
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ln(III)-complexes of a DOTA analogue with an ethylenediamine pendant arm as pH-responsive PARACEST contrast agents†
T.
Krchová
a,
A.
Gálisová
b,
D.
Jirák
bc,
P.
Hermann
a and
J.
Kotek
*a
aDepartment of Inorganic Chemistry, Faculty of Science, Universita Karlova (Charles University), Hlavova 2030, 128 43 Prague 2, Czech Republic. E-mail: modrej@natur.cuni.cz; Fax: +420-221951253; Tel: +420-221951261
bDepartment of Radiodiagnostic and Interventional Radiology, Magnetic Resonance Unit, Institute for Clinical and Experimental Medicine, Vídeňská 1958/9, Prague 4, 140 21, Czech Republic
cInstitute of Biophysics and Informatics, 1st Faculty of Medicine, Universita Karlova (Charles University), Salmovská 1, 120 00 Prague 2, Czech Republic
First published on 11th January 2016
Abstract
A novel macrocyclic DO3A derivative containing a linear diamine pendant arm, H3do3aNN, was prepared and its protonation and complexation properties were studied by means of potentiometry. It determined ligand consecutive protonation constants log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) KAn = 12.62, 10.28, 9.67, 8.30, 3.30 and 1.58 and stability constants of selected lanthanide (Eu(III), Yb(III)) complexes logKEuL = 23.16 and logKYbL = 22.76. The complexes could be protonated on the pendant amino group(s) with logK(HLM) ≈ 5.6 and logK(H2LM) ≈ 4.8. Solution structures of both complexes were studied by NMR spectroscopy. The study revealed that the complex species exist exclusively in the form of twisted-square-antiprismatic (TSA) isomers. The complexes show significant pH dependence of the Chemical Exchange Saturation Transfer (CEST) between their amino groups and the bulk water molecules in the pH range of 5–8. Thus, the pH dependence of the magnetization transfer ratio of CEST signals can be used for pH determination using magnetic resonance imaging techniques in a pH range relevant for in vivo conditions.
KAn = 12.62, 10.28, 9.67, 8.30, 3.30 and 1.58 and stability constants of selected lanthanide (Eu(III), Yb(III)) complexes logKEuL = 23.16 and logKYbL = 22.76. The complexes could be protonated on the pendant amino group(s) with logK(HLM) ≈ 5.6 and logK(H2LM) ≈ 4.8. Solution structures of both complexes were studied by NMR spectroscopy. The study revealed that the complex species exist exclusively in the form of twisted-square-antiprismatic (TSA) isomers. The complexes show significant pH dependence of the Chemical Exchange Saturation Transfer (CEST) between their amino groups and the bulk water molecules in the pH range of 5–8. Thus, the pH dependence of the magnetization transfer ratio of CEST signals can be used for pH determination using magnetic resonance imaging techniques in a pH range relevant for in vivo conditions.
Introduction
Magnetic resonance imaging (MRI), due to its non-invasive character and spatial resolution (down to a mm3 at clinical magnetic fields), is currently one of the most important diagnostic methods used in clinical medicine.1 Relevant diagnostic information from MRI images can be obtained even with the natural contrast between various tissues. However, for further improvement of image contrast and resolution, exogenous contrast agents (CAs) based on complexes of highly paramagnetic metal ions or superparamagnetic nanocrystalline materials altering the relaxation times of bulk water are widely used.1,2 In addition to common MRI T1- and T2-contrast agents, which shorten the longitudinal (T1) and transversal (T2) relaxation times,2 a new class of CAs based on a Chemical Exchange Saturation Transfer (CEST) mechanism was introduced in the past decade.3,4The principle of the CEST effect is based on saturation of the proton signal of the contrast agent molecule by a selective radiofrequency pulse. This saturation is transferred to the surrounding water molecules via chemical exchange of the labile protons between the contrast agent and bulk water resulting in a decrease in the water signal intensity and, therefore, darkening of the corresponding area in the MR image. To reduce any nonspecific water proton irradiation and to increase the sensitivity of the CEST CAs, paramagnetic complexes (PARACEST agents) are used to shift the resonance frequency of the exchangeable protons far away from that of bulk water.4–6 These agents contain a paramagnetic metal ion chelated by a multidentate ligand. Most often, Ln(III) complexes of ligands derived from DOTA (thus ensuring high stability and kinetic inertness of the complexes) have been used.6,7 As alternatives, complexes of transition metal ions having suitable magnetic properties, such as Ni(II), Fe(II) or Co(II), with ligands based on cyclam, cyclen, 1,4,7-triazacyclononane or 1,4,10-trioxa-7,13-diazacyclopenta-decane, etc. have also been reported (Fig. 1).8
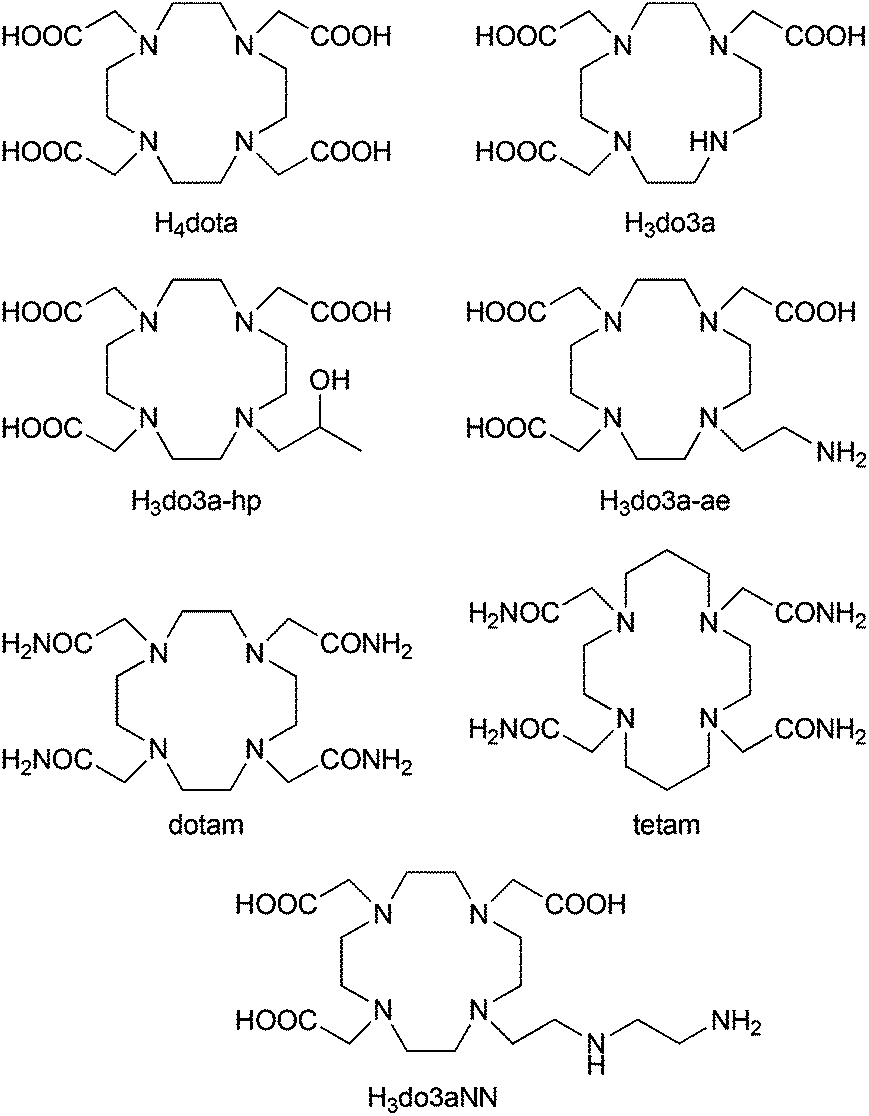 | ||
| Fig. 1 Structural formulas of the ligands discussed in the text. | ||
One of the major advantages of CEST agents is the possibility to modulate water signal intensity by a selective presaturation pulse and, therefore, image contrast produced by these CAs can be switched “on” or “off” at will by selecting the appropriate irradiation frequency. This fact makes it possible to detect several agents in the same sample.9 Another advantage lies in the sensitivity of the proton exchange rate (kex) to a number of external factors and, thus, the CEST complexes are suitable for measuring various physiological parameters, such as temperature, pH, metabolite or metal ion concentration, etc.4c,5,6
Recently, a lot of effort has been invested into developing MRI CAs capable of reporting on in vivo changes of pH in a tissue as they could serve as valuable biomarkers of disease progression or indicators for the choice of treatment.10 Several studies have demonstrated the unique ability of PARACEST CAs to act as pH sensors and nowadays ratiometric methods are being explored to make the assessments independent of the local concentration of the CAs.11 For example, a Yb(III) analogue of the clinically approved MRI CA [Gd(do3a-hp)] (ProHance®, ligand shown in Fig. 1) shows two independent well-resolved PARACEST peaks at 71 and 99 ppm originating from the protons of the coordinated alcohol group of individual complex isomers.11a The ratio of these two PARACEST signals is pH-dependent, which can be used to develop a concentration-independent method of pH measurement, and the Yb(III) complex has been already tested for measuring extracellular pH in murine melanoma.11b Similarly, the PARACEST peaks of a Co(II) complex of tetam (Fig. 1) have distinct pH dependencies and the two most shifted signals (at 95 and 112 ppm) were shown to be suitable for pH mapping.8e
It was shown that Ln(III) complexes of cyclen derivatives with pendant arms containing an amido-amine pendant arm,11g,h or a (semi)coordinating amino group12 produced a pH-sensitive PARACEST effect in the pH region relevant for living systems. Based on these findings, we decided to synthesize a new macrocyclic ligand H3do3aNN (Fig. 1) containing a semi-labile coordinating pendant arm with two (primary and secondary) amino groups (as two potentially independent proton exchanging pools), and to investigate the PARACEST properties of its Ln(III) complexes.
Results and discussion
Synthesis
The synthesis of H3do3aNN is shown in Scheme 1. The alkylation agent 3 was prepared by CBr4/PPh3 bromination of ethyl-carbamate-protected N-(2-aminoethyl)ethanolamine 2. The alkylation of tBu3do3a·HBr was performed using a slight excess of the alkylation agent as, under the reaction conditions, the alkylation agent undergoes elimination of HBr. The tBu-ester groups were removed by reflux in a CF3CO2H:CHCl3 1:1 mixture and the ethyl-carbamate protection groups were removed by hydrolysis in 10% aq. NaOH. Surprisingly, in this reaction step, preferential formation of the urea-derivative 6 was observed, with only trace amounts of the required compound H3do3aNN. However, the intermediate 6 can be isolated by crystallization in a zwitterionic form with 42% overall yield (based on tBu3do3a). The identity of the intermediate 6 was confirmed by a single-crystal X-ray diffraction study (see ESI and Fig. S1†). Hydrolysis of 6 with aq. HCl produced H3do3aNN with a high yield. The best way to obtain the product in the solid form was trituration of the evaporated reaction mixture in dry THF or EtOH overnight. However, the resulting off-white solid is very hygroscopic and has to be stored in a desiccator over P2O5. All other attempts (different organic solvents used for trituration or crystallization) led to the isolation of the title ligand as oil. To prevent possible esterification by EtOH, the use of THF was preferred for trituration.
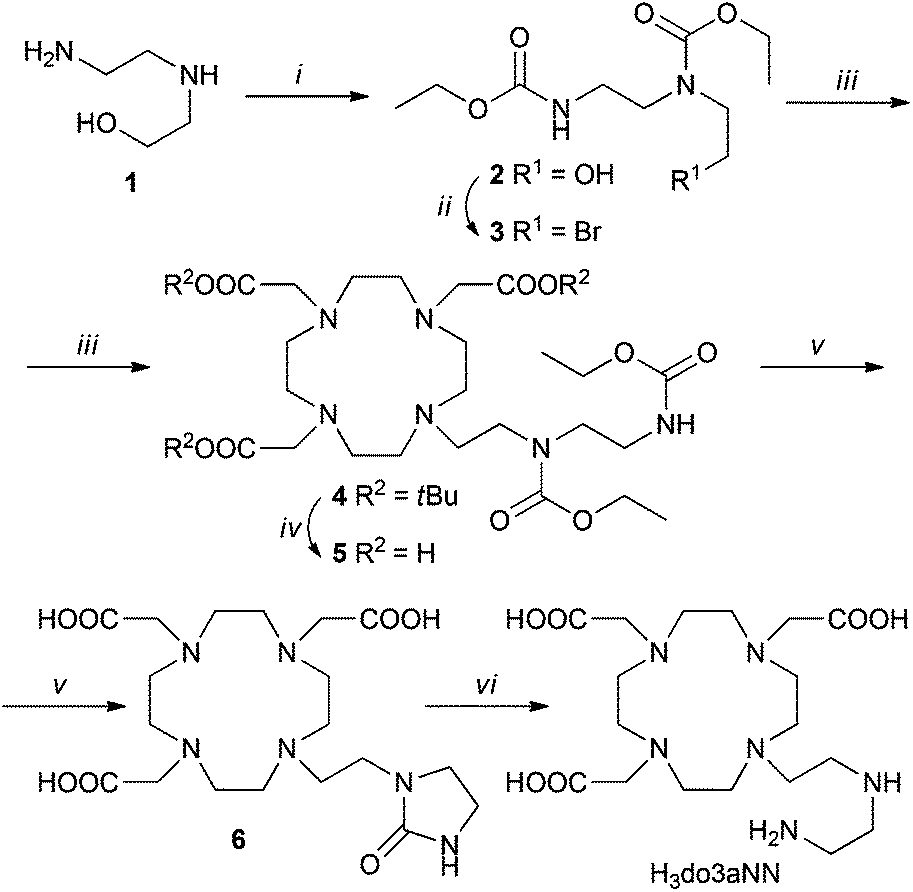 | ||
| Scheme 1 Synthesis of H3do3aNN. (i) CH3CH2OC(O)Cl, dioxane:H2O (1:1), RT, 2 h; (ii) CBr4, PPh3, THF, RT, 1 h; (iii) tBu3do3a·HBr, K2CO3, MeCN, 60 °C, 24 h; (iv) CF3CO2H:CHCl3 (1:1), reflux 18 h; (v) 10% aq. NaOH, 90 °C, 24 h; (vi) aq. HCl (1:1), 95 °C, 7 d. | ||
Thermodynamic behaviour of H3do3aNN and its Ln(III) complexes
Potentiometric titrations of the ligand performed in the pH range of 1.6–12.2 revealed six consecutive protonation processes in this region (Tables 1 and S1†). Based on comparison with the literature data,12,13 the first protonation step (logKP(HL) = 12.6) can be attributed to the protonation of one of (or to sharing of a proton over several of) the macrocycle amino groups. The next three protonation steps proceed in part simultaneously due to the similarity of the constants (logKP(H2L) = 10.3, logKP(H3L) = 9.7 and logKP(H4L) = 8.3) and occur on one other macrocycle amino group and two amino groups of the pendant N-(2-aminoethyl)-2-aminoethyl moiety (the value reported for analogous protonation of a 2-aminoethyl pendant moiety for H3do3a-ae is logKP = 8.9).12 Further protonations of H3do3aNN proceed on the carboxylate groups and lie in the usual range.
KP and logKML)a of H3do3aNN (0.1 M NMe4Cl, 25 °C) and its complexes, and the comparison with corresponding constants reported for related ligands
| Equilibrium | H3do3aNN | H3do3a-aeb | H4dota | H3do3a-hpc | H3do3ac |
|---|---|---|---|---|---|
| a K P = [HhL]/{[H]·[Hh–1L]} or [HhLM]/{[H]·[Hh–1LM]}; KML = [ML]/{[L]·[M]}. b Ref. 12. c Ref. 13. d Ref. 14. | |||||
| H+ + Ln− ↔ HL1–n | 12.62(2) | 13.19 | 11.74c | 11.17 | 12.46 |
| H+ + L1−n ↔ H2L2–n | 10.28(2) | 10.51 | 9.76c | 9.33 | 9.49 |
| H+ + L2−n ↔ H3L2−n | 9.67(2) | 8.90 | 4.68c | 4.99 | 4.26 |
| H+ + L3−n ↔ H4L2−n | 8.30(3) | 3.87 | 4.11c | 3.80 | 3.51 |
| H+ + L4−n ↔ H5L2−n | 3.30(3) | 1.27 | 2.37c | 2.84 | 1.97 |
| H+ + L5−n ↔ H6L2−n | 1.58(3) | — | — | — | — |
| Eu3+ + Ln− ↔ [Eu(L)]3−n | 23.16(5) | — | 24.2d | — | — |
| H+ + [Eu(L)]3−n ↔ [Eu(HL)]4−n | 6.03(5) | — | — | — | — |
| H+ + [Eu(HL)]4−n ↔ [Eu(H2L)]5−n | 5.09(7) | — | — | — | — |
| Yb3+ + Ln− ↔ [Yb(L)]3−n | 22.76(4) | — | 24.0d | — | — |
| H+ + [Yb(L)]3−n ↔ [Yb(HL)]4−n | 6.22(4) | — | — | — | — |
| H+ + [Yb(HL)]4−n ↔ [Yb(H2L)]5−n | 5.07(4) | — | — | — | — |
| Gd3+ + Ln− ↔ [Gd(L)]3−n | — | 22.23 | 24.67c | 24.5 | 22.02 |
Stability constants of [Ln(do3aNN)] (23.16 and 22.76 for the Eu(III) and Yb(III) complexes, respectively, Tables 1 and S2†) were obtained by the out-of-cell titration technique. The values are slightly lower compared with those reported for H4dota itself, but lie in the expected range, as can be seen from a comparison with the values reported for the related ligand H3do3a-ae – although, in that case, stability constants were determined for other lanthanides: La(III), logKLaL = 20.02, and Gd(III), logKGdL = 22.23.12 However, according to the distribution diagram of the Eu(III)–H3do3aNN system shown in Fig. 2, the metal complexation is not quantitative until pH ≈ 6 due to a combination of low affinity of the amino groups for Ln(III) ions and high donor-site basicity.
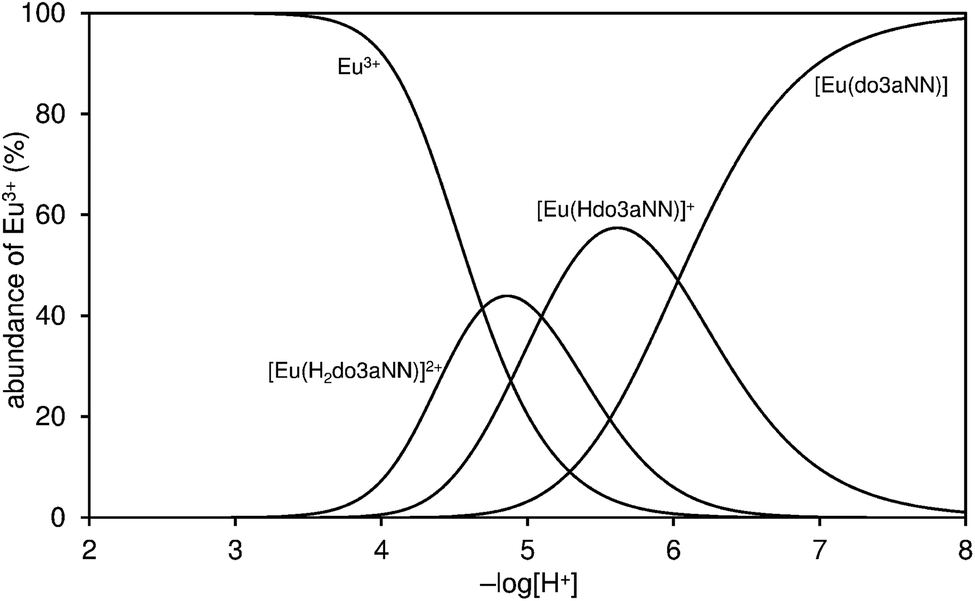 | ||
| Fig. 2 Distribution diagram of metal-containing species in the Eu(III)–H3do3aNN system (cM = cL = 0.004 M, 25 °C, I = 0.1 NMe4Cl). | ||
Equilibrated protonation steps of [Ln(do3aNN)] proceed with logKP(HLM) = 6.03/6.22 and those of the [Ln(Hdo3aNN)]+ species occur with logKP(H2LM) = 5.09/5.07 for the Eu(III)/Yb(III) complexes, respectively (Tables 1 and S2†). They are close to the values of analogous protonation constants reported for [Ln(do3a-ae)] complexes (logKP(HLM) = 6.06 and 5.83 for La(III)/Gd(III) systems).12 The observed values are slightly higher than the protonation constants found for the pre-formed complexes under “non-equilibrium” conditions: in such experiments, complexes were pre-formed at pH ≈ 7 and were titrated employing the standard (“fast”) acid–base titration method. The corresponding observed protonation constants were logKP(HLM) = 5.57 and 5.67, and logKP(H2LM) = 4.84 and 4.85 for Eu(III)/Yb(III), respectively (Table S3†). From these slight differences between the protonation constants, one can conclude that, during out-of-cell titrations, protons in the [Ln(Hdo3aNN)]+ and [Ln(H2do3aNN)]2+ species are probably located not only on the amine pendant arm but, at least partly, also on the macrocycle amino groups. The suggested structures of individual species with tentative protonation sites are shown in Schemes S1 and S2.†
Unfortunately, the [Ln(do3aNN)] complexes are not fully kinetically inert, and slowly dissociate at pH < 6. This was confirmed by a xylenol orange test: after the addition of a solution of the pre-formed complex (at pH = 7.5) to a buffered solution of xylenol orange at pH = 5.5, the colour gradually changed on standing from orange to orange-violet as a result of free metal appearance in the solution. A quantitative measurement revealed the dissociation of about 9–10% of the complex after standing for one week at room temperature (compare Fig. S16 and S17†). From a thermodynamic point of view, the extent of complex dissociation should be less than 20% at pH = 5 (see the distribution diagram shown in Fig. 2). It was confirmed by independent experiments that neither the free metal aqua ion nor the free ligand interferes with the 1H NMR or CEST measurements. Therefore, conclusions drawn from PARACEST experiments (see below) are fully valid even at pH 5–6.
Solution structure of the [Ln(Hndo3aNN)]‡ complexes
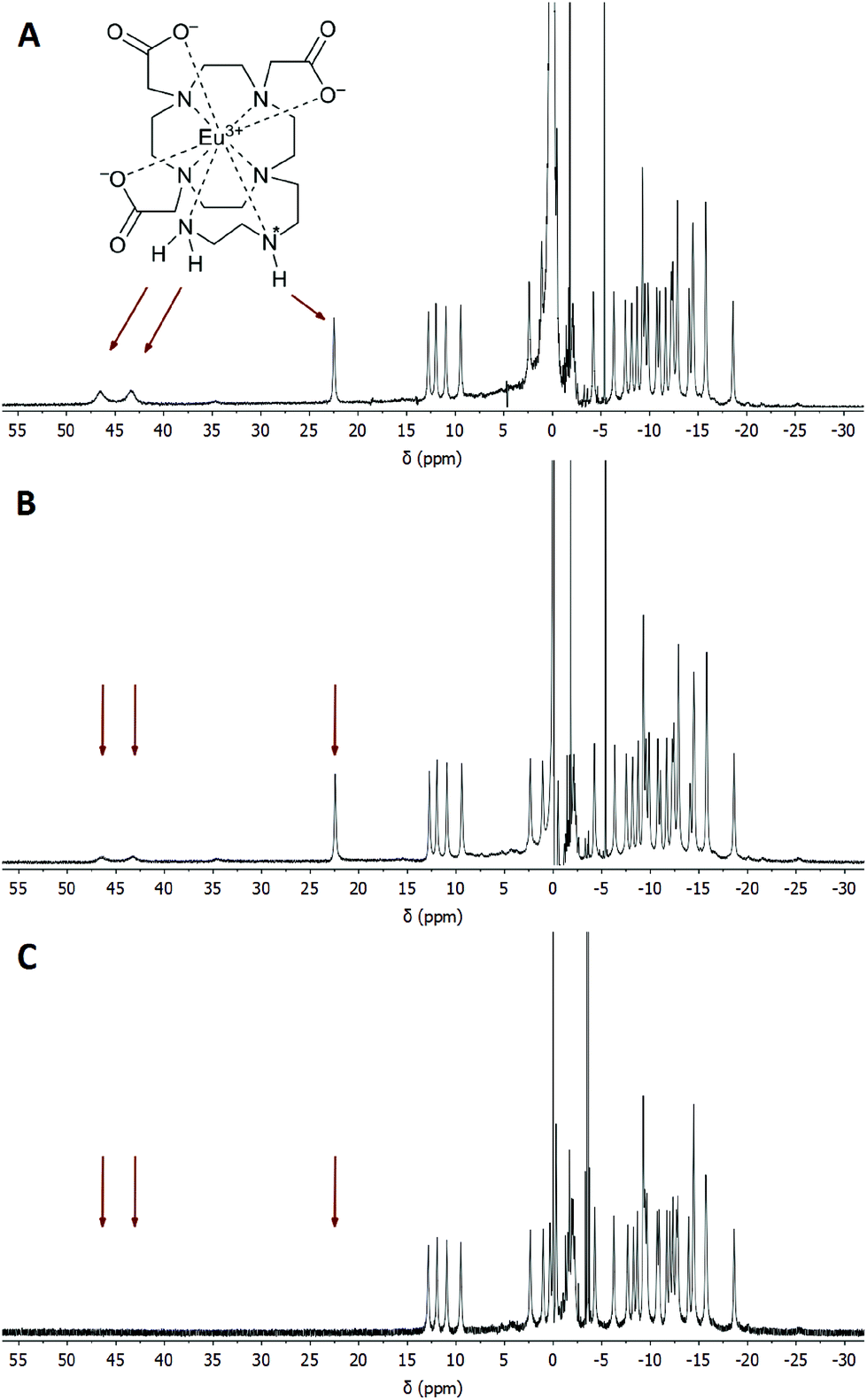
It is well-known that in Ln(III) complexes of DOTA-like ligands the central Ln(III) is coordinated between two planes – one formed by the macrocycle amino groups (N4-plane), and the other by the oxygen atoms of the carboxylate pendant moieties (O4-plane), and these species exhibit two types of isomerisms in solution.15 The first type is connected with the conformation of the macrocycle ethylene bridges, i.e. with the sign of the torsion angle around the C–C bond (δ/λ), and the second one is related to the direction of rotation of the pendant arms (Δ/Λ). A combination of these isomerisms leads to the formation of two diastereomeric pairs of enantiomers (i.e. four isomers): Δλλλλ/Λδδδδ (SA, square-antiprismatic) and Δδδδδ/Λλλλλ (TSA, twisted-square-antiprismatic).2 The isomer ratio in solution can be determined from the 1H NMR spectra using the “axial” protons of the macrocyclic chelate ring, which are the ones closest to the Ln(III) ion and to the principal magnetic axis, and usually can be easily found in the 1H NMR spectra.16 Therefore, the solution structures of the [Eu(do3aNN)] and [Yb(do3aNN)] complexes were investigated by variable-temperature 1H NMR spectroscopy (Fig. S2 and S3†). The pD of the samples in D2O was adjusted to the alkaline region to ensure full deprotonation and coordination of the pendant amino group. In both complexes, only one set of signals was detected pointing to the presence of only one diastereomer. The signals of “axial” protons appear in the range typical for the TSA isomers (Eu(III): 9–13 ppm, Yb(III): 45–62 ppm; with respect to the signal of bulk water referenced to 0 ppm). No 1H NMR signals of “axial” CH2 protons attributable to an SA isomer were observed (such signals typically lie in the chemical shift regions of 25–40 ppm and 100–150 ppm for Eu(III) and Yb(III) complexes, respectively).16,17 Thus, based on the 1H NMR data, exclusive formation of the TSA isomer is expected. With increasing temperature, the 1H NMR signals become broader, pointing to the occurrence of a conformational change of the complex molecules (Fig. S2 and S3†).To identify the signals of exchangeable (N–H) protons in the 1H NMR spectra, samples of the [Eu(Hndo3aNN)] and [Yb(Hndo3aNN)] complexes were investigated in H2O at 25 and 5 °C (Fig. 3, 4, S4 and S5†). In the 1H NMR spectra of the [Eu(Hndo3aNN)] complex recorded in H2O at pH = 6.75 (Fig. 3A), three main signals of exchangeable protons (one narrow signal at 22.2 and two broad signals at 43.3 and 46.5 ppm; with respect to the bulk water signal) can be observed, which disappear upon bulk water presaturation (Fig. 3B). Of these, only the signals at 43.3 and 46.5 ppm are influenced by water presaturation at pH = 11.7, whilst the signal at 22.2 ppm remains unaffected (Fig. 4A and B). When recording 1H NMR spectra in a D2O solution (pD = 10.7), none of the three signals are observable (Fig. 4C and S2†). Based on this behaviour, the narrow signal at 22.2 ppm is attributed to the coordinated secondary amino group. The assignment is supported by the similarity of the chemical shift of this signal to that of one of the –NH2 protons of the [Eu(do3a-ae)] complex (19.5 ppm).12 The two broad signals are attributed to the coordinated primary amino group and this assignment is supported by their coalescence at higher temperatures (Fig. S6†).
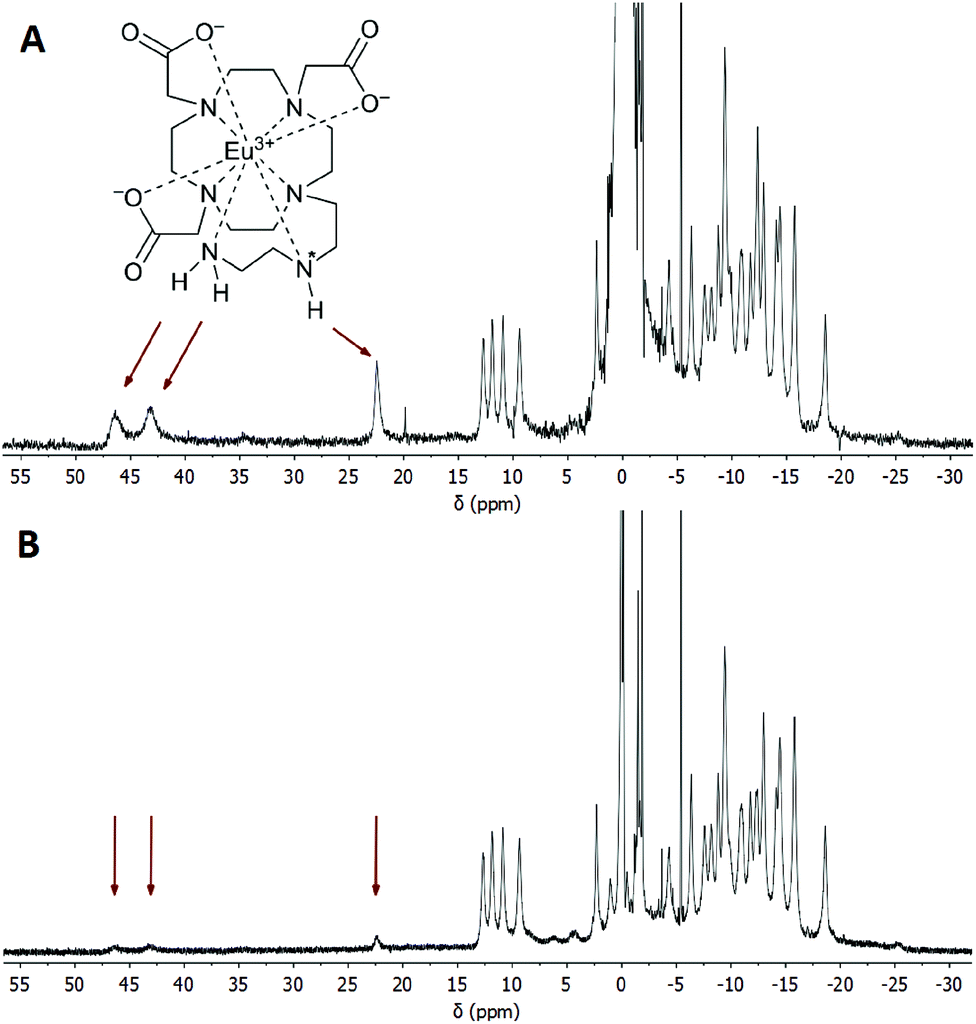 | ||
| Fig. 3 (A) 1H NMR spectrum of the [Eu(Hndo3aNN)] complex (0.09 M solution in H2O, B0 = 7.05 T, 25 °C, pH = 6.75). (B) The same sample, the signal of water was saturated. Arrows show the positions of exchangeable (N–H) protons. The chemical shift of H2O in the sample solution was referenced to 0 ppm. | ||
 | ||
| Fig. 4 (A) 1H NMR spectrum of the [Eu(do3aNN)] complex in H2O (0.09 M, B0 = 7.05 T, 25 °C, pH = 11.70). (B) The same sample, the signal of water was saturated. (C) 1H NMR spectrum of the [Eu(do3aNN)] complex in D2O (0.04 M, B0 = 7.05 T, 25 °C, pD = 10.73). Arrows show the positions of exchangeable (N–H) protons. The chemical shifts of H2O/HDO in the sample solutions were referenced to 0 ppm. | ||
The primary amino group is expected to be coordinated in a position capping the O3N-plane formed by the pendant donor atoms and, thus, close to the magnetic axis of the complex. Therefore, the corresponding protons are markedly influenced by the paramagnetic ion and their chemical shifts lie in the range typical for a coordinated water molecule.5,18 However, the presence of these signals in 1H NMR spectra also in an alkaline solution (and the presence of a corresponding CEST effect in Z-spectra at alkaline pH, see below) clearly excludes the possibility that these signals belong to a coordinated water molecule, the signal of which disappears in the alkaline region.19 In slightly acidic solutions where protonation of the uncoordinated primary amino group (and thus, its decoordination) is expected, even a proton of the secondary amino group is exchanged with bulk water on an NMR time scale. In contrast, in an alkaline solution, only the exchange of the terminal primary amino group protons is observable.
Besides the three signals of exchangeable protons of the [Eu(do3aNN)] complex discussed above, a small signal at ≈35 ppm was found in the 1H NMR spectra (Fig. 3 and 4), better seen at a lower temperature (37.5 ppm, Fig. S5A†).
At this chemical shift, a minor exchangeable pool of protons was found also in the CEST experiments (see discussion of Z-spectra below), accompanied by two other peaks in Z-spectra lying at 10 and 15 ppm, which are visible especially at low saturation power (Fig. 5 and S7A†). Due to the absence of any 1H NMR signals of “axial” CH2 protons attributable to the SA isomer, the presence of this isomer in the solution can be excluded. Therefore, these minor exchangeable proton pools were attributed to another TSA isomer that originates from the chirality of the nitrogen atom of the secondary amino group caused by coordination of this group. Judging by the similarity of the chemical shift of the exchangeable proton pool at 35 ppm to one of the signals attributed to the coordinated amino group in [Eu(do3a-ae)] (34 ppm),12 one can suggest that this signal belongs to the secondary amino group of the TSA isomer with reverse orientation of H vs. the CH2CH2NH2 substituents (i.e. with opposite chirality of the coordinated secondary amine). The results of simple molecular modelling shown in Fig. S9† suggest that apical coordination of the primary amino group is possible only in the Δδδδδ-S/Λλλλλ-R enantiomeric pair, and thus, this isomer is suggested to be the major one, leaving the Δδδδδ-R/Λλλλλ-S species as the minor isomer. In the case of this low-abundance isomer, the position of the primary amino group is not suitable for coordination close to the magnetic axis and, therefore, the signals of the primary amino group in Z-spectra are significantly closer (at 10 and 15 ppm, Fig. S7†) to the free water signal. As both protons of the primary amino group have individual signals, their resolution triggered by coordination or by fixing in an intramolecular hydrogen bond system is expected.
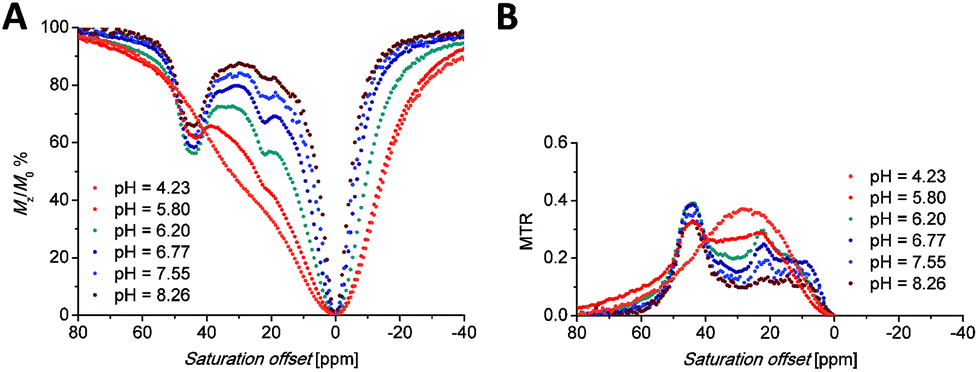 | ||
| Fig. 5 (A) Z-Spectra of an 83 mM aqueous solution of the [Eu(Hndo3aNN)] complex (B0 = 7.05 T, B1 = 21.7 μT (920 Hz), RF presaturation pulse applied for 2 s, 25 °C). (B) Corresponding MTR spectra. | ||
A similar behaviour was observed also for the [Yb(do3aNN)] complex. In an alkaline solution, there are two signals disappearing in D2O, see Fig. S4† – a narrow signal of the proton of the secondary amino group at 35 ppm (this assignment is supported by the similarity of the chemical shift of the analogous 1H NMR signal of [Yb(do3a-ae)], 42 ppm)12 and a very broad signal of NH2 protons at 82–104 ppm (the signals cannot be distinguished at 25 °C, but split at 5 °C, Fig. S5B†). As in the previous case, only the signal attributable to the primary amino group is affected by water presaturation in an alkaline solution, Fig. S4B†. Minor signals of another TSA isomer are also observable in 1H NMR spectra, and minor exchangeable pools of protons were found in CEST experiments at low saturation power (three other peaks in Z-spectra at 17, 26 and 57 ppm, Fig. S7B†).
CEST experiments
Saturation transfer experiments in solutions ranging from slightly acidic to slightly alkaline (pH 5.7–8.3) revealed two signals in the 1H Z-spectra of each complex. These signals are centred at +22.2 and +44.4 ppm for the Eu(III) complex (Fig. 5A) and +35 and +95 ppm for the Yb(III) complex (Fig. 6A).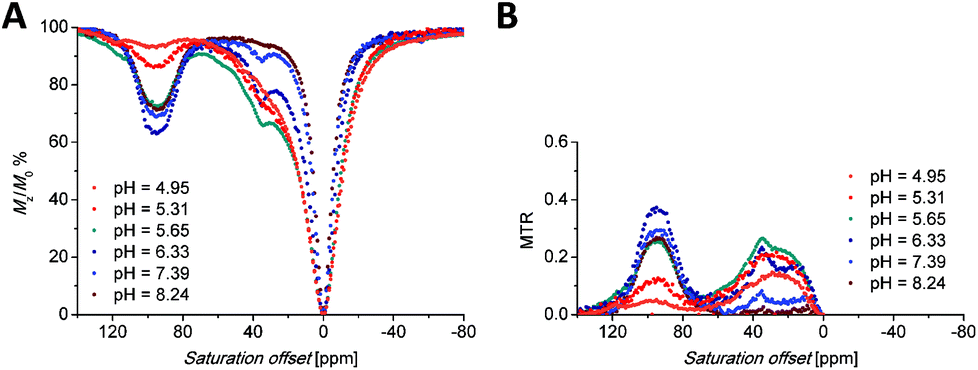 | ||
| Fig. 6 (A) Z-Spectra of an 87 mM aqueous solution of the [Yb(Hndo3aNN)] complex (B0 = 7.05 T, B1 = 21.7 μT (920 Hz), RF presaturation pulse applied for 2 s, 25 °C). (B) Corresponding MTR spectra. | ||
The broad signals at the higher chemical shifts (44.4 and 95 ppm for the Eu(III) and Yb(III) complex, respectively) correspond to the averaged signals of the primary amino group. This broad signal splits into two distinct signals of magnetically non-equivalent protons at a lower intensity of presaturation pulses and at low temperatures (Fig. S7A†), similar to the behaviour of this group found in 1H NMR spectra (Fig. 3, 4, S5 and S6†). The Z-spectra signals with lower chemical shifts (22.2 and 35 ppm for the Eu(III) and Yb(III) complex, respectively) were attributed to the signal of the proton of the secondary amino group. Thus, the Z-spectra of both complexes clearly confirm the presence of proton-exchanging pools that belong to the protons of the primary and secondary amino groups as they were identified in the 1H NMR spectra (see above).
Besides the signals attributable to the major isomer, a set of minor signals (at 10, 15 and 35 ppm, distinguishable especially when low saturation power was applied) appears in the Z-spectra of the [Eu(Hndo3aNN)] complex (Fig. S7A†). At slightly acidic to neutral pH, all three Z-signals are apparent. In contrast, in the alkaline region only the signals at 10 and 15 ppm remain in the Z-spectra, implying their assignment to the primary amino group, with the last one (at 35 ppm) belonging to the secondary amino group. These signals belong to the less abundant isomer with opposite chirality of the coordinated secondary amino group (see discussion of 1H NMR spectra above). A similar set of minor signals (at 17, 26 and 57 ppm) appears also in the Z-spectra of the [Yb(Hndo3aNN)] complex (Fig. S7B†).
The shape of the Z-spectra of the [Ln(Hndo3aNN)] complexes (Ln = Eu, Yb) has significant pH dependence in slightly acidic to neutral regions (Fig. 5A and 6A). To see the differences in Z-spectra more clearly, the magnetization transfer ratio (MTR) spectra were constructed (Fig. 5B and 6B). At pH < 5.5, the CEST effect of the coordinated primary amino group gradually disappears as a consequence of protonation and decoordination of the group. Simultaneously, a new, very broad CEST signal appears centred at ≈25–30 ppm for both complexes. Although partial dissociation of the complexes occurs in this pH region (see above for discussion of thermodynamic properties), the free metal aqua ions as well as the free ligand are CEST-silent (as proved by an independent experiment) and, therefore, these new signals can be attributed to the chemical exchange of the protonated primary amino group of the complex. Such a group – whilst uncoordinated – is still paramagnetically shifted, but not as much as when the group is coordinated. On the other hand, an effective CEST of the secondary amino group was detected for the Eu(III) and Yb(III) complexes in the pH region of ≈5.5–8.5. At higher pH values, the chemical exchange of the NH proton becomes too slow to transfer saturation to bulk water and, thus, the CEST effect of the secondary amino group is not observable (Fig. 5A, 6A and S8†). It is consistent with the 1H NMR spectra of the studied complexes (Fig. 4 and S4†), where the signals of secondary amino groups are observable even in alkaline solutions (pH > 11) (i.e. their chemical exchange with bulk water is slow) and remain unaffected after water presaturation. A graphical representation of the suggested processes giving rise to the peaks in Z-spectra is shown in Scheme 2.
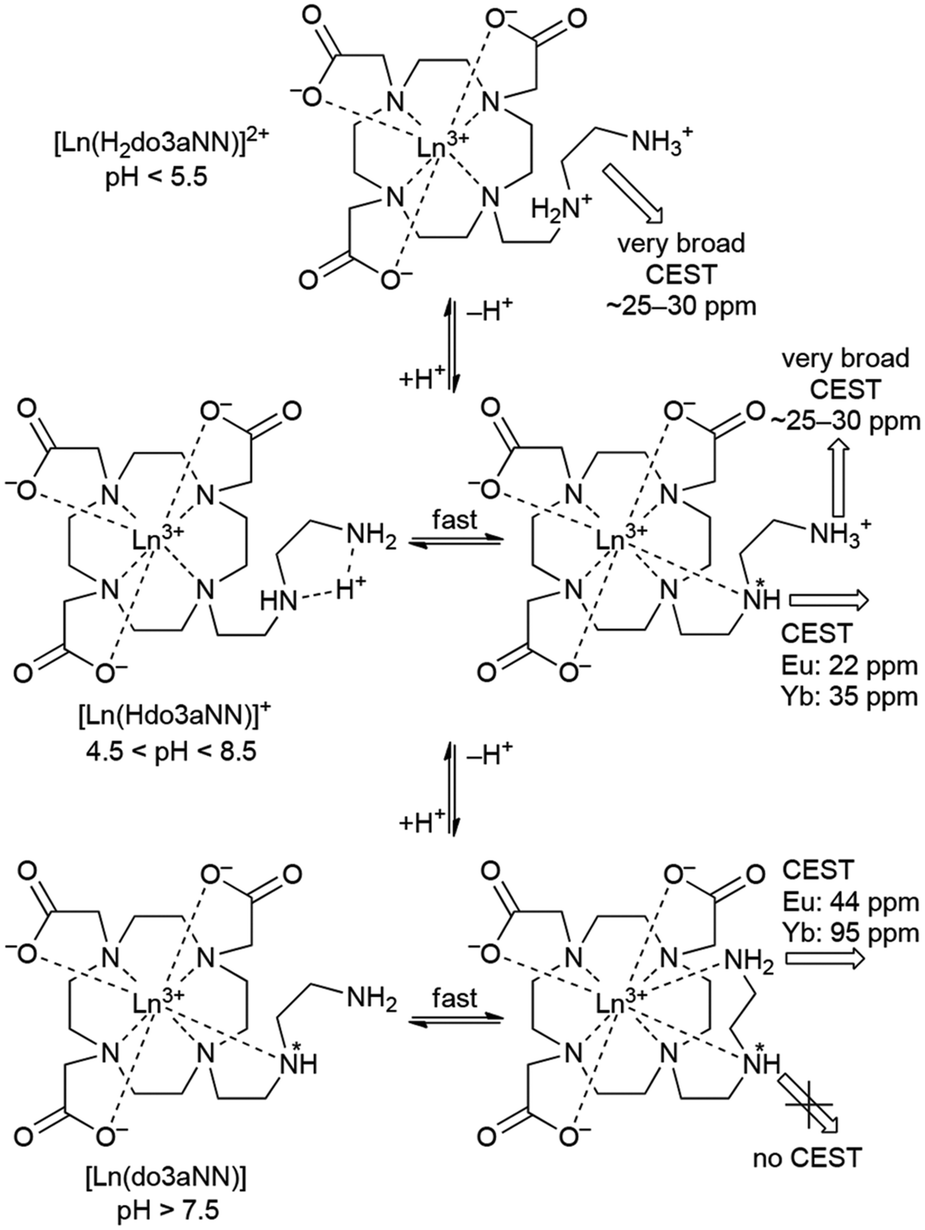 | ||
| Scheme 2 A suggested mechanism of origin of pH-dependent CEST effects on [Ln(Hndo3aNN)] complexes. In hepta/octa-coordinated species, binding of a water molecule(s) to the central ion giving the coordination number to 8–9 is expected, but it is not shown for the sake of clarity. | ||
It is evident that the two pools of exchanging amine protons show different dependences of their CEST effects in the pH range relevant to the physiological conditions. The applicability of the [Eu(Hndo3aNN)] and [Yb(Hndo3aNN)] complexes as pH-sensitive MRI probes was tested for solutions with different pH values (HEPES/MES buffers) and concentrations of the complex (Fig. 7).
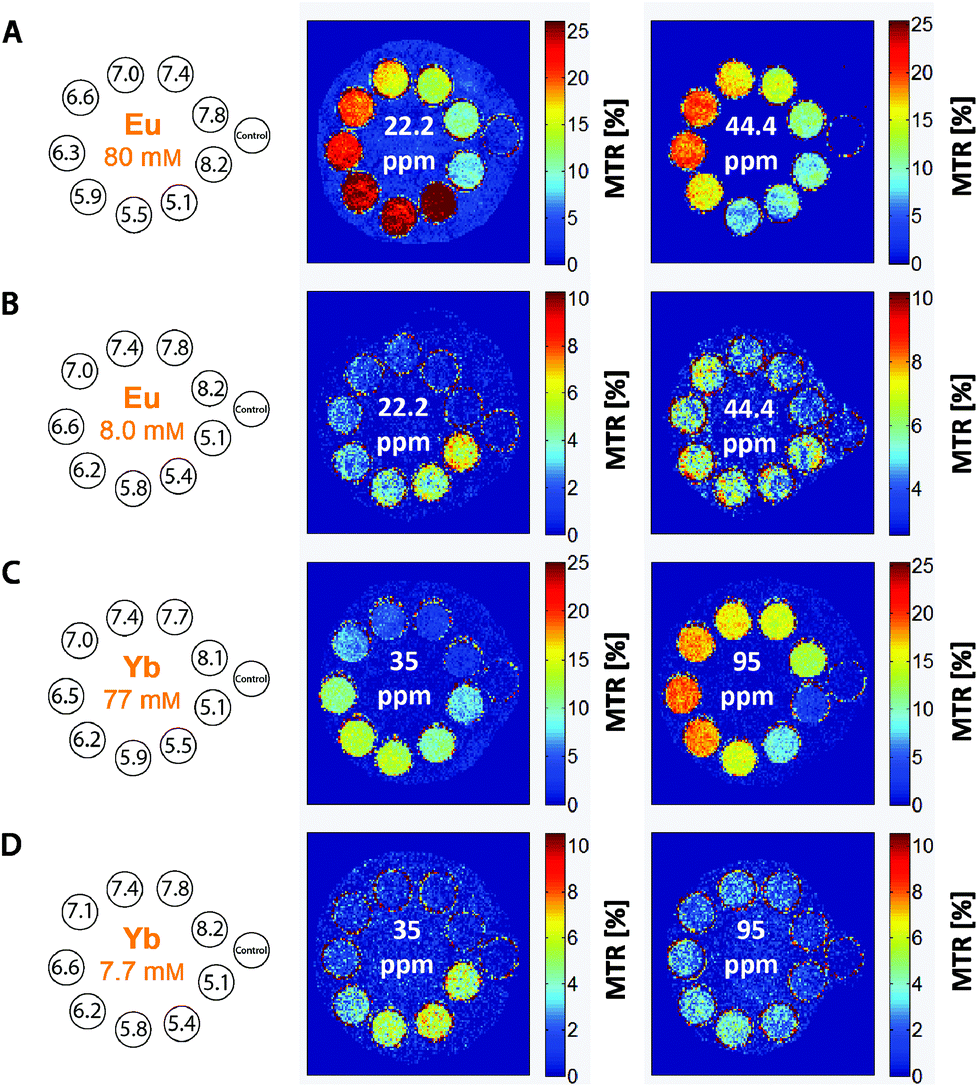 | ||
| Fig. 7 MRI-CEST images of phantoms consisting of one vial containing an aq. solution of MES and HEPES (1:1, 50 mM) as a standard and nine vials containing solutions of (A, B) the [Eu(Hndo3aNN)] or (C, D) [Yb(Hndo3aNN)] complexes in the same buffer with different pH values and concentrations. Experimental conditions: RARE pulse sequence, B0 = 4.7 T, RF presaturation pulse applied for 2 s, T = 298 K, TR = 5 s, TE = 8.9 ms, (B) B1 = 35 μT (1490 Hz), (A, C, D) B1 = 25 μT (1060 Hz). | ||
To define the pH-dependent but concentration-independent function, the ratio of MTR intensities was calculated. However, this ratio can be defined reasonably only for the Yb(III) complex (35/95 ppm), as in the case of the Eu(III) complex there is a significant overlap of the low-shift signal of the coordinated secondary amino group (22.2 ppm) with the new signal appearing in the acidic region (attributable to the protonated and uncoordinated primary amine, Fig. 5B).
To prove the suggested concept of ratiometric pH determination, samples of [Yb(Hndo3aNN)] with different complex concentrations and different pH values were measured by both NMR and MRI techniques. The concentration range used covers about one order of magnitude (7.7–8.7 mM). All calibration curves were very similar (see Fig. 8 and S10†), although standard deviations of the data points from MRI experiments acquired for the low concentration were relatively high due to a high background noise and, thus, a low signal-to-noise ratio was obtained under these conditions. The final curves are compiled in Fig. 8. Although the method has high ESDs with respect to the determination of an exact pH value, the shape of calibration curves enables distinguishing between samples with pH > 7 and those with pH < 6. Such a finding is relevant for the design of contrast agents useful e.g. for distinguishing between normal and hypoxic tissues.
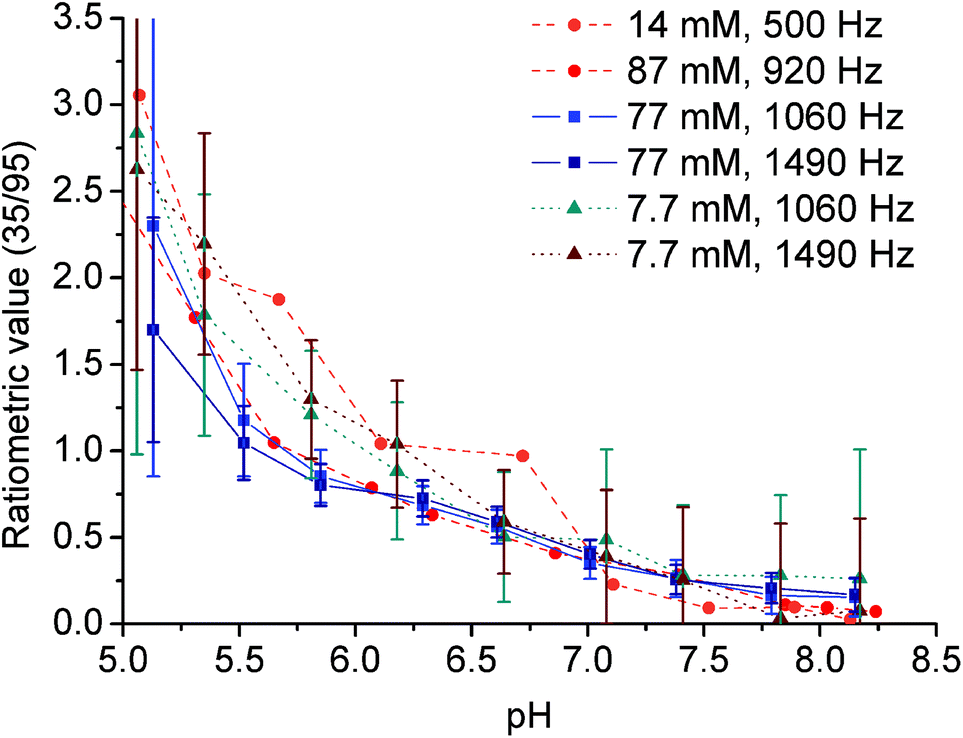 | ||
| Fig. 8 Ratiometric plots of the 7.7–8.7 mM [Yb(Hndo3aNN)] complex, 25 °C; RF presaturation pulse applied for 2 s. Circles: aq. solution, B0 = 7.05 T (NMR), B1 = 500 Hz (11.8 μT) or 920 Hz (21.7 μT). Squares and triangles: 50 mM HEPES–MES, B0 = 4.7 T (MRI), B1 = 1060 Hz (25 μT) or 1490 Hz (35 μT). The ratiometric value (35/95) is the ratio of MTR intensity at 35 ppm to MTR intensity at 95 ppm. | ||
Experimental
Materials and methods
All reagents and solvents were commercially available, had synthetic purity and were used as received. Water used for potentiometric titrations was deionized by using a Milli-Q (Millipore).1,4,7-Tris(tert-butylcarboxymethyl)-1,4,7,10-tetraazacyclododecane hydrobromide (tBu3do3a·HBr) was prepared according to the published procedure.20 THF was dried by the standard method21 and stored over molecular sieves under an argon atmosphere. Anhydrous MeCN and EtOH were from commercial sources.
NMR characterization data (1D: 1H, 13C{1H}; 2D: HSQC, HMBC, 1H–1H COSY) were recorded on a VNMRS300 or Bruker Avance III 600, using 5-mm sample tubes. Chemical shifts are reported as δ values and are given in ppm. Coupling constants J are reported in Hz. Unless stated otherwise, NMR experiments were performed at 25 °C. For samples dissolved in D2O, the pD value was calculated by correcting the pH-electrode reading by +0.4, i.e. pD = pH reading +0.4. For the 1H and 13C NMR measurements of diamagnetic compounds in D2O, tBuOH was used as an internal standard (δH = 1.25, δC = 30.29). For the measurements in CDCl3, TMS was used as an internal standard (δH = 0.00, δC = 0.00). In the case of paramagnetic complexes, chemical shifts were referenced to the water signal of the sample (δH = 0.00) to keep the chemical shift values in 1H NMR spectra consistent with the scale of Z-spectra. The abbreviations s (singlet), t (triplet), q (quartet), m (multiplet) and br (broad) are used in order to express the signal multiplicities. Lanthanide(III) concentrations in solutions were determined by measurement of the bulk magnetic susceptibility (BMS) shifts.22 The ESI-MS spectra were recorded on a Bruker ESQUIRE 3000 spectrometer equipped with an electrospray ion source and ion-trap detection. Measurements were carried out in both the positive and negative modes. UV-Vis solution spectra were recorded using a SPECORD® 50 PLUS (ANALYTIC JENA AG) spectrophotometer at 25 °C in the range of 300–1000 nm with data intervals of 0.2 nm and integration time of 0.04 s. Elemental analysis was performed at the Institute of Macromolecular Chemistry of the Czech Academy of Sciences (Prague, Czech Republic).
Synthesis
1H NMR (600 MHz, CDCl3): δ 1.15–1.19 (6H, m, CH2CH3); 3.28 (2H, br, NHCH2); 3.36 (4H, br, CH2NCH2); 3.63–3.69 (2H, br, CH2OH); 4.02–4.08 (4H, m, OCH2).
13C{1H} NMR (150.9 MHz, CDCl3): δ 14.61, 14.65 (2C, CH2CH3); 40.03, 40.18 (1C, NHCH2); 48.45, 48.74 (1C, CH2); 51.11, 51.51 (1C, CH2); 60.91 (1C, OCH2); 61.15, 61.40 (1C, CH2OH); 61.66 (1C, OCH2); 157.30 (2C, CO). All four backbone carbon atoms show two 13C NMR signals due to rigid conformations of the molecule locked by different orientations of the amide groups.
MS-ESI: (+) 270.8 ([M + Na]+, calcd 271.1).
:CH2Cl2 (1:9). Fractions containing the pure product 3 (as checked by NMR) were combined and evaporated to give compound 3 (2.24 g, 87%) as a colourless oil.
1H NMR (600 MHz, CDCl3): δ 1.16–1.18 (3H, br, CH2CH3); δ 1.22 (3H, t, 3JHH = 7.2, CH2CH3); 3.28 (2H, br, CH2); 3.39–3.45 (4H, br, CH2); 3.58 (2H, br, CH2Br); 4.04–4.08 (2H, m, CH2CH3); 4.10 (2H, q, 3JHH = 7.2, CH2CH3).
13C{1H} NMR (150.9 MHz, CDCl3): δ 14.78, 14.84 (2C, CH2CH3); 29.28, 29.53 (1C, CH2Br); 40.04 (1C, NHCH2); 48.11 (1C, CH2); 49.74, 50.41 (1C, CH2); 61.05, 62.02 (2C, OCH2); 156.41, 157.09 (2C, CO). Two of the backbone carbon atoms show two 13C NMR signals due to rigid conformations of the molecule locked by different orientations of the amide groups.
MS-ESI: (+) 745.3 ([M + H]+, calcd 745.5); 767.2 ([M + Na]+; calcd 767.5).
:1, 30 ml). The resulting solution was refluxed for 18 h and evaporated in a rotary evaporator. The oily residue was dissolved in a small amount of distilled water and evaporated (this procedure was then repeated three more times) to produce a yellow oil (3.10 g) containing compound 5, which was used in the next step without purification.
MS-ESI: (+) 577.0 ([M + H]+, calcd 577.3). (−) 574.9 ([M − H]−; calcd 575.3).
:MeOH mixture (1:5, v:v, ≈5 ml) and overlaid with EtOH (≈5 ml) and the mixture was left to stand for 2 d. After this period, the solid product was filtered off and dried under vacuum to yield 6·2.5H2O (900 mg, 42% based on tBu3do3a) as a white powder.
1H NMR (600 MHz, D2O, pD = 5.88): δ 3.05 (2H, br, CH2CH2NCO); 3.16 (4H, br, (CH2)2NCH2CH2NCO); 3.20–3.29 (4H, m, (CH2)2NCH2CO); 3.33–3.44 (12H, br, CH2CH2NCO, CH2CH2NH, (CH2)2NCH2CO); 3.52 (2H, br s, CH2CO); 3.57 (2H, t, 3JHH = 8.1, CH2NH); 3.70 (4H, br s, CH2CO).
13C{1H} NMR (150.9 MHz, D2O, pD = 5.88): δ 38.69 (1C, NCH2CH2NH); 39.45 (1C, CH2CH2NCO); 46.05 (1C, CH2NH); 49.05 (2C, (CH2)2NCH2CH2NCO); 49.95 (2C, (CH2)2NCH2CO); 50.17 (1C, CH2CH2NCO); 51.14 (2C, (CH2)2NCH2CO); 51.79 (2C, (CH2)2NCH2CO); 56.21 (1C, CH2CO); 57.43 (2C, CH2CO); 165.09 (1C, NCONH); 173.25 (2C, CH2CO); 175.78 (1C, CH2CO).
MS-ESI: (+) 480.9 ([M + Na]+, calcd 481.2). (−) 456.8 ([M − H]−, calcd 457.3).
Elemental analysis: found (calcd for 6·2.5H2O, C19H39N6O9.5, Mr = 503.6) C: 45.30 (45.32), H: 7.85 (7.81), N: 16.12 (16.69).
:1) and the resulting solution was stirred for 7 d at 95 °C and evaporated in a rotary evaporator. The oily residue was dissolved in a small amount of distilled water and evaporated to dryness, leaving a glassy solid, which was triturated in dry THF overnight. Next, the product was collected by filtration, and stored in a desiccator (P2O5) to give H3do3aNN in the form of a hydrochloride hydrate (500 mg, 95%) as a white powder.
Mother liquors after crystallization of the intermediate 6 can be also used for the preparation of the title ligand. After acid hydrolysis of impure 6, the crude H3do3aNN was converted to its ammonium salt by chromatography on a strong cation exchanger (Dowex 50, 50–100 mesh, H+-form). Acids were eluted by water and the crude product was collected by 5% aq. ammonia. After evaporation of volatiles, the oily residue was dissolved in water and poured onto a column of a weak cation exchanger (Amberlite CG50, 200–400 mesh, H+-form). Impurities were eluted with water and the H3do3aNN compound was collected by 3% aq. HCl. Fractions containing the product were combined and evaporated to dryness leaving a glassy solid, which was triturated as described above.
1H NMR (600 MHz, D2O, pD = 6.07, 55 °C, Fig. S11†): δ 2.85–2.87 (4H, m, macrocyclic CH2); 3.01–3.09 (6H, br m, NCH2CH2NH, macrocyclic CH2); 3.11 (2H, s, CH2CO2); 3.19 (2H, t, 3JHH = 5.1, NCH2CH2NH); 3.30–3.36 (2H, br m, macrocyclic CH2); 3.42–3.45 (2H, br m, macrocyclic CH2); 3.47–3.49 (2H, br t, NHCH2CH2NH2); 3.54–3.56 (2H, br t, NHCH2CH2NH2); 3.57–3.61 (2H, m, macrocyclic CH2); 3.74–3.80 (2H, br, macrocyclic CH2); 3.92 (2H, AB-multiplet, CH2CO2).
13C{1H} NMR (150.9 MHz, D2O, pD = 6.07, 55 °C, Fig. S12†): δ 37.07 (1C, NHCH2CH2NH2); 46.04 (1C, NCH2CH2NH); 46.99 (1C, NHCH2CH2NH2); 48.84 (4C, macrocyclic CH2); 50.45 (1C, NCH2CH2NH); 51.23 (2C, macrocyclic CH2); 53.30 (2C, macrocyclic CH2); 55.55 (1C, CH2CO2); 58.34 (2C, CH2CO2); 171.24 (2C, CH2CO2); 179.41 (1C, CH2CO2).
MS-ESI: (+) 432.9 ([M + H]+, calcd 433.3). (−) 430.8 ([M − H]−, calcd 431.3).
Elemental analysis: found (calcd for H3do3aNN·5HCl·1.5H2O, C18H44Cl5N6O7.5, Mr = 641.8) C: 34.18 (33.68), H: 7.13 (6.91), N: 12.75 (13.09), Cl: 27.57 (27.62).
All the prepared samples were checked using a xylenol orange test (acetate buffer, pH = 5.7) to exclude the presence of free Ln(III) ions. The exact concentration of the Ln(III) complexes in the solution was determined using Evans's method.22
[Eu(Hndo3aNN)]
MS-ESI: (+) 588.9 ([M + Li]+, calcd 589.2). (−) 616.7 ([M + Cl]−, calcd 617.2).
[Yb(Hndo3aNN)]
MS-ESI: (+) 609.9 ([M + Li]+, calcd 610.2). (−) 637.7 ([M + Cl]−, calcd 638.2).
PARACEST experiments
All Z-spectra were recorded using a VNMRS300 spectrometer operating at 299.9 MHz (B0 = 7.05 T); 5 mm sample tubes and a coaxial capillary with D2O and tBuOH as an external standard were used. Solutions of the complexes for PARACEST NMR experiments were prepared in pure water with the pH adjusted using aq. HCl/LiOH solutions and had concentrations in the range of 14–87 mM. Standard pulse sequences for presaturation experiments were used. Saturation offsets were set using the array function (increment 200–250 Hz). Data from the PARACEST experiments were plotted as the dependence of normalized water signal intensity (Mz/M0%) on saturation offset. Here, M0 represents the magnetization (i.e. intensity) of the water signal without RF saturation and Mz corresponds to the water signal when a presaturation pulse is applied. Other experimental parameters are specified in the figure captions.The magnetization transfer ratio (MTR) was calculated using MTR = MΔω/M0 – M–Δω/M0 in which M±Δω is the magnetization (i.e. intensity) of the water signal with the use of a presaturation frequency ±Δω away from the bulk water signal.
MRI PARACEST images were measured with a phantom consisting of one vial containing an aqueous solution of buffers [a mixture of 0.025 M 2-(N-morpholino)ethanesulfonic acid (MES) and 0.025 M 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)] as a standard and nine vials containing solutions of the Eu3+ or Yb3+-complexes dissolved in buffers (0.025 M MES and 0.025 M HEPES) with different pH values and concentrations. The innocence of the chosen buffers was confirmed by silence of pure buffer solutions in the CEST experiment (i.e., no signal in the Z-spectra of the pure buffers was found). All MRI PARACEST images were acquired on a 4.7 T scanner (Bruker BioSpec, Germany) using a modified spin-echo sequence (Rapid Acquisition with Refocused Echoes – RARE). Experimental conditions: repetition time (TR) = 5000 ms, echo time (TE) = 8.9 ms, resolution 0.35 × 0.35 × 2 mm3, turbo factor = 4. Other experimental parameters are specified in the figure captions. For all MR experiments, a resonator coil with an inner diameter of 70 mm was used. MTR maps were normalized to the signal acquired at a frequency offset –Δω and reconstructed from a manually outlined region of interest on a pixelwise basis using a custom script written in Matlab (Mathworks, Natick, MA, USA). MTR maps were visualized on a false-colour scale in percentage units.
Potentiometry
Potentiometric titrations23 were carried out in a thermostatted vessel at 25.0 ± 0.1 °C at a constant ionic strength I(NMe4Cl) = 0.1 M. The measurements were taken with an HCl excess added to the initial mixture and the mixtures were titrated with a stock NMe4OH solution. An inert atmosphere was maintained by constant passage of argon saturated with water vapour. The ligand concentration in the titration vessel was ≈0.004 M.Ligand protonation constants were determined by standard potentiometric titrations performed in the pH range of 1.6–12.2 (80 points per titration, titrations were carried out four times).
In the cases of the Ln(III)–H3do3aNN systems, the equilibria were established slowly and, therefore, the out-of-cell technique was used in the pH range of 1.6–7.2 (two titrations per system, 25 points per titration). The metal:ligand ratio was 1:1 in all cases. The waiting time was 7 weeks. Then, the potential at each titration point (tube) was determined with a freshly calibrated electrode.
Pre-formed complexes for the determination of their protonation constants were prepared in the following way: in an ampoule, equimolar molar amounts of the ligand and metal stock solutions were mixed and a calculated amount (based on the out-of-cell titration data) of a stock solution of NMe4OH was gradually added to reach pH ≈ 7, which corresponds to full complexation according to the out-of-cell titration. Ampoules were flame-sealed and left at 55 °C for 3 d. Aliquots were taken from the final solution, a defined amount of an HCl stock solution was added into these samples and the mixtures were immediately titrated by an NMe4OH stock solution in a way analogous to the procedure described above for the determination of ligand protonation constants in the pH range of 2.3–12.1. The initial volumes were ≈5 cm3 for the conventional titrations and ≈1 cm3 for the out-of-cell ones, respectively.
The constants with their standard deviations were calculated by using the OPIUM program package.24 Overall protonation constants are defined as βh = [HhL]/{[H]h·[L]}, and they can be transferred to the consecutive protonation constants logKP by logKP(HhL) = logβh − logβ(h−1); it should be noted that logKP = pKA of the corresponding protonated species HhL. The overall stability constants βhlm are concentration constants defined as βhlm = [HhLlMm]/{[H]h·[L]l·[M]m}. The water ion product used in the calculations was pKw = 13.81. Stability constants of metal hydroxido complexes were taken from the literature.14 In the text, pH means −log[H+]. The best fits of experimental data are shown in Fig. S13–S15† and the results are compiled in Tables S1–S3.†
Conclusions
The present study revealed significant pH dependence of the Chemical Exchange Saturation Transfer (CEST) effect of selected Ln(III) complexes with the novel macrocyclic ligand H3do3aNN containing a linear diamine pendant arm. The pH dependence is substantial in the pH range relevant for biological systems (pH ≈ 5.5–8.5). Based on these findings, we have shown that the magnetization transfer ratio of CEST signals of the complexes can be used for pH determination by MRI, and it is independent of the concentration of the probes.Unfortunately, the studied complexes are not fully kinetically inert in acidic solutions and slowly release the free metal ions, which excludes their direct use in medical applications. However, the study brings proof-of-principle of possibility to use a linear diaminic fragment for pH determination using MRI ratiometry.
Acknowledgements
This work was supported by the Czech Science Foundation (P207-11-1437), by the Grant Agency of the Charles University (no. 110213) and by the project of the Ministry of Health, Czech Republic, for the development of a research organization IN00023001 (Institutional support, Institute for Clinical and Experimental Medicine). We thank Z. Böhmová for potentiometric measurements, I. Císařová for collection of X-ray diffraction data and nH. Blahut for his help with some NMR experiments and for Fig. S9.†References
- The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging, ed. A. Merbach, L. Helm and É. Tóth, 2nd edn, Wiley, Chichester, United Kingdom, 2013 Search PubMed.
- (a) P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293–2352 CrossRef CAS PubMed; (b) P. Hermann, J. Kotek, V. Kubíček and I. Lukeš, Dalton Trans., 2008, 3027–3047 RSC; (c) C. F. G. C. Geraldes and S. Laurent, Contrast Media Mol. Imaging, 2009, 4, 1–23 CrossRef CAS PubMed.
- K. M. Ward, A. H. Aletras and R. S. Balaban, J. Magn. Reson., 2000, 143, 7987 CrossRef PubMed.
- (a) J. Zhou and P. C. M. van Zijl, Prog. Nucl. Magn. Reson. Spectrosc., 2006, 48, 109–136 CrossRef CAS; (b) Y. Wu, M. Evbuomwan, M. Melendez, A. Opina and A. D. Sherry, Future Med. Chem., 2010, 2, 351–366 CrossRef CAS PubMed; (c) E. Terreno, D. D. Castelli and S. Aime, Contrast Media Mol. Imaging, 2010, 5, 78–98 CAS; (d) P. C. M. van Zijl and N. N. Yadav, Magn. Reson. Med., 2011, 65, 927–948 CrossRef CAS PubMed; (e) G. Liu, X. Song, K. W. Y. Chan and M. T. McMahon, NMR Biomed., 2013, 26, 810–828 CrossRef CAS PubMed.
- S. Zhang, M. Merritt, D. E. Woessner, R. E. Lenkinski and A. D. Sherry, Acc. Chem. Res., 2003, 36, 783–790 CrossRef CAS PubMed.
- (a) M. Woods, D. E. Woessner and A. D. Sherry, Chem. Soc. Rev., 2006, 35, 500–511 RSC; (b) S. Viswanathan, Z. Kovács, K. N. Green, S. J. Ratnakar and A. D. Sherry, Chem. Rev., 2010, 110, 2960–3018 CrossRef CAS PubMed; (c) T. C. Soesbe, Y. Wu and A. D. Sherry, NMR Biomed., 2013, 26, 829–838 CrossRef CAS PubMed.
- (a) M. Woods, D. E. Woessner, R. Zhao, A. Pasha, M.-Y. Yang, C.-H. Huang, O. Vasalitiy, J. R. Morrow and A. D. Sherry, J. Am. Chem. Soc., 2006, 128, 10155–10162 CrossRef CAS PubMed; (b) C.-H. Huang and J. R. Morrow, Inorg. Chem., 2009, 48, 7237–7243 CrossRef CAS PubMed.
- (a) S. J. Dorazio, P. B. Tsitovich, K. E. Sisters, J. A. Spernyak and J. R. Morrow, J. Am. Chem. Soc., 2011, 133, 14154–14156 CrossRef CAS PubMed; (b) A. O. Olatunde, S. J. Dorazio, J. A. Spernyak and J. R. Morrow, J. Am. Chem. Soc., 2012, 134, 18503–18505 CrossRef CAS PubMed; (c) S. J. Dorazio, A. O. Olatunde, P. B. Tsitovich and J. R. Morrow, J. Biol. Inorg. Chem., 2014, 19, 191–205 CrossRef CAS PubMed; (d) A. O. Olatunde, J. M. Cox, D. Daddarion, J. A. Spernyak, J. B. Benedict and J. R. Morrow, Inorg. Chem., 2014, 53, 8311–8321 CrossRef CAS PubMed; (e) S. J. Dorazio, A. O. Olatunde, J. A. Spernyak and J. R. Morrow, Chem. Commun., 2013, 49, 10025–10027 RSC; (f) P. B. Tsitovich and J. R. Morrow, Inorg. Chim. Acta, 2012, 393, 3–11 CrossRef CAS; (g) S. J. Dorazio, P. B. Tsitovich, S. A. Gardina and J. R. Morrow, J. Inorg. Biochem., 2012, 117, 212–219 CrossRef CAS PubMed.
- (a) S. Aime, C. Carrera, D. D. Castelli, S. G. Crich and E. Terreno, Angew. Chem., Int. Ed., 2005, 44, 1813–1815 CrossRef CAS PubMed; (b) G. Ferrauto, D. D. Castelli, E. Terreno and S. Aime, Magn. Reson. Med., 2013, 69, 1703–1711 CrossRef CAS PubMed; (c) E. Gianolio, R. Stefania, E. D. Gregorio and S. Aime, Eur. J. Inorg. Chem., 2012, 1934–1944 CrossRef CAS.
- D. V. Hingorani, A. S. Bernstein and M. D. Pagel, Contrast Media Mol. Imaging, 2015, 10, 245–265 CrossRef CAS PubMed.
- (a) D. D. Castelli, E. Terreno and S. Aime, Angew. Chem., 2011, 123, 1838–1840 CrossRef; (b) D. D. Castelli, G. Ferrauto, J. C. Cutrin, E. Terreno and S. Aime, Magn. Reson. Med., 2014, 71, 326–332 CrossRef PubMed; (c) S. Aime, A. Barge, D. D. Castelli, F. Fedeli, A. Mortillaro, F. U. Nielsen and E. Terreno, Magn. Reson. Med., 2002, 47, 639–648 CrossRef CAS PubMed; (d) Y. Wu, T. C. Soesbe, G. E. Kiefer, P. Zhao and A. D. Sherry, J. Am. Chem. Soc., 2010, 132, 14002–14003 CrossRef CAS PubMed; (e) D. L. Longo, P. Z. Sun, L. Concolino, F. C. Michelotti, F. Uggeri and S. Aime, J. Am. Chem. Soc., 2014, 136, 14333–14336 CrossRef CAS PubMed; (f) D. L. Longo, W. Dastrú, G. Diglio, J. Keupp, S. Langereis, S. Lanzardo, S. Prestigio, O. Steinbach, E. Terreno, F. Uggeri and S. Aime, Magn. Reson. Med., 2011, 65, 202–211 CrossRef CAS PubMed; (g) V. R. Sheth, Y. Li, L. Q. Chen, C. M. Howison, C. A. Flask and M. D. Pagel, Magn. Reson. Med., 2012, 67, 760–768 CrossRef PubMed; (h) G. Liu, Y. Li, V. R. Sheth and M. D. Pagel, Mol. Imaging, 2012, 11, 47–57 CAS; (i) N. McVicar, A. X. Li, M. Suchý, R. H. E. Hudson, R. S. Menon and R. Bartha, Magn. Reson. Med., 2013, 70, 1016–1025 CrossRef CAS PubMed.
- T. Krchová, J. Kotek, D. Jirák, J. Havlíčková, I. Císařová and P. Hermann, Dalton Trans., 2013, 42, 15735–15747 RSC.
- A. Bianchi, L. Calabi, C. Giorgi, P. Losi, P. Mariani, P. Paoli, P. Rossi, B. Valtancoli and M. Virtuani, J. Chem. Soc., Dalton Trans., 2000, 697–705 RSC.
- A. E. Martell, R. M. Smith and R. J. Motekaitis, NIST Critically Selected Stability Constants of Metal Complexes, Version 7, Texas A&M University, College Station, TX, 2003 Search PubMed.
- S. Aime, M. Botta and G. Ermondi, Inorg. Chem., 1992, 31, 4291–4299 CrossRef CAS.
- S. Aime, M. Botta, M. Fasano, M. P. M. Marques, C. F. G. C. Geraldes, D. Pubanz and A. E. Merbach, Inorg. Chem., 1997, 36, 2059–2068 CrossRef CAS PubMed.
- (a) T. Vitha, V. Kubíček, J. Kotek, P. Hermann, L. V. Elst, R. N. Muller, I. Lukeš and J. A. Peters, Dalton Trans., 2009, 3204–3214 RSC; (b) M. Polášek, J. Kotek, P. Hermann, I. Císařová, K. Binnemans and I. Lukeš, Inorg. Chem., 2009, 48, 455–465 CrossRef PubMed.
- (a) S. Viswanathan, S. J. Ratnakar, K. N. Green, Z. Kovács, L. M. De Leónrodríguez and A. D. Sherry, Angew. Chem., Int. Ed., 2009, 48, 9330–9333 CrossRef CAS PubMed; (b) S. J. Ratnakar, M. Woods, A. J. M. Lubag, Z. Kovács and A. D. Sherry, J. Am. Chem. Soc., 2008, 130, 6–7 CrossRef CAS PubMed.
- M. Woods, A. Pasha, P. Zhao, G. Tircso, S. Chowdhury, G. Kiefer, D. E. Woessner and A. D. Sherry, Dalton Trans., 2011, 40, 6759–6764 RSC.
- B. Jagadish, G. L. Brickert-Albrecht, G. S. Nichol, E. A. Mash and N. Raghunand, Tetrahedron Lett., 2011, 52, 2058–2061 CrossRef CAS PubMed.
- D. D. Perrin and W. L. F. Armarego, Purification of Laboratory Chemicals, Pergamon Press, Oxford, 3rd edn, 1988 Search PubMed.
- D. M. Corsi, C. Platas-Iglesias, H. van Bekkum and J. A. Peters, Magn. Reson. Chem., 2001, 39, 723–726 CrossRef CAS.
- (a) P. Táborský, P. Lubal, J. Havel, J. Kotek, P. Hermann and I. Lukeš, Collect. Czech. Chem. Commun., 2005, 70, 1909–1942 CrossRef; (b) M. Försterová, I. Svobodová, P. Lubal, P. Táborský, J. Kotek, P. Hermann and I. Lukeš, Dalton Trans., 2007, 535–549 RSC.
- (a) M. Kývala and I. Lukeš, International Conference Chemometrics 1995 (Pardubice, Czech Republic), July 3–7, 1995, p. 63; (b) M. Kývala, P. Lubal and I. Lukeš, IX. Solution Equilibria Analysis with the OPIUM Computer Program; Spanish-Italian and Mediterranean Congress on Thermodynamics of Metal Complexes (SIMEC 98) (Girona, Spain), June 2–5, 1998, The full version of the OPIUM program is available (free of charge) on http://www.natur.cuni.cz/~kyvala/opium.html Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: X-ray diffraction data, temperature- and pH-dependence of NMR spectra of studied compounds, additional Z-spectra and magnetization transfer ratio spectra of studied complexes, protonation and stability constants of studied ligand and its complexes, tentative structures of isomeric complex species with different protonation. CCDC 1430249. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt04443j |
| ‡ Such a formula is used when more species differing in protonation are present in a solution. For the range of n refer to the distribution diagram shown in Fig. 2. |
| This journal is © The Royal Society of Chemistry 2016 |