
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A functional mesoporous ionic crystal based on polyoxometalate†
Ryosuke
Kawahara
a,
Kazuma
Niinomi
a,
Junko N.
Kondo
bc,
Mitsuhiro
Hibino
d,
Noritaka
Mizuno
d and
Sayaka
Uchida
*ab
aDepartment of Basic Science, School of Arts and Sciences, The University of Tokyo, 3-8-1 Komaba, Meguro-ku, Tokyo 153-8902, Japan. E-mail: csayaka@mail.ecc.u-tokyo.ac.jp
bJST-PRESTO, 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
cChemical Resources Laboratory, Tokyo Institute of Technology, 4259-R1-10 Nagatsuta, Midori-ku, Yokohama 226-8503, Japan
dDepartment of Applied Chemistry, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
First published on 15th January 2016
Abstract
A mesoporous ionic crystal is synthesized with a polyoxometalate and a macrocation with polar cyano groups. The compound possesses one-dimensional mesopores with an opening of 3.0 × 2.0 nm. The compound shows high proton conductivity and catalytic activity, which are due to the water molecules in the mesopores.
Porous materials have attracted wide attention in chemistry due to their well-defined pore structures, which are useful in gas storage, separation, catalysis, proton conduction, and so on. According to the IUPAC definition, porous materials are divided into three classes: microporous (pore size <2 nm), mesoporous (2–50 nm), and macroporous (>50 nm) materials. Zeolites and metal–organic frameworks (MOFs) are representatives of microporous materials and are utilized as adsorbents and catalysts.1 However, microporous materials have limitations in diffusion especially when large guest molecules are involved, so an important line of research has focused on the enlargement of pore sizes into the mesopore range.2
Among the mesoporous materials, MCM-41 and FSM-16 with a highly ordered array of one-dimensional pores and amorphous silica pore walls have been extensively researched.2 In addition, mesoporous organosilicates,3 non-silica metal oxides,4 metals,5 and semiconductors6 have been synthesized. Recently, mesoporous MOFs have been synthesized by the design and modification of organic linkers and applied to asymmetric diethylzinc and alkynylzinc additions,7 co-oxidation of 4-aminoantipyrine and phenol,8 cycloaddition reaction of CO2 with carbonates,9 proton conduction,10 and so on.
On the other hand, ionic crystals (e.g., NaCl) are normally non-porous because of the isotropic and long-range Coulomb interaction.11 We and other research groups have reported that the use of molecular ions with appropriate elements, charges, sizes, shapes, or ligands enables anisotropic packing and/or utilization of anisotropic interactions such as π–π interactions and hydrogen-bonding, resulting in the formation of micropores.12 Strong electrostatic fields are generated at the internal surfaces of micropores due to the constituent ions, and the micropores are suitable for sorptive separation of small molecules such as CO2.12e,f
Polyoxometalates (POMs) are nano-sized metal-oxide macroanions with unique catalytic, electrochemical, magnetic, and luminescence properties, and have stimulated research in broad fields of science.13 Particularly, giant polyoxomolybdates with nano-sized cavities can be obtained by the aggregation of simple clusters in water under controlled-pH conditions, reducing reagents, and templates.14 Recently, nano-structured materials have been fabricated by the combination of these giant polyoxomolybdates with surfactant molecules or nanocrystals.15,16 Since POMs bear negative charges, they are potentially useful as building blocks of mesoporous compounds in combination with appropriate macrocations. However, in order to construct mesopores, a strategy is required to prevent the close-packing of ionic building blocks. In supramolecular chemistry, it is well known that hydrogen-bonded water clusters (H2O)n serve as templates in the formation of complexes with nano-sized cavities, and polar functional groups or hydrogen-bonding sites are often required to stabilize the water clusters.17
Based on these considerations, we synthesized a mesoporous ionic crystal Cr[Cr3O(OOCCH2CN)6(H2O)3]3[α-PW12O40]2·69H2O [I] with a α-Keggin-type POM [α-PW12O40]3− and a macrocation [Cr3O(OOCCH2CN)6(H2O)3]+ with polar cyano groups (CN). M(III)-carboxylates (M = Cr, Fe, Mn, Ru, etc.) with a general formula of [M3O(OOCR)6(L)3]+ have been studied as building blocks of solid materials because of the versatile selection of the ligands (R, L) and unique acid properties.18,19 Compound I possessed one-dimensional mesopores with an opening of 3.0 × 2.0 nm, and the mesoporous structure was maintained by water molecules as templates.
Compound I was obtained as green crystals via the in situ formation of [Cr3O(OOCCH2CN)6(H2O)3]+ from Cr(NO3)3 and cyano acetic acid in acetone, followed by the addition of an aqueous solution containing Cr(NO3)3 and H3[α-PW12O40].‡ Single crystal X-ray diffraction (XRD) analysis§ showed that POMs and macrocations were arranged alternately in the ab-plane, and formed a ring and a mesopore with an opening of 3.0 × 2.0 nm (Fig. 1a). The distances between adjacent ions in the ring were in the range of 3.142–3.330 Å (C(H)–O), showing a weak hydrogen-bonding between POMs and macrocations. CN groups of macrocations pointed toward the center of the mesopore and interacted with the water of crystallization (see below). While the existence of Cr3+ as a counter cation was suggested by the charge balance and elemental analysis of I, Cr3+ sites could not be determined by single crystal XRD analysis.¶ POMs and macrocations lined up along the c-axis, and CN groups of adjacent macrocations were aligned parallel to each other with distances of 3.593 Å, suggesting that a weak dispersion force existed between the CN groups (Fig. 1b). The void volume of I was 44.2% of the crystal lattice (11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 285 Å3 per unit cell) and was the largest among the porous ionic crystals reported to date (Fig. 1c, d and Table S1†).
285 Å3 per unit cell) and was the largest among the porous ionic crystals reported to date (Fig. 1c, d and Table S1†).
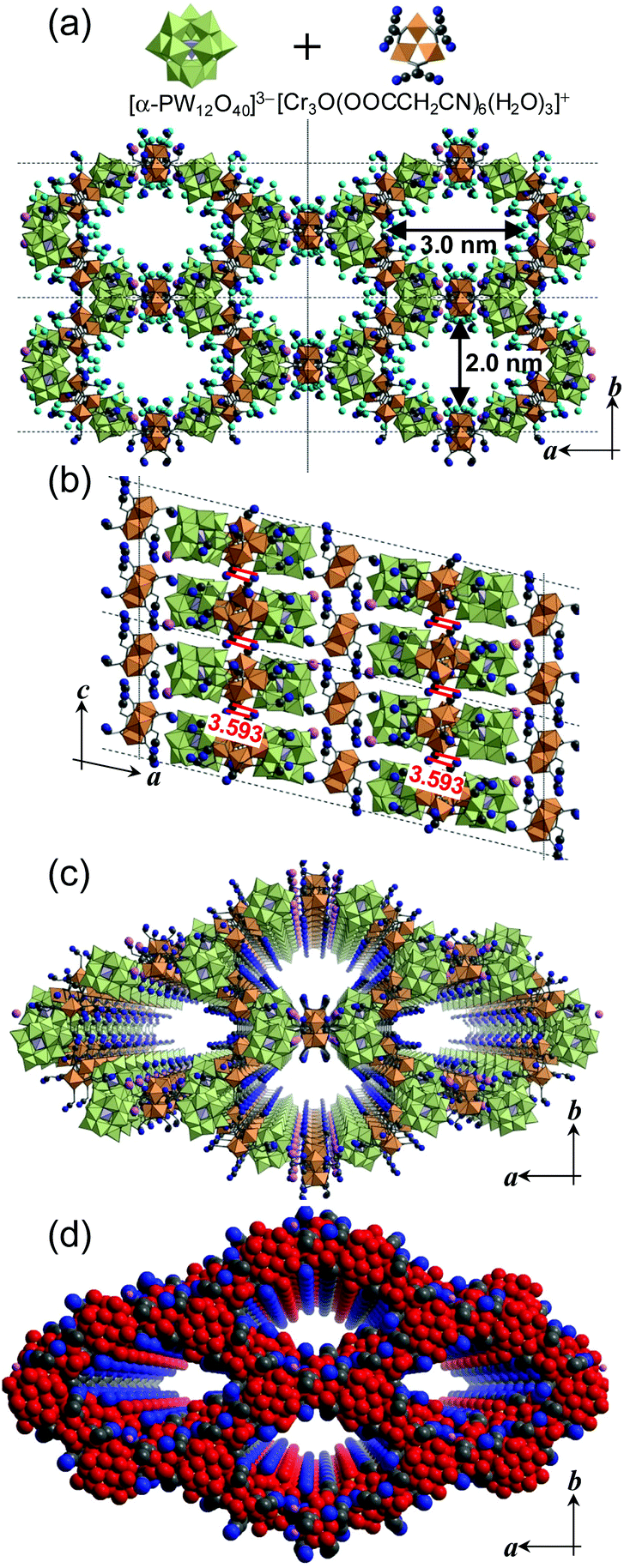 | ||
| Fig. 1 (a) Molecular structures of the constituent ions and crystal structure of I (ab-plane). (b) Crystal structure of I (ac-plane). Mesoporous structure of I (ab-plane) represented in (c) polyhedral and (d) space filling models. Hydrogen atoms are omitted. Size of the mesopores (3.0 × 2.0 nm) is shown in (a). Red lines in (b) show the possible dispersion force between the cyano groups. Blue and gray spheres in (a)–(c) show the nitrogen and carbon atoms, respectively, of the CN groups of the macrocation. Blue, gray, and red spheres in (d) are the nitrogen (r = 1.5 Å), carbon (r = 1.7 Å), and oxygen (r = 1.7 Å) atoms, respectively, depicted in their van der Waals radii. Pink and light blue spheres show the chromium ion as a counter cation and the water of crystallization, respectively. The water of crystallization is omitted for clarity in (b)–(d). | ||
Thermogravimetry (TG) (Fig. S1†) and elemental analysis suggested the existence of 69 mol per mol of water of crystallization. The water molecules desorbed in two steps: about 54 and 15 mol mol−1 desorbed by the treatment with dry N2 at room temperature (rt) and between rt−423 K, respectively. Single crystal XRD analysis could locate 14 mol per mol of water molecules at the periphery of the mesopore. These water molecules were hydrogen-bonded to the constituent ions (i.e., oxygen atoms of POM, water ligands or CN groups of macrocation (O–O: 2.660–3.060 Å, N–O: 3.111–3.119 Å)). The solid state 1H-MASNMR spectrum of I showed a sharp signal at 4.8 ppm (Fig. S2†). Since hydrogen-bonding causes a low-field shift in the 1H-NMR signal of water (cf. H2O in C6D6 0.40 ppm, H2O in D2O 4.79 ppm),20 a hydrogen-bonding network of water molecules is probably formed in the mesopore. Moreover, the signal was sharp like that of liquid water, suggesting that the water molecules are highly mobile.
The powder XRD pattern of I is shown in Fig. 2. The experimental data of I (Fig. 2b) well agreed with that calculated from the crystal structure of I (Fig. 2a), showing that the mesoporous structure is stable under ambient conditions. The pattern of I broadened after evacuation at rt (Fig. S3†), showing that water molecules are necessary to maintain the mesoporous structure. These results indicate that I shows a low porosity and BET surface area based on N2 adsorption,|| while it is well known that such kinds of host–guest systems also show interesting properties in guest-exchange,21 proton conduction,22etc. In order to characterize the states of guests (i.e., water molecules) in the mesopores, proton conductivity measurements and heterogeneous acid-catalyzed reactions were carried out.
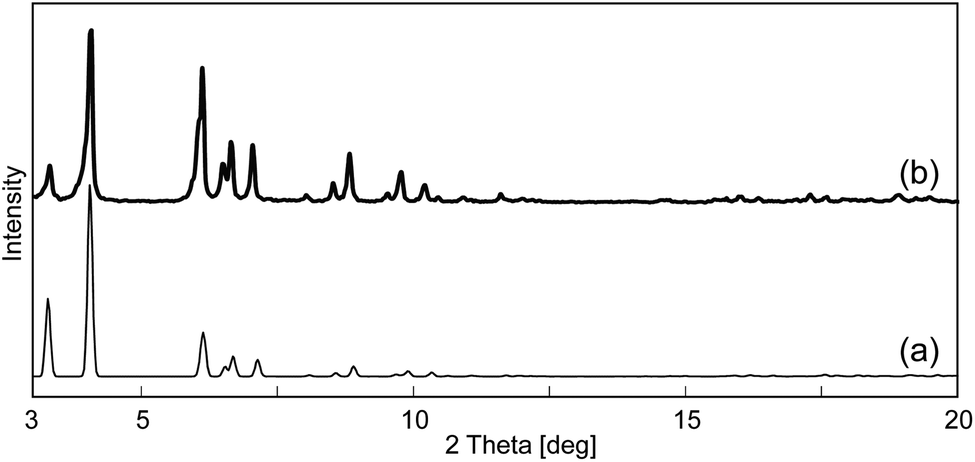 | ||
| Fig. 2 (a) Calculated and (b) experimental powder XRD patterns of I. | ||
Fig. 3a shows the Nyquist plots of the impedance spectra of I at 303–323 K under 95% relative humidity (95% RH).** The proton conductivity estimated from the spectrum at 303 K was 1.0 × 10−4 S cm−1, and reached up to 2.2 × 10−4 S cm−1 by increasing the temperature to 323 K. On the other hand, the proton conductivity dropped to 6.7 × 10−5 S cm−1 by a decrease in RH to 55% (Fig. S4†). The activation energy determined from the proton conductivities at 95% RH as a function of temperature using the Arrhenius equation was 0.34 eV (32.6 kJ mol−1) (Fig. 3b). While the proton conductivity (1.0 × 10−4 S cm−1) was lower than acid hydrates of POMs (e.g., H3PW12O40·29H2O:23 0.18 S cm−1 at 298 K, 80% RH), the value was higher than those of POM-based salts (Table S2†). The activation energy (0.34 eV) was relatively lower than those of POM-based salts (Table S2†). The relatively high proton conductivity and low activation energy can be explained by the hydrogen-bonding network of water molecules in the one-dimensional mesopores, which facilitates the exchange and/or diffusion of protons throughout the whole bulk solid.
 | ||
| Fig. 3 (a) Nyquist plots of the impedance spectra of I at 303–323 K. (b) Arrhenius plot of the conductivities at 95% RH. | ||
Porous materials based on POMs24 (POM-MOFs) have been recently studied as catalysts in aerobic oxidation,25 asymmetric dihydroxylation of olefins,26 visible-light-driven H2 production,27 and so on. The allylation of an aldehyde with allyltin (Scheme 1) is a typical acid-catalyzed reaction performed in water and is a useful method for C–C bond formation.28Fig. 4 shows the time course of changes in the amounts of the reactant (benzaldehyde) and the product (1-phenyl-3-butene-1-ol). The reaction proceeded smoothly, and the conversion and yield after 48 h were 91% and 76% (turn over number (TON) = 39), respectively.†† The activity of I was comparable to that of scandium triflate-grafted mesoporous silica with Lewis acid sites (yield 81–91% at 353 K in water).29‡‡ The existence of acid sites in I was confirmed by IR spectroscopy: the difference spectrum of I treated with pyridine as a basic probe molecule (Fig. S5†) suggested that the pyridine molecules were adsorbed on Brønsted acid sites (1534 cm−1), Lewis acid sites and/or experienced hydrogen-bonding (1440 cm−1).30
 | ||
| Scheme 1 | ||
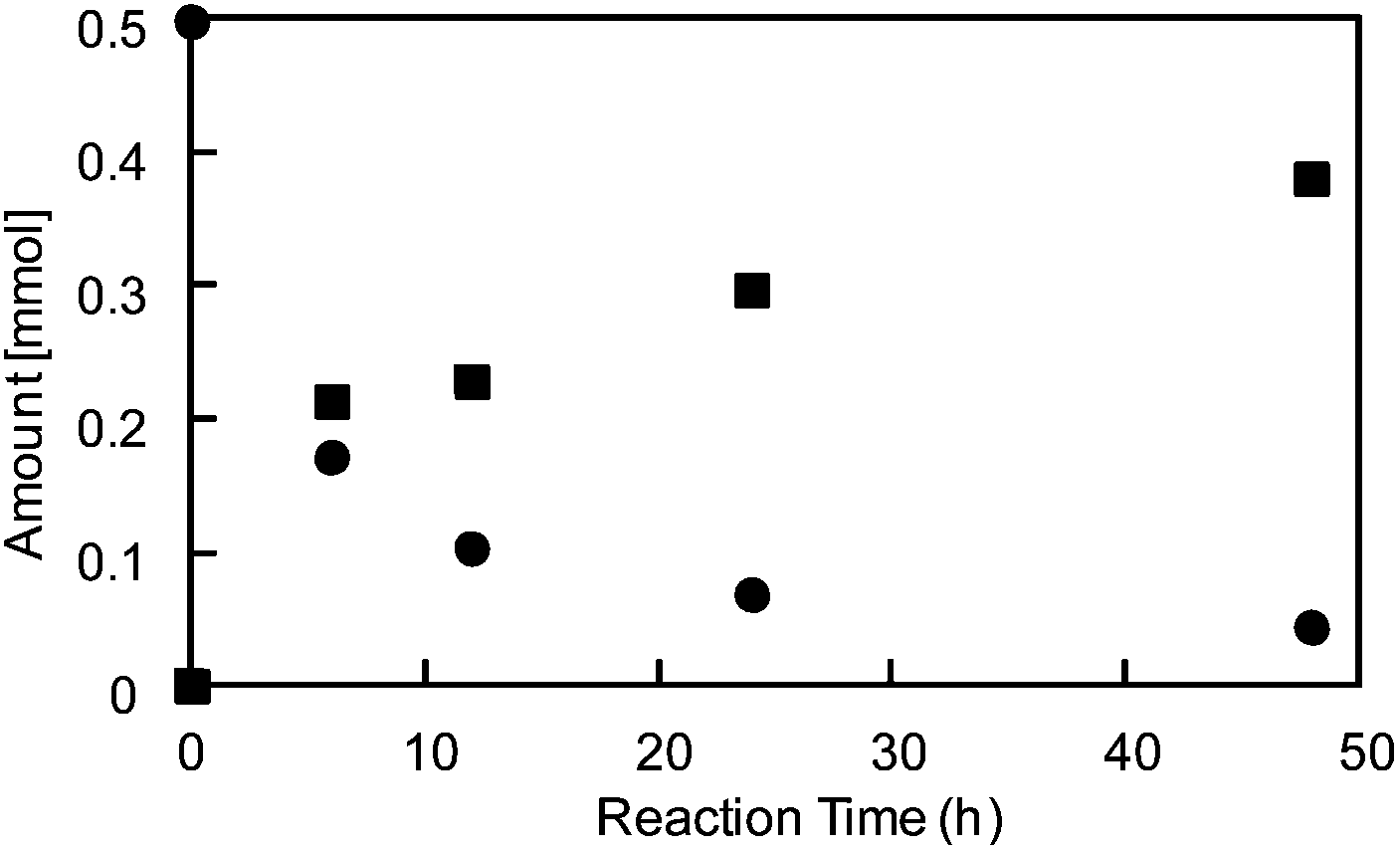 | ||
| Fig. 4 Time course of the allylation of benzaldehyde with allyltin catalyzed by I (353 K). Circle and square plots show the amounts of reactants and products, respectively. | ||
This work was supported by JST-PRESTO and Grant-in-Aids for Scientific Research from the Ministry of Education, Culture, Science, Sports, and Technology of Japan. Prof. Motoyuki Matsuo and Dr Katsumi Shozugawa (Univ. of Tokyo) are acknowledged for providing access to the ICP instrument.
Notes and references
- (a) A. Corma, Chem. Rev., 1997, 97, 2373–2420 CrossRef CAS PubMed; (b) J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- (a) A. Taguchi and F. Schüth, Microporous Mesoporous Mater., 2005, 77, 1–45 CrossRef CAS; (b) K. Ariga, A. Vinu, Y. Yamauchi, Q. Ji and J. P. Hill, Bull. Chem. Soc. Jpn., 2012, 85, 1–32 CrossRef CAS.
- (a) N. Mizoshita, T. Tani and S. Inagaki, Chem. Soc. Rev., 2011, 40, 789–800 RSC; (b) B. J. Melde, B. T. Holland, C. F. Blanford and A. Stein, Chem. Mater., 1999, 11, 3302–3308 CrossRef CAS.
- (a) P. Yang, D. Zhao, D. I. Margolese, B. F. Chmelka and G. D. Stucky, Nature, 1998, 396, 152–155 CrossRef CAS; (b) Z. Zhang and T. J. Pinnavaia, J. Am. Chem. Soc., 2002, 124, 12294–12301 CrossRef CAS PubMed; (c) B. Lee, D. Lu, J. N. Kondo and K. Domen, J. Am. Chem. Soc., 2002, 124, 11256–11257 CrossRef CAS PubMed.
- Y. Yamauchi and K. Kuroda, Chem. – Asian. J., 2008, 3, 664–676 CrossRef CAS PubMed.
- Y. G. S. Armatas and M. G. Kanatzidis, Nature, 2006, 441, 1122–1125 CrossRef PubMed.
- L. Ma, J. M. Falkowski, C. Abney and W. Lin, Nat. Chem., 2010, 2, 838–846 CrossRef CAS PubMed.
- K. Wang, D. Feng, T. F. Liu, J. Su, S. Yuan, Y. P. Chen, M. Bosch, X. Zou and H. C. Zhou, J. Am. Chem. Soc., 2014, 136, 13983–13986 CrossRef CAS PubMed.
- M. H. Beyzavi, R. C. Klet, S. Tussupbayev, J. Borycz, N. A. Vermeulen, C. J. Cramer, J. F. Stoddart, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2014, 136, 15861–15864 CrossRef CAS PubMed.
- V. G. Ponomareva, K. A. Kovalenko, A. P. Chupakhin, D. N. Dybtsev, E. S. Shutova and V. P. Fedin, J. Am. Chem. Soc., 2012, 134, 15640–15643 CrossRef CAS PubMed.
- D. F. Shriver, P. W. Atkins and C. H. Langford, Inorganic Chemistry, Oxford University Press, Oxford, 2nd edn, 1994 Search PubMed.
- (a) M. V. Bennett, L. G. Beauvais, M. P. Shores and J. R. Long, J. Am. Chem. Soc., 2001, 123, 8022–8032 CrossRef CAS PubMed; (b) S. Takamizawa, T. Akatsuka and T. Ueda, Angew. Chem., Int. Ed., 2008, 47, 1689–1692 CrossRef CAS PubMed; (c) X. N. Cheng, W. Xue, J. B. Lin and X. M. Chen, Chem. Commun., 2010, 46, 246–248 RSC; (d) Z. Zhang, M. Sadakane, T. Murayama, N. Sakaguchi and W. Ueda, Inorg. Chem., 2014, 53, 7309–7318 CrossRef CAS PubMed; (e) R. Eguchi, S. Uchida and N. Mizuno, Angew. Chem., Int. Ed., 2012, 51, 1635–1639 CrossRef CAS PubMed; (f) R. Kawahara, S. Uchida and N. Mizuno, Inorg. Chem., 2014, 53, 3665–3661 CrossRef PubMed.
- C. L. Hill , ed. Thematic Issue on Polyoxometalates, Chem. Rev., 1998, 98, 1–390.
- (a) U. Kortz, A. Müller, J. van Slageren, J. Schnack, N. S. Dalal and M. Dressel, Coord. Chem. Rev., 2009, 253, 2315–2327 CrossRef CAS; (b) A. Müller, S. Q. N. Shah, H. Bögge and M. Schmidtmann, Nature, 1999, 397, 48–50 CrossRef; (c) T. Liu, Q. Wan, Y. Xie, C. Burger, L. Z. Liu and B. Chu, J. Am. Chem. Soc., 2001, 123, 10966–10972 CrossRef CAS PubMed; (d) S. Shishido and T. Ozeki, J. Am. Chem. Soc., 2008, 130, 10588–10595 CrossRef CAS PubMed; (e) B. Botar, P. Kögerler and C. L. Hill, J. Am. Chem. Soc., 2006, 128, 5336–5337 CrossRef CAS PubMed; (f) L. Cronin, C. Beugholt, E. Krickemeyer, M. Schmidtmann, H. Bögge, P. Kögerler, T. Kim, K. Luong and A. Müller, Angew. Chem., Int. Ed., 2002, 41, 2805–2808 CrossRef CAS.
- (a) D. G. Kurth, P. Lehmann, D. Volkmer, H. Cölfen, M. J. Koop, A. Müller and A. Du Chesne, Chem. – Eur. J., 2000, 6, 385–393 CrossRef CAS PubMed; (b) S. Noro, R. Tsunashima, Y. Kamiya, K. Uemura, H. Kita, L. Cronin, T. Akutagawa and T. Nakamura, Angew. Chem., Int. Ed., 2009, 48, 8703–8706 CrossRef CAS PubMed.
- M. I. Bodnarchuk, R. Erni, F. Krumeich and M. V. Kovalenko, Nano Lett., 2013, 13, 1699–1705 CAS.
- (a) L. J. Barbour, G. W. Orr and J. L. Atwood, Nature, 1998, 393, 671–673 CrossRef CAS; (b) S. K. Ghosh and P. K. Bharadwaj, Angew. Chem., Int. Ed., 2004, 43, 3577–3580 CrossRef CAS PubMed.
- T. Fujihara, J. Aonahata, S. Kumakura, A. Nagasawa, K. Murakami and T. Ito, Inorg. Chem., 1998, 37, 3779–3784 CrossRef CAS PubMed.
- A. Vimont, J. M. Goupil, J. C. Lavalley, M. Daturi, S. Surblé, C. Serre, F. Millange, G. Férey and N. Audebrand, J. Am. Chem. Soc., 2006, 128, 3218–3227 CrossRef CAS PubMed.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS PubMed.
- K. Biradha, D. Dennis, N. K. R. MacKinnon, V. K. Sharma and M. J. Zaworotko, J. Am. Chem. Soc., 1998, 120, 11894–11903 CrossRef CAS.
- H. Wang, X. Xu, N. M. Johnson, N. K. R. Dandala and H. F. Ji, Angew. Chem., Int. Ed., 2011, 50, 12538–12541 CrossRef CAS PubMed.
- O. Nakamura, T. Kodama, I. Ogino and Y. Miyake, Chem. Lett., 1979, 8, 17–18 CrossRef.
- (a) C. Zhao, E. N. Glass, B. Chica, D. G. Musaev, J. M. Sumliner, R. B. Dyer, T. Lian and C. L. Hill, J. Am. Chem. Soc., 2014, 136, 12085–12091 CrossRef CAS PubMed; (b) W. Salomon, C. Roch-Marchal, P. Mialane, P. Rouschmeyer, C. Serre, M. Haouas, F. Taulelle, S. Yang, L. Ruhlmann and A. Dolbecq, Chem. Commun., 2015, 51, 2972–2975 RSC.
- J. Song, Z. Luo, D. K. Britt, H. Furukawa, O. M. Yaghi, K. I. Hardcastle and C. L. Hill, J. Am. Chem. Soc., 2011, 133, 16839–16846 CrossRef CAS PubMed.
- Q. Han, C. He, M. Zhao, B. Qi, J. Niu and C. Duan, J. Am. Chem. Soc., 2013, 135, 10186–10189 CrossRef CAS PubMed.
- Z.-M. Zhang, T. Zhang, C. Wang, Z. Lin, L.-S. Long and W. Lin, J. Am. Chem. Soc., 2015, 137, 3197–3200 CrossRef CAS PubMed.
- (a) S. Pratihar and S. Roy, Organometallics, 2011, 30, 3257–3269 CrossRef CAS; (b) A. Yanagisawa, M. Morodome, H. Nakashima and H. Yamamoto, Synlett, 1997, 1309–1311 CrossRef CAS.
- M. Chen, C. Liang, F. Zhang and H. Li, ACS Sustainable Chem. Eng., 2014, 2, 486–492 CrossRef CAS.
- T. Barzetti, E. Selli, D. Moscotti and L. Forni, J. Chem. Soc., Faraday Trans., 1996, 92, 1401–1407 RSC.
- M. E. Grillo and J. Carrazza, J. Phys. Chem., 1996, 100, 12261–12264 CrossRef CAS.
- C.-Y. Chen, H.-X. Li and M. E. Davis, Microporous Mater., 1993, 2, 17–26 CrossRef CAS.
- (a) N. Dupré, P. Rémy, K. Micoine, C. Boglio, S. Thorimbert, E. Lacôte, B. Hasenknopf and M. Malacria, Chem. – Eur. J., 2010, 16, 7256–7264 CrossRef PubMed; (b) B. Riflade, D. Lachkar, J. Oble, J. Li, S. Thorimbert, B. Hasenknopf and E. Lacôte, Org. Lett., 2014, 16, 3860–3863 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Pore sizes and volumes of porous ionic crystals (Table S1). Proton conductivities of POM-based compounds (Table S2). Thermogravimetry (Fig. S1). Solid state 1H-MASNMR (Fig. S2). Powder XRD pattern after evacuation (Fig. S3). Nyquist plots of the impedance spectra under 55–95% RH (Fig. S4). Difference IR spectrum of I treated with pyridine (Fig. S5). CCDC 1405050. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt04556h |
‡ Synthesis of Cr[Cr3O(OOCCH2CN)6(H2O)3]3[α-PW12O40]2·69H2O [I]: Cr(NO3)3·9H2O (6.0 g, 15 mmol) and cyano acetic acid (3.0 g, 35 mmol) were dissolved in 30 mL of acetone, and the solution was stirred at 323 K for 18 h (Solution A). Solution A was concentrated by evaporation followed by the addition of 280 mL of water. 40 mL of the aqueous solution was placed in a screw cap storage bottle (50 mL) followed by the addition of Cr(NO3)3·9H2O (6.0 g, 15 mmol) and H3[α-PW12O40]·nH2O (0.6 g, 0.2 mmol) in 1 mL of water. Green crystals of I were obtained after 24 h (yield 60%). IR absorption peaks: 1655 νasym(OCO), 1437 νasym(CH), 1383 νsym(OCO), 1080 νasym(P–O), 981 νasym(W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 898 νasym(W–Oc–W), 818 νasym(W–Oe–W), 597 νasym(Cr3–O). Elemental analysis (%) calcd for I: C 7.02, H 2.09, N 2.73, Cr 5.63, P 0.67, W 47.75; found: C 6.80, H 1.80, N 2.42, Cr 5.97, P 0.64, W 47.79. O), 898 νasym(W–Oc–W), 818 νasym(W–Oe–W), 597 νasym(Cr3–O). Elemental analysis (%) calcd for I: C 7.02, H 2.09, N 2.73, Cr 5.63, P 0.67, W 47.75; found: C 6.80, H 1.80, N 2.42, Cr 5.97, P 0.64, W 47.79. |
| § Single-crystal X-ray structure analysis: X-ray diffraction data of I were collected at 93 K with a CCD 2D detector by using a Rigaku Mercury diffractometer with graphite-monochromated MoKα radiation (λ = 0.71069 Å). The structure was solved by direct methods (SHELX97), expanded by Fourier techniques, and refined by full-matrix least-squares against F2 with the SHELXL-2014 package. CCDC 1405050 contains the supplementary crystallographic data. Crystal data for I: monoclinic C2/c (#15), a = 57.01(2), b = 24.477(11), c = 18.797(7), β = 103.494(11), V = 25506(18), Z = 4, R1 = 0.0636, wR2 = 0.1841, GOF = 1.064. |
| ¶ There are four Cr3+ in a unit cell because Z is 4. On the other hand, there are eight equivalent sites in the space group C2/c. Therefore, Cr3+ sites are intrinsically disordered. One of the oxygen atoms of the water of crystallization showed a relatively small isotropic temperature factor, and may be a possible candidate for the Cr3+ site. Notably, in the case of zeolites, the extraframework cations are also frequently unlocated because of severe disordering, and the locations have been suggested by computational methods.31 |
| || Theoretical surface area can be estimated from the crystal structure of I. There are two channel openings (3.0 × 2.0 nm) in the ab-plane per unit cell of I. The circumference and length of the channel are 1.6 × 102 Å and 1.9 × 101 Å, respectively. The formula weight of I without guests is 8.0 × 103 g mol−1. Therefore, the theoretical surface area [m2 g−1] of I can be estimated by the following equation: {2 × (1.6 × 102) × (1.9 × 101) × 10−20}/{8.0 × 103 × 4/(6.0 × 1023)} = 1.1 × 103 m2 g−1. This value is comparable to the BET surface area of mesoporous silica MCM-41 (9.8 × 102 m2 g−1).32 |
| ** Alternating current (AC) impedance spectroscopy: impedance measurements were carried out in a thermo-hygrostat chamber (ESPEC) with a VMP3 multi-channel potentiostat/galvanostat (BioLogic Science Instruments) over the frequency range of 5 Hz to 500 kHz. About 100 mg of the compounds were compressed at 100 kgf cm−2 into pellets of 10 mm in diameter and ca. 0.5 mm in thickness. Gold electrodes were attached on both sides of the pellet. Bulk conductivities were estimated by a semicircle fitting of Nyquist plots. Activation energies were determined from the conductivities as a function of temperature using the Arrhenius equation. |
| †† Acid-catalyzed heterogeneous reaction: The reaction was carried out in a glass reactor equipped with a magnetic stirrer. In a typical run of Barbier reaction, a mixture of benzaldehyde (0.5 mmol), allyltributyltin (1.0 mmol), 1,4-dioxane (0.5 mmol, internal standard), and I (0.1 g, 11 μmol) in water (1 mL) was stirred under air at 353 K. The reaction progress was followed by gas chromatography using a GC-2014 (Shimadzu) fitted with a TC-WAX capillary column and a flame ionization detector. |
| ‡‡ It has been reported that Dawson and Keggin-type POMs grafted with Lewis acid sites catalyze various C–C bond formation reactions under mild conditions in organic solvents.33 In contrast, the allylation of benzaldehyde with allyltin and I was probably promoted by Brønsted acid sites as reported in ref. 28b, which originated from the water of crystallization. |
| This journal is © The Royal Society of Chemistry 2016 |