DOI:
10.1039/C5DT04506A
(Paper)
Dalton Trans., 2016,
45, 3627-3634
Cationic ruthenium alkylidene catalysts bearing phosphine ligands†
Received
16th November 2015
, Accepted 19th January 2016
First published on 28th January 2016
Abstract
The discovery of highly active catalysts and the success of ionic liquid immobilized systems have accelerated attention to a new class of cationic metathesis catalysts. We herein report the facile syntheses of cationic ruthenium catalysts bearing bulky phosphine ligands. Simple ligand exchange using silver(I) salts of non-coordinating or weakly coordinating anions provided either PPh3 or chelating Ph2P(CH2)nPPh2 (n = 2 or 3) ligated cationic catalysts. The structures of these newly reported catalysts feature unique geometries caused by ligation of the bulky phosphine ligands. Their activities and selectivities in standard metathesis reactions were also investigated. These cationic ruthenium alkylidene catalysts reported here showed moderate activity and very similar stereoselectivity when compared to the second generation ruthenium dichloride catalyst in ring-closing metathesis, cross metathesis, and ring-opening metathesis polymerization assays.
Introduction
Olefin metathesis is a convenient and powerful methodology for the construction of carbon–carbon double bonds.1 Due to their high activity and functional group tolerance, ruthenium-based catalysts have been utilized in a variety fields including natural product synthesis,2 biochemistry,3 green chemistry4 and polymer chemistry.5 Along with the expansion of these applications, the catalysts themselves have dramatically evolved. In particular, the introduction of an N-heterocyclic carbene (NHC) ligand in place of a phosphine ligand led to a large enhancement in catalyst activity and stability.6 These types of catalysts are widely used in both academic and industrial laboratories.
The modification of X-type ligands, which are generally chlorides in the original catalyst systems, is becoming a popular avenue of investigation because of their easy substitution and large impact on catalyst performance. Buchmeiser et al. first utilized silver(I) salts to remove a chloride ligand and simultaneously introduce an anionic ligand in its place.7 They expanded this methodology to form solid supported systems where a catalyst is bound to a monolith through a polymeric X-type ligand.7a,b Additionally, Hoveyda et al. formed a bidentate chiral NHC ligated complex through similar ligand exchange reactions which enabled highly enantioselective asymmetric ring-opening cross metathesis (AROM) and cross metathesis (CM).8 We recently reported a family of NHC chelated catalysts which were produced by coordination of two pivalate ligands and subsequent intramolecular C–H bond activation.9 This family of catalysts has showed very high activity and Z-selectivity in a variety of metathesis reactions.10
The formation of catalysts that are cationic at the metal center or contain pendant positively charged groups has been previously reported (1–6 in Fig. 1). One of the remarkable examples are ionic liquid tagged catalysts, like 1 and 2,11 that have an oligomeric tether capped with a cationic imidazolium group. Due to their cationic charge, they can be immobilized in an ionic liquid phase and behave as supported catalysts, achieving efficient catalyst recyclability. Several cationic catalysts, in which the ruthenium center is positively charged, have been also reported.12 Among these catalysts, 3 shows much higher activity in ring-opening metathesis polymerization (ROMP) reactions of cyclooctene compared to standard NHC-ligated catalyst.12a In ring-closing metathesis (RCM) reactions of a tetra-substituted olefin, which is one of the toughest olefin metathesis transformations, 5 surpasses the second generation catalyst.12c
 |
| | Fig. 1 Examples of cationic ruthenium alkylidene catalysts. | |
Relying on the recent successes mentioned above, modification of the X-type ligand to produce cationic ruthenium-based catalysts is highly promising in improving catalyst efficiency and expanding their applications. Therefore, the discovery of facile methodologies to prepare these types of catalysts is highly desired. Here we report the convenient syntheses of cationic catalysts bearing bulky phosphine ligands, and explore their reactivity in standard metathesis reactions.
Results and discussion
Catalyst syntheses
We chose [H2IMes2]RuCl2[![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH-o-(OiPr)C6H4] (7, H2I = imidazolidinylidene, Mes = mesityl) as a starting complex due to its high activity in various metathesis reactions.13 It was previously reported that AgBF4 is capable of extracting a single chloride ligand from complex 7.12d When 7 was reacted with AgBF4 in the presence of PPh3, a cationic catalyst bearing a bulky PPh3 ligand (8a) was obtained in high yield. In the same manner, other silver salts of non-coordinating or weekly coordinating anions also provided similar cationic catalysts (8b–d) (Scheme 1). In the 1H NMR spectra of 8, signals for the methyl groups of the two N-mesityl substituents appear as all inequivalent, suggesting that the bulky PPh3 ligand hinders rotation of the NHC ligand at room temperature. X-ray quality crystals of 8c were grown and the crystal structure indicates steric repulsion between a phenyl group of the PPh3 ligand and the N-mesityl group of the NHC ligand (Fig. 2). Compared to 7,138c has smaller C1–Ru1–O1 angle (96.73(5)° versus 176.2(14)°) and larger C1–Ru1–Cl1 angle (154.88(4)° versus 96.6(12)° and 90.9(12)°). The latter structural feature is also observed in previously reported complexes 4
CH-o-(OiPr)C6H4] (7, H2I = imidazolidinylidene, Mes = mesityl) as a starting complex due to its high activity in various metathesis reactions.13 It was previously reported that AgBF4 is capable of extracting a single chloride ligand from complex 7.12d When 7 was reacted with AgBF4 in the presence of PPh3, a cationic catalyst bearing a bulky PPh3 ligand (8a) was obtained in high yield. In the same manner, other silver salts of non-coordinating or weekly coordinating anions also provided similar cationic catalysts (8b–d) (Scheme 1). In the 1H NMR spectra of 8, signals for the methyl groups of the two N-mesityl substituents appear as all inequivalent, suggesting that the bulky PPh3 ligand hinders rotation of the NHC ligand at room temperature. X-ray quality crystals of 8c were grown and the crystal structure indicates steric repulsion between a phenyl group of the PPh3 ligand and the N-mesityl group of the NHC ligand (Fig. 2). Compared to 7,138c has smaller C1–Ru1–O1 angle (96.73(5)° versus 176.2(14)°) and larger C1–Ru1–Cl1 angle (154.88(4)° versus 96.6(12)° and 90.9(12)°). The latter structural feature is also observed in previously reported complexes 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12b and 512c (Fig. 1).
12b and 512c (Fig. 1).
 |
| | Scheme 1 | |
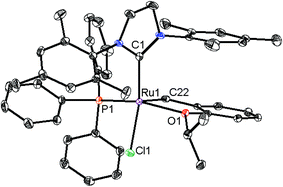 |
| | Fig. 2 X-ray crystal structure of 8c. Displacement ellipsoids are drawn at 50% probability. For clarity, hydrogen atoms and the counter anion PF6 have been omitted. Selected bond length (Å) for 8c: C1–Ru1 2.0723(15), C22–Ru1 1.8349(15), O1–Ru1 2.3442(10), Cl1–Ru1 2.3517(4), P1–Ru1 2.2788(4). | |
Next, we were able to synthesize cationic diphosphine chelated catalysts derived from [H2IMes2]RuCl2(PPh3)(CHPh) (9)14 (Scheme 2). First, 9 was exposed to TMS(OTf) causing the substitution of one chloride ligand with an OTf anion, yielding mono-triflate complex 10. Next, diphosphines DPPE and DPPP were added, subsequently replacing both the labile PPh3 and OTf ligands, and afforded cationic diphosphine chelated complexes 11a and 11b, respectively. As a result of coupling with two unequivalent phosphorus atoms, the signal of the benzylidene proton appeared as doublet of doublets in the 1H NMR spectrum. Additionally, a large difference in the chemical shifts of the two phosphorus atoms in the 31P NMR spectrum suggests that the diphosphine ligand coordinates to the ruthenium center with one phosphorus atom at the equatorial position and the other one at the axial position in the catalyst.
 |
| | Scheme 2 | |
Metathesis assays
Ring closing metathesis (RCM).
First, the standard RCM reaction of diethyldiallyl malonate (12)15 was carried out using catalysts 8a–c and 11a–b in order to evaluate their activities (Scheme 3). As shown in Fig. 3, mono-phosphine catalysts 8a–c showed moderate activities and the conversion to reach ∼90% after 6 hours. The lower activity of PPh3-substituted complexes 8a–c compared to 7 may be due to the steric bulkiness of the phosphine ligand that possibly hinders olefin coordination. It should be noted that no significant dependence of the counter anions on metathesis activity was observed. The anions, which potentially could coordinate to the ruthenium center and compete with olefin coordination did not affect catalyst reactivity under the presented conditions. Alternatively, diphosphine-substituted catalysts 11a–b exhibited poor activity, providing significantly low to no conversion of 12. Considering the mechanism of initiation, it is seemingly necessary to dissociate one phosphorous arm from the ruthenium center in order to make a vacant site for coordination of incoming olefin (Fig. 4). Because the chelated form seems like a dormant species, the high energy barrier of the dechelation of the diphosphine ligand is thought to decelerate the catalyst initiation and overall metathesis reaction.
 |
| | Scheme 3 | |
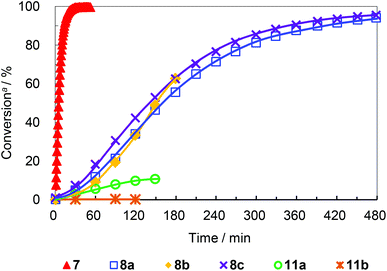 |
| | Fig. 3 Plots for conversion vs. time for RCM of 12. All reactions were carried out using 0.080 mmol of 12 and 0.80 μmol of catalyst in 0.8 ml of CD2Cl2 at 30 °C. Data for 7 is from ref. 15. aConversion of 12 to 13 determined by 1H NMR analysis. | |
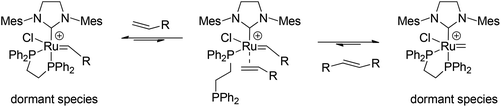 |
| | Fig. 4 One plausible mechanism of catalyst initiation for 11a. | |
Cross metathesis (CM).
In order to evaluate the activity and stereoselectivity of these new catalysts, the standard CM reaction of allylbenzene (14) and cis-1,4-diacetoxy-2-butene (15)15 was carried out (Scheme 4). Selected data for the cationic catalysts are summarized in Table 1 and plotted in Fig. 5. Similar to the RCM assay above, while 8a–b and 11a showed moderate activities, no conversion was observed when 11b was used. It has been reported that some asymmetric ruthenium-based catalysts substituted with one bulky X-type ligand tend to give lower E/Z ratio of the products compared to catalyst 7.16 In one case, a ruthenium catalyst bearing a single bulky thiolate ligand (18) provided an E/Z ratio of 0.20 in the homocoupling of substrate 14 (Fig. 6).16b However, the E/Z ratio of product 16 formed by the cationic catalysts reported here are very similar to the one by the dichloro analogue 7 (Fig. 5(b)). This possibly indicates that the phosphine ligands are too far from the reaction center to influence the stereoselectivity (Fig. 6).
 |
| | Scheme 4 | |
 |
| | Fig. 5 Plots for (a) conversion vs. time and (b) E/Z ratio vs. conversion for CM of 14 and 15. aConversion of 14 to 16 determined by GC analysis.17bMolar ratio of E isomer and Z isomer of 16 determined by GC analysis. | |
 |
| | Fig. 6 Plausible intermediates for (a) 18 and (b) 8. | |
Table 1 Selected data for the CM of 14 and 15a
| Entry |
Cat. |
Cat. loadb, mol% |
Solvent |
Time, min |
16
|
17
|
| Conv.c, % |
E/Zd |
Conv.c, % |
E/Zd |
|
All reactions were carried out using 0.20 mmol of 14, 0.40 mmol of 15 and 0.10 mmol of tridecane (internal standard for GC analysis) in 1.0 ml of solvent at 23 °C.
Based on 14.
Conversion of 14 to the product determined by GC analysis.
Molar ratio of E isomer and Z isomer of the product determined by GC analysis.
Ref. 15.
GC signal of the product was too small to quantify.
|
| 1e |
7
|
2.5 |
CH2Cl2 |
2 |
75 |
8.4 |
4.0 |
4.40 |
|
|
|
|
30 |
72 |
10.1 |
5.0 |
5.9 |
| 2 |
8a
|
2.5 |
CH2Cl2 |
30 |
34 |
3.3 |
0.0 |
(NA)f |
|
|
|
|
120 |
74 |
6.1 |
2.5 |
(NA)f |
| 3 |
8b
|
2.5 |
CH2Cl2 |
30 |
28 |
3.2 |
0.0 |
(NA)f |
|
|
|
|
120 |
73 |
5.7 |
2.8 |
(NA)f |
| 4 |
11a
|
5.0 |
CH2Cl2 |
30 |
6.2 |
2.2 |
0.0 |
(NA)f |
|
|
|
|
120 |
34 |
3.2 |
0.0 |
(NA)f |
| 5 |
11b
|
5.0 |
CH2Cl2 |
30 |
0.0 |
(NA)f |
0.0 |
(NA)f |
|
|
|
|
120 |
0.0 |
(NA)f |
0.0 |
(NA)f |
Ring opening metathesis polymerization (ROMP).
Next, the ROMP of norbornene (19) was tested with the presented cationic catalysts (Scheme 5). In all cases, an immediate increase in the viscosity of the reaction solution was observed after stirring substrate 19 with the catalysts, indicating rapid polymerization. The conversion and E/Z ratio of the product poly-norbornene 20 are summarized in Table 2. 8a–b and 11a were able to complete the reaction within 30 min at the presented condition. Even 11b, which showed negligible activity in the RCM and CM reactions above, provided 70% yield of 20 after 30 min.
 |
| | Scheme 5 | |
Table 2 Data for the ROMP of 19a
| Entry |
Cat. |
Cat. loadb, mol% |
Solvent |
Time, min |
20
|
| Conv.c, % |
E/Zd |
|
All reactions were carried out using 0.20 mmol of 19 and 0.002 mmol of catalyst in 0.8 ml of solvent at 23 °C.
Based on 19.
Conversion of 19 to 20 determined by 1H NMR analysis.
Molar ratio of E isomer and Z isomer of 20 determined by 1H NMR analysis.
|
| 1 |
7
|
1.0 |
CD2Cl2 |
30 |
100 |
0.69 |
| 2 |
8a
|
1.0 |
CD2Cl2 |
30 |
100 |
0.70 |
| 3 |
8b
|
1.0 |
CD2Cl2 |
30 |
100 |
0.69 |
| 4 |
11a
|
1.0 |
CD2Cl2 |
30 |
100 |
0.68 |
| 5 |
11b
|
1.0 |
CD2Cl2 |
30 |
70 |
0.65 |
Experimental section
General information
Atmosphere.
All reactions were carried out in dry glassware under an argon atmosphere using standard Schlenk techniques or in a Vacuum Atmospheres Glovebox under a nitrogen atmosphere unless otherwise specified.
Solvents.
CD2Cl2 was dried over CaH2 and vacuum transferred to a dry Schlenk flask and subsequently degassed with argon. Ethyl vinyl ether and isopropyl alcohol were used as received. All the other solvents were purified by passage through solvent purification columns and further degassed with argon.18
Materials.
[H2IMes2]RuCl2[CH-o-(OiPr)C6H4] (7, H2I = imidazolidinylidene, Mes = mesityl) was obtained from Materia, Inc. [H2I(Mes)2]RuCl2(PPh3)(CHPh) (9) was synthesized according to the literature procedure.14 Diethyldiallyl malonate (12), allylbenzene (14), cis-1,4-diacetoxy-2-butene (15) and tridecane were distilled over CaH2 and stored under nitrogen in Schlenk flasks. Norbornene (19) was purified by sublimation before use. All the other commercially available reagents were used as received without further purification.
Instruments.
1H, 13C and 31P NMR spectra were recorded on a Varian 500 MHz spectrometer or a Varian 300 MHz spectrometer. High-resolution mass spectra were provided by the California Institute of Technology Mass Spectrometry Facility using JEOL JMS-600H High Resolution Mass Spectrometer. X-ray crystallographic data were collected by the California Institute of Technology Beckman Institute X-ray Crystallography Facility using Bruker KAPPA APEXII X-ray diffractometer. Gas chromatography data were obtained using Agilent 6850 FID gas chromatograph equipped with a DB-Wax polyethylene glycol capillary column (Agilent).
Catalyst syntheses
General procedure for the synthesis of {[H2IMes2]RuCl(PPh3)[![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) CH-o-(OiPr)C6H4]}X (8).
In a glove box, 7, the corresponding silver salt, triphenylphosphine and dichloromethane were added into a 20 ml screw-cap vial equipped with a magnetic stir bar. The reaction mixture was stirred at room temperature for 2 h under dark. The resulting slurry was filtered and the filtrate was evaporated. The crude product was recrystallized from CH2Cl2–pentane or THF at −20 °C.
CH-o-(OiPr)C6H4]}X (8).
In a glove box, 7, the corresponding silver salt, triphenylphosphine and dichloromethane were added into a 20 ml screw-cap vial equipped with a magnetic stir bar. The reaction mixture was stirred at room temperature for 2 h under dark. The resulting slurry was filtered and the filtrate was evaporated. The crude product was recrystallized from CH2Cl2–pentane or THF at −20 °C.
{[H2IMes2]RuCl(PPh3)[CH-o-(OiPr)C6H4]}BF4 (8a).
Starting from 7 (300 mg, 479 μmol), AgBF4 (103 mg, 527 μmol) and triphenylphosphine (151 mg 575 μmol) in 10 ml of CH2Cl2, 8a was obtained as a red-orange crystalline solid (397 mg, 422 μmol, 88.1% yield based on 7). 1H NMR (500 MHz, CD2Cl2): δ/ppm 15.16 (d, J = 7.0 Hz, 1H), 8.0–6.3 (br, 15H), 7.55–7.51 (m, 1H), 7.24 (d, J = 8.5 Hz, 1H), 7.17 (br s, 1H), 6.95–6.92 (m, 1H), 6.69–6.67 (m, 1H), 6.53 (br s, 1H), 6.42 (br s, 1H), 6.04 (br s, 1H), 5.74 (sep, J = 6.7 Hz, 1H), 4.20 (br s, 1H), 4.05 (br s, 1H), 3.97 (br s, 1H), 3.63 (br s, 1H), 2.93 (br s, 3H), 2.35 (br s, 3H), 2.07 (br s, 3H), 1.97 (br s, 3H), 1.88–1.86 (br m, 6H), 1.59 (d, J = 6.7 Hz, 3H), 1.30 (br s, 3H). 13C{1H} NMR (125.7 MHz, CD2Cl2): δ/ppm 289.4 (d), 208.9 (d), 154.1, 142.0, 141–140 (br m), 137–136 (br m), 136.0 (br s), 135.1 (br s), 135–134 (br m), 133–132 (br m), 132.7, 132–131 (br m), 130.0 (br s), 130–128 (br m), 126.9, 122.9, 118.0, 81.8, 23.4, 22.8, 22–21 (br, m), 20.7 (br s), 20–19 (br m), 17.7 (br s). 31P{1H} NMR (121.4 MHz, CD2Cl2): δ/ppm 53.6. HRMS (FAB+): Calculated for {[H2IMes2]RuCl(PPh3)[CH-o-(OiPr)C6H4]}+: 853.2628, Found: 853.2617.
{[H2IMes2]RuCl(PPh3)[CH-o-(OiPr)C6H4]}OTf (8b).
Starting from 7 (300 mg, 479 μmol), AgOTf (135 mg, 527 μmol) and triphenylphosphine (151 mg 575 μmol) in 10 ml of CH2Cl2, 8b was obtained as a red-orange crystalline solid (277 mg, 276 μmol, 57.6% yield based on 7). 1H NMR (500 MHz, CD2Cl2): δ/ppm 15.15 (d, J = 7.3 Hz, 1H), 8.0–6.3 (br, 15H), 7.54–7.51 (m, 1H), 7.22 (d, J = 8.2 Hz, 1H), 7.17 (br s, 1H), 6.94–6.91 (m, 1H), 6.68–6.66 (m, 1H), 6.53 (br s, 1H), 6.42 (br s, 1H), 6.04 (br s, 1H), 5.74 (sep, J = 6.7 Hz, 1H), 4.19 (br s, 1H), 4.04 (br s, 1H), 3.96 (br s, 1H), 3.64 (br s, 1H), 2.92 (br s, 3H), 2.35 (br s, 3H), 2.07 (br s, 3H), 1.97 (br s, 3H), 1.87–1.86 (br m, 6H), 1.58 (d, J = 6.7 Hz, 3H), 1.30 (br s, 3H). 13C{1H} NMR (125.7 MHz, CD2Cl2): δ/ppm 209.1 (d), 154.2, 154.2, 142.0, 141–140 (br m), 137–136 (br m), 136.0 (br s), 135.1 (br s), 135–134 (br m), 133–132 (br m), 132.7, 132–131 (br m), 130.1 (br s), 130–128 (br m), 127.0, 122.9, 118.0, 81.8, 23.5, 22.8, 22–21 (br, m), 20.7 (br s), 20–19 (br m), 17.8 (br s). 31P{1H} NMR (121.4 MHz, CD2Cl2): δ/ppm 53.5. HRMS (FAB+): Calculated for {[H2IMes2]RuCl(PPh3)[CH-o-(OiPr)C6H4]}+: 853.2628, Found: 853.2666.
{[H2IMes2]RuCl(PPh3)[CH-o-(OiPr)C6H4]}PF6 (8c).
Starting from 7 (300 mg, 479 μmol), AgPF6 (133 mg, 527 μmol) and triphenylphosphine (151 mg 575 μmol) in 10 ml of CH2Cl2, 8c was obtained as a red-orange crystalline solid (406 mg, 406 μmol, 84.7% yield based on 7). 1H NMR (500 MHz, CD2Cl2): δ/ppm 15.16 (d, J = 7.3 Hz, 1H), 8.0–6.3 (br, 15H), 7.55–7.51 (m, 1H), 7.23 (d, J = 8.5 Hz, 1H), 7.18 (br s, 1H), 6.95–6.92 (m, 1H), 6.69–6.68 (m, 1H), 6.53 (br s, 1H), 6.42 (br s, 1H), 6.05 (br s, 1H), 5.74 (sep, J = 6.7 Hz, 1H), 4.18 (br s, 1H), 4.03 (br s, 1H), 3.96 (br s, 1H), 3.63 (br s, 1H), 2.93 (br s, 3H), 2.35 (br s, 3H), 2.07 (br s, 3H), 1.98 (br s, 3H), 1.88–1.86 (br m, 6H), 1.59 (d, J = 6.7 Hz, 3H), 1.30 (br s, 3H). 13C{1H} NMR (125.7 MHz, CD2Cl2): δ/ppm 289.5 (d), 209.0 (d), 154.2, 154.2, 142.0, 141–140 (br m), 137–136 (br m), 136.0 (br s), 135.2 (br s), 135–134 (br m), 133–132 (br m), 132.7, 132–131 (br m), 130.0 (br s), 130–128 (br m), 127.0, 122.9, 118.0, 81.8, 23.5, 23.4, 22.8, 22–21 (br, m), 20.7 (br s), 20–19 (br m), 17.7 (br s). 31P{1H} NMR (121.4 MHz, CD2Cl2): δ/ppm 53.5. HRMS (FAB+): Calculated for {[H2IMes2]RuCl(PPh3)[CH-o-(OiPr)C6H4]}+: 853.2628, Found: 853.2647.
{[H2IMes2]RuCl(PPh3)[CH-o-(OiPr)C6H4]}SbF6 (8d).
Starting from 7 (300 mg, 479 μmol), AgSbF6 (181 mg, 527 μmol) and triphenylphosphine (151 mg 575 μmol) in 10 ml of CH2Cl2, 8d was obtained as a red-orange crystalline solid (490 mg, 450 μmol, 93.9% yield based on 7). 1H NMR (500 MHz, CD2Cl2): δ/ppm 15.17 (d, J = 7.3 Hz, 1H), 8.0–6.3 (br, 15H), 7.55–7.51 (m, 1H), 7.22 (d, J = 8.5 Hz, 1H), 7.18 (br s, 1H), 6.95–6.92 (m, 1H), 6.69–6.67 (m, 1H), 6.54 (br s, 1H), 6.42 (br s, 1H), 6.05 (br s, 1H), 5.74 (sep, J = 6.7 Hz, 1H), 4.16 (br s, 1H), 4.02 (br s, 1H), 3.96 (br s, 1H), 3.62 (br s, 1H), 2.92 (br s, 3H), 2.35 (br s, 3H), 2.07 (br s, 3H), 1.98 (br s, 3H), 1.87–1.86 (br m, 6H), 1.59 (d, J = 6.7 Hz, 3H), 1.31 (br s, 3H). 13C{1H} NMR (125.7 MHz, CD2Cl2): δ/ppm 289.5 (d), 209.1 (d), 154.2, 154.2, 142.0, 141–140 (br m), 137–136 (br m), 136.0 (br s), 135.2 (br s), 135–134 (br m), 133–132 (br m), 132.7, 132–131 (br m), 130.1 (br s), 130–128 (br m), 127.0, 122.9, 118.0, 81.8, 23.5, 23.4, 22.8, 22–21 (br, m), 20.7 (br s), 20–19 (br m), 17.7 (br s). 31P{1H} NMR (121.4 MHz, CD2Cl2): δ/ppm 53.5. HRMS (FAB+): Calculated for {[H2IMes2]RuCl(PPh3)[CH-o-(OiPr)C6H4]}+: 853.2628, Found: 853.2643.
General procedure for the synthesis of {[H2IMes2]RuCl[Ph2P(CH2)nPPh2](CHPh)}(OTf) (11).
In a glove box, 9 and CH2Cl2 were added into a 20 ml screw-cap vial equipped with a magnetic stir bar. With stirring, trimethylsilyl trifluoromethanesulfonate was added slowly. After stirring at room temperature for 1 h, the corresponding diposphine was added. Then the reaction solution was stirred at room temperature for 2 h, and evaporated. The crude product was dissolved in a small amount of CH2Cl2. The solution was added dropwise with vigorous stirring into a large amount of pentane (for 11a) or Et2O (for 11b). The appeared precipitate was corrected on a filter, washed with appropriate solvent, and dried under reduced pressure.
{[H2IMes2]RuCl[Ph2P(CH2)2PPh2](CHPh)}(OTf) (11a).
Starting from 9 (100 mg, 120 μmol), TMS(OTf) (24.0 μl, 29.5 mg, 133 μmol) and DPPE (57.5 mg 144 μmol) in 5.0 ml of CH2Cl2, 11a was obtained as a yellow-brown solid (76.7 mg, 71.0 μmol, 53.4% yield based on 9). 1H NMR (500 MHz, CD2Cl2): δ/ppm 16.23 (dd, J = 26.9 Hz, J = 1.2 Hz, 1H), 7.87–7.76 (m, 5H), 7.69–7.56 (m, 1H), 7.53–7.19 (m, 12H), 7.04 (s, 1H), 6.92–6.88 (m, 2H), 6.85–6.76 (m, 2H), 6.62–6.59 (m, 4H), 6.43 (s, 1H), 5.60 (s, 1H), 4.06–4.00 (m, 1H), 3.93–3.87 (m, 1H), 3.81–3.70 (m, 2H), 2.76 (s, 3H), 2.42 (s, 3H), 2.28 (s, 3H), 2.19–2.13 (br m, 1H), 2.10–2.03 (br m, 1H), 1.94 (s, 3H), 1.82 (s, 3H), 1.40–1.34 (br m, 1H), 1.17 (s, 3H), 0.68–0.64 (br m, 1H). 13C{1H} NMR (125.7 MHz, CD2Cl2): δ/ppm 213.1 (dd), 149.0, 141.5, 140.0, 139.6, 139.4, 138.3, 137.7, 136.8, 135.9, 135.6, 135.3, 135.2, 135.0, 133.9, 133.9, 133.7, 133.3, 132.5–132.4 (m), 132.0, 131.9, 131.9, 131.3–131.2 (m), 130.9, 130.8–130.8 (m), 130.6–130.6 (m), 130.5, 130.4, 130.2–130.2 (m), 129.9, 129.0, 128.9, 128.9, 128.9, 128.5, 128.5, 128.4, 128.2, 41.4–41.0 (m), 21.8–21.4 (m), 21.0–20.9 (m), 20.6–20.5 (m), 18.9–18.9 (m), 18.0–18.0 (m), 15.9. 31P{1H} NMR (121.4 MHz, CD2Cl2): δ/ppm 53.4 (d), 52.4 (d). HRMS (FAB+): Calculated for {[H2IMes2]RuCl[Ph2P(CH2)2PPh2](CHPh)}+: 931.2651, Found: 931.2671.
{[H2IMes2]RuCl[Ph2P(CH2)3PPh2](CHPh)}(OTf) (11b).
Starting from 9 (100 mg, 120 μmol), TMS(OTf) (24.0 μl, 29.5 mg, 133 μmol) and DPPP (59.6 mg 144 μmol) in 5.0 ml of CH2Cl2, 11b was obtained as a yellow solid (78.7 mg, 71.9 μmol, 59.7% yield based on 9). 1H NMR (500 MHz, CD2Cl2): δ/ppm 16.83 (dd, J = 26.5 Hz, J = 1.2 Hz, 1H), 7.68–7.60 (m, 7H), 7.50–7.44 (m, 3H), 7.39–7.36 (m, 2H), 7.32–7.28 (m, 3H), 7.17–7.14 (m, 3H), 7.11–7.08 (br m, 2H), 6.87–6.84 (m, 2H), 6.67–6.64 (br m, 2H), 6.62 (s, 1H), 6.48 (s, 1H), 6.41–6.37 (br m, 2H), 5.73 (s, 1H), 4.02–3.95 (m, 1H), 3.84–3.78 (m, 1H), 3.71–3.66 (m, 2H), 2.83 (s, 3H), 2.42 (s, 3H), 2.31–2.19 (br m, 3H), 2.12 (s, 3H), 1.95 (s, 3H), s1.89 (s, 3H), 1.78–1.73 (br m, 1H), 1.57–1.50 (br m, 1H), 1.15 (s, 3H), 0.93–0.80 (br m, 1H). 13C{1H} NMR (125.7 MHz, CD2Cl2): δ/ppm 211.0 (dd), 150.2, 150.2, 141.5, 140.1, 139.4, 139.0, 138.3, 138.1, 137.5–137.5 (m), 137.2–137.2 (m), 137.1, 136.0, 135.9, 135.6, 134.7, 134.7, 133.7, 133.5, 133.4, 133.2, 133.1, 131.7–131.7 (m), 131.6–131.6 (m), 131.3, 131.2, 131.0, 130.6, 130.6, 130.2, 130.1, 130.0, 129.8, 129.8, 129.4, 129.0, 128.7. 128.5, 128.4, 127.6, 127.5, 36.5–36.2 (m), 23.4, 23.2, 21.5–21.0 (m), 19.4–19.4 (m), 18.3, 18.0–18.0 (m), 16.8. 31P{1H} NMR (121.4 MHz, CD2Cl2): δ/ppm 18.9 (d), 3.5 (d). HRMS (FAB+): Calculated for {[H2IMes2]RuCl[Ph2P(CH2)3PPh2](CHPh)}+: 945.2808, Found: 945.2801.
Crystal structure determination of 8c
Single crystals of 8c were recrystallised by diffusion of pentane into a saturated CH2Cl2 solution of 8c; a suitable crystal was mounted on a hand-crafted glass fiber with Paratone oil (Exxon) and transferred to the 100 K cold gas stream of an OxfordCryosystems 700 Series Cryostream Cooler on the Bruker KAPPA APEXII 02B diffractometer.
Crystal data. C52H59Cl7F6N2OP2Ru, M = 1253.17, monoclinic, a = 10.1739(4), b = 23.3587(10), c = 23.0472(9) Å, U = 5463.3(4) Å3, T = 100 K, space group P21/n (no. 14), Z = 4, 160797 reflections measured, 21852 unique (Rint = 0.056), which were used in all calculations. Refinement of F2 against all reflections (except the omitted (0 1 1)) with the weights w = 1/σ2(Fo2). All non-hydrogen atoms were refined anisotropically. The cation and solvent hydrogen atoms were treated differently. All hydrogen atoms in the cation were freely refined with four parameters, three positional and one isotropic displacement. The six hydrogen atoms on the three dichloromethane molecules were included into the model at geometrically calculated positions and refined using a riding model. The isotropic displacement parameters of these hydrogen atoms were fixed to 1.2 times the Ueq value of the carbon atoms to which they are bonded. The final wR(F2) was 0.059 (all data).
Metathesis assays
Representative procedure for RCM of diethyldiallyl malonate (12)15.
In a glove box, a 1.0 ml volumetric flask was charged with 8a (7.5 mg, 8.0 μmol) and CD2Cl2 was added to prepare 1.0 ml of stock solution (0.008 M). CD2Cl2 (700 μl) and the stock solution (100 μl, 0.80 μmol) were added into an NMR tube with a screw-cap septum top. The sample was equilibrated at 30 °C in the NMR probe before 12 (19.3 μl, 19.2 mg, 80 μmol) was added via syringe. Data points were collected over an appropriate period of time using the Varian array function. The conversion of 12 to 13 was determined by comparing the ratio of the integrals of the methylene protons in the starting material with those in the product in the 1H NMR spectra.
Representative procedure for CM of allylbenzene (14) and cis-1,4-diacetoxy-2-butene (15)15.
Preparation of a substrate mixture.
In a glove box, tridecane (92.0 μl, 69.6 mg, 377 μmol), 14 (100 μl, 89.2 mg, 755 μmol) and 15 (240 μl, 259 mg, 1.51 mmol) were combined in a 5 ml vial with a screw-cap septum top and a magnetic stir bar; this mixture was allowed to stir for 5 min.
Preparation of reaction solution and CM of 14 and 15.
In a glove box, a 5 ml vial with a screw-cap septum top and a magnetic stir bar was charged with 8a (4.7 mg, 5.0 μmol) and CH2Cl2 (1.0 ml). The catalyst solution was removed from the glove box and then stirred at 23 °C under argon. To the catalyst solution, the substrate mixture (115 μl; tridecane: 24.5 μl, 18.5 mg, 100 μmol; 14: 26.6 μl, 23.7 mg, 201 μmol; 15: 63.9 μl, 69.0 mg, 401 μmol) was added via syringe. The reaction solution was allowed to stir at 23 °C and reaction aliquots (ca. 40 μl) were taken at the specific time points.
GC analysis.
Samples for GC analysis were obtained by adding the reaction aliquot to 400 μl of a 3 M solution of ethyl vinyl ether in isopropyl alcohol. The sample was shaken and allowed to stand for 10 min. 100 μl of 1 M slurry of tris(hydroxymethyl)phosphine in isopropyl alcohol was added.19 The sample was heated at 50 °C for 30 min, cooled to room temperature, passed through a pad of silica gel using CH2Cl2 as eluent and then analyzed by GC. GC response factor and retention time for each substrate were summarized in Table S6.† The amounts of the substrates in each sample were determined by previously reported method using response factors.15
Representative procedure for ROMP of norbornene (19).
A 2 ml volumetric flask was charged with 19 (125 mg, 1.33 mmol), and CD2Cl2 was added to prepare 2.0 ml of stock solution (0.665 M). In a glove box, 8a (1.9 mg, 2.0 μmol) and CD2Cl2 (500 μl) were added into an NMR tube with a screw-cap septum top. The stock solution (300 μl; 19: 18.8 mg, 200 μmol) was added via syringe and the sample was vigorously shaken for 30 seconds. Then the sample was allowed to stand for 30 min at 23 °C and analyzed by 1H NMR. The conversion of 19 to 20 was determined by comparing the ratio of the integrals of the olefinic protons in the starting material with those in the product in the 1H NMR spectrum.
Conclusion
While aiming to discover a convenient methodology for the preparation of cationic ruthenium alkylidene catalysts, simple ligand exchange using silver(I) salt of non-coordinating or weakly coordinating anion was investigated. When NHC-substituted catalyst 7 was reacted with a variety of silver(I) salt in the presence of PPh3, cationic catalysts bearing a single PPh3 ligand 8a–d were afforded selectively in good yield. Catalysts 8a–d were shown to exhibit moderate activity in the standard metathesis reactions. In contrast to reported neutral catalysts bearing a single bulky ligand, the stereoselectivities of 8a–d were very similar to that of dichloride substituted catalyst 7. Additionally, diphosphines DPPE or DPPP were reacted with complex 9, causing formation of cationic diphosphine-chelated catalysts 11a–b. 11a–b exhibited poor activity in the standard RCM and CM compared to 8a–d, most likely due to slow initiation derived from the high energy barrier of dechelation of the diphosphine ligand. Modification of the charge of ruthenium alkylidene catalyst is a promising avenue of investigation in improving catalyst efficiency and expanding their applications. The simple methods presented in this report will enable easy access to similar cationic catalysts and facilitate further investigations.
Acknowledgements
We thank Dr M. B. Herbert for helpful discussions and suggestions for this work, Materia, Inc. for the generous donation of catalysts and Dr M. W. Day and Mr L. M. Henling for X-ray crystallography. The Bruker KAPPA APEXII X-ray diffractometer was purchased via an NSF CRIF:MU award to the California Institute of Technology, CHE-0639094. This work was financially supported by NIH (NIH 5R01GM031332), NSF (CHE-0410425 and CHE-0809418) and Mitsui Chemicals, Inc.
References
- Selected reviews on olefin metathesis:
(a) A. Fürstner, Angew. Chem., Int. Ed., 2000, 39, 3012 CrossRef;
(b) T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18 CrossRef CAS PubMed;
(c) R. R. Schrock, Chem. Rev., 2002, 102, 145 CrossRef CAS PubMed;
(d) R. R. Schrock and A. H. Hoveyda, Angew. Chem., Int. Ed., 2003, 42, 4592 CrossRef CAS PubMed;
(e) C. Samojłowicz, M. Bieniek and K. Grela, Chem. Rev., 2009, 109, 3708 CrossRef PubMed.
-
J. Cossy, S. Arseniyadis and C. Meyer, Metathesis in Natural Product Synthesis: Strategies, Substrates, and Catalysts, Wiley-VCH, Weinheim, 1st edn, 2010 Search PubMed.
- J. B. Binder and R. T. Raines, Curr. Opin. Chem. Biol., 2008, 12, 767 CrossRef CAS PubMed.
- Y. Schrodi, T. Ung, A. Vargas, G. Mkrtumyan, C. W. Lee, T. M. Champagne, R. L. Pederson and S. H. Hong, Clean: Soil, Air, Water, 2008, 36, 669 CrossRef CAS.
-
(a) A. Leitgeb, J. Wappel and C. Slugovc, Polymer, 2010, 51, 2927 CrossRef CAS;
(b) S. Sutthasupa, M. Shiotsuki and F. Sanda, Polym. J., 2010, 42, 905 CrossRef CAS;
(c) X. Liu and A. Basu, J. Organomet. Chem., 2006, 691, 5148 CrossRef CAS.
- G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 1746 CrossRef CAS PubMed and references cited therein.
-
(a) J. O. Krause, S. Lubbad, O. Nuyken and M. R. Buchmeiser, Adv. Synth. Catal., 2003, 345, 996 CrossRef CAS;
(b) J. O. Krause, S. H. Lubbad, O. Nuyken and M. R. Buchmeiser, Macromol. Rapid Commun., 2003, 24, 875 CrossRef CAS;
(c) J. O. Krause, O. Nuyken, K. Wurst and M. R. Buchmeiser, Chem. – Eur. J., 2004, 10, 777 CrossRef CAS PubMed;
(d) J. O. Krause, O. Nuyken and M. R. Buchmeiser, Chem. – Eur. J., 2004, 10, 2029 CrossRef CAS PubMed.
-
(a) J. J. van Veldhuizen, S. B. Garber, J. S. Kingsbury and A. H. Hoveyda, J. Am. Chem. Soc., 2002, 124, 4954 CrossRef CAS PubMed;
(b) J. J. van Veldhuizen, D. G. Gillingham, S. B. Garber, O. Kataoka and A. H. Hoveyda, J. Am. Chem. Soc., 2003, 125, 12502 CrossRef CAS PubMed;
(c) D. G. Gillingham, O. Kataoka, S. B. Garber and A. H. Hoveyda, J. Am. Chem. Soc., 2004, 126, 12288 CrossRef CAS PubMed;
(d) J. J. van Veldhuizen, J. E. Campbell, R. E. Giudici and A. H. Hoveyda, J. Am. Chem. Soc., 2005, 127, 6877 CrossRef CAS PubMed.
-
(a) K. Endo and R. H. Grubbs, J. Am. Chem. Soc., 2011, 133, 8525 CrossRef CAS PubMed;
(b) B. K. Keitz, K. Endo, P. R. Patel, M. B. Herbert and R. H. Grubbs, J. Am. Chem. Soc., 2012, 134, 693 CrossRef CAS PubMed;
(c) L. E. Rosebrugh, M. B. Herbert, V. M. Marx, B. K. Keitz and R. H. Grubbs, J. Am. Chem. Soc., 2013, 135, 1276 CrossRef CAS PubMed;
(d) K. Endo, M. B. Herbert and R. H. Grubbs, Organometallics, 2013, 32, 5128 CrossRef CAS PubMed;
(e) J. S. Cannon, L. Zou, P. Liu, Y. Lan, D. J. O'Leary, K. N. Houk and R. H. Grubbs, J. Am. Chem. Soc., 2014, 136, 6733 CrossRef CAS PubMed;
(f) M. B. Herbert, B. A. Suslick, P. Liu, L. Zou, P. K. Dornan, K. N. Houk and R. H. Grubbs, Organometallics, 2015, 34, 2858 CrossRef CAS.
-
(a) B. K. Keitz, K. Endo, M. B. Herbert and R. H. Grubbs, J. Am. Chem. Soc., 2011, 133, 9686 CrossRef CAS PubMed;
(b) B. K. Keitz, A. Fedorov and R. H. Grubbs, J. Am. Chem. Soc., 2012, 134, 2040 CrossRef CAS PubMed;
(c) M. B. Herbert, V. M. Marx, R. L. Pederson and R. H. Grubbs, Angew. Chem., Int. Ed., 2013, 52, 310 CrossRef CAS PubMed;
(d) V. M. Marx, M. B. Herbert, B. K. Keitz and R. H. Grubbs, J. Am. Chem. Soc., 2013, 135, 94 CrossRef CAS PubMed;
(e) H. Miyazaki, M. B. Herbert, P. Liu, X. Dong, X. Xu, B. K. Keitz, T. Ung, G. Mkrtumyan, K. N. Houk and R. H. Grubbs, J. Am. Chem. Soc., 2013, 135, 5848 CrossRef CAS PubMed;
(f) L. E. Rosebrugh, V. M. Marx, B. K. Keitz and R. H. Grubbs, J. Am. Chem. Soc., 2013, 135, 10032 CrossRef CAS PubMed;
(g) J. Hartung and R. H. Grubbs, J. Am. Chem. Soc., 2013, 135, 10183 CrossRef CAS PubMed;
(h) J. S. Cannon and R. H. Grubbs, Angew. Chem., Int. Ed., 2013, 52, 9001 CrossRef CAS PubMed;
(i) J. Hartung and R. H. Grubbs, Angew. Chem., Int. Ed., 2014, 53, 3885 CrossRef CAS PubMed;
(j) S. L. Mangold, D. J. O'Leary and R. H. Grubbs, J. Am. Chem. Soc., 2014, 136, 12469 CrossRef CAS PubMed;
(k) J. Hartung, P. K. Dornan and R. H. Grubbs, J. Am. Chem. Soc., 2014, 136, 13029 CrossRef CAS PubMed;
(l) M. B. Herbert and R. H. Grubbs, Angew. Chem., Int. Ed., 2015, 54, 5018 CrossRef CAS PubMed;
(m) P. K. Dornan, Z. K. Wickens and R. H. Grubbs, Angew. Chem., Int. Ed., 2015, 54, 7134 CrossRef CAS PubMed.
-
(a) N. Audic, H. Clavier, M. Mauduit and J.-C. Guillemin, J. Am. Chem. Soc., 2003, 125, 9248 CrossRef CAS PubMed;
(b) H. Clavier, N. Audic, M. Mauduit and J.-C. Guillemin, Chem. Commun., 2004, 2282 RSC;
(c) H. Clavier, N. Audic, J.-C. Guillemin and M. Mauduit, J. Organomet. Chem., 2005, 690, 3585 CrossRef CAS;
(d) Q. Yao and Y. Zhang, Angew. Chem., Int. Ed., 2003, 42, 3395 CrossRef CAS PubMed;
(e) Q. Yao and M. Sheets, J. Organomet. Chem., 2005, 690, 3577 CrossRef CAS.
-
(a) M. A. O. Volland, S. M. Hansen, F. Rominger and P. Hofmann, Organometallics, 2004, 23, 800 CrossRef CAS;
(b) M. Zirngast, E. Pump, A. Leitgeb, J. H. Albering and C. Slugovc, Chem. Commun., 2011, 47, 2261 RSC;
(c) O. Songis, A. M. Z. Slawin and C. S. J. Cazin, Chem. Commun., 2012, 48, 1266 RSC;
(d) B. Autenrieth, W. Frey and M. R. Buchmeiser, Chem. – Eur. J., 2012, 18, 14069 CrossRef CAS PubMed;
(e) B. Autenrieth, E. B. Anderson, D. Wang and M. R. Buchmeiser, Macromol. Chem. Phys., 2013, 214, 33 CrossRef CAS;
(f) A. M. McKinty, C. Lund and D. W. Stephan, Organometallics, 2013, 32, 4730 CrossRef CAS.
- S. B. Garber, J. S. Kingsbury, B. L. Gray and A. H. Hoveyda, J. Am. Chem. Soc., 2000, 122, 8168 CrossRef CAS.
- M. S. Sanford, J. A. Love and R. H. Grubbs, J. Am. Chem. Soc., 2001, 123, 6543 CrossRef CAS PubMed.
- T. Ritter, A. Hejl, A. G. Wenzel, T. W. Funk and R. H. Grubbs, Organometallics, 2006, 25, 5740 CrossRef CAS.
-
(a) P. Teo and R. H. Grubbs, Organometallics, 2010, 29, 6045 CrossRef CAS;
(b) G. Occhipinti, F. R. Hansen, K. W. Törnroos and V. R. Jensen, J. Am. Chem. Soc., 2013, 135, 3331 CrossRef CAS PubMed;
(c)
V. R. Jensen, G. Occhipinti and F. R. Hansen, Int. Patent Appl, WO2012/032131A1, 2012 Search PubMed.
- Conversion of 14 to 16 slightly decreased between 240 min and 480 min. We speculate that this was caused by secondary metathesis involving 16 as a reactant. Similar phenomenon has been observed in other metathesis reactions by ruthenium alkylidene catalysts. See ref. 15 for the data.
- A. B. Pangborn, M. A. Giardello, R. H. Grubbs, R. K. Rosen and F. J. Timmers, Organometallics, 1996, 15, 1518 CrossRef CAS.
- R. L. Pederson, I. M. Fellows, T. A. Ung, H. Ishihara and S. P. Hajela, Adv. Synth. Catal., 2002, 344, 728–735 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra and metathesis data. CCDC 784488. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt04506a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence









