Hz) confirming the presence of two different phosphorus environments. Resonances at δ −143.4 (app. dt, 3JPF = 48.6 Hz; 3JHF = 16.9 Hz; 3JPF = 16.2 Hz) in the 19F NMR spectrum, δ 4.69 (ddd, 3JHF = 16.9 Hz; 2JPH = 5.0 Hz; 3JPH = 1.7 Hz) in the 1H NMR spectrum and 113.7 (dt, 1JCF = 243.0 Hz; 2JCP = 5.6 Hz) and 25.9 (ddd, 1JCP = 75.1 Hz; 2JCF = 7.8 Hz; 2JCP = 2.7 Hz) in the 13C{1H} spectrum demonstrated that the fluorinated alkene ligand was present.
CPh.
The facile syntheses of complexes [3]BF4 has allowed an exploration of the reactivity of these species to evaluate the effects of fluorine incorporation into vinylidene ligands. In the case of half-sandwich ruthenium complexes with either mono-62,63 or di-substituted vinylidene ligands64 reaction with NCMe has been shown to reverse the alkyne-vinylidene tautomerisation and generate [Ru(η5-L)(NCMe)(PPh3)2]+, (L = C5H5, [9]+) liberating the corresponding alkynes HCCR and PhCCR respectively. These studies have permitted detailed kinetic insight into the nature of the alkyne-vinylidene interconversion.65 However, heating a sample of [3a]+ in NCMe-d3 did not result in an analogous reaction to form [9]+ and FCCPh. Instead, the major product from the reaction was [10a]BF4, in which a coordinated alkene ligand has been formed from a combination of vinylidene and phosphine ligands, the latter having undergone an orthometallation process (Scheme 3). The 31P NMR spectrum of [10a]BF4 exhibits two resonances at δ 46.6 (dd, 3JPF = 48.6 Hz; 3JPP = 4.4 Hz) and δ 42.3 (dd, 3JPF = 16.2 Hz; 3JPP = 4.4 Hz) confirming the presence of two different phosphorus environments. Resonances at δ −143.4 (app. dt, 3JPF = 48.6 Hz; 3JHF = 16.9 Hz; 3JPF = 16.2 Hz) in the 19F NMR spectrum, δ 4.69 (ddd, 3JHF = 16.9 Hz; 2JPH = 5.0 Hz; 3JPH = 1.7 Hz) in the 1H NMR spectrum and 113.7 (dt, 1JCF = 243.0 Hz; 2JCP = 5.6 Hz) and 25.9 (ddd, 1JCP = 75.1 Hz; 2JCF = 7.8 Hz; 2JCP = 2.7 Hz) in the 13C{1H} spectrum demonstrated that the fluorinated alkene ligand was present.
 | ||
| Scheme 3 (i) NCMe, 50 °C, 14 d. X− = BF4− or N(SO2Ph)2−. | ||
Other products form on heating at higher temperature (80 °C), but after ca. two weeks at 50 °C, the reaction was reasonably selective (70% conversion based on 31P NMR spectroscopy). A complex related to [10a]BF4 has been reported previously from the nucleophilic attack of PPh3 at a coordinated (protio)vinylidene.66 It is proposed that [10a]BF4 is formed by initial loss of PPh3, followed by attack of the free phosphine at the metal-bound carbon of the fluorovinylidene. C–H activation and hydride migration subsequently leads to [10a]BF4. This is supported by the fact that the dppe-containing fluorovinylidene complex [Ru(η5-C5H5)(CC{F}Ph)(dppe)]N(SO2Ph)2, [3e]N(SO2Ph)2 (prepared from [Ru(η5-C5H5)(–CCPh)(dppe)] and 2b) is unchanged on heating at 100 °C for 14 days in NCMe-d3. These data indicate that the conversion of the fluorovinylidene to its alkyne form does not proceed under any of these conditions.
C{F}Ph)(PPh3)2]BF4, (py = pyridine), [11]BF4 (Scheme 4), demonstrating that the incorporation of the fluorine substituent does not inhibit nucleophilic attack at the metal-bound carbon. In CD2Cl2 solution the 19F NMR spectrum of [11]BF4 exhibited a doublet resonance at δ −71.1 (4JPF = 20.1 Hz) and the 31P{1H} a corresponding resonance at δ 44.0 (d, 4JPF = 20.3 Hz): the ESI-MS demonstrated that a single pyridine had added to the ruthenium complex. The structure of [11]BF4 was confirmed by X-ray crystallography (Fig. 3), which showed that the alkenyl ligand had adopted a Z-configuration. We have previously proposed that this type of alkenyl complex was a key intermediate in the direct C–H functionalization of the pyridine,68 and the previous inability to observe such a species was principally owing to its low intrinsic concentration due to competitive deprotonation of the vinylidene ligand. Evidently, the presence of the fluorine atom allows for the observation and characterization of this species.
 | ||
| Scheme 4 (i) Pyridine, 20 °C, <5 min. (ii) Pyridine, –HF, 20 °C, 10 days. | ||
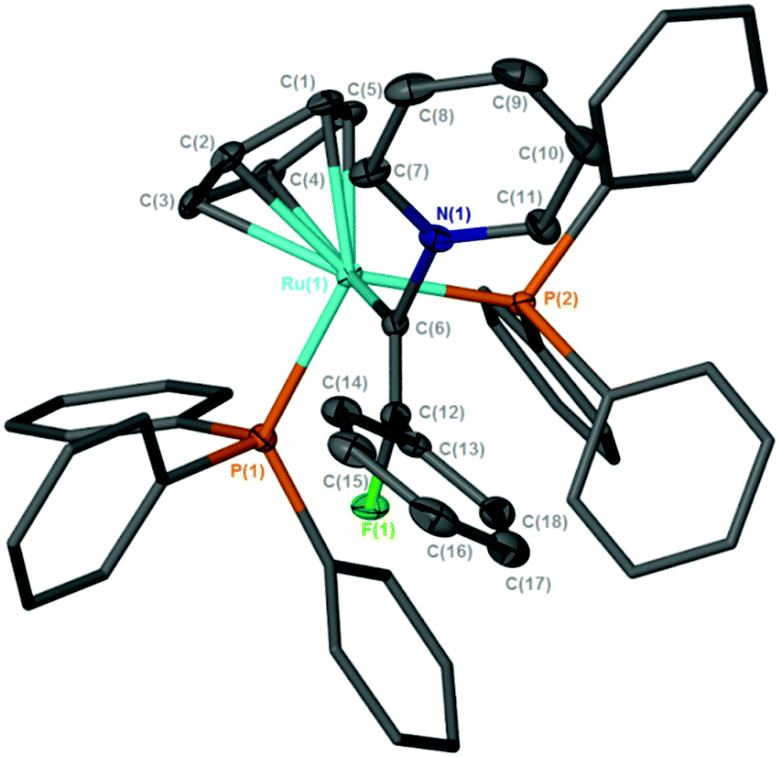 | ||
| Fig. 3 Solid-state structure of the cation [11]+, hydrogen atoms omitted for clarity, thermal ellipsoids (where shown) are at the 50% probability level. | ||
On standing in pyridine solution, [11]BF4 underwent a further reaction to give [12]BF4 and free PPh3. Monitoring this reaction by NMR spectroscopy indicated that this process proceeds through a number of phosphorus- and fluorine-containing intermediates. The molecular structure of [12]BF4 (Fig. 4 as its pyridine solvate) showed that this species was a novel pyridylidene complex had been formed by activation of a C–H bond of the pyridine group in [11]BF4, incorporation of a second pyridine molecule and formal loss of HF, the fate of which could not be determined.
 | ||
| Fig. 4 Solid-state structure of one of the two crystallographically independent cations in the structure of [12]BF4·pyridine, hydrogen atoms omitted for clarity, thermal ellipsoids (where shown) are at the 50% probability level. | ||
A potential mechanism for the conversion of [3a]BF4 to [12]BF4 was investigated by a DFT study. Given the changes in molecularity during the proposed mechanism there are significant differences in the relative zero-point-energy-corrected electronic energies (ESCF+ZPE) and Gibbs energies of different states. The following discussion refers solely to the Gibbs energies at 298 K, although both are presented in Fig. 5. In the first step, nucleophilic attack of pyridine occurs at the metal-bound carbon atom of the vinylidene in [3a]+ (taken as the reference point for the calculations) to give [11-Z]+ through transition state TS[3a]+[11-Z]+. The fact that [11]+ is predicted to be lower in energy than [3a]+ and TS[3a]+[11-Z]+ is reasonably accessible (ΔG‡298 + 100 kJ mol−1) is consistent with the experimental observation of fast formation of [11]BF4.
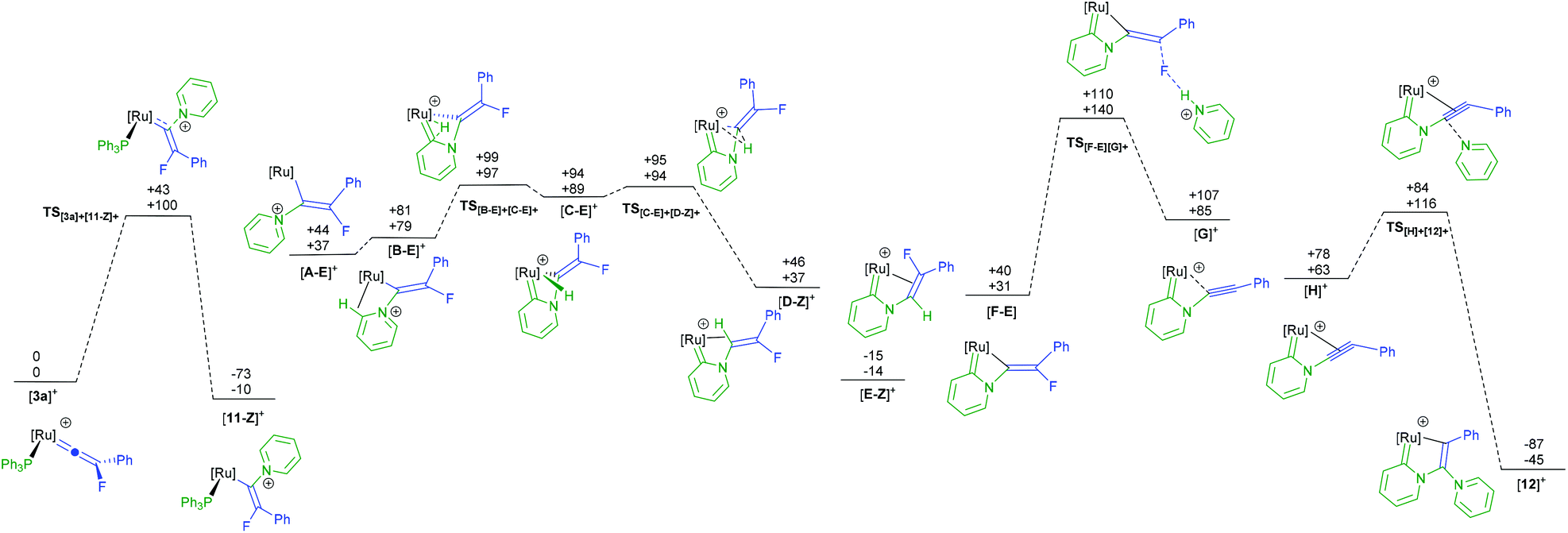 | ||
| Fig. 5 Potential energy surface for the proposed mechanism of formation of [12]+ from [3a]+. ESCF+ZPE (top) and Gibbs energies at 298.15 K (bottom), relative to [3a]+ and two molecules of pyridine, are shown in kJ mol−1. Calculations are at the (RI-)PBE0-D3/def2-TZVPP//(RI-)BP86/SV(P) level with COSMO solvation in pyridine. [Ru] = [Ru(η5-C5H5)(PPh3)]. | ||
The next stages in the reaction pathway are directly related to previous studies on pyridine C–H activation.68,69 Loss of PPh3 from [11]+ leads to intermediate [A]+. Formation of agostic complex [B]+ followed by C–H activation viaTS[B]+[C]+ leads to hydride complex [C]+ which may then undergo a subsequent migration to afford [D]+. A subsequent isomerisation to the lower energy isomer [E]+ then occurs. Formal elimination of HF must then occur to give [12]+ although attempts to model elimination of HF from [E]+via a pyridine-induced E2-process were unsuccessful. However, deprotonation of [E]+ (or hydride complex [C]+) may occur in pyridine solution to afford [F] and [HNC5H5]+. Defluorination may now occur through TS[F][G]+ in which the [HNC5H5]+ aids the loss of the fluorine to give (after coordination of the triple bond in [G]+ to the metal) alkyne complex [H]+. Nucleophilic attack by pyridine through TS[H]+[12]+ then gives the observed product [12]+, which is also the lowest point on the potential energy surface (−45 kJ mol−1).
The calculations indicate that the nucleophilic attack by pyridine of [3a]+ to give the E-isomer ([11-E]+) of the complex is both kinetically (TS[3a]+[11-E]+ +155 kJ mol−1) and thermodynamically unfavorable (+65 kJ mol−1) when compared to the formation of the Z-isomer. This appears to be due to an unfavourable interaction between the PPh3 ligands and the Ph-group of the alkenyl ligand in the E-isomer. In contrast, however, defluorination from [F] is far more accessible from the E-isomer of the alkene complex ([F-E]) than from the Z-isomer (TS[F-E][G]+ +140 kJ mol−1; TS[F-Z][G]+ +173 kJ mol−1, see ESI† for full E- and Z-pathways). There are a number of possible points on the PES where interconversion of the E- and Z-isomers may occur. For example, previous calculations indicate that the vinylidene complexes, [I-Z]+ and [I-E]+ (Fig. 6a) would be readily accessible from complexes such as [A]+.39 Rotation of vinylidene ligands, which will interchange the E- and Z-isomers, is a low energy process.70,71 We tentatively propose that at some point following state [11-Z]+ the system isomerises and follows the E-isomer pathway.
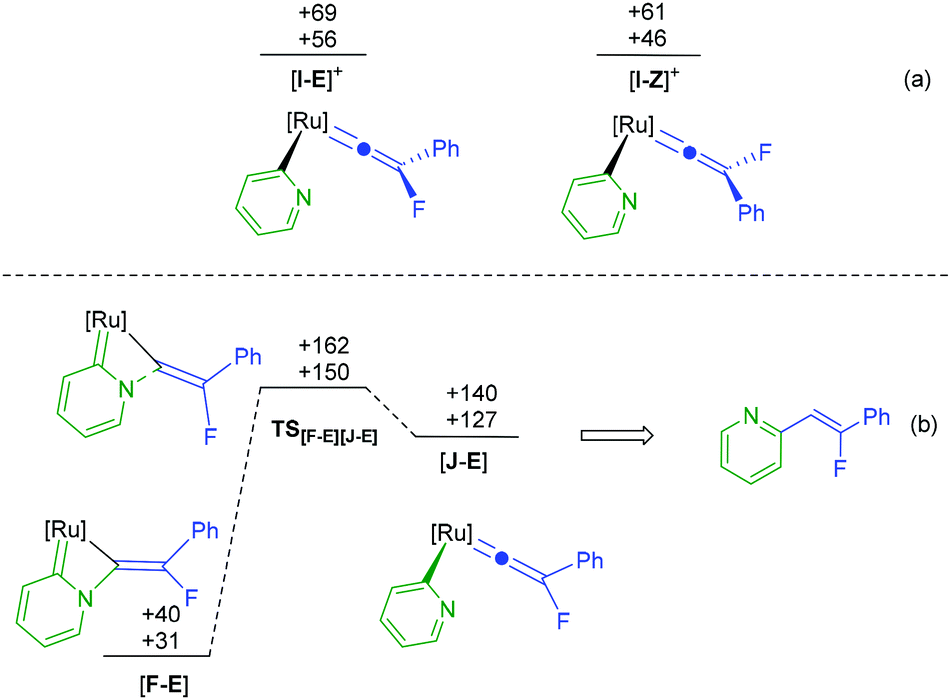 | ||
| Fig. 6 Calculations at the (RI-)PBE0-D3/def2-TZVPP//(RI-)BP86/SV(P) level with COSMO solvation in pyridine. [Ru] = [Ru(η5-C5H5)(PPh3)]. ESCF+ZPE (top) and Gibbs energies at 298.15 K (bottom), relative to [3a]+, pyridine and PPh3. | ||
The formation of [11]+ is rapid and a second, slower, reaction subsequently gives [12]+. In addition to supporting our previous observation that the [Ru(η5-C5H5)(PPh3)]+ fragment may selectively activate pyridine in the 2-position,68,72 the DFT study also provides an explanation as to why no alkenylated pyridine is observed in the fluorine-containing system. The key transition state in this process involves C–N bond cleavage in [F-E] viaTS[F-E][J-E] which at +150 kJ mol−1 lies higher in energy than TS[F-E][G]+ (Fig. 6b) implying that defluorination is preferred to C–N bond cleavage.
It appears that C–F bond cleavage in this system occurs by a pyridinium-assisted fluoride abstraction. This process is related to Hughes’ studies on half-sandwich iridium complexes in which α-fluoride abstraction leads to a cationic carbene complex.73,74
In this case, a cationic alkyne complex is the product of the fluoride loss from the β-position of complex [F], which then undergoes nucleophilic attack by pyridine.
CPh)(PPh3)2], [1a] the latter corresponding to the formal loss of “F+” from the fluorovinylidene.83 Monitoring the reactions over an extended period (ca. two weeks) at room temperature showed almost total conversion to [13]. Heating the reactions at 50 °C increased the rate of conversion but not the final product distribution. This should be contrasted with the reaction of [4a]+ with water which results in the formation of [Ru(η5-C5H5)(CO)(PPh3)2]+ and toluene.76
In contrast, hydrolysis of [3a]BF4 in the presence of chloride as an additive (introduced as NnBu4Cl·H2O§) results in the selective, if slow, formation of [Ru(η5-C5H5)Cl(PPh3)2], [13] and the acyl-fluoride 14 (Scheme 5a), which has resulted from formal hydration of the vinylidene ligand and isomerisation of the resulting α-fluoroaldehyde (vide infra). Compound 14 was identified on the basis of characteristic resonances in the 19F (δ 43.7, t, 3JHF = 1.5 Hz), 1H (δ 3.84, d, 3JHF = 1.5 Hz), and 13C{1H} NMR spectra (δ 39.2, d, 2JCF = 54 Hz, CH2; 162.1, d, 2JCF = 362.3 Hz, COF) matching those previously reported.84 Monitoring the reaction by NMR spectroscopy demonstrated that [13] and 14 were formed at approximately the same rate. Compound 14 was isolated by sublimation and proved to be highly moisture sensitive.
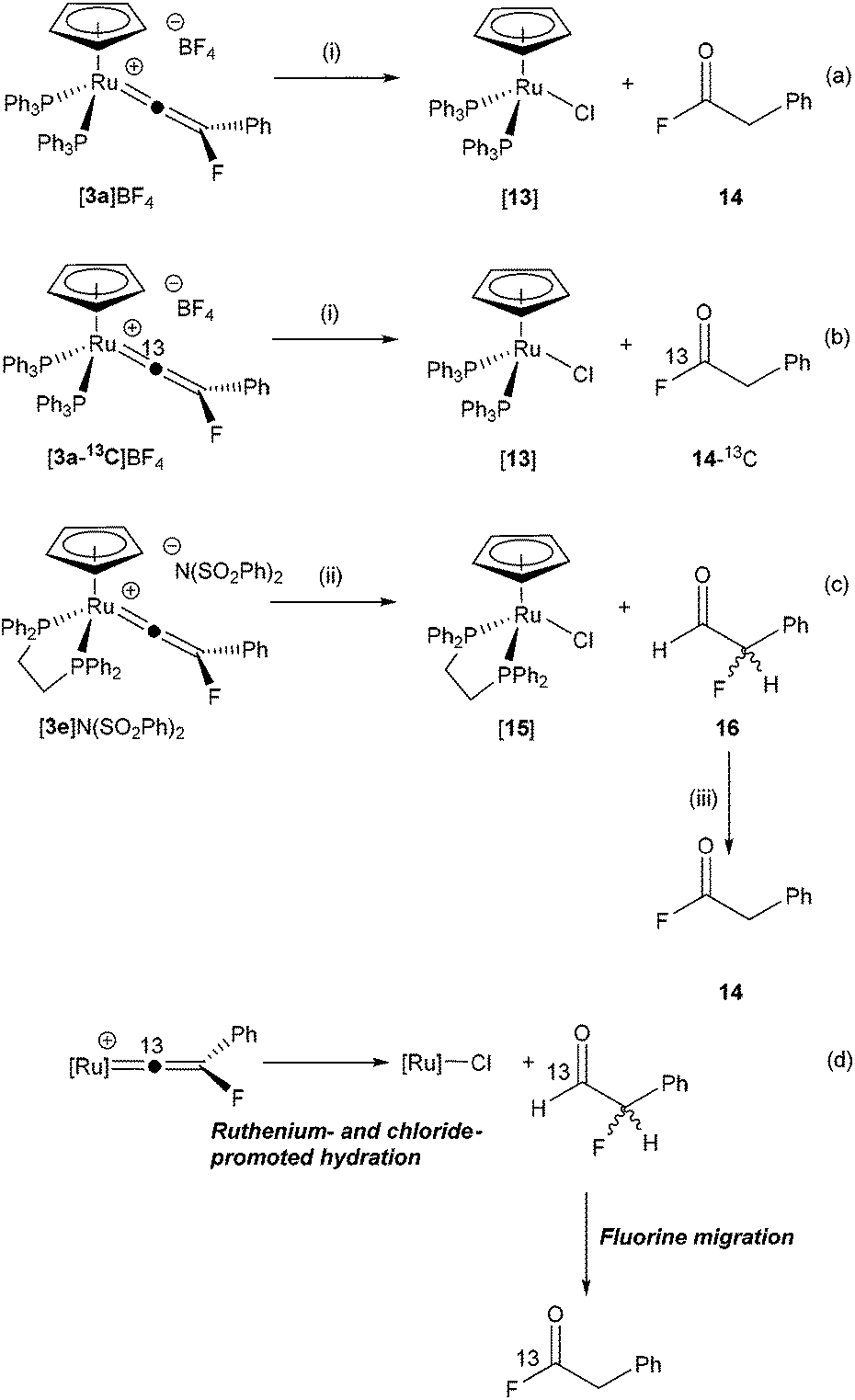 | ||
| Scheme 5 (i) CD2Cl2, +NnBu4Cl, –NnBu4BF4, R. T., 14 d. (ii) CD2Cl2, +NnBu4Cl, –NnBu4(SO2Ph)2, R. T. (iii) 6 d, R. T. | ||
The formation of 14 is somewhat surprising as it appears that either fluoro- or phenyl-migration must have occurred. Given that the reaction of [3a]BF4 with NCMe did not give rise to the alkyne FCCPh and [9]BF4 (which would also require F or Ph migration) it might be presumed that these are high-energy pathways in this system, although phenyl-migration in di-substituted vinylidene complexes is well established.64,85–88 In order to gain further mechanistic insight into this process, the 13C-enriched vinylidene complex [3a-13C]BF4 was prepared and subjected to an identical reaction with NnBu4Cl·H2O (Scheme 5b). Analysis of the resulting 19F NMR spectra demonstrated that the resonance for 14 now appeared as a doublet (1JCF = 361.4 Hz) with no evidence of a species with the labelled atom in the alternative position. Repeating the reaction with [3a]+ as the benzenesulfonimide salt (prepared from [1a] and NFSI) also gave 14, indicating that the source of the fluorine was the vinylidene ligand not the [BF4]− counteranion.
Hydrolysis of the dppe-containing complex [3e]N(SO2Ph)2 in the presence of chloride (as NnBu4Cl·H2O) is much faster than the PPh3-containing analogue. At room temperature rapid formation of [Ru(η5-C5H5)Cl(dppe)], [15] and the α-fluoroaldehyde 16 (identified by resonances in the 1H and 19F NMR spectra at δ 9.72 (dd, 3JHF = 7.6 Hz, 5JHH = 0.7 Hz and δ −191.7 (dd, 2JHF = 47.0 Hz, 3JHF = 7.6 Hz) respectively) was observed, Scheme 5c.89 Over the course of 6 days the resonances for 16 decreased in intensity to be replaced by those for 14. It is proposed that anti-Markovnikov hydration of the vinylidene77–79,82 occurs to give 16 which then undergoes a slower isomerisation to 14. DFT calculations indicate that 16 is 97 kJ mol−1 higher in energy (ΔG298) than 14 so this conversion is thermodynamically favoured and aldehydes such as 16 have been reported to be unstable.90 The overall process is summarised in Scheme 5d.
The reaction of metal alkynyl complexes (derived from terminal alkynes) with “F+” followed by hydrolysis and liberation of α-fluoroaldehydes represents a stepwise functionalization of alkynes with the incorporation of both new functionality and the generation of a new stereocentre. This type of reactivity is highly desirable as the fluorinated vinylidene ligands are effectively acting as synthetic equivalents of 1-fluoroalkynes. Work is underway in our groups to exploit these stoichiometric observations for the development of catalytic protocols for combined alkyne functionalization and fluorination.
Conclusions
The reaction between alkynyl complexes [1] and electrophilic fluorinating and trifluoromethylating agents offers a highly selective and simple route for the direct formation of C–F and C–CF3 bonds and the construction of novel fluorinated vinylidene ligands. Although the addition of other electrophiles to alkynyl complexes has been reported, it is remarkable that the highly oxidising sources of “F+” result in selective C–F bond formation rather than direct fluorination of the metal complexes. These data demonstrate that OSEF (or analogous trifluoromethylation) represents a complementary approach to current metal-mediated routes for electrophilic C–F bond formation and may be applicable to a range of organometallic systems.As the new fluorinated ligands were retained within the coordination sphere of ruthenium it was possible to evaluate the effects of incorporation of the fluorine and explore subsequent reactivity. Reactions of fluorovinylidene complexes with nucleophiles such as pyridine and water show many similarities to the reactivity of non-fluorinated vinylidenes. As these25 are important intermediates in a range of metal-catalysed reactions, this suggests that access to fluorinated analogues offers new opportunities for the formation of novel fluorinated organic compounds without re-writing the rule book in terms of their reactivity. Indeed, our observation of anti-Markovnikov hydration of fluorovinylidenes, a key reaction of non-fluorinated analogues, to form both new functionality and a new stereocentre in the α-fluoroaldehyde products supports this argument. In addition to applications in synthesis, the introduction of fluorine into ligands of organometallic complexes may significantly alter metal–ligand bonding and, for example, may be used to blue-shift the emission from luminescent iridium complexes.91–94 In summary, it has been demonstrated how OSEF allows for the facile formation of C–F (and C–CF3 bonds) in metal alkynyl complexes to be achieved and that C–F bond cleavage, assisted by a pyridinium salt, may occur from the β-position of an organic ligand.
Acknowledgements
We thank the EPSRC (DTA studentship to LMM and computational equipment EP/H011455/1 and EP/K031589/1) and the University of York (PhD Studentship to LMH and summer studentship to MKS) for funding. The insightful comments of Professors Duncan Bruce and Robin Perutz on this work are gratefully acknowledged.Notes and references
- X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826–870 CrossRef CAS PubMed.
- Z. Jin, G. B. Hammond and B. Xu, Aldrichimica Acta, 2012, 45, 67–83 CAS.
- T. Besset, T. Poisson and X. Pannecoucke, Chem. – Eur. J., 2014, 20, 16830–16845 CrossRef CAS PubMed.
- T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470–477 CrossRef CAS PubMed.
- T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed.
- X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826–870 CrossRef CAS PubMed.
- T. Furuya, J. E. M. N. Klein and T. Ritter, Synthesis, 2010, 1804–1821 CAS.
- C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2015, 54, 3216–3221 CrossRef CAS PubMed.
- M. G. Campbell and T. Ritter, Org. Process Res. Dev., 2014, 18, 474–480 CrossRef CAS PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- E. Lee, A. S. Kamlet, D. C. Powers, C. N. Neumann, G. B. Boursalian, T. Furuya, D. C. Choi, J. M. Hooker and T. Ritter, Science, 2011, 334, 639–642 CrossRef CAS PubMed.
- D. O'Hagan and H. Deng, Chem. Rev., 2015, 115, 634–649 CrossRef PubMed.
- S. M. Senaweera, A. Singh and J. D. Weaver, J. Am. Chem. Soc., 2014, 136, 3002–3005 CrossRef CAS PubMed.
- T. Ahrens, J. Kohlmann, M. Ahrens and T. Braun, Chem. Rev., 2015, 115, 931–972 CrossRef CAS PubMed.
- M. G. Campbell and T. Ritter, Chem. Rev., 2015, 115, 612–633 CrossRef CAS PubMed.
- M. Reinhold, J. E. McGrady and R. N. Perutz, J. Am. Chem. Soc., 2004, 126, 5268–5276 CrossRef CAS PubMed.
- E. Clot, O. Eisenstein, N. Jasim, S. A. Macgregor, J. E. McGrady and R. N. Perutz, Acc. Chem. Res., 2011, 44, 333–348 CrossRef CAS PubMed.
- B. Procacci, Y. Jiao, M. E. Evans, W. D. Jones, R. N. Perutz and A. C. Whitwood, J. Am. Chem. Soc., 2015, 137, 1258–1272 CrossRef CAS PubMed.
- M. E. Evans, C. L. Burke, S. Yaibuathes, E. Clot, O. Eisenstein and W. D. Jones, J. Am. Chem. Soc., 2009, 131, 13464–13473 CrossRef CAS PubMed.
- E. Clot, C. Mégret, B. M. Kraft, O. Eisenstein and W. D. Jones, J. Am. Chem. Soc., 2004, 126, 5647–5653 CrossRef CAS PubMed.
- M. F. Kuehnel, D. Lentz and T. Braun, Angew. Chem., Int. Ed., 2013, 52, 3328–3348 CrossRef PubMed.
- M. K. Whittlesey and E. Peris, ACS Catal., 2014, 4, 3152–3159 CrossRef CAS.
- G. Meier and T. Braun, Angew. Chem., Int. Ed., 2009, 48, 1546–1548 CrossRef CAS PubMed.
- A. J. Cresswell, S. G. Davies, P. M. Roberts and J. E. Thomson, Chem. Rev., 2015, 115, 566–611 CrossRef CAS PubMed.
- A. G. Gilicinski, G. P. Pez, R. G. Syvret and G. S. Lal, J. Fluorine Chem., 1992, 59, 157–162 CrossRef CAS.
- E. W. Oliver and D. H. Evans, J. Electroanal. Chem., 1999, 474, 1–8 CrossRef CAS.
- T. Furuya and T. Ritter, J. Am. Chem. Soc., 2008, 130, 10060–10061 CrossRef CAS PubMed.
- T. Furuya, D. Benitez, E. Tkatchouk, A. E. Strom, P. Tang, W. A. Goddard and T. Ritter, J. Am. Chem. Soc., 2010, 132, 3793–3807 CrossRef CAS PubMed.
- N. D. Ball and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 3796–3797 CrossRef CAS PubMed.
- P. Tang, T. Furuya and T. Ritter, J. Am. Chem. Soc., 2010, 132, 12150–12154 CrossRef CAS PubMed.
- C–F reductive elimination from a Au(III) fluoride, generated by oxdiation of a Au(I) complex by XeF2 has also been reported, N. P. Mankad and F. D. Toste, Chem. Sci., 2012, 3, 72–76 RSC.
- C. R. Pitts, S. Bloom, R. Woltornist, D. J. Auvenshine, L. R. Ryzhkov, M. A. Siegler and T. Lectka, J. Am. Chem. Soc., 2014, 136, 9780–9791 CrossRef CAS PubMed.
- K. M. Engle, T.-S. Mei, X. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2011, 50, 1478–1491 CrossRef CAS PubMed.
- A. R. Mazzotti, M. G. Campbell, P. Tang, J. M. Murphy and T. Ritter, J. Am. Chem. Soc., 2013, 135, 14012–14015 CrossRef CAS PubMed.
- J. R. Brandt, E. Lee, G. B. Boursalian and T. Ritter, Chem. Sci., 2014, 5, 169–179 RSC.
- L. Hintermann and A. Togni, Angew. Chem., Int. Ed., 2000, 39, 4359–4362 CrossRef CAS.
- S. Piana, I. Devillers, A. Togni and U. Rothlisberger, Angew. Chem., Int. Ed., 2002, 41, 979–982 CrossRef CAS.
- M. Althaus, C. Becker, A. Togni and A. Mezzetti, Organometallics, 2007, 26, 5902–5911 CrossRef CAS.
- J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650–682 CrossRef CAS PubMed.
- L. M. Milner, N. E. Pridmore, A. C. Whitwood, J. M. Lynam and J. M. Slattery, J. Am. Chem. Soc., 2015, 137, 10753–10759 CrossRef CAS PubMed.
- L. M. Hall, J. M. Lynam, L. M. Milner and J. M. Slattery, UK Pat, GB1421598.2, 2014 Search PubMed.
- C. Testa, J. Roger, S. Scheib, P. Fleurat-Lessard and J.-C. Hierso, Adv. Synth. Catal., 2015, 357, 2913–2923 CrossRef CAS.
- J. Berger, T. Braun, R. Herrmann and B. Braun, Dalton Trans., 2015, 44, 19553–19565 RSC.
- M. E. Slaney, D. J. Anderson, M. J. Ferguson, R. McDonald and M. Cowie, J. Am. Chem. Soc., 2010, 132, 16544–16558 CrossRef CAS PubMed.
- M. E. Slaney, M. J. Ferguson, R. McDonald and M. Cowie, Organometallics, 2012, 31, 1384–1396 CrossRef CAS.
- M. E. Slaney, D. J. Anderson, M. J. Ferguson, R. McDonald and M. Cowie, Organometallics, 2012, 31, 2286–2301 CrossRef CAS.
- D. J. Anderson, R. McDonald and M. Cowie, Angew. Chem., Int. Ed., 2007, 46, 3741–3744 CrossRef CAS PubMed.
- W. Schulze and K. Seppelt, Inorg. Chem., 1988, 27, 3872–3873 CrossRef CAS.
- B. M. Trost, M. U. Frederiksen and M. T. Rudd, Angew. Chem., Int. Ed., 2005, 44, 6630–6666 CrossRef CAS PubMed.
- B. M. Trost and A. McClory, Chem. – Asian J., 2008, 3, 164–194 CrossRef CAS PubMed.
- C. Bruneau and P. H. Dixneuf, Acc. Chem. Res., 1999, 32, 311–323 CrossRef CAS.
- C. Bruneau and P. H. Dixneuf, Angew. Chem., Int. Ed., 2006, 45, 2176–2203 CrossRef CAS PubMed.
- M. I. Bruce and R. C. Wallis, Aust. J. Chem., 1979, 32, 1471–1485 CrossRef CAS.
- M. I. Bruce, G. A. Koutsantonis, M. J. Liddell and B. K. Nicholson, J. Organomet. Chem., 1987, 320, 217–227 CrossRef CAS.
- L. Ciano, N. Fey, C. J. V. Halliday, J. M. Lynam, L. M. Milner, N. Mistry, N. E. Pridmore, N. S. Townsend and A. C. Whitwood, Chem. Commun., 2015, 51, 9702–9705 RSC.
- M. I. Bruce, A. G. Swincer and R. C. Wallis, J. Organomet. Chem., 1979, 171, C5–C8 CrossRef CAS.
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed.
- M. I. Bruce, Chem. Rev., 1991, 91, 197–257 CrossRef CAS.
- O. J. S. Pickup, I. Khazal, E. J. Smith, A. C. Whitwood, J. M. Lynam, K. Bolaky, T. C. King, B. W. Rawe and N. Fey, Organometallics, 2014, 33, 1751–1761 CrossRef CAS.
- E. F. van der Eide, P. Yang and R. M. Bullock, Angew. Chem., Int. Ed., 2013, 52, 10190–10194 CrossRef CAS PubMed.
- R. M. Bullock, J. Chem. Soc., Chem. Commun., 1989, 0, 165–167 RSC.
- M. Bassetti, V. Cadierno, J. Gimeno and C. Pasquini, Organometallics, 2008, 27, 5009–5016 CrossRef CAS.
- Y. Mutoh, K. Imai, Y. Kimura, Y. Ikeda and Y. Ishii, Organometallics, 2010, 30, 204–207 CrossRef.
- J. M. Lynam, Chem. – Eur. J., 2010, 16, 8238–8247 CrossRef CAS PubMed.
- K. Onitsuka, M. Nishii, Y. Matsushima and S. Takahashi, Organometallics, 2004, 23, 5630–5632 CrossRef CAS.
- B. M. Trost and R. C. Livingston, J. Am. Chem. Soc., 2008, 130, 11970–11978 CrossRef CAS PubMed.
- D. G. Johnson, J. M. Lynam, N. S. Mistry, J. M. Slattery, R. J. Thatcher and A. C. Whitwood, J. Am. Chem. Soc., 2013, 135, 2222–2234 CrossRef CAS PubMed.
- A. Škríba, J. Schulz and J. Roithová, Organometallics, 2014, 33, 6868–6878 CrossRef.
- M. J. Cowley, J. M. Lynam, A. C. Whitwood, M. J. Cowley, J. M. Lynam and A. C. Whitwood, Dalton Trans., 2007, 4427–4438 RSC.
- G. Consiglio and F. Morandini, Inorg. Chim. Acta, 1987, 127, 79–85 CrossRef CAS.
- J. M. Lynam, L. M. Milner, N. S. Mistry, J. M. Slattery, S. R. Warrington and A. C. Whitwood, Dalton Trans., 2014, 43, 4565–4572 RSC.
- R. P. Hughes, R. B. Laritchev, J. Yuan, J. A. Golen, A. N. Rucker and A. L. Rheingold, J. Am. Chem. Soc., 2005, 127, 15020–15021 CrossRef CAS PubMed.
- R. P. Hughes, Eur. J. Inorg. Chem., 2009, 2009, 4591–4606 CrossRef.
- C. Bianchini, J. A. Casares, M. Peruzzini, A. Romerosa and F. Zanobini, J. Am. Chem. Soc., 1996, 118, 4585–4594 CrossRef CAS.
- M. I. Bruce and A. G. Swincer, Aust. J. Chem., 1980, 33, 1471–1483 CrossRef CAS.
- B. Breit, U. Gellrich, T. Li, J. M. Lynam, L. M. Milner, N. E. Pridmore, J. M. Slattery, A. C. Whitwood, B. Breit, U. Gellrich, T. Li, J. M. Lynam, L. M. Milner, N. E. Pridmore, J. M. Slattery and A. C. Whitwood, Dalton Trans., 2014, 43, 11277–11285 RSC.
- D. B. Grotjahn, C. D. Incarvito and A. L. Rheingold, Angew. Chem., Int. Ed., 2001, 40, 3884–3887 CrossRef CAS.
- D. B. Grotjahn and D. A. Lev, J. Am. Chem. Soc., 2004, 126, 12232–12233 CrossRef CAS PubMed.
- T. Suzuki, M. Tokunaga and Y. Wakatsuki, Org. Lett., 2001, 3, 735–737 CrossRef CAS PubMed.
- M. Tokunaga, T. Suzuki, N. Koga, T. Fukushima, A. Horiuchi and Y. Wakatsuki, J. Am. Chem. Soc., 2001, 123, 11917–11924 CrossRef CAS PubMed.
- F. Chevallier and B. Breit, Angew. Chem., Int. Ed., 2006, 45, 1599–1602 CrossRef CAS PubMed.
- A similar conceptual loss of “F+” was observed in the case of the metallocyclic complex [III]+, although it does appear to be coupled to a hydrolysis reaction.
- R. T. C. Brownlee and D. J. Craik, Org. Magn. Reson., 1981, 15, 248–256 CrossRef CAS.
- Y. Ikeda, T. Yamaguchi, K. Kanao, K. Kimura, S. Kamimura, Y. Mutoh, Y. Tanabe and Y. Ishii, J. Am. Chem. Soc., 2008, 130, 16856–16857 CrossRef CAS PubMed.
- Y. Mutoh, Y. Ikeda, Y. Kimura and Y. Ishii, Chem. Lett., 2009, 38, 534–535 CrossRef CAS.
- M. Otsuka, N. Tsuchida, Y. Ikeda, Y. Kimura, Y. Mutoh, Y. Ishii and K. Takano, J. Am. Chem. Soc., 2012, 134, 17746–17756 CrossRef CAS PubMed.
- K. Kanao, Y. Ikeda, K. Kimura, S. Kamimura, Y. Tanabe, Y. Mutoh, M. Iwasaki and Y. Ishii, Organometallics, 2013, 32, 527–537 CrossRef CAS.
- S. T. Purrington, N. V. Lazaridis and C. L. Bumgardner, Tetrahedron Lett., 1986, 27, 2715–2716 CrossRef CAS.
- D. D. Steiner, N. Mase and C. F. Barbas, Angew. Chem., Int. Ed., 2005, 44, 3706–3710 CrossRef CAS PubMed.
- A. B. Tamayo, B. D. Alleyne, P. I. Djurovich, S. Lamansky, I. Tsyba, N. N. Ho, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2003, 125, 7377–7387 CrossRef CAS PubMed.
- S. J. Lee, K.-M. Park, K. Yang and Y. Kang, Inorg. Chem., 2009, 48, 1030–1037 CrossRef CAS PubMed.
- C.-H. Yang, M. Mauro, F. Polo, S. Watanabe, I. Muenster, R. Fröhlich and L. De Cola, Chem. Mater., 2012, 24, 3684–3695 CrossRef CAS.
- F. Kessler, Y. Watanabe, H. Sasabe, H. Katagiri, M. K. Nazeeruddin, M. Gratzel and J. Kido, J. Mater. Chem., 2013, 1, 1070–1075 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: CIF data, experimental details, DFT methods and coordinates. CCDC 1040309–1040312 and 1419777–1419780. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt04596g |
| ‡ [8aCl]BF4 was prepared by a new method from [1a] and N-chlorosuccinamide, [8aBr]BF4 was synthesised from one equivalent of Br2 and NaBF4, see ESI. |
§ The ratio of NnBu4Cl to H2O was shown to be ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by elemental analysis. :1 by elemental analysis. |
| This journal is © The Royal Society of Chemistry 2016 |