
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The synthesis and unexpected solution chemistry of thermochromic carborane-containing osmium half-sandwich complexes†
Anaïs
Pitto-Barry
 ,
Amy
South
,
Alison
Rodger
and
Nicolas P. E.
Barry
*
,
Amy
South
,
Alison
Rodger
and
Nicolas P. E.
Barry
*
Department of Chemistry, University of Warwick, Coventry CV4 7AL, UK. E-mail: N.Barry@warwick.ac.uk
First published on 24th December 2015
Abstract
The functionalisation of the 16-electron complex [Os(η6-p-cymene)(1,2-dicarba-closo-dodecarborane-1,2-dithiolato)] (1) with a series of Lewis bases to give the 18-electron complexes of general formula [Os(η6-p-cymene)(1,2-dicarba-closo-dodecarborane-1,2-dithiolato)(L)] (L = pyridine (2), 4-dimethylaminopyridine (3), 4-cyanopyridine (4), 4-methoxypyridine (5), pyrazine (6), pyridazine (7), 4,4′-bipyridine (8) and triphenylphosphine (9)) is reported. All 18-electron complexes are in equilibrium in solution with the 16-electron precursor, and thermochromic properties are observed in some cases (2, 3, 5, 8, and 9). The binding constants and Gibbs free energies of the equilibria are determined using UV-visible titrations and their stabilities investigated. Synthetic routes for forcing the formation of the 18-electron species are proposed, and analytical methods to characterise the equilibria are described.
Introduction
Half-sandwich complexes generally follow the 18-electron rule, although some stable 16-electron (16-e) complexes have been isolated.1,2 The latter are generally coordinatively unsaturated leading to potential applications in catalysis and as precursors for 18-electron (18-e) complexes.3 We recently reported the solution chemistry4,5 of the electron-deficient half-sandwich ruthenium complex [Ru(η6-p-cymene)(1,2-dicarba-closo-dodecarborane-1,2-dithiolato)]. In this 16-e complex, first synthesised by Herberhold and co-workers in 2000,6 the carborane ligand is bound to the ruthenium atom via sulfur atoms.7–12 This complex is stable owing to the steric hindrance of the bulky carborane, which prevents the dimerisation of the compound and the formation of a more electronic favoured 18-e species. We have recently developed a strong interest in the osmium analogue [Os(η6-p-cym)(1,2-dicarba-closo-dodecarborane-1,2-dithiolato)] (1) of this ruthenium 16-e complex, particularly with regards to applications in biology13–16 and in nanotechnology.17–19 Osmium, the heavier congener of ruthenium and a third row transition metal, commonly exhibits slower kinetics than ruthenium, and is often considered to be relatively inert. It has been shown that it is possible to tune the chemical reactivity of osmium complexes through understanding their solution chemistry.20–24 Similarly to the ruthenium analogue, the 16-e Os complex 1 is highly stable and does not dimerise to form an 18-e species. Currently, there is a lack of knowledge of the chemistry of such electron-deficient Os organometallics, and understanding the reactivity of complex 1 towards ligands is important not only in order to synthesise more structurally-sophisticated carborane-containing Os complexes but also to tune their reactivity by addition of suitable ligands. There is also a need for the development of analytical techniques in order to assess the functionalisation of complex 1. In this report, we describe the functionalisation of 1 by addition of a series of pyridine and phosphine derivatives acting as Lewis bases. Complexes with the general formula [Os(η6-p-cym)(1,2-dicarba-closo-dodecarborane-1,2-dithiolato)(L)] (where L = pyridine (2), 4-dimethylaminopyridine (3), 4-cyanopyridine (4), 4-methoxypyridine (5), pyrazine (6), pyridazine (7), 4,4-bipyridine (8) and triphenylphosphine (9)) have been prepared (Scheme 1) and characterised by UV-visible and 1H NMR spectroscopies. Their unusual chemistry in solution has been studied, and routes for forcing the formation of 18-e species are suggested.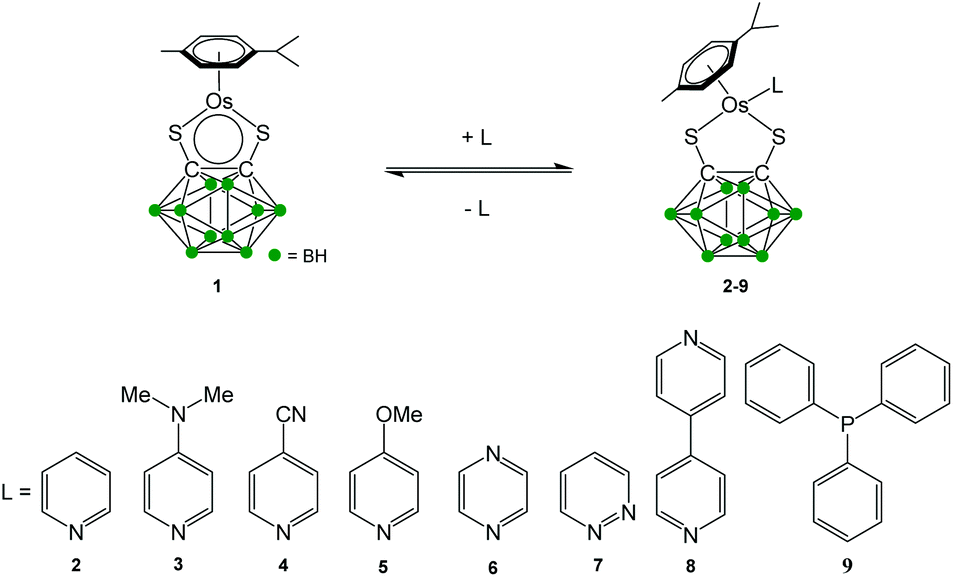 | ||
| Scheme 1 Molecular structures of the 16- and 18-electron complexes investigated. | ||
Results
The new 18-e complexes 2–9 were prepared following a one-step strategy using the 16-e complex [Os(η6-p-cym)(1,2-dicarba-closo-dodecarborane-1,2-dithiolato)] (1)6 as precursor. Addition of 1 mol equiv. of ligand to complex 1 in dry dichloromethane at ambient temperature for six hours gives the corresponding 18-e complex (Scheme 1); the binding of the ligands occurs via coordination of donor atoms N and P to the metal centre, as with the ruthenium analogue of 1.4 Complexes 2–9 were characterised by 1H NMR spectroscopy (the aromatic region of the 1H NMR spectra of isolated complexes 1–9 in CDCl3 at ambient temperature is shown in Fig. 1, along with the assigned 1H NMR spectrum of complex 2). The shifts and broadenings of the peaks suggest the presence of equilibria between 16- and 18-electron species at ambient temperature in CDCl3, with particularly dramatic upfield shifts and broadening of the aromatic arene and ligand resonances in the 1H NMR spectra of complexes 2, 3, 5, 8. The presence of such an equilibrium in solution is consistent with previously reported results on the ruthenium analogue of 1,4 and is presumably due to the high stability of the 16-e metal species. The BH proton signals of the carborane cluster in the region 0.0 to 3.0 ppm are broad and unresolved owing to the quadrupolar nature of 10B and 11B, and due to chemical exchange between 16- and 18-electron species.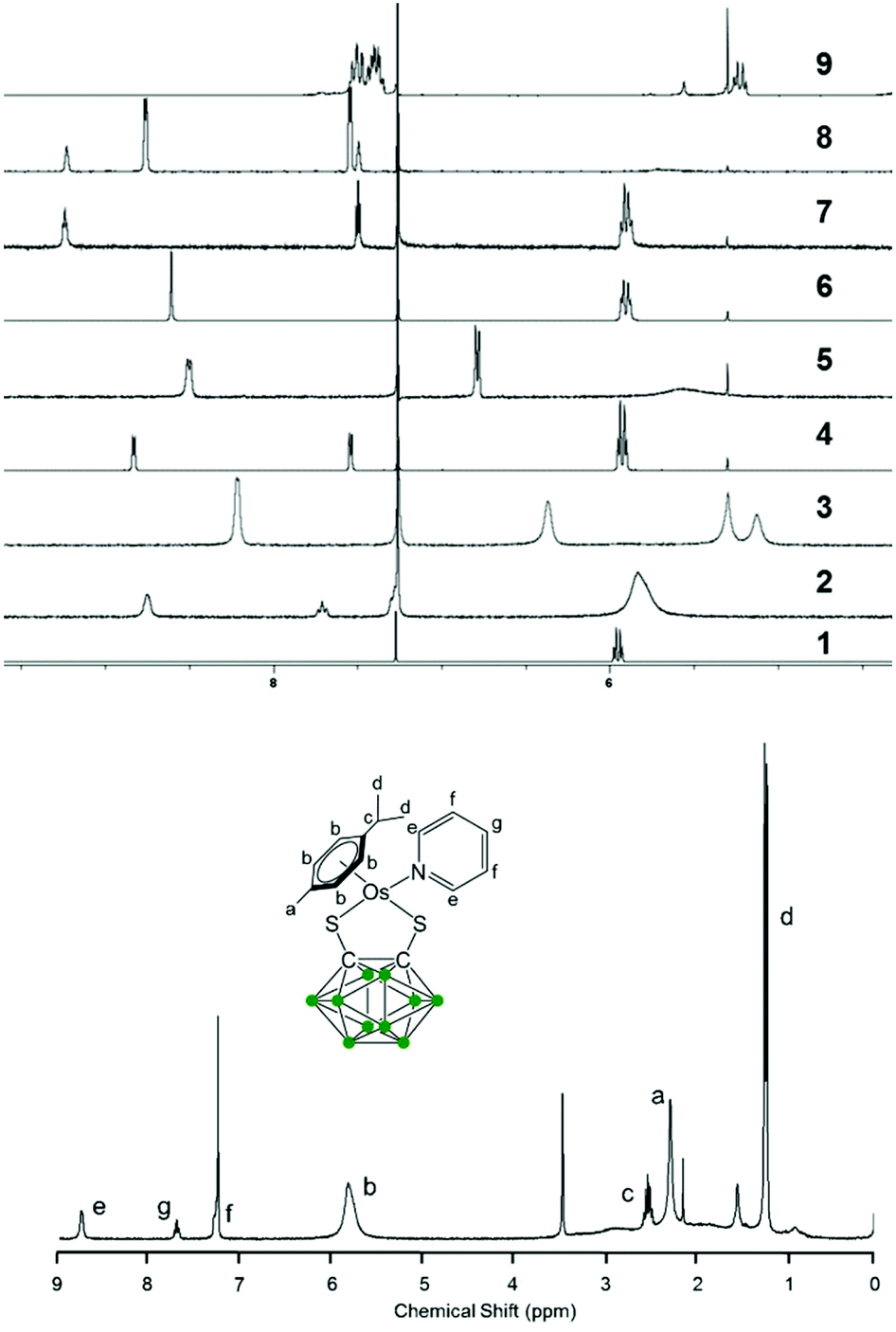 | ||
| Fig. 1 Aromatic region of the 1H NMR spectra of 5 mM solutions of complexes 1–9 in CDCl3, 298 K, and assigned 1H NMR spectrum of complex 2. | ||
Variable temperature 1H NMR spectra were measured from 228 K to 298 K in CDCl3 for complex 3 (Fig. 2). The peaks for the p-cymene ligand become sharper as the temperature decreases, whilst the dimethylaminopyridine (DMAP) resonances are upfield shifted upon coordination compared to the free ligand, and become sharper and more defined as the temperature decreases. The peaks shift even further upfield as the temperature decreases, indicating that there is more DMAP coordinating to the metal centre at lower temperatures and that the equilibrium shifts towards the 18-e complex as the temperature decreases.
 | ||
| Fig. 2 Low temperature 1H NMR spectra of the aromatic region in CDCl3 for complex 3 at 298 K (c), 278 K (d), 258 K (e), and 228 K (f). The aromatic regions of the 1H NMR spectra of DMAP (a) and complex 1 (b) at 298 K are shown as a reference. | ||
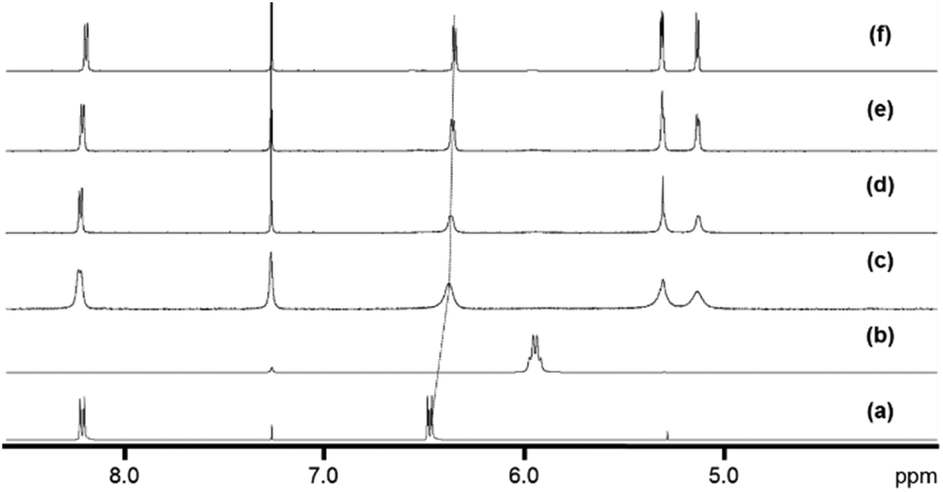
UV-visible absorption spectra were measured for complexes 2–9 in dichloromethane solutions (10−4 M) at 298 K (Fig. 3; Fig. S1† for the spectrum of complex 1). All spectra present two bands, one at 520 nm and a second one of lower intensity at 460 nm. Based on calculations reported on Ru analogues,4 we hypothesise that these bands are due to a mixture of ligand-to-metal charge-transfer (LMCT) from sulfur σ and π orbitals to osmium, plus d–d transitions, plus metal-to-ligand charge-transfer (MLCT) from Os–S π orbitals to Os-p-cymene δ* molecular orbitals. Calculations will be performed in future work to fully assign these various transitions.
 | ||
| Fig. 3 UV-visible spectra of complexes 2–9 (a–h) in dichloromethane (10−4 M) at 298 K. | ||
The effect of temperature on the equilibrium between the 16- (1) and 18-e complexes with nitrogen donors (2–8) was investigated by variable temperature UV-visible spectroscopy from 263 to 293 K in dichloromethane. The UV-visible spectra of a solution of complex 2 in a dichloromethane solution (10−4 M) at four different temperatures (263 K to 293 K; Fig. 4) clearly highlight the decreases of absorption at 520 nm with temperature decrease. The ratio of intensities between the two bands of the spectra (I460/I520) evolves from 0.83 at 293 K to 1.33 at 263 K, following the equilibrium shifts towards the 18-e complex as the temperature decreases. The intensity of the absorption band at 360 nm also significantly increases as temperature increases. These changes in the absorption spectra of the solution are reversible and result in thermochromic properties as illustrated in Fig. 5 (colours of the solutions of complex 8 at four different temperatures).
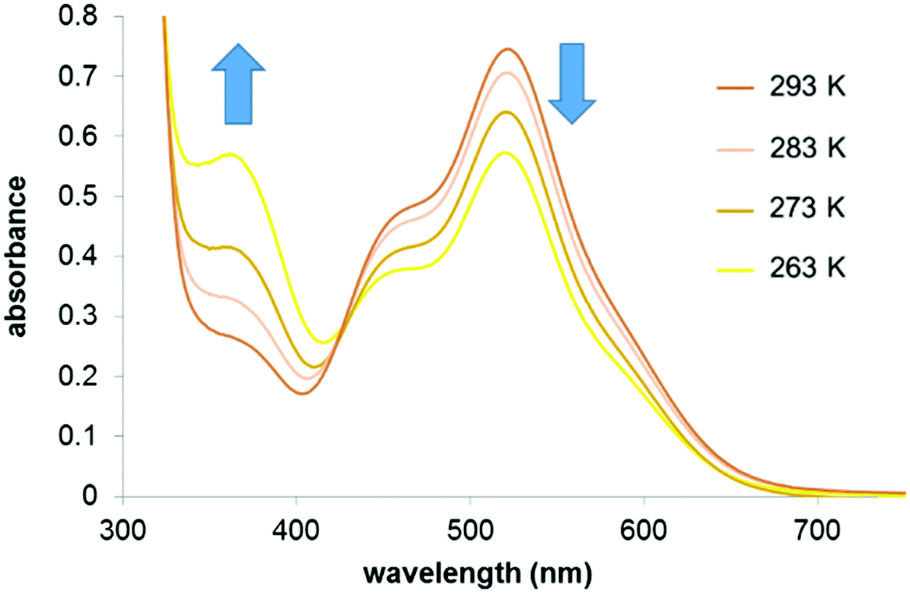 | ||
| Fig. 4 UV-visible absorbance spectra of a solution of complex 2 in dichloromethane (10−4 M) at different temperatures. | ||
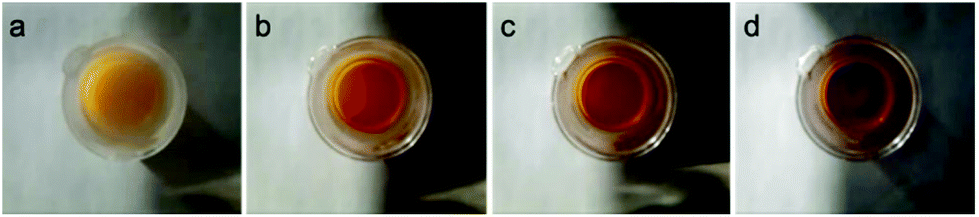 | ||
| Fig. 5 Images showing colour of solution of complex 8 in dichloromethane (10−3 M) at 258 K (a), 268 K (b), 278 K (c) and 288 K (d). | ||
The colours of complexes 1–9 in dichloromethane solutions (10−3 M) at 298 K, and whether they exhibit thermochromic properties are shown in Table 1. The thermochromic properties of complexes 2, 3, 5, and 9 are illustrated in Fig. S2–S5.†
| Colour in solution 298 K, CH2Cl2 | Thermo-chromism | K 103 M−1 | ΔG° kcal mol−1 | pKa ligand30 | |
|---|---|---|---|---|---|
| 2 | Red | Yes | 6.6 ± 1.5 | −5.2 ± 0.2 | 5.2 |
| 3 | Orange | Yes | 2.9 ± 1.1 | −4.7 ± 0.2 | 9.2 |
| 4 | Purple | No | 38.4 ± 5.6 | −6.2 ± 0.1 | 1.9 |
| 5 | Orange | Yes | 1.1 ± 0.5 | −4.1 ± 0.4 | 6.6 |
| 6 | Purple | No | 17.7 ± 2.8 | −5.8 ± 0.1 | 0.6 |
| 7 | Purple | No | 24.8 ± 3.9 | −6.0 ± 0.1 | 2.3 |
| 8 | Red | Yes | 4.3 ± 2.0 | −5.0 ± 0.3 | 4.8 |
| 9 | Orange | Yes | 51.2 ± 7.0 | −6.4 ± 0.1 | 2.7 |
The stoichiometry of the equilibrium in dichloromethane was determined by UV-visible absorption spectroscopy, with the method of continuous variations (Job's plot). The value of X(A − A0) was plotted against X where X is the mole fraction of complex 1, A is the absorbance and A0 is the absorbance when X = 0. All of the Job's plots had a maximum at X = 0.5 demonstrating that the stoichiometry is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mol equiv. for 1:ligand. Unexpectedly the 1:1 stoichiometry even holds true for the bidentate ligands 4,4′-bipyridine and pyrazine (Fig. S6†). An attempt was made to synthesise the 2:1 complex with 4,4′-bipyridine by changing the ratio of 1:4,4′-bipyridine in the reaction mixture to 2:1 instead of 1:1, but this was unsuccessful.
:1 mol equiv. for 1:ligand. Unexpectedly the 1:1 stoichiometry even holds true for the bidentate ligands 4,4′-bipyridine and pyrazine (Fig. S6†). An attempt was made to synthesise the 2:1 complex with 4,4′-bipyridine by changing the ratio of 1:4,4′-bipyridine in the reaction mixture to 2:1 instead of 1:1, but this was unsuccessful.
UV-visible titrations in dichloromethane were then carried out to determine the relative binding strengths of the 16-e complex and the ligands for complexes 2–9. For each titration a solution of the ligand was gradually added (0 to 20 mol equiv.) to a solution of complex 1 with a constant concentration (10−4 M). In order to minimise the dilution of the solution of complex 1, the ligand solution was prepared by adding 20 mol equiv. of the ligand to a 10−4 M solution of complex 1. The titration of complex 1 by 4,4′-bipyridine in dichloromethane, forming 8, is shown in Fig. 6. The UV-visible absorption spectra were characterised by two main bands at 520 nm and 360 nm as noted above. The band at 520 nm decreased in intensity upon addition of the pyridine whereas the band at 360 nm increased in intensity upon addition of the ligand.
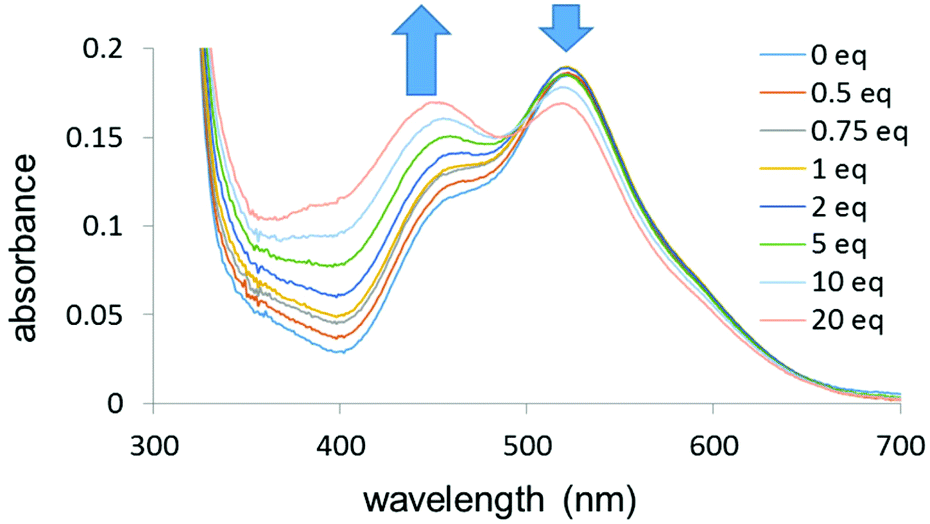 | ||
| Fig. 6 UV-visible titration of complex 1 by 4,4′-bipyridine (0–20 mol equiv.) in dichloromethane (10−4 M) at 298 K. | ||
From the UV-visible titrations, the binding constants K between complex 1 and the ligands were calculated using the non-linear ThordarsonFittingProgram (Table 1).25 All the titrations were repeated three times and the standard deviation for the calculated values of K are given in Table 1. The magnitude of the binding constants (103–104 M−1) is low as compared to the usually observed complexation constants in coordination chemistry (≫106 M−1),26 and is in the range of binding constants observed in host–guest inorganic chemistry (e.g. via non-covalent interactions between a metalla-cage and an aromatic planar guest molecule27–29), which is also consistent with the presence of equilibria. The experimental Gibbs free energy (ΔG°) was obtained from the Gibbs equation using the calculated value of K. The calculated values of the binding constants are reported in Table 1, along with the pKa (acid dissociation constant) of the corresponding ligand.
The choice of solvent can have a significant effect on the equilibrium between two species and on the reactions of the complexes in solution. For example the alkyne–azide cycloaddition using [RuCp*(PiPr3)Cl] has a higher conversion in dichloromethane than in tetrahydrofuran.2 The effect of the solvent on the stability of complexes 2–8 (pyridine derivatives) was investigated. We found that reversible thermochromism also occurs when tetrahydrofuran is used as the solvent for the same complexes as in dichloromethane (Table S1† and Fig. 7). The colours are very similar to the dichloromethane solution (with very similar UV-vis absorption spectra, see Fig. S7†), suggesting a very weak or non-existent solvatochromism.
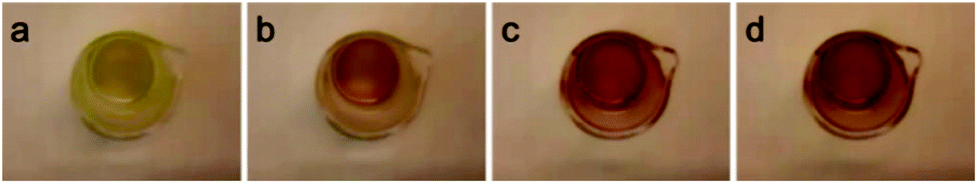 | ||
| Fig. 7 Images showing the colour of complex 2 in tetrahydrofuran (10−3 M) at 263 K (a), 268 K (b), 273 K (c) and 278 K (d). | ||
As in dichloromethane, the stoichiometry of the complexes in tetrahydrofuran solutions was determined by the method of continuous variations for complexes 2–8 so that the effect of the solvent on the binding constants could be determined. The titrations were carried out using the same procedure as above (Fig. S7† for the UV-visible titration of complex 1 by 4-dimethylaminopyridine in tetrahydrofuran). The binding constants for complex 1 with the ligands were then estimated from these titrations and are shown in Table S1.† We were particularly interested in complexes 6 and 8 which both contain potentially bidentate ligands, in order to investigate whether the unexpected 1:1 stoichiometry observed in dichloromethane holds true in tetrahydrofuran. Both Job's plots confirms that the choice of solvent makes no difference to the 1:1 stoichiometry. UV-visible titrations were also carried out in tetrahydrofuran.
Discussion
We sought to rationalise the observed stability of the various 18-electron amine adducts (2–8) by correlating the Gibbs free energies of the 18-e complexes with the acidity of the ligands (pKa). The plot of experimental free energies versus pKa values of the ligands is reasonably linear, Fig. 8. This suggests that the electron donor strength of the amine nitrogen plays a major role in determining the stability of the 18-e adduct: as the pKa of the ligands increases, ΔG° also increases, and the stability of the complexes decreases. This is unexpected and is the opposite from what has previously been observed with the Ru analogue.4 Although the analogy between Os2+ and Ru2+ with H+ does provide some degree of rationality in the observations, there are limitations in using Mn+/H+ analogy to rationalise the formation of a metal–ligand bond (e.g. differences in acidity strengths, ({Ru–OH2} being ca. 1.5 pKa × more basic than {Os–OH2}31). Furthermore, the stability of the 18-electron adducts is not only dependent on the acidity of the ligand, but also depends on the electronic structure of the latter, and for example on its ability to back-π accept electrons, as exemplified with complex 9 (with triphenylphosphine as ligand) which does not follow the same trend as pyridine-containing complexes 2–8. The PPh3 adduct has a much larger binding constant (phosphorous radius: 195 pm; phosphine: σ-donor, π-acceptor) than all of the complexes containing pyridine derivatives although it has a similar pKa value (Table 1).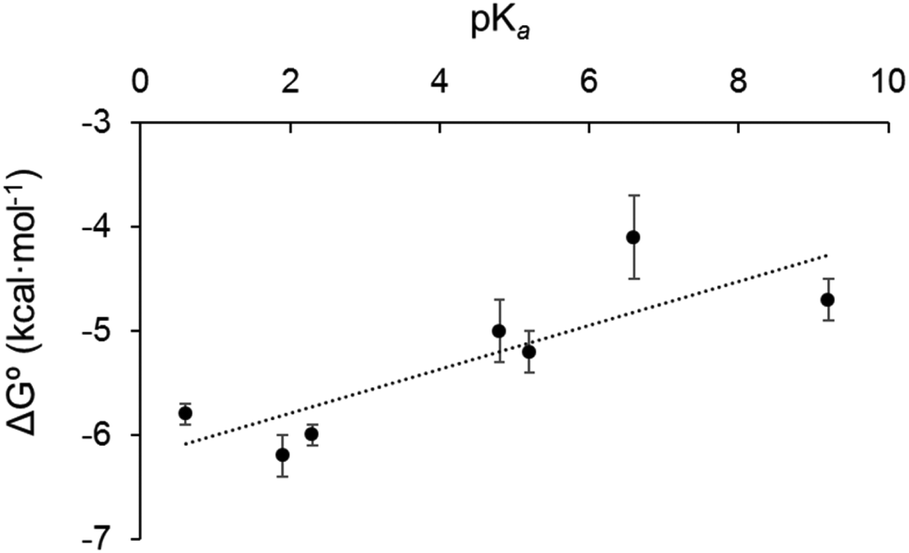 | ||
| Fig. 8 Plot of Gibbs free energy versus the pKa of the ligand in dichloromethane for complexes 2–8. | ||
The formation of the mononuclear Os organometallic complexes 6 and 8 by addition of bidentate pyridinic ligands (pyrazine and bipyridine) is another surprising result, since the two binding sites for these ligands are expected to be identical, and therefore the formation of a dinuclear complex was expected to take place. More surprisingly, the 2:1 complexes had been previously reported with the Ru analogue of the Os 16-electron complex 1 (with pyrazine and bipyridine acting as bridging ligands between two Ru metal centres6). Hence, it seems clear that possible electrostatic repulsion between the two carborane ligands can be ruled out. Ruthenium and osmium possess similar atomic radii (178, and 185 pm, respectively – the lanthanide contraction),30,31 so the difference of reactivity between the Ru/Os analogues does not seem to arise from steric constraints. Relativistic effects (stronger with Os than with Ru – Os being heavier) are of importance in metal–metal bonds, but are less significant for metal–nitrogen bonds; furthermore, if existing, they should favour the formation of an Os-bridging ligand–Os complex analogous to a Ru-bridging ligand–Ru complex. We hypothesise that differences in electron distribution in the complexes [Os(η6-p-cymene)(1,2-dicarba-closo-dodecarborane-1,2-dithiolato)(pyrazine/bipyridine)] and [Ru(η6-p-cymene)(1,2-dicarba-closo-dodecarborane-1,2-dithiolato)(pyrazine/bipyridine)] possibly lead to a difference in acidity for the second nitrogen site of the bidentate ligand and might explain the different stoichiometries observed with both metals. Density functional theory calculations will be performed on both systems in future work to determine the pKa of the second nitrogen atom after coordination of the metal centre to the first nitrogen site of the bidentate ligands in order to test this hypothesis.
Interestingly, the Gibbs free energies of the complexes in tetrahydrofuran are of the same magnitude as in dichloromethane. This confirms that the solvent seems to have very little effect on the strength of the binding in the complexes and the stability of the complexes, and that only weak interaction (as compared to N-donor and P-donor ligands) between metal centre and solvent take place with solvents we had chosen assuming they were non-coordinative solvents (as compared to dimethylsulfoxide or acetonitrile for example which do coordinate the metal centre to form the 18-e solvate-adduct). We also plotted the Gibbs free energy against the pKa of the ligand for complexes 2–8 in tetrahydrofuran (Fig. S8†), and obtained a similar reasonably linear trend as observed in dichloromethane, demonstrating that the Mn+/H+ analogy offers a similar rationalisation in both solvents.
Conclusions
We synthesised a range of 18-electron Os complexes by addition of aromatic amines to the 16-electron precursor [Os(p-cym)(1,2-dicarba-closo-dodecarborane-1,2-dithiolato)] (1). In non-coordinating solvent solutions at ambient temperature, the corresponding yellow or red 18-electron complexes [Os(p-cym)(1,2-dicarba-closo-dodecarborane-1,2-dithiolato)(L)] (L = pyridine (2), 4-dimethylaminopyridine (3), 4-cyanopyridine (4), 4-methoxypyridine (5), pyrazine (6), pyridazine (7), 4,4′-bipyridine (8) and triphenylphosphine (9)) are in equilibrium with the red 16-electron precursor 1. The thermal displacement of this equilibrium results in marked thermochromic properties for complexes 2, 3, 5, 8, and 9. An attempt to correlate the stability of each 18-electron system accordingly to the basicity of the corresponding ligand unveiled that the more acidic the ligand is, the more stable the 18-electron species is. This trend was found both in dichloromethane and in tetrahydrofuran, and is the opposite of what we previously reported with the Ru analogues. The values of the Gibbs free energy were found to be of the same order of magnitude in both dichloromethane and tetrahydrofuran, indicating that the solvent has very little influence on the stability of the complexes formed, and that the thermochromic complexes do not seem to exhibit any solvatochromic properties. We demonstrated in this piece of research that it is possible to force the formation of carborane-containing arene Os 18-e species by decreasing the temperature of the solution, by choosing ligands with low acid dissociation constant (typically inferior at 2), and by using ligands with strong donor atom strength, such as phosphorous. Utilisation of absorption techniques such as UV-visible spectroscopy also allowed us to gauge the displacement of a reaction in solution, and offers ways to characterise equilibria involving complex 1 and to monitor the completion of the synthesis of 18-e carborane-containing Os organometallics.Acknowledgements
We thank the Leverhulme Trust (Early Career Fellowship No. ECF-2013-414 to NPEB), and the University of Warwick (Grant No. RD14102 to NPEB).Notes and references
- R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97–102 CrossRef CAS.
- M. Lamberti, G. C. Fortman, A. Poater, J. Broggi, A. M. Z. Slawin, L. Cavallo and S. P. Nolan, Organometallics, 2012, 31, 756–767 CrossRef CAS.
- V. V. Krishna Mohan Kandepi, J. M. S. Cardoso, E. Peris and B. Royo, Organometallics, 2010, 29, 2777–2782 CrossRef.
- N. P. E. Barry, R. J. Deeth, G. J. Clarkson, I. Prokes and P. J. Sadler, Dalton Trans., 2013, 42, 2580–2587 RSC.
- N. P. E. Barry, T. F. Kemp, P. J. Sadler and J. V. Hanna, Dalton Trans., 2014, 43, 4945–4949 RSC.
- M. Herberhold, H. Yan and W. Milius, J. Organomet. Chem., 2000, 598, 142–149 CrossRef CAS.
- S. Liu, G. L. Wang and G. X. Jin, Dalton Trans., 2008, 425–432 RSC.
- S. Liu, J. S. Zhang, X. Wang and G. X. Jin, Dalton Trans., 2006, 5225–5230 RSC.
- Y. F. Han, J. S. Zhang, Y. J. Lin, J. Dai and G. X. Jin, J. Organomet. Chem., 2007, 692, 4545–4550 CrossRef CAS.
- H. B. Zhu, G. X. Jin and N. H. Hu, J. Organomet. Chem., 2002, 655, 167–171 CrossRef CAS.
- X. Meng, F. S. Wang and G. X. Jin, Coord. Chem. Rev., 2010, 254, 1260–1272 CrossRef CAS.
- X. Wang, S. Liu, L. H. Weng and G. X. Jin, Chem. – Eur. J., 2007, 13, 188–195 CrossRef CAS PubMed.
- I. Romero-Canelón, B. Phoenix, A. Pitto-Barry, J. Tran, J. J. Soldevila-Barreda, N. Kirby, S. Green, P. J. Sadler and N. P. E. Barry, J. Organomet. Chem., 2015, 796, 17–25 CrossRef.
- A. Pitto-Barry and N. P. E. Barry, Polym. Chem., 2014, 5, 3291–3297 RSC.
- N. P. E. Barry, A. Pitto-Barry, I. Romero-Canelon, J. Tran, J. J. Soldevila-Barreda, I. Hands-Portman, C. J. Smith, N. Kirby, A. P. Dove, R. K. O'Reilly and P. J. Sadler, Faraday Discuss., 2014, 175, 229–240 RSC.
- N. P. E. Barry and P. J. Sadler, ACS Nano, 2013, 7, 5654–5659 CrossRef CAS PubMed.
- A. Pitto-Barry, L. M. A. Perdigao, M. Walker, J. Lawrence, G. Costantini, P. J. Sadler and N. P. E. Barry, Dalton Trans., 2015, 44, 20308–20311 RSC.
- N. P. E. Barry, A. Pitto-Barry, J. Tran, S. E. F. Spencer, A. M. Johansen, A. M. Sanchez, A. P. Dove, R. K. O'Reilly, R. J. Deeth, R. Beanland and P. J. Sadler, Chem. Mater., 2015, 27, 5100–5105 CrossRef CAS PubMed.
- N. P. E. Barry, A. Pitto-Barry, A. M. Sanchez, A. P. Dove, R. J. Procter, J. J. Soldevila-Barreda, N. Kirby, I. Hands-Portman, C. J. Smith, R. K. O'Reilly, R. Beanland and P. J. Sadler, Nat. Commun., 2014, 5, 3851 CAS.
- M. Patra and G. Gasser, ChemBioChem, 2012, 13, 1232–1252 CrossRef CAS PubMed.
- K. J. Kilpin, S. Crot, T. Riedel, J. A. Kitchen and P. J. Dyson, Dalton Trans., 2014, 43, 1443–1448 RSC.
- Y. Fu, R. Soni, M. J. Romero, A. M. Pizarro, L. Salassa, G. J. Clarkson, J. M. Hearn, A. Habtemariam, M. Wills and P. J. Sadler, Chem. – Eur. J., 2013, 19, 15199–15209 CrossRef CAS PubMed.
- P. Govender, F. Edafe, B. C. E. Makhubela, P. J. Dyson, B. Therrien and G. S. Smith, Inorg. Chim. Acta, 2014, 409, 112–120 CrossRef CAS.
- G. Gupta, N. Nagesh, B. S. Murray, P. J. Dyson and B. Therrien, Inorg. Chim. Acta, 2014, 423, 31–35 CrossRef CAS.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- O. Gutten and L. Rulíšek, Inorg. Chem., 2013, 52, 10347–10355 CrossRef CAS PubMed.
- N. P. E. Barry, J. Furrer and B. Therrien, Helv. Chim. Acta, 2010, 93, 1313–1328 CrossRef CAS.
- N. P. E. Barry, J. Furrer, J. Freudenreich, G. Süss-Fink and B. Therrien, Eur. J. Inorg. Chem., 2010, 725–728 CrossRef CAS.
- N. P. E. Barry and B. Therrien, Eur. J. Inorg. Chem., 2009, 4695–4700 CrossRef CAS.
- S. Kotrly and L. Šůcha, Handbook of Chemical Equilibria in Analytical Chemistry, ed. R. A. Chalmers and M. Masson, John Wiley & Sons, Chichester, 1985, pp. 87–108 Search PubMed.
- A. F. A. Peacock, M. Melchart, R. J. Deeth, A. Habtemariam, S. Parsons and P. J. Sadler, Chem. – Eur. J., 2007, 13, 2601–2613 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5dt04398k |
| This journal is © The Royal Society of Chemistry 2016 |