DOI:
10.1039/C5DT04563K
(Paper)
Dalton Trans., 2016,
45, 4843-4853
Spectroscopic investigations into the binding of hydrogen sulfide to synthetic picket-fence porphyrins†
Received
20th November 2015
, Accepted 1st February 2016
First published on 1st February 2016
Abstract
The reversible binding of hydrogen sulfide (H2S) to hemeprotein sites has been attributed to several factors, likely working in concert, including the protected binding pocket environment, proximal hydrogen bond interactions, and iron ligation environment. To investigate the importance of a sterically-constrained, protected environment on sulfide reactivity with heme centers, we report here the reactivity of H2S and HS− with the picket-fence porphyrin system. Our results indicate that the picket-fence porphyrin does not bind H2S in the ferric or ferrous state. By contrast, reaction of the ferric scaffold with HS− results in reduction to the ferrous species, followed by ligation of one equivalent of HS−, as evidenced by UV-vis, NMR spectroscopy and mass spectrometry studies. Measurement of the HS− binding affinities in the picket-fence or tetraphenyl porphyrin systems revealed identical binding. Taken together, these results suggest that the protected, sterically-constrained binding pocket alone is not the primary contributor for stabilization of ferric H2S/HS− species in model systems, but that other interactions, such as hydrogen bonding, must play a critical role in facilitation of reversible interactions in ferric hemes.
Introduction
Interactions between heme-containing metalloproteins and gas molecules play important roles in biological systems. For example, dioxygen (O2) ligation to Myoglobin (Mb) and Hemoglobin (Hb) constitutes a critical mechanism of oxygen transport associated with aerobic respiration.1 Consistent with the importance of a protected binding pocket, not only to provide protection for the bound O2 but also to prevent unwanted μ-oxo bridged dimer formation, preparation and use of the all-cis-tetra-pivaloylamide-ortho-substituted phenyl porphyrin (“picket-fence porphyrin”) by Collman and co-workers provided the first example of reversible O2 binding to a synthetic heme model almost 40 years ago.2,3
In addition to O2, the last few decades have witnessed the acceptance of other small molecule gases, namely carbon monoxide (CO), nitric oxide (NO), and hydrogen sulfide (H2S), as important endogenously-produced gaseous signalling molecules involved in physiological functions, including smooth muscle relaxation, neurotransmission, and vasoregulation.4,5 Extending the use of the picket-fence porphyrin model beyond O2-binding investigations, treatment with CO leads to irreversible heme-CO formation and inhibition of O2 binding, both of which are consistent with the effects of CO poisoning.3 Similarly, the reaction of NO and related oxidized species including nitrite (NO2−) has also been investigated in the picket-fence porphyrin, leading to an greater understanding of interactions involved in the heme-mediated oxidation of NO to NO2−.6–9 For example, reaction of atmospheric oxygen in pyridine with heme-nitrosyl produces a stable bis-ligated NO2− adduct in which the pocket-bound NO2− is stabilized by a weak hydrogen bond to one of the amides in the porphyrin scaffold.9,10 Despite the important insights gained from use of the picket-fence porphyrin model to investigate O2, CO, and NO interactions with heme centers, analogous investigations with H2S remain absent.
Hydrogen sulfide is the most recently discovered endogenously produced gasotransmitter, with identified roles in diverse aspects of biological signalling and human health.11,12 Adding to the challenges of unravelling this important biomolecule, H2S exhibits more complex reactivity than that of NO or CO. In addition to being a good reductant, highly metallophilic, and oxygen sensitive, H2S exists in various protonation states at biological pH, thus complicating whether H2S or HS− is the active species when reacting with bioinorganic centers.13–15 One of the first recognized reactions of sulfide with heme iron was sulfheme formation, in which the periphery of the porphyrin is irreversibly modified by sulfide in the presence of an oxidant, such as H2O2, iron hydroperoxo, or iron(IV) oxo species.16–20 The resulting compound is characterized by a green color and dominant absorption band near 620 nm in ferrous Mb.17,21,22 Sulfheme formation reduces the affinity for other gasses at the heme center, as evidenced by a 2500- and 1500-fold reduction in O2 and CO affinity, respectively.16–18 By contrast to the detrimental effects of sulfheme formation, H2S/HS− binds reversibly to heme centers in many organisms that live in sulfide-rich environments, such as the HbI center of the Lucina Pectinata clam found in sulfide-rich mangrove swamps.23,24 The reversible binding of sulfur to hemeprotein sites has been attributed to several factors, likely working in concert, including the protected binding pocket environment and proximal glutamate residue forming a hydrogen bond with bound sulfide, and iron ligation environment.20,23,24
As a whole, investigations of sulfide with synthetic hemes have focused on relatively simple porphyrin platforms. In one of the first such studies, Scheidt and co-workers reported the structure of an iron hydrosulfido complex from a tetra(methoxyphenyl)porphyrin system in 1984.25 Studies by Collman and co-workers aimed at reactions of sulfide with a cytochrome c oxidase model renewed the field in 2012.26,27 Recent investigations into heme/sulfide interactions have included work on the binding of HS− to open-faced ferrous porphyrin complexes28 and use of silyl-protected sulfur species to overcome Fe–SH instabilities.29 The importance of distal histidine/pyridine coordination has also been investigated using a microperoxidase and a cyclodextrin-functionalized porphyrin system; both of which suggest that distal ligation is important for stable ferric hydrosulfido formation.30,31 As a whole, these investigations have focused on open-faced synthetic porphyrins, which do not probe effects such as a protected binding site or the importance of proximal hydrogen bonding interactions. Motivated by the utility of the picket-fence porphyrin in investigating and understanding O2,3 CO,3,32,33 and NO34–37 reactivity with synthetic heme systems, we sought to extend the utility of this model compound to investigate whether the presence of a protected binding pocket alone would be sufficient to bind H2S or HS−. In addition, we hypothesized that the protected pocket in the picket fence porphyrin system could provide useful insight into the different factors impacting bound sulfide stability in synthetic heme centers.
Results and discussion
To investigate the chemistry of H2S with synthetic heme platforms and to determine whether protected axial binding environments affect H2S/HS− binding, we chose to use the “picket-fence porphyrin” (PFP) model because of its protected axial cavity. Furthermore, the PFP model allows for direct sulfide binding comparison in both low and high polarity solvents, as well as with the parent tetraphenyl porphyrin (TPP) complexes, which lack the axial pocket.
We prepared the picket-fence porphyrin as described in the literature using minor modifications (see Experimental section) to afford isomerically-pure all-cis ligand (Fig. 1). To compare the charge and the importance of axial ligands, we prepared FeIII(TPivPP)Br (1), imidazole-free FeII(TPivPP) (2), and FeII(TPivPP)(N-MeIm)2 (3).3,38,39 To compare the results of these protected systems, we also used the parent TPP complexes FeIII(TPP)Cl (4), FeII(TPP) (5), and FeII(TPP)(N-MeIm)2 (6). Although such model compounds are only soluble in organic solvents, the broad solubility in solvents ranging from toluene to DMF provides the unique opportunity to investigate the role of solvent polarity in concert with the protected PFP pocket. Additionally, the ability to isolate different protonation states of sulfide using H2S gas or tetrabutylammonium hydrosulfide (NBu4SH) allows for differentiation of H2S versus HS− reactivity, which is otherwise not possible in aqueous systems.13,40–42
 |
| | Fig. 1 Model Fe(II) and Fe(III) scaffolds used to study sulfide binding. The protected pocket in picket-fence porphyrin systems (1–3) provides a distinct ligand environment from the parent tetraphenyl porphyrin systems (4–6). | |
Reaction of FeII(TPivPP)(N-MeIm)2 with sulfide
To determine the reactivity of different sulfide sources with Fe(II) picket-fence systems, we first treated toluene solutions of 3 with stoichiometric as well as saturated solutions of H2S gas. In all experiments, excess N-methylimidazole (N-MeIm) was present to ensure blockage of the bottom face of 3. No changes in the UV-Vis or NMR spectra of 3 were observed upon treatment with H2S, suggesting that H2S does not bind to the ferrous system. Similarly, treatment with elemental sulfur (S8) failed to change the UV-Vis spectrum of 3. Treatment of 3 in toluene (Fig. 2a) with NBu4SH, an organic-soluble source of HS−,43 results in clean formation of [FeII(TPivPP)(SH)]− (7), as evidenced by a hypsochromic shift of the Soret band from 429 to 419 nm with a concomitant increase in intensity and appearance of a shoulder at 455 nm. Additionally, the principle Q-band absorbance of 3 at 534 nm decreased in intensity with the associated formation of new bands at 534, 578, and 620 nm (Fig. 2a, inset). Similar spectroscopic changes in the Soret band, including the shoulder near 450 nm, have been observed previously in the binding of HS− to FeII(octaethylporphyrinate) (FeII(OEP)), FeII(tetra-p-methoxyphenylporphyrinate) (FeII(p-MeOPP)), and FeII(tetramesitylpophyrinate) (FeII(TMP)).28
 |
| | Fig. 2 (a) Titration of 3 (black) with 0.25 equiv. increments of NBu4SH leads to clean conversion to [Fe(TPivPP)(SH)]−, 7, (red). Conditions: MePh solution of 10.8 μM 3 with 367 μM N-methylimidazole titrated with 10 equiv. of NBu4SH in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 MeCN:MePh. (b) Titration of 3 (black) with 0.25 equiv. increments of NBu4SH leads to clean conversion to 7 (red). Conditions: DMF solution of 5.2 μM 3 with 36.5 μM N-methylimidazole titrated with 10 equiv. of NBu4SH in DMF. All titrations were performed under anaerobic conditions with sufficient time to allow for complete reaction between aliquot additions. :9 MeCN:MePh. (b) Titration of 3 (black) with 0.25 equiv. increments of NBu4SH leads to clean conversion to 7 (red). Conditions: DMF solution of 5.2 μM 3 with 36.5 μM N-methylimidazole titrated with 10 equiv. of NBu4SH in DMF. All titrations were performed under anaerobic conditions with sufficient time to allow for complete reaction between aliquot additions. | |
Because toluene (2.38) has a low dielectric constant, we also investigated whether a solvent with a higher dielectric, such as DMF (38.25), would lead to similar observed reactivity with HS−. Much like in toluene, treatment of a DMF solution of 3 with H2S gas or S8 failed to produce spectral changes, however treatment with NBu4SH resulted in a hypsochromic shift of the Soret band from 443 to 419 nm, with concurrent appearance of a shoulder at 455 nm (Fig. 2b). The Q-band region of the spectrum also parallels the observed reactivity in toluene with new absorbances at 538, 575, and 621 nm corresponding to 7. As observed in toluene, conversion of 3 to 7 proceeds cleanly, with well-anchored isosbestic points at 428 and 457 nm. The identical reactivity between NBu4SH in toluene and DMF suggests that the sulfide bound in the PFP cavity of 3 is unaffected by changes to bulk solvent polarity.
To confirm that excess imidazole was not impacting HS− binding, we prepared imidazole-free FeII(TPivPP) (2). Addition of 0.25 equiv. of NBu4SH to 2 in toluene in the absence of N-methylimidazole resulted in splitting of the Soret band from 414 nm to new peaks at 445 and 419 nm (Fig. 3), which is consistent with disaggregation of the porphyrin.44 Further addition of HS− cleanly converts the split Soret band to a final absorbance at 419 nm and results in three Q-bands at 534, 577, and 621 nm, all of which match the spectrum generated from reaction of 3 with NBu4SH (Fig. 2a), suggesting that the imidazole is not bound in the final product. Formation of a five-coordinate, SH-ligated product is also consistent with previous work with FeII(OEP), FeII(p-MeOPP), and FeII(TMP).28 In addition, no reaction was observed when N-methylimidazole-free 2 was treated with H2S and S8, suggesting that N-methylimidazole does not out-compete sulfide binding. When taken together with the previous experiments using 3, as well as the observation that addition of N-methylimidazole at the end of the titration does not significantly alter the spectrum, these results suggest that the presence of N-methylimidazole does not interfere with sulfide binding to the iron center.3,45
 |
| | Fig. 3 Titration of 2 (black) with 0.25 equiv. increments of NBu4SH leads to conversion to 7 (red) after disaggregation of the porphyrin (blue). Conditions: MePh solution of 5.7 μM 2 titrated with 5 equiv. of NBu4SH in a 1:9 MeCN:MePh solution. All titrations were performed under anaerobic conditions with sufficient time to allow for complete reaction between aliquot additions. | |
Reactivity of FeIII(TPivPP)Br with sulfide
Based on our results with 2 and 3, we expected that treatment of ferric 1 with HS− would result in initial reduction from FeIII to FeII producing various oxidized polysulfide species, followed by HS− binding to form 7. As expected, titration of 1 in toluene with NBu4SH initially produces a spectrum similar to that of 3 with an increase in intensity of the Soret band and a bathochromic shift from 419 nm to 429 nm and also a shift in the Q-band from 510 to 534 nm (Fig. 4a). Further addition of NBu4SH produces a spectrum identical to that of 7, consistent with initial reduction of FeIII to FeII, followed by binding of HS− to form [FeII(TPivPP)(SH)]−. Identical reactivity was observed in DMF solution, which upon titration of 1 with NBu4SH (Fig. 4b) produced a spectrum similar to that of 3 with Q-band peaks at 536, 567, and 609 nm. Further addition of NBu4SH produces an identical spectrum to 7 with a Soret band at 419 nm, a prominent shoulder at 455 nm, and Q-band peaks at 538, 575, and 621 nm. Similar to the ferrous system, treatment of 1 in MePh or DMF, in the presence or absence of N-methylimidazole, with S8 failed to produce any changes in the UV-Vis spectrum. In addition to reaction with HS−, we reasoned that the protected binding pocket in the PFP could potentially allow for observation of bound H2S to the ferric scaffold; however, addition of stoichiometric H2S or a saturated H2S solution to 1 in the presence or absence of N-methylimidazole failed to produce any changes in the UV-vis spectrum in either toluene or DMF.
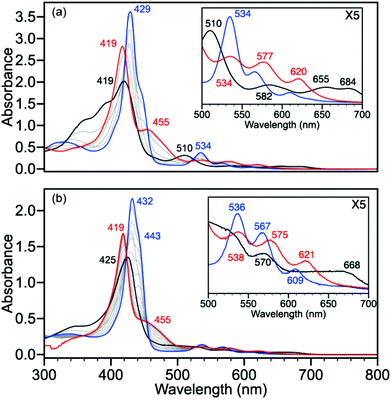 |
| | Fig. 4 (a) Titration of 1 (black) with 0.25 equiv. increments of NBu4SH leads to reduction of Fe(III) to Fe(II) (blue trace shows 0.75 equiv. NBu4SH) followed by binding of HS− to form 7 (red). Conditions: MePh solution of 20 μM 1 with 69 μM N-methylimidazole titrated with 3 equiv. of NBu4SH in 1:9 MeCN:MePh. (b) Titration of 1 (black) with 0.25 equiv. increments of NBu4SH leads to reduction of Fe(III) to Fe(II) (blue trace shows 0.5 equiv. NBu4SH) followed by binding of HS− to form 7 (red). Conditions: DMF solution of 12 μM 1 with 80 μM N-methylimidazole titrated with 5 equiv. of NBu4SH. | |
Oxygen sensitivity of [FeII(TPivPP)(SH)]−
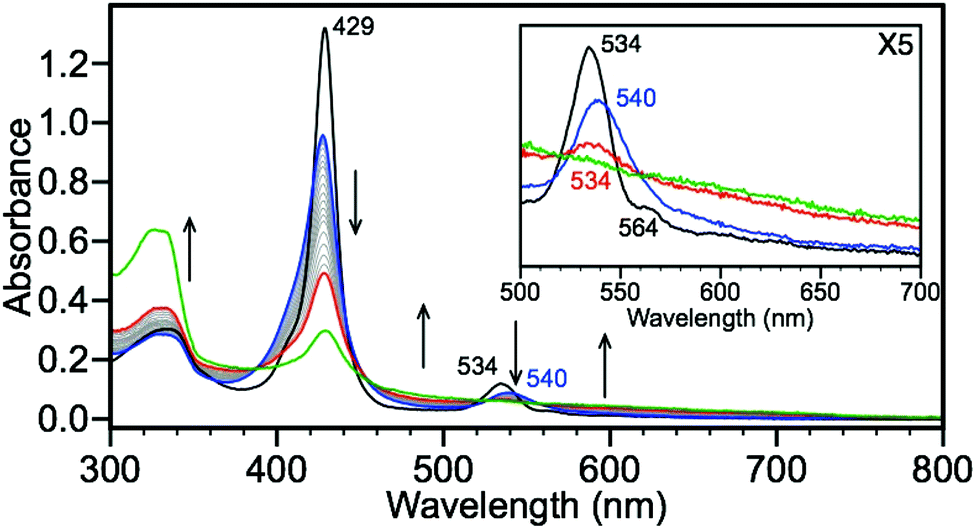
Unlike the TPP systems, the FeII(TPivPP)(N-MeIm)2 scaffolds can bind O2 reversibly, thus providing a unique opportunity to directly investigate the interaction of HS− and O2 at the heme center. Compound 3 readily binds O2, either by exposure of 3 to the atmosphere or by direct injection of O2, and the presence of Fe(TPivPP)(O2)(N-MeIm) (9) is characterized by a decrease in intensity and hypsochromic shift of the Soret band from 429 to 427 nm with a concomitant attenuation and bathochromic shift of the Q-band from 534 to 540 nm (Fig. 5).3 To remove any excess O2, the headspace of the cuvette was purged with dry N2 prior to subsequent NBu4SH addition. Upon treatment of 9 with NBu4SH in toluene, the intensity of the porphyrin absorbances are attenuated with an increased baseline absorbance, suggesting the formation of particulates in the solution. Additionally, an increase in absorbance at 320 nm is observed, which is consistent with formation of sulfur oxidation and polysulfide formation.46–49 Treatment of 3 with NBu4SH to form 7, followed by exposure to the atmosphere afforded the same reactivity. These data suggest that the porphyrin ring is irreversibly oxidized in the presence of oxygen and HS−, matching earlier observations of irreversible oxidation of O2-bound PFP compounds in the presence of acids.3 Mass spectrometric data of the oxidized product showed a peak at 1160.4010 m/z, which matches the exact mass (1160.4097 m/z) and isotope pattern for addition of three O2 molecules to the PFP system, consistent with porphyrin oxidation.50
 |
| | Fig. 5 Exposure of 3 to atmospheric oxygen leads to conversion to 7. Titration of 7 (blue) with 0.25 equiv. increments of NBu4SH leads to irreversible oxidation (red, 20 equiv. and green, 220 equiv.). Conditions: MePh solution of 5.6 μM 3 and 192 μM N-methylimidazole, exposed to atmosphere for 5 minutes, followed by purging headspace for 10 minutes with N2, and titrated with NBu4SH in a 1:39 MeCN–MePh solution. | |
NMR reactivity of picket-fence complexes with sulfide
To complement the UV-Vis spectroscopic studies, and to determine any changes in the iron spin state upon HS− binding, we used 1H NMR spectroscopy to probe the reaction of 3 with NBu4SH. The 1H NMR spectrum of 3 in toluene-d8 exhibits sharp features consistent with an FeII complex and allow for its reactivity to be monitored by NMR spectroscopy.51,52 Treatment of 3 in toluene-d8 with 5 equiv. of NBu4SH in CD3CN results in a clean downfield shift of the pyrrole protons to 63 ppm, consistent with formation of a five-coordinate high-spin FeII complex ligated by a sulfur (Fig. 6).53 The loss of the resonance at −14 ppm is consistent with dissociation of N-methylimidazole from the complex (Fig. S22 and S30†).53,54 These spectral changes, as well as the upfield shift of the phenyl protons to the 7–12 ppm region are all consistent with formation of 7. Similarly, 1H NMR spectroscopy of 1 with NBu4SH also confirmed the reduction to 2, followed by binding of HS− to form 7. The 80 ppm pyrrole resonance, indicative of a high spin FeIII complex,54 shifts upon treatment of 1 with 5 equiv. of NBu4SH, consistent with reduction to FeII followed by formation of 7.53
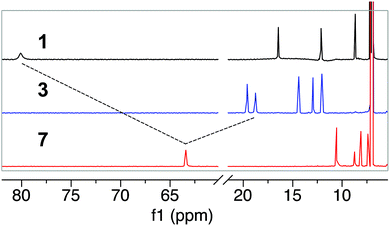 |
| | Fig. 6
1H NMR (600 MHz, toluene-d8) spectra of 1 (4 mM, black), 3 (4 mM, blue) and 7 (red) in CD3CN. The spectrum of 7 was recorded after addition of 5 equiv. of NBu4SH to either 1 or 3. | |
To confirm the spin and charge change from 1 to 7, we measured the magnetic susceptibility of both complexes using the Evans method.55,56 The magnetic susceptibility of 1 was measured to be μeff = 5.6μB, supporting a high-spin (S = 5/2) FeIII complex, which is consistent with solid state measurements.38 Upon treatment of 1 with NBu4SH, the magnetic susceptibility changes to μeff = 5.0μB, supporting the formation of a high-spin (S = 2) FeII complex, matching previously reported Fe–SH bound structures.53,57,58 Taken together, the NMR data supports the reaction sequence in which HS− initially reduces FeIII to FeII, after which an additional equivalent of HS− can bind to the metal center forming a ferrous hydrosulfide product.
Reaction of Fe(TPP) with sulfide
Based on the identical reactivity of 1 and 3 in toluene and DMF, we reasoned that the protected axial binding pocket of the PFP compounds does not provide additional thermodynamic stability or protection from solvent over the un-protected systems. To provide a direct comparison for the experimental sulfide reactivity reactions of the picket-fence heme analogues, we performed analogous experiments with the parent tetraphenylporphyrin (FeII(TPP)(N-MeIm)2, 6). Upon treatment of 6 with NBu4SH (Fig. 7a), similar spectral changes to those observed with 3 were observed, which are consistent with formation of [FeII(TPP)(SH)]− (8). The Soret band decreases in intensity and shifts to 418 nm with concomitant formation of a prominent shoulder at 463 nm with a well-anchored isosbestic point at 444 nm. The Q-band shows characteristic change from a prominent peak at 532 nm to three peaks at 532, 573, and 622 nm. Similarly, treatment of FeIII(TPP)Cl (4) with NBu4SH (Fig. 7b) in DMF shows an initial change in the spectrum consistent with the formation of 6 with features at 432 and 563 nm. Further addition of NBu4SH results in clean conversion to 8 with an associated Soret band shift from 432 nm to 418 nm and shoulder at 463 nm. The Q-bands adopt characteristic peaks at 532, 573, and 622 nm, decreasing in intensity with lower energy (Fig. 7b, inset). Similar to 1, 2, and 3, treatment of 4, 5, or 6 in either DMF or toluene, in the presence or absence of N-methylimidazole, with S8 or H2S fails to perturb the UV-vis spectrum of the iron complexes.
 |
| | Fig. 7 (a) Titration of 6 (black) with 0.25 equivalent increments of NBu4SH to form 8 (red). Conditions: MePh solution of 3.8 μM 6 with 35 μM N-methylimidazole titrated with 10 equivalents of NBu4SH. (b) Titration of 4 (black) with 0.25 increments of NBu4SH leads to reduction of FeIII to FeII (6) (blue trace shows 0.75 equiv. NBu4SH) followed by binding of HS− to form 8 (red). Conditions: DMF solution of 4.5 μM 4 with 30 μM N-methylimidazole titrated with 10 equivalents NBu4SH. | |
Sulfide binding affinities
To confirm a 1:1 Fe:SH binding stoichiometry, we constructed a competitive continuous variation (CCV) plot of 3 and NBu4SH by varying the molar ratios of 3 and HS− while keeping the N-methylimidazole concentration constant. These experiments resulted in a plot with a clean break centered at 0.5, which is consistent with a 1:1 binding stoichiometry (Fig. 8).59–61 Based on this binding stoichiometry, we titrated solutions of 2 and 3 with NBu4SH to determine the apparent association constants of sulfide and fit all titration data to a 1:1 Fe:SH model using the Thordarson method.62,63 The magnitude of these measurements matches binding constants associated with Lucina Pectinata HbI, and several synthetic porphyrin compounds including a cyclodextrin pyridine coordinated porphyrin, Fe(OEP), Fe(p-MeOPP), and Fe(TMP) (Table 1).28,30,40 Furthermore, the observed Ka values show that the binding affinity of HS− for the porphyrin complexes are similar in DMF and toluene, and that the presence or absence of N-methylimidazole does not appreciably impact the observed sulfide binding affinity. Comparison of the picket-fence 3 with the parent 6 reveals that the presence of the protected binding pocket does not provide a significant thermodynamic stabilization of sulfide binding.
 |
| | Fig. 8 A competitive continuous variation plot of 3 and NBu4SH supports 1:1 binding. Conditions: total concentration: 11.1 μM in 1:9 MePh:MeCN solution with 0.33 mM N-methylimidazole. The molar ratio of Fe was varied from 0.1 to 1 and the absorbance was corrected for the concentration of N-methylimidazole. | |
Table 1 Comparison of HS− binding constants for Fe-porphyrin systems
| Species |
Solvent |
log(Ka) |
Source |
| Fe(TPivPP)(N-MeIm)2 |
MePh |
4.5 ± 0.1 |
This work |
| Fe(TPivPP)(N-MeIm)2 |
DMF |
5.0 ± 0.1 |
This work |
| Fe(TPivPP) |
MePh |
4.2 ± 0.1 |
This work |
| Fe(TPP)(N-MeIm)2 |
MePh |
4.8 ± 0.2 |
This work |
|
L. Pectinata HbI |
Water |
4.7 |
30 and 40
|
| Fe(OEP) |
PhCl |
5.0 ± 0.2 |
28
|
| Fe(p-MeOPP) |
PhCl |
4.7 ± 0.4 |
28
|
| Fe(TMP) |
PhCl |
4.6 ± 0.7 |
28
|
| Met-hemoCD3 |
Buffer |
4.9 |
30
|
Mass spectrometry
To gain further insight into sulfide binding, and to determine whether the picket-fence system provides a kinetic barrier to sulfide dissociation, we used HRMS to investigate HS− binding and dissociation in the gas phase. Based on the 1:1 Fe:SH stoichiometry determined from the CCV plot, we expected to only observe ligation of one HS− ligated to the metal center. Mass spectrometric analysis of ferrous 3 treated with 15 equiv. of NBu4SH confirmed this expectation with the appearance of a parent ion peak at 1097.4294 m/z, which matches the expected mass (1097.4122 m/z) and isotope pattern of anionic 7 (Fig. 9a). We also observed a mass peak at 1063.4426 m/z corresponding to [2 − H+]− (calculated 1063.4402 m/z), which we attribute to the loss of HS− as well as H+ (likely from the amide NH in the PFP scaffold) from 7. No peaks corresponding to ligation of two HS− ligands were observed (Fig. S33†). Consistent with the results obtained from 3, treatment of ferric 1 with 15 equiv. of NBu4SH in THF produces an identical mass spectrum as experiments with the ferrous species, which supports initial reduction followed by HS− ligation (Fig. 9b). Taken with the CCV plot, this is consistent with a 1:1 binding ratio of Fe:SH.
 |
| | Fig. 9 (a) Mass spectrum of 3 with 15 equiv. of NBu4SH added in THF results in formation of 2 (blue) and 7 (red), [FeII(TPivPP) − H+]− and [Fe(TPivPP)(SH)]− respectively. (b) Mass spectrum of 1 with 15 equiv. of NBu4SH added in THF results in the formation of 2 (blue) and 7 (red), [FeII(TPivPP) − H+]− and [Fe(TPivPP)(SH)]− respectively. (c) Mass spectrum of 6 with 15 equiv. of NBu4SH added in THF results in formation of 5 (blue) and 8 (red), which ionize as [FeII(TPP) − H+]− and [FeII(TPP)(SH) − H˙]− respectively. (d) Mass spectrum of 4 with 15 equiv. of NBu4SH added in THF results in formation of 5 (blue) and 8 (red), which ionize as [FeII(TPP) − H+]− and [FeII(TPP)(SH) − H˙]− respectively. Conditions: electrospray in negative ion mode with [Fe] = 1 mM in THF with 15 equiv. NBu4SH added in 1:1 THF–acetonitrile. | |
Similar to 1 and 3, treatment of ferrous 6 in THF with 15 equiv. of NBu4SH resulted in formation of mono-sulfur ligated 8 at 700.1522 m/z (ionized as [8 − H˙]−, calculated 700.1385 m/z) (Fig. 9c). As observed in the PFP system, the second major peak at 668.1656 m/z corresponds with unligated 5 (calculated m/z 668.1664). Treatment of ferric 4 under identical conditions affords an identical spectrum as the reaction of the ferrous TPP 6 with NBu4SH (Fig. 9d). Interestingly, the [M − H2S]− peak observed in the PFP system was not observed in the TPP system ([M − SH˙]− was observed), suggesting that the proximity of the acidic N–H groups on the PFP ligand were involved in the H2S ionization. A second notable difference between the PFP and parent TPP system is the relative intensity of the HS− ligated and unligated species. With both mass spectrometric experiments measured under identical instrumental conditions, these relative stabilities suggest that the protective pocket in the PFP system provides additional stability to the bound hydrosulfide ligand. Based on the similar binding thermodynamics measured in solution, this increased stability is likely due to an increased kinetic barrier for sulfide dissociation from the protected pocket in the PFP system rather than a thermodynamic ground state stabilization of bound sulfide. Variation of the collisional energy and comparison to the Fe(TPP) scaffold showed kinetic stabilization of the bound sulfide, suggesting it is bound inside the porphyrin pocket (see ESI, Fig. S38†).64,65
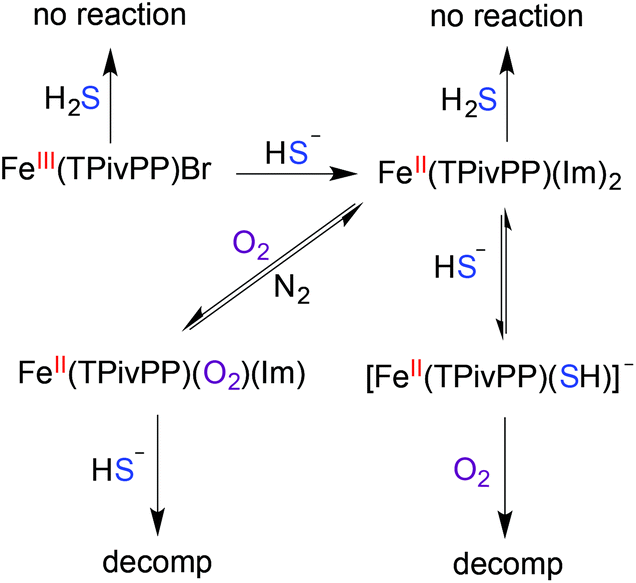
Conclusions
Motivated to extend the utility of synthetic heme structures we used the picket-fence porphyrin as a model to investigate sulfide binding. Our spectroscopic investigations revealed that the FeIII or FeII PFP scaffolds do not react with H2S, but rather with HS−. Upon treatment with HS−, FeIII(TPivPP)Br is reduced to its FeII form (Scheme 1), after which a single HS− ligand binds to the heme iron. This binding stoichiometry was confirmed by UV-Vis and NMR spectroscopic studies as well as mass spectrometric investigations. Comparison of the sulfide binding affinity to FeII in the PFP system to that observed in the less hindered TPP system revealed similar sulfide association constants, suggesting that the protected axial pocket of the PFP system did not provide significant thermodynamic stabilization of the bound sulfide. Mass spectrometric studies using variable collisional energy experiments established that sulfide dissociation from the PFP system had a larger kinetic barrier than ionization of sulfide from the TPP system, which is consistent with a kinetic, rather than thermodynamic, stabilization of bound sulfide within sterically constrained PFP scaffold. Taken together, these results suggest that the sterically-constraining environment alone is not the primary contributor for stabilization of ferric H2S/HS− species in model systems, but that other interactions, such as hydrogen bonding or the interplay between hydrogen-bonding and a protected binding pocket must play a critical role. We are currently investigating other systems that probe these different interactions, which will be reported in due course.
 |
| | Scheme 1 Overall reactivity of the FeIII/II(TPivPP) scaffold with H2S, HS−, and O2. | |
Experimental details
Materials and methods
All manipulations were performed under an inert atmosphere using standard Schlenk techniques or an Innovative Atmospheres N2-filled glove box unless otherwise noted. All chemicals were used as received unless otherwise noted. Pyrrole, 2-nitrobenzaldehyde, and N-methylimidazole were purchased from TCI Chemicals. Pivaloyl chloride, 1,2-dimethoxyethane, tetrabutylammonium chloride, and 1,3,5-trimethoxybenzene were purchased from Sigma-Aldrich. SnCl2·2H2O was purchased from Strem chemicals. FeBr2 (anhydrous) and NaSH (anhydrous) were purchased from Strem chemicals and handled under nitrogen. Hydrogen sulfide gas was purchased from Sigma Aldrich and transferred through a custom-built stainless steel transfer line into a glass storage bulb prior to use. Tetrabutylammonium hydrosulfide (NBu4SH) was synthesized as previously reported.43 Note: hydrogen sulfide and its salts are highly toxic and should be handled carefully to avoid exposure. Spectroscopic grade toluene, acetonitrile, and tetrahydrofuran were degassed by sparging with argon followed by passage through a Pure Process Technologies solvent purification system to remove water and stored over 4 Å molecular sieves in an inert atmosphere glove box. Heptane was passed through an alumina column, dried and distilled over calcium hydride then deoxygenated by three freeze–pump–thaw cycles and stored in an inert atmosphere glove box over 4 Å molecular sieves. Spectroscopic grade N,N-dimethylformamide was dried and distilled over calcium hydride, deoxygenated by three freeze–pump–thaw cycles, and stored in an inert atmosphere glove box over 4 Å molecular sieves. Toluene-d8 and acetonitrile-d3 were purchased from Cambridge Isotope laboratories and distilled from calcium hydride, deoxygenated by three freeze–pump–thaw cycles, and stored in an inert atmosphere glove box. Porphyrin stock solutions were prepared in dry toluene or DMF and stored in an inert atmosphere glove box until immediately prior to use.
Spectroscopic methods
UV-Vis measurements were acquired on an Agilent Cary 100 UV-Vis spectrophotometer equipped with a QNW dual cuvette temperature controller at 25.00 ± 0.05 °C. All spectroscopic samples were prepared under an inert atmosphere in septum-sealed cuvettes obtained from Starna Scientific. NMR spectra were acquired on a Brüker Avance-III-HD 600 spectrometer with a Prodigy multinuclear broadband cryoProbe at 25.0 °C. Chemical shifts are reported in parts per million (δ) and are referenced to residual solvent resonances. IR spectra were acquired on a Thermo Scientific Nicolet 6700 spectrometer equipped with a diamond crystal Smart ATR Attachment. Mass spectra were acquired on a Synapt G2-Si from Waters Corporation and introduced by nanoelectrospray using a platinum wire at 0.4 kV potential. All data were acquired with an ESI voltage of 500 V using “resolution” mode.
General procedure for UV-Vis spectroscopic studies
In a glove box, the porphyrin stock solutions were diluted to the desired concentration by addition to 3.0 mL of solvent in a septum-sealed cuvette and removed from the glove box. Non-porphyrin reagents were prepared approximately 105 times more concentrate than the porphyrin to provide minimal dilution during addition. NBu4SH, H2S, and other reagents were added to the septum-sealed cuvettes by gas-tight syringe. Titration studies were performed using 0.25 equiv. increments unless otherwise noted. Binding constants (Kassoc) were determined from UV-Vis titration data and fit to a 1:1 model.62,63
General procedure for NMR studies
In a glove box, 1.6 μmol of the different Fe(TPivPP) species were added to ∼0.4 mL of toluene-d8 in a septum-capped NMR tube. Stock solution of NBu4SH (0.688 M in acetonitrile-d3) was prepared, and ∼5 equiv. was added to the NMR tube using a gas-tight syringe.
General procedure for mass spectrometry studies
Solutions of Fe species (1 mM) containing 1.5 mM N-methylimidazole were prepared in THF. To this solution, 15 equiv. of NBu4SH in 1:1 THF–acetonitrile were added, and negative ion mode nano-electrospray mass spectra were acquired with a Synapt G2-Si quadrupole time-of-flight mass spectrometer (under identical conditions for each species).
General procedure for Evans’ method magnetic susceptibility measurements
A stock solution containing 1,3,5-trimethoxybenzene (20.2 mg, 120 μmol) in 500 μL of toluene-d8 and 100 μL of CD3CN was prepared. Fe(TPivPP)Br (2.7 mg, 2.3 μmol) was added to 450 μL of the 1,3,5-trimethoxybenzene stock solution. NBu4SH (36.5 mg, 132 μmol) was added to 100 μL of the stock solution and an additional 100 μL of CD3CN was added to ensure complete solubility. The remaining 50 μL of 1,3,5-trimethoxybenzene stock solution was added to a capillary tube. A septum-sealed NMR tube was charged with 400 μL of the Fe(TPivPP)Br stock solution and the capillary tube containing the 1,3,5-trimethoxybenzene standard. The 1H NMR spectrum was recorded, after which 16 μL (5 equiv.) of the NBu4SH stock solution was added. The NMR tube was sonication for 20 minutes to ensure complete mixing, after which another 1H NMR spectrum was recorded. The chemical shift difference between the 1,3,5-trimethoxybenzene resonances in the capillary tube and the Fe(TPivPP)Br solution were measured and corrected using standard diamagnetic corrections.55
Synthesis of Cr(OAc)2 and Cr(acac)2
A 20 mL scintillation vial equipped with a stir bar was charged with K2Cr2O7 (2.0 g, 6.8 mmol), powdered zinc (5.0 g, 76 mmol), and 5 mL of water under nitrogen. Concentrated hydrochloric acid (20 mL) was then added drop wise to the stirred reaction mixture for approximately 20 minutes, and the solution was stirred until the color of the reaction mixture changed from yellow to green and finally to blue. The supernatant was transferred by filter cannula to a 100 mL flask containing 20 mL of a saturated NaOAc solution, which resulted in formation of a red precipitate. The solid product was filtered, washed with EtOH and Et2O, and then dried under vacuum overnight to yield the desired product (1.06 g, 41%). Cr(acac)2 was prepared from the crude Cr(OAc)2 product in a glove box as described in the literature.66 Note: Cr(acac)2 is a pyrophoric powder and should be handled carefully under an inert atmosphere.
Synthesis of iron porphyrin complexes
The synthesis of the iron porphyrin complexes was conducted according to published procedures using the modifications noted below.3,38,39 Spectroscopic properties of the isolated product are provided to aid future preparations of these complexes.
5,10,15,20-Tetrakis(2-nitrophenyl)porphyrin (H2TNPP)
2-Nitrobenzaldehyde (25.0 g, 165 mmol) was dissolved in 500 mL of glacial acetic acid in a 3-neck 1-L round bottom flask fitted with a reflux condenser and a syringe pump inlet. The solution was then heated to reflux and stirred vigorously. Pyrrole (12 mL, 170 mmol) was added drop-wise via the syringe pump over 30 minutes. (Note: this is a highly exothermic reaction and care is needed to keep the reaction under control. We have found that uniform addition of pyrrole using a syringe pump facilitates maintaining a vigorous, but controllable, reaction and increases the reproducibility). After the addition of pyrrole was completed, the solution had turned black and was refluxed for 30 minutes. The oil bath was then allowed to cool to 35 °C and CHCl3 (65 mL) was added to prevent tar formation. (Note: fast cooling or cooling below 35 °C can lead to intractable tar formation, as noted previously.)38 After stirring for 1 h, the reaction mixture was filtered through an extra course frit, and the collected solid was washed with 5 × 100 mL of CHCl3. Additional product can be obtained by pouring the filtrate through a medium porosity frit and washing the collected solid with 5 × 50 mL of CHCl3. The purple solids obtained by filtration were combined and dried overnight at 100 °C under vacuum to yield the desired product (2.89 g, 8.7%). FTIR (ATR, neat) cm−1: 3316 (w, C–H), 1518 (s, NO2), 1341 (s, NO2); 1H NMR (300 MHz, DMSO-d6) δ: 8.98–7.95 (m, 24H), −2.80 (s, 2H); λmax (DMF): 420, 516, 550, 593, 650 nm.
5,10,15,20-Tetrakis(2-aminophenyl)porphyrin (H2TAPP)
A 1-L round bottom flask was charged with H2TNPP (2.5 g, 3.14 mmol) and 125 mL of concentrated HCl. Reagent grade SnCl2·2H2O (10.6 g, 47.0 mmol, 15 equiv.) dissolved in 10 mL of HCl was added to the H2TNPP solution and stirred for 1 h at room temperature, after which the flask was transferred to an oil bath that had been preheated to 65 °C. (Note: the activity of the SnCl2 can be tested by mixing 1 g of SnCl2·2H2O in 1.5 mL of HCl with 0.25 g of 3-nitrobenzaldehyde. The mixture should become warm and turn red-orange within 10 minutes.)38 After heating with vigorous stirring for 25 min, the flask was removed from the oil bath and cooled to room temperature in an ice bath. The reaction mixture was then neutralized carefully with 250 mL concentrated NH4OH over 25 minutes. After the pH was adjusted to >10 with NH4OH, 200 mL of CHCl3 was added and the biphasic solution was stirred for at least 12 hours, after which the organic layer was separated. The aqueous layer was transferred to a 1-L separatory funnel, diluted with water, and extracted with CHCl3 (3 × 150 mL). The combined organic layers were washed with 400 mL of 10% NH4OH and concentrated to ∼50 mL and filtered through celite to remove any remaining tin compounds. The celite was washed with CHCl3 until the filtrate had faded in color, and the combined filtrates were concentrated to ∼50 mL. EtOH (34 mL) and NH4OH (2 mL) were added, and the solution was further concentrated to ∼40 mL, after which an additional 20 mL of EtOH was added and the solution was concentrated to ∼16 mL. The resultant solution was filtered through a medium porosity frit and the purple-black crystals were rinsed with small amounts of ethanol and dried in a 140 °C oven for 1 h to afford 1.65 g (78%) of the desired product. FTIR (ATR, neat) cm−1: 3350 (w, N–H), 3312 (w, N–H), 3023 (w); 1H NMR (600 MHz, CDCl3) δ 8.91 (s, 8H), 7.91–7.81 (m, 4H), 7.59 (t, J = 7.9 Hz, 4H), 7.16 (q, J = 6.4 Hz, 4H), 7.09 (d, J = 8.0 Hz, 4H), 3.51 (d, J = 17.3 Hz, 8H), −2.67 (s, 2H); 13C{1H} NMR (151 MHz, CDCl3) δ: 146.9, 134.9, 131.4, 129.8, 127.0, 117.7, 116.0, 115.4; λmax (CHCl3): 420, 515, 550, 590, 650 nm.
Separation of all-cis-H2TAPP
The separation was carried out in a one-column procedure that enriches the desired all-cis isomer as described in the literature.39 To separate the mixture, 45 g of silica, 100 mL of benzene, and a stir bar were added to a 250 mL 3-neck round bottom flask equipped with a reflux condenser and a benzene-saturated nitrogen stream. After heating at 75 °C for 2 h, 1.25 g of H2TAPP in 6 mL of benzene was added via syringe and the resultant mixture was stirred at 75 °C for 20 h. The resultant dark slurry was cooled to room temperature and poured into a 50 mm OD column. The undesired isomers were eluted with 1:1 C6H6:Et2O until the eluent became a pale red color (∼400 mL). The solvent was then switched to 1:1 Et2O:acetone and the desired isomer was eluted. The collected fractions were checked for purity by TLC (SiO2, 4:1 CHCl3:Et2O, Rf = 0.57) and stored at 4 °C for no longer than 12 hours prior to use. Isolated fractions were not reduced in volume in order to minimize isomerization.
(All-cis)-5,10,15,20-tetrakis[2-(2,2-dimethylpropionamido)phenyl]porphyrin (H2TPivPP)
All of the pure collected fractions of the all-cis isomer were placed in a 1-L round bottom flask under N2. Pyridine (3 mL) and pivaloyl chloride (3 mL) were added and the reaction mixture was stirred for 2.5 h, after which 5 mL of MeOH was added to quench any residual acid chloride. The reaction mixture was evaporated to dryness, dissolved in 125 mL CHCl3, and washed with 80 mL of 10% NH4OH and 2 × 80 mL of water. The combined aqueous washes were combined and extracted with 2 × 30 mL of CHCl3. The combined organic layers were dried with Na2SO4, filtered, and evaporated to dryness under reduced pressure. The crude residue was purified by column chromatograph (SiO2, 4:1 CHCl3:Et2O). Further purification was achieved by dissolving the product in CHCl3, adding EtOH and heptane, reducing the volume under reduced pressure, and filtering the purple crystalline product. The crystals were dried overnight under vacuum to yield 0.72 g (39%) of the desired isomerically-pure product. FTIR (ATR, neat) cm−1: 3430 (m, N–H), 3315 (m, N–H), 3060 (w, C–H), 2956 (m, C–H), 2867 (m, C–H), 1686 (s, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 1H NMR (600 MHz, CDCl3) δ: 8.83 (s, 8H), 8.73 (d, J = 8.45 Hz, 4H), 7.90 (d, J = 6.43 Hz, 4H), 7.85 (t, J = 8.59 Hz, 4H), 7.50 (t, J = 7.84, 4H), 7.21 (s, 4H), 0.07 (s, 36H), −2.59 (s, 2H); 13C{1H} NMR (151 MHz, CDCl3) δ: 175.6, 138.6, 134.4, 131.0, 130.3, 123.2, 121.0, 115.0, 39.1, 26.6; λmax (CHCl3): 418, 512, 545, 588, 641 nm.
O); 1H NMR (600 MHz, CDCl3) δ: 8.83 (s, 8H), 8.73 (d, J = 8.45 Hz, 4H), 7.90 (d, J = 6.43 Hz, 4H), 7.85 (t, J = 8.59 Hz, 4H), 7.50 (t, J = 7.84, 4H), 7.21 (s, 4H), 0.07 (s, 36H), −2.59 (s, 2H); 13C{1H} NMR (151 MHz, CDCl3) δ: 175.6, 138.6, 134.4, 131.0, 130.3, 123.2, 121.0, 115.0, 39.1, 26.6; λmax (CHCl3): 418, 512, 545, 588, 641 nm.
Bromo{(all-cis)-5,10,15,20-tetrakis-[2-(2,2-dimethylpropionamido)phenyl]porphyrinato(2-)}-iron(III) (Fe(TPivPP)Br)
In an inert atmosphere glove box, H2TPivPP (0.54 g, 0.53 mmol), anhydrous FeBr2 (0.54 g, 2.5 mmol) 1,2-dimethoxyethane (35 mL), pyridine (0.25 mL) and a stir bar were added to a 250 mL round bottom flask equipped with a reflux condenser. The apparatus was then removed from the glove box, placed under positive pressure of N2, and refluxed at a bath temperature of 100 °C for 1 h. The progress of the reaction was monitored by UV-Vis spectroscopy by removing small aliquots, exposing them to the atmosphere and acidifying the solution with a few drops of concentrated HBr. Any remaining free base porphyrin is readily detected by its characteristic absorption at 450 nm (Fig. S19†). When no remaining free porphyrin was detected, the reaction mixture was cooled to room temperature, exposed to the atmosphere, and brought to dryness under reduced pressure. The crude residue was dissolved in 16 mL of CHCl3 and purified by column chromatograph (basic Al2O3, CHCl3). The black residue obtained after removing the solvent from the combined fractions was suspended in 10 mL of MeOH and 0.1 mL of HBr. The resultant reaction mixture was heated to 70 °C for 5 minutes and cooled to room temperature. CH2Cl2 (5 mL) was added to dissolve any remaining solid while warm, and the flask was placed in a 20 °C refrigerator overnight to afford a crystalline product. The crystals were filtered, washed with MeOH, and dried at 70 °C under vacuum overnight. A second crop of crystals was obtained from the filtrate, total: 0.15 g (24%). FTIR (ATR, neat) cm−1: 3430 (m, N–H), 3315 (m, N–H), 3060 (w, C–H), 2956 (m, C–H), 2867 (m, C–H), 1686 (s, CO); 1H NMR (600 MHz, toluene-d8) δ: 80.09, 16.45, 12.13, 8.68, 1.35, 0.93, 0.28, −0.16; λmax (CHCl3): 359(sh), 391(sh), 419, 509, 584, 655, 685 nm.
Bis(N-methylimidazole)[all-cis)-5,10,15,20-tetrakis-[2-(2,2-dimethylpropionamido)phenyl]porphyrinato(2-)]-iron(II) (Fe(TPivPP)(N-MeIm)2)
In a glove box, a 20 mL scintillation vial was charged with Fe(TPivPP)Br (30 mg, 26 μmol), Cr(acac)2 (19.7 mg, 110 μmol), THF (10 mL), N-methylimidazole (21 μL, 260 μmol), and a stir bar. The stirred reaction mixture was heated to boiling for 5 minutes, filtered hot, then cooled to room temperature, while layering with heptane, and cooled to −25 °C overnight. The resultant microcrystalline solution was filtered through a glass wool filter, washed with heptane, extracted from the filter with THF, and evaporated to dryness under vacuum to yield the desired product (7.1 mg, 54%). FTIR (ATR, neat) cm−1: 3425 (m, N–H), 2658 (m, C–H), 2868 (m, C–H), 1686 (s, CO); 1H NMR (600 MHz, toluene-d8) δ: 19.60, 18.75, 14.40, 12.94, 12.04, 3.35, 1.35, 0.29, −14.01; λmax (toluene): 333, 429, 535, 564, 606 nm.
Acknowledgements
This work was supported by the Oregon Medical Research Foundation, the National Science Foundation (CHE-1454747), and the Sloan Foundation. The NMR facilities at the University of Oregon are supported by NSF/ARRA (CHE-0923589).
Notes and references
-
J. M. Berg, J. L. Tymoczko and S. Lubert, Biochemistry, W. H. Freeman and Company, 41 Madison Avenue, New York, NY 10010, 6th edn, 2007 Search PubMed.
- J. P. Collman, R. R. Gagne, T. R. Halbert, J. C. Marchon and C. A. Reed, J. Am. Chem. Soc., 1973, 95, 7868–7870 CrossRef CAS PubMed.
- J. P. Collman, R. R. Gagne, C. A. Reed, T. R. Halbert, G. Lang and W. T. Robinson, J. Am. Chem. Soc., 1975, 97, 1427–1439 CrossRef CAS PubMed.
- A. K. Mustafa, M. M. Gadalla and S. H. Snyder, Sci. Signaling, 2009, 2, re2 Search PubMed.
- K. Abe and H. Kimura, J. Neurosci., 1996, 16, 1066–1071 CAS.
- J. Heinecke and P. C. Ford, Coord. Chem. Rev., 2010, 254, 235–247 CrossRef CAS.
- M. D. Lim, I. M. Lorkovic and P. C. Ford, J. Inorg. Biochem., 2005, 99, 151–165 CrossRef CAS PubMed.
-
M. W. Wolf, in Nitrosyl Complexes in Inorganic Chemistry, Biochemistry and Medicine II, ed. P. D. M. Mingos, Springer, Berlin, Heidelberg, 2014, pp. 155–223 Search PubMed.
- L. Cheng, M. A. Khan, G. B. Richter-Addo and D. R. Powell, Chem. Commun., 2000, 2301–2302 RSC.
- M. K. Ellison, C. E. Schulz and W. R. Scheidt, Inorg. Chem., 1999, 38, 100–108 CrossRef CAS.
- R. Wang, Physiol. Rev., 2012, 92, 791–896 CrossRef CAS PubMed.
- J. L. Wallace and R. Wang, Nat. Rev. Drug Discovery, 2015, 14, 329–345 CrossRef CAS PubMed.
- M. D. Hartle, S. K. Sommer, S. R. Dietrich and M. D. Pluth, Inorg. Chem., 2014, 53, 7800–7802 CrossRef CAS PubMed.
- R. Pietri, E. Roman-Morales and J. Lopez-Garriga, Antioxid. Redox Signaling, 2011, 15, 393–404 CrossRef CAS PubMed.
- C. C. Tsou, W. C. Chiu, C. H. Ke, J. C. Tsai, Y. M. Wang, M. H. Chiang and W. F. Liaw, J. Am. Chem. Soc., 2014, 136, 9424–9433 CrossRef CAS PubMed.
- J. A. Berzofsky, J. Peisach and J. O. Alben, J. Biol. Chem., 1972, 247, 3774–3782 CAS.
- J. A. Berzofsky, J. Peisach and W. E. Blumberg, J. Biol. Chem., 1971, 246, 3367–3377 CAS.
- J. A. Berzofsky, J. Peisach and W. E. Blumberg, J. Biol. Chem., 1971, 246, 7366–7372 CAS.
- R. Pietri, A. Lewis, R. G. Leon, G. Casabona, L. Kiger, S. R. Yeh, S. Fernandez-Alberti, M. C. Marden, C. L. Cadilla and J. Lopez-Garriga, Biochemistry, 2009, 48, 4881–4894 CrossRef CAS PubMed.
- B. B. Rios-Gonzalez, E. M. Roman-Morales, R. Pietri and J. Lopez-Garriga, J. Inorg. Biochem., 2014, 133, 78–86 CrossRef CAS PubMed.
- R. O. Beauchamp Jr., J. S. Bus, J. A. Popp, C. J. Boreiko and D. A. Andjelkovich, CRC Crit. Rev. Toxicol., 1984, 13, 25–97 CrossRef CAS PubMed.
- T. J. Walsh and C. Beehler, Arch. Intern. Med., 1969, 124, 377–378 CrossRef CAS PubMed.
- M. Rizzi, J. B. Wittenberg, A. Coda, M. Fasano, P. Ascenzi and M. Bolognesi, J. Mol. Biol., 1994, 244, 86–99 CrossRef CAS PubMed.
- M. Rizzi, J. B. Wittenberg, A. Coda, P. Ascenzi and M. Bolognesi, J. Mol. Biol., 1996, 258, 1–5 CrossRef CAS PubMed.
- D. R. English, D. N. Hendrickson, K. S. Suslick, C. W. Eigenbrot and W. R. Scheidt, J. Am. Chem. Soc., 1984, 106, 7258–7259 CrossRef CAS.
- E. Blackstone, M. Morrison and M. B. Roth, Science, 2005, 308, 518 CrossRef CAS PubMed.
- J. P. Collman, S. Ghosh, A. Dey and R. A. Decreau, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 22090–22095 CrossRef CAS PubMed.
- J. W. Pavlik, B. C. Noll, A. G. Oliver, C. E. Schulz and W. R. Scheidt, Inorg. Chem., 2010, 49, 1017–1026 CrossRef CAS PubMed.
- D. J. Meininger, J. D. Caranto, H. D. Arman and Z. J. Tonzetich, Inorg. Chem., 2013, 52, 12468–12476 CrossRef CAS PubMed.
- K. Watanabe, T. Suzuki, H. Kitagishi and K. Kano, Chem. Commun., 2015, 51, 4059–4061 RSC.
- S. A. Bieza, F. Boubeta, A. Feis, G. Smulevich, D. A. Estrin, L. Boechi and S. E. Bari, Inorg. Chem., 2014, 54, 527–533 CrossRef PubMed.
- J. P. Collman, L. Fu, A. Zingg and F. Diederich, Chem. Commun., 1997, 193–194 RSC.
- E. A. Kerr, H. C. Mackin and N. T. Yu, Biochemistry, 1983, 22, 4373–4379 CrossRef CAS PubMed.
- H. Nasri, K. J. Haller, Y. Wang, H. Huynh Boi and W. R. Scheidt, Inorg. Chem., 1992, 31, 3459–3467 CrossRef CAS.
- J. L. Heinecke, C. Khin, J. C. Pereira, S. A. Suarez, A. V. Iretskii, F. Doctorovich and P. C. Ford, J. Am. Chem. Soc., 2013, 135, 4007–4017 CrossRef CAS PubMed.
- L. Cheng, D. R. Powell, M. A. Khan and G. B. Richter-Addo, Chem. Commun., 2000, 2301–2302 RSC.
- J. Conradie and A. Ghosh, Inorg. Chem., 2006, 45, 4902–4909 CrossRef CAS PubMed.
- T. N. Sorrell, C. M. Bump and J. Jordan, Inorg. Synth., 1980, 20, 161–169 CrossRef.
- J. Lindsey, J. Org. Chem., 1980, 45, 5215–5215 CrossRef CAS.
- D. W. Kraus and J. B. Wittenberg, J. Biol. Chem., 1990, 265, 16043–16053 CAS.
- T. Brittain, Y. Yosaatmadja and K. Henty, IUBMB Life, 2008, 60, 135–138 CrossRef CAS PubMed.
- Although most protein-based H2S/heme investigations have assumed that HS− is the active sulfide species at physiological pH, sulfide binding in Mb decreases in alkaline solution. This observation suggests that H2S may be the active species or that pH changes significantly influence the protonation state of important residues that stabilize sulfide binding interactions. ( D. W. Kraus and J. B. Wittenberg, J. Biol. Chem., 1990, 265, 16043–16053 Search PubMed and T. Brittain, Y. Yosaatmadja and K. Henty, IUBMB Life, 2008, 60, 135–138 CrossRef CAS PubMed ).
- M. D. Hartle, D. J. Meininger, L. N. Zakharov, Z. J. Tonzetich and M. D. Pluth, Dalton Trans., 2015, 44, 19782–19785 RSC.
- An alternative possibility is that two porphyrins are coming together to form a μ-sulfido bridged complex through initial sulfide binding to the bottom face of the porphyrin. Although we are unaware of μ-sulfido species in similar systems, the μ-oxo bridged Fe(III) PFP exhibits a single, well-resolved band at 408 nm. ( J. P. Collman, R. R. Gagne, T. R. Halbert, J. C. Marchon and C. A. Reed, J. Am. Chem. Soc., 1973, 95, 7868–7870 CrossRef CAS PubMed .) In addition, the parent Fe(II)(TPP) ( L. Cheng, D. R. Powell, M. A. Khan and G. B. Richter-Addo, Chem. Commun., 2000, 2301–2302 RSC .) exhibits a split Soret band at 418 and 443 nm for the unligated species, and single absorbance at 409 nm is associated with the μ-oxo bridged species. ( J. Conradie and A. Ghosh, Inorg. Chem., 2006, 45, 4902–4909 CrossRef PubMed ).
- J. P. Collman, F. Basolo, E. Bunnenberg, T. J. Collins, J. H. Dawson, P. E. Ellis, M. L. Marrocco, A. Moscowitz, J. L. Sessler and T. Szymanski, J. Am. Chem. Soc., 1981, 103, 5636–5648 CrossRef CAS.
- K. Y. Chen and S. K. Gupta, Environ. Lett., 1973, 4, 187–200 CrossRef CAS PubMed.
- D. J. O'Brien and F. B. Birkner, Environ. Sci. Technol., 1977, 11, 1114–1120 CrossRef.
- G. Schwarzenbach and A. Fischer, Helv. Chim. Acta, 1960, 43, 1365–1390 CrossRef CAS.
- F. Seel, H. J. Guttler, G. Simon and A. Wieckowski, Pure Appl. Chem., 1977, 49, 45–54 CrossRef CAS.
- This mass increase also suggests against the formation of verdoheme derivatives, which are another form of oxidized porphryins in which the meso position of the porphyrin ring is oxidized. Y. Du, G. Liu, Y. Yan, D. Huang, W. Luo, M. Martinkova, P. Man and T. Shimizu, BioMetals, 2013, 26, 839–852 CrossRef CAS PubMed; F. Yan, V. Fojtikova, P. Man, M. Stranava, M. Martinkova, Y. Du, D. Huang and T. Shimizu, BioMetals, 2015, 28, 637–652 CrossRef PubMed.
- L. Latos-Grazynski, R. J. Cheng, G. N. La Mar and A. L. Balch, J. Am. Chem. Soc., 1982, 104, 5992–6000 CrossRef CAS.
- G. N. Sinyakov and A. M. Shulga, J. Mol. Struct., 1993, 295, 1–14 CrossRef CAS.
- R. C. Parmely and H. M. Goff, J. Inorg. Biochem., 1980, 12, 269–280 CrossRef CAS PubMed.
- F. A. Walker, J. Buchler, J. T. West and J. L. Hinds, J. Am. Chem. Soc., 1983, 105, 6923–6929 CrossRef CAS.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532 CrossRef CAS.
- D. F. Evans, J. Chem. Soc., 1959, 1959, 2003–2005 RSC.
- M. Dhifet, M. S. Belkhiria, J. C. Daran and H. Nasri, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m967–m968 CAS.
- M. Schappacher, L. Ricard, J. Fischer, R. Weiss, R. Montiel-Montoya, E. Bill and A. X. Trautwein, Inorg. Chem., 1989, 28, 4639–4645 CrossRef CAS.
- D. Brault and M. Rougee, Biochemistry, 1974, 13, 4591–4597 CrossRef CAS PubMed.
- D. Landy, F. Tetart, E. Truant, P. Blach, S. Fourmentin and G. Surpateanu, J. Inclusion Phenom. Macrocyclic Chem., 2007, 57, 409–413 CrossRef CAS.
- C. J. Martin and M. A. Marini, J. Biol. Chem., 1967, 242, 5736–5743 CAS.
-
MATLAB and Statistics Tollbox Release 2014b, The MathWorks Inc., Natick, Massachusettes, United States, 2014 Search PubMed.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
-
R. Schäfer and P. C. Schmidt, Methods in Physical Chemistry, John Wiley & Sons, Somerset, NJ, USA, 2nd edn, 2012 Search PubMed.
-
D. Schröder, Mass Spectrometry for Ion Chemistry and Links from the Gas Phase to “Real” Processes, in Methods in Physical Chemistry ed. R. Schäfer and P. C. Schmidt, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim,
Germany, 2012 Search PubMed.
- L. R. Ocone, B. P. Block, J. P. Collman and D. A. Buckingham, Inorg. Synth., 1966, 8, 125–132 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic schemes, spectroscopic characterization data, full 1H-NMR spectra for NBu4SH addition to 1 and 3, representative plot of data fit to a 1:1 model, mass spectrometry data, tabulated spectroscopic properties. See DOI: 10.1039/c5dt04563k |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence