
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multiscale structural characterizations of mixed U(IV)–An(III) oxalates (An(III) = Pu or Am) combining XAS and XRD measurements
B.
Arab-Chapelet
*a,
P. M.
Martin
b,
S.
Costenoble
a,
T.
Delahaye
a,
A. C.
Scheinost
c,
S.
Grandjean
a and
F.
Abraham
d
aCEA, DEN, DRCP, SERA/ Laboratoire de Conversion des Actinides et Radiolyse, BP 17171, F-30207 Bagnols sur Cèze Cedex, France. E-mail: benedicte.arab-chapelet@cea.fr
bCEA, DEN, DEC, 13108 St Paul Lez Durance, France
cHelmholtz-Zentrum Dresden-Rossendorf, Institute of Resource Ecology, D-01314 Dresden, Germany
dUniv. Lille Nord de France, Unité de Catalyse et de Chimie du Solide, UCCS, UMR CNRS 8181, ENSCL-USTL, BP 90108, 59652 Villeneuve d'Ascq Cedex, France
First published on 2nd March 2016
Abstract
Mixed actinide(III,IV) oxalates of the general formula M2.2UIV1.8AnIII0.2(C2O4)5·nH2O (An = Pu or Am and M = H3O+ and N2H5+) have been quantitatively precipitated by oxalic precipitation in nitric acid medium (yield >99%). Thorough multiscale structural characterization using XRD and XAS measurements confirmed the existence of mixed actinide oxalate solid solutions. The XANES analysis confirmed that the oxidation states of the metallic cations, tetravalent for uranium and trivalent for plutonium and americium, are maintained during the precipitation step. EXAFS measurements show that the local environments around U+IV, Pu+III and Am+III are comparable, and the actinides are surrounded by ten oxygen atoms from five bidentate oxalate anions. The mean metal–oxygen distances obtained by XAS measurements are in agreement with those calculated from XRD lattice parameters.
Introduction
Considering the risk of dependence on uranium importation, the necessity to save uranium resources and the desire to reduce the volume and radiotoxicity of eventual nuclear wastes, French policy has been oriented towards the closing of the nuclear fuel cycle with the (multi)reprocessing of plutonium, present in spent fuel, in MOX ((U,Pu)O2 mixed oxide) fuel fabrication. In particular, towards this end, Pu, which is the most important contributor to the long term radiotoxicity of eventual radioactive wastes is removed from the waste inventory to fabricate MOX fuel usable in Pressurized Water Reactors (PWR) and Fast Neutron Reactors (FNRs).With the ongoing nuclear transition towards 4th generation reactors, France has initiated a project called ASTRID (Advanced Sodium Technological Reactor for Industrial Demonstration), which is a French prototype of a sodium-cooled FNR.1 Although the main objective of this project is to assess the ability of fast reactors to burn plutonium isotopes, spurred on by the French Bataille Act, it will also be of great interest to evaluate minor actinide (MA = Np, Am or Cm) partitioning and transmutation.2 Two types of transmutation are currently considered: homogeneous and heterogeneous.3 In homogeneous mode, MAs are directly added to the fuel to form MA-MOX.4 Only a small percentage of MAs will be included in the fuel to avoid changing the core neutronics. In the heterogeneous mode, minor actinides are introduced into a UO2 based matrix located in the core periphery as a minor actinide bearing blanket (MABB).5,6 This last option is preferred due to its low influence on the core behavior and the higher quantity of MAs transmuted. Among the MAs, americium has been given priority due to its high radiotoxicity after Pu recycling and its proportion in spent fuel, with the irradiation of U1−xAmxO2±δ pellets called Am bearing blankets (AmBB).
FNR MOX fuel integrates higher quantities of Pu than is in the MOX currently used in PWR or AmBB blankets, and so fuel fabrication routes based on pulverulent precursors for fuel pelletization steps should be modified. Conventional fuel fabrication7 is currently based on multistep powder metallurgy processes, which involve mixtures of simple oxides, ball milling, pelletizing and reactive sintering, and could be limited for several reasons.
These methods, due to the number of steps, generate large quantities of very fine, and thus contaminating, particles. Also, they can lead to very significant chemical and microstructural heterogeneities in the sintered MOX or AmBB that may be accentuated by the presence of a miscibility gap in the U–Pu–O phase diagram or the higher oxygen potential of the americium oxides compared to UO2.
For the pellet fabrication of MOX and AmBB intended to be irradiated in FNRs and thus promote the transition to 4th generation reactors, it would be advantageous to develop new non-proliferative synthesis routes, which allow the obtaining of precursor powders of homogeneous mixed oxide that could be directly pelletized and sintered.8–10 Using actinides from spent fuel dissolution purified in nitric acid media, these synthesis routes are based on a co-conversion step, which permits the quantitative transfer of the actinides from solution into mixed actinide oxides. Various co-conversion methods such as the oxalate precipitation route have been investigated. Since the 1990s, several works have provided evidence that this process, based on the precipitation of an oxalate compound followed by its thermal treatment into an oxide, is a particularly convenient route for actinide co-conversion into mixed oxalate compounds, suitable as the precursors of mixed actinide oxides.11–17 Thus, oxalic precipitation of different actinide(IV)–actinide(III) oxalates characterized by different crystallographic structures and morphologies can be achieved. Moreover, a strong relationship between the microstructure and properties of the mixed actinide oxides and the morphology and crystallographic structure of the oxalate precursors has been established.18,19 The influence of oxide morphology during the fuel pellet formation is an important factor in the process, and therefore, the thorough structural characterization of the mixed oxalate precursor is a key step. Up until now, there is only one structural determination from single-crystal X-ray diffraction data concerning mixed actinide(IV)–actinide(III) oxalate compounds.20 In many works U(IV)–Ln(III) systems are considered as good structural analogs of An(IV)/An(III) systems due to their similar ionic radii.21–23 Consequently, the structure of precipitated actinide oxalates14,16,18 has often been identified by comparison with the structural data of a three mixed U(IV)–Ln(III) oxalate series, named hexagonal, triclinic and tetragonal, with the formula M2+xUIV2−xLnIIIx(C2O4)5·nH2O for the first and M1−xLnIII1−xUIVx(C2O4)2·nH2O for the last two,24–26 where M is a monovalent cation. These three structural series are characterized by a mixed crystallographic site which accommodates both U(IV) and Ln(III) ions despite their charge difference. The charge compensation due to the substitution of the trivalent cation for the tetravalent cation, or vice versa, is ensured by the presence of single-charged cations, M, located in the interspaces of the structure.
This article aims to present multiscale characterizations combining X-ray Absorption Spectroscopy (XAS) and powder X-Ray Diffraction (XRD) measurements performed on mixed U(IV)–An(III) (An(III) = Pu or Am) oxalates.
Experimental section
Precursors
Because of the radioactive nature of actinides, especially plutonium and americium, the experiments involving these elements were carried out in glove boxes with very restrictive protocols. Actinide(IV) and actinide(III) solutions were prepared using specific procedures, either from purified mono-metallic solutions or by dissolving mono-metallic oxides. U(IV) nitrate solution was prepared by catalytic reduction of U(VI) nitrate by H2 on a Pt/Si backing. Pu(IV) and Am(III) solutions were prepared by dissolving the corresponding oxides, PuO2 and 241AmO2, with concentrated HNO3 in a glove box. Am(III) is stable in nitric acid solution whereas Pu(III) solution was prepared by the reduction of the Pu(IV) solution by hydrazinium nitrate (N2H5+, NO3−). Hydrazinium nitrate was also used as an anti-nitrous agent to stabilize the lowest oxidation states (typically IV for U, and III for Pu) and also as a source of monocharged cations. The concentration, purity and oxidation state in these primary solutions were essentially determined by UV-visible spectroscopy. The concentration of actinides in these solutions is close to 1 mol L−1 and the concentration of hydrazinium cations in the uranium(IV) or plutonium(III) solutions is around 0.2 mol L−1.Lanthanide nitrate salts (Aldrich, 99.9% Reagent Grade Ln(NO3)3·6H2O, Ln = Ce, Nd or Gd) were used when appropriate to prepare Ln(III) solutions to simulate An(III) solutions in pseudo-active experiments, considering the analogies between Ln(III) and An(III) ions.21–23
Oxalate co-precipitation experiments
The oxalate co-precipitates were synthesized in a vortex reactor (V = 50 mL) under ambient conditions, by mixing a solution of U(IV) and An(III)/Ln(III) (An(III) = Pu or Am and Ln(III) = Ce, Nd or Gd; AnIII/(UIV + AnIII) ∼ 10% and [U + An/Ln] = 0.12 M) and a concentrated H2C2O4 solution (with a slight excess of oxalic acid [H2C2O4] = 0.1 M) in nitric medium ([HNO3] = 1 mol L−1).13,14,18 The time of addition of the actinide solution and oxalic acid solution was fixed at 10 minutes.The resulting crystallized powders, whose color depends on the elements involved, were filtered off and dried at room temperature.
XAFS reference compounds and sample preparation
The two oxide references UO2 and PuO2 were obtained by calcination under an inert atmosphere or under air, respectively, of the corresponding oxalate precursors AnIV(C2O4)2·6H2O (An = U or Pu). The PuF3 sample is synthesized by the precipitation of a trivalent plutonium solution using concentrated HF. The trivalent plutonium is obtained after the reduction of tetravalent plutonium by hydrazinium nitrate. Their crystalline structures were checked using XRD.For XAS measurements, samples were fabricated using a standard method for transmission geometry/configuration: ∼2 mg of mixed oxalate or oxide powder was mixed with 15 mg of cellulose using an agate mortar. The resulting mixture was pelletized and then confined in two independent sealed sample holders.
XRD acquisition
X-ray powder diffraction data for all mixed oxalates were obtained with a BRUKER D8 Advanced diffractometer (LynxEye detector) using Cu-Kα radiation (λ = 1.5418 Å). Each powder pattern was recorded within an angular range of 5°–60° in 2θ, with steps of 0.02° and a counting time of 0.5 s per step. The stripping of the Kα2 radiation according to the Rachinger method as well as the baseline and angle shift corrections was done using empirical calculations and DIFFRACplus EVA software.27Silicon was added to all samples as an internal standard to calibrate the angular positions of the observed XRD lines. Actinide oxalates were mixed with an epoxy resin to prevent contamination spreading and their diffraction patterns were compared with the recently built M+–U(IV)–Ln(III) oxalate structures database20,24–26 to detect isomorphic similitude.
The refinement of the lattice parameters was carried out using the “pattern matching” option of the Fullprof software28 where only the profile parameters (cell dimensions, peak shapes, background, zero point correction and asymmetry) were refined. The peak shape was described by a pseudo-Voigt function with an asymmetry correction at low angles.
UV-visible analysis
Actinide and lanthanide concentrations in solution were determined by UV-visible spectroscopy using a VARIAN Cary 6000 spectrophotometer between 350 and 900 nm.The hydrazinium content was determined by colorimetric analysis after dissolution of the precipitated solids in concentrated nitric acid solution. In a 1 M nitric medium, hydrazinium cations react with dimethylamino-4-benzaldehyde to form a yellow complex characterized by the presence of a peak around 455 nm.
Thermogravimetric analysis
Thermogravimetric analyses (TGA) were carried out with a NETZSCH STA 409C thermal analysis system with an alumina crucible up to 950 °C, under an argon flow and with a heating rate of 20 °C min−1 to determine the water content of the freshly synthesized powders.TIMS analysis
TIMS (Thermal Ionization Mass Spectrometer) analyses were performed with a VG-54 magnetic sector mass spectrometer. The precipitated solids were quantified after the dissolution of the oxalate compounds in nitric acid medium ([HNO3] = 8 M). An internal standard whose isotopic composition and concentration are both known was added to the sample. The actinide content has been determined from the measured sample’s final concentration and the known internal standard concentration.ICP/AES analysis
Lanthanide cations (Ce, Nd and Gd) inserted in the solid were quantified after dissolution of the oxalates in nitric acid media ([HNO3] = 8 M) using Inductively Coupled Plasma-Atomic Emission Spectrometry (ICP-AES) with a Perkin Elmer Optima 3000 DL system.XAS analysis
XAS measurements were performed at the Rossendorf Beamline (BM20)29 located at the European Synchrotron Radiation Facility (ESRF, Grenoble, France). Storage ring operation conditions were 6.0 GeV and 70–90 mA. The beam was conditioned using a double crystal monochromator mounted with Si (111). Samples were held at 15 K using a closed-cycle helium cryostat in order to avoid the thermal vibration contributing to the Debye–Waller factors of EXAFS spectra, which can then be considered as only influenced by static structural disorder. XANES and EXAFS spectra at the U-LIII edge (17166 eV) for each mixed oxalate sample ((U,An) and (U,Ln)) were collected in transmission mode. Spectra at the plutonium (LIII (18057 eV) and LII (22266 eV)) and americium (Am LIII (18510 eV) XANES and EXAFS) edges were recorded in fluorescence mode using a 13-element Ge solid state detector (Canberra) equipped with a digital spectrometer (XIA). Fluorescence spectra were corrected for dead time using a measured relationship between incoming count rate (ICR) and selected channel analyzer (SCA) readings for each channel using the SixPack software package.30 For plutonium, the Pu-LII edge (22266 eV) was preferred for EXAFS analysis because, as we previously observed,31 EXAFS oscillations collected at Pu-LIII which were distorted due to the U-LIII EXAFS signal. Nevertheless, the XANES data collected at Pu-LIII could be used as both the white line intensity and the edge jump are much stronger than U-LIII EXAFS oscillations. Energy calibrations were performed using a Y foil (17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 038 eV) for data at the U-LIII edge, a Zr (foil 17998.0 eV) for data at the Pu and Am LIII edges or a Mo foil (20000.0 eV) for data at the Pu-LII edge. The metallic foils were positioned after the second ionization chamber. The E0 values were taken as the first inflection point using the first zero-crossing value of the second derivative. The first zero-crossing of the first derivative was used to determine the position of the white line (WL) maximum. A minimum of two spectra were collected per edge and per sample. For each spectrum, XANES spectra of the reference foil detailed previously was systematically collected simultaneously. The acquisition conditions were typically 0.8 eV steps for XANES (U-LIII, Pu-LIII and Am-LIII) and 0.05 Å−1 steps for EXAFS (U-LIII, Pu-LII and Am-LIII). Before averaging scans, each scan was aligned using the XANES spectra collected on the reference foils. The ATHENA software32 was used for XANES spectra normalization using linear functions for both pre- and post-edge regions.
038 eV) for data at the U-LIII edge, a Zr (foil 17998.0 eV) for data at the Pu and Am LIII edges or a Mo foil (20000.0 eV) for data at the Pu-LII edge. The metallic foils were positioned after the second ionization chamber. The E0 values were taken as the first inflection point using the first zero-crossing value of the second derivative. The first zero-crossing of the first derivative was used to determine the position of the white line (WL) maximum. A minimum of two spectra were collected per edge and per sample. For each spectrum, XANES spectra of the reference foil detailed previously was systematically collected simultaneously. The acquisition conditions were typically 0.8 eV steps for XANES (U-LIII, Pu-LIII and Am-LIII) and 0.05 Å−1 steps for EXAFS (U-LIII, Pu-LII and Am-LIII). Before averaging scans, each scan was aligned using the XANES spectra collected on the reference foils. The ATHENA software32 was used for XANES spectra normalization using linear functions for both pre- and post-edge regions.
To determine the oxidation states of U, Pu and Am cations, XANES spectra were compared to data collected on several reference compounds at the same beam line (BM20) using the same experimental set-up. The reference compounds were U+IVO2 for uranium, Am+IVO2 for americium,33 Pu+IVO2 and Pu+IIIF3 for plutonium31 and the data were collected at U-LIII, Am-LIII and Pu-LIII, respectively.
The ATHENA software32 was used for extracting EXAFS oscillations. The experimental EXAFS spectra collected on the (U,Pu) and (U,Am) mixed oxalate samples were Fourier-transformed using a Hanning window over the full k space range available for each edge: 2.6–13.5 Å−1 for U and Am LIII; 2.6–12.8 Å−1 for Pu-LII. Curve fitting of the EXAFS spectra was performed simultaneously on k1, k2 and k3-weighted data in an R-space between 1.5 and 4.4 Å using the ARTEMIS software.32 Phases and amplitudes for the interatomic scattering paths were calculated with the ab initio code FEFF8.40.34 Spherical 7.5 Å clusters of atoms built using the hexagonal crystallographic structure (space group = P6/3mmc) determined by Chapelet-Arab et al.24 were used for the FEFF8.40 calculations. The cell parameters used for theoretical calculations are those determined experimentally in this work by X-ray diffraction. In this crystallographic structure, each cation (U, Pu and Am) is surrounded by five oxalate ions (C2O4). Three single-scattering (two-legged) paths are then used in the EXAFS fitting process. These three paths correspond to 10 oxygen atoms (O1) located at ∼2.46 Å, 10 carbon atoms at ∼3.29 Å and 10 oxygen atoms (O2) at ∼4.52 Å. These mean distances have been obtained by averaging the different distances given by the crystallographic structure for each coordination shell. Furthermore, two three-legged and one four-legged multiple-scattering paths, based on their relatively high magnitudes, were included to fit the spectra. The three-legged paths are Abs-C(1)–O-Abs (Abs representing the absorbing atoms) and Abs-O(2)–C-Abs. The four legged one is Abs-C–O(1)–C-Abs. Without the addition of these three multiple-scattering paths the third coordination corresponding to O(2) atoms cannot be properly fitted as its relative amplitude is lower relative to these paths. Due to the reduced spectral domain available for Pu-LII, it was not necessary to consider the three-legged path Abs-C–O(1)-Abs during the fit. Furthermore, in order not to limit the introduction of free parameters, the N and σ2 values for each multiple-scattering path were not individually fitted but linked to the values used for the included single-scattering paths. Only the distances were fitted because, as mentioned for the single-scattering paths, the distances obtained are an average of the different equivalent contributions. As often employed for An LIII/LII31–37 the amplitude factor (S02) was set at 0.90 for U-LIII and Am-LIII and 1.0 for Pu-LII. The shift in threshold energy (ΔE0) was varied as a global parameter. The goodness of fit is estimated using the Rfactor.32
Results and discussion
Elemental composition and crystalline structure
In nitric acid medium, actinide and lanthanide cations form very insoluble complexes with oxalic acid. Tetravalent uranium and trivalent actinides or lanthanides co-precipitate quantitatively and without the modification of either the U(IV)–An(III)/Ln(III) ratio or the oxidation state of the metallic cations (for example IV for uranium and III for plutonium; Fig. 1). Actinide and lanthanide concentrations in the nitric supernatant after oxalic co-precipitation are very weak (<10 mg L−1 for the trivalent cations and <50 mg L−1 for uranium(IV)). Under controlled conditions, these concentrations are comparable to or even lower than the values published for AnIV(C2O4)2·6H2O38 and AnIII2(C2O4)3·10H2O oxalates. Thus, the co-precipitation yields exceeded 99% using the experimental method described above. This property was used to target a 10% mass U(IV)–An(III)/Ln(III) ratio in the co-precipitate (Table 1) from the formulation of the initial U(IV)–An(III)/Ln(III) mixture prepared in solution.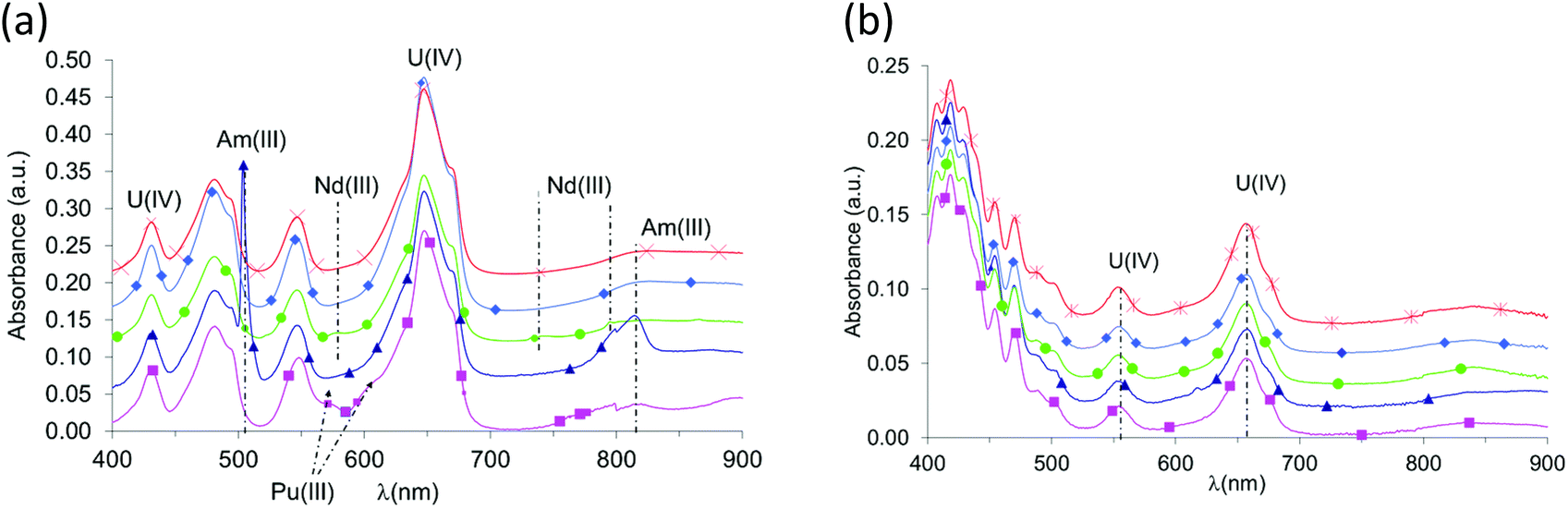 | ||
Fig. 1 UV-Vis spectra of solutions obtained during U(IV) – An(III)/Ln(III) oxalic co-precipitation tests: (a) nitric acid solutions containing U(IV)–An(III)/Ln(III) mixtures with a dilution factor for spectrum acquisition equal to 1:20; (b) nitric acid supernatants after oxalic co-precipitation of metallic cations without any dilution for spectrum acquisition; ■ U90Pu10; ▲ U90Am10; ◆ U90Ce10; ● U90Nd10;  U90Gd10. U90Gd10. | ||
| System | U(IV) | An(III) or Ln(III) | AnIII/(AnIII + UIV) (%) | ||
|---|---|---|---|---|---|
| Experimental method | [U(IV)] (mol L−1) | Experimental method | [An(III)] or [Ln(III)] (mol L−1) | ||
| U(IV)–Ce(III) | TIMS | 3.19 × 10−3 | ICP/AES | 3.4 × 10−4 | 9.6 ± 0.9 |
| U(IV)–Nd(III) | TIMS | 3.53 × 10−3 | ICP/AES | 3.8 × 10−4 | 9.7 ± 0.9 |
| U(IV)–Gd(III) | TIMS | 3.33 × 10−3 | ICP/AES | 3.8 × 10−4 | 10.3 ± 1.0 |
| U(IV)–Pu(III) | TIMS | 2.77 × 10−3 | TIMS | 2.7 × 10−4 | 8.8 ± 0.2 |
| U(IV)–Am(III) | TIMS | 3.38 × 10−3 | TIMS | 4.1 × 10−4 | 10.8 ± 0.3 |
For each U(IV)–An(III)/Ln(III) (An(III) = Pu or Am and Ln(III) = Ce, Nd or Gd) oxalic co-precipitation experiment, the resulting solid was characterized from the powder diffraction pattern (Fig. 2) by analogy with uranium(IV)–lanthanide(III)24–26 and uranium(IV)–plutonium(III)20 oxalates whose structures were solved using single-crystal X-ray diffraction data. Isomorphic similitudes have been detected and permit the identification of the precipitation of a homeotype single-phase mixed U(IV)–An(III)/Ln(III) oxalate characterized using the general formula M2+xUIV2−xAn/LnIIIx(C2O4)5·nH2O (M = single charged cation which can be H3O+ and/or N2H5+). This mixed oxalate crystallizes in a hexagonal symmetry and its structural arrangement is based on a mixed crystallographic site which can accept either a tetravalent cation or a trivalent one, with the charge balance being ensured by monovalent cations (H3O+ and/or N2H5+) located in the cavities of the structure. The crystal network is built on honeycomb-like hexagonal rings leading to a three-dimensional structure. Considering the U(IV)–An(III)/Ln(III) pairs (An(III) = Pu or Am and Ln(III) = Ce, Nd or Gd) and a similar An(III)/(U(IV) + An(III)) ratio close to 10% for all compounds, no modification of the global structure of the precipitate is observed.
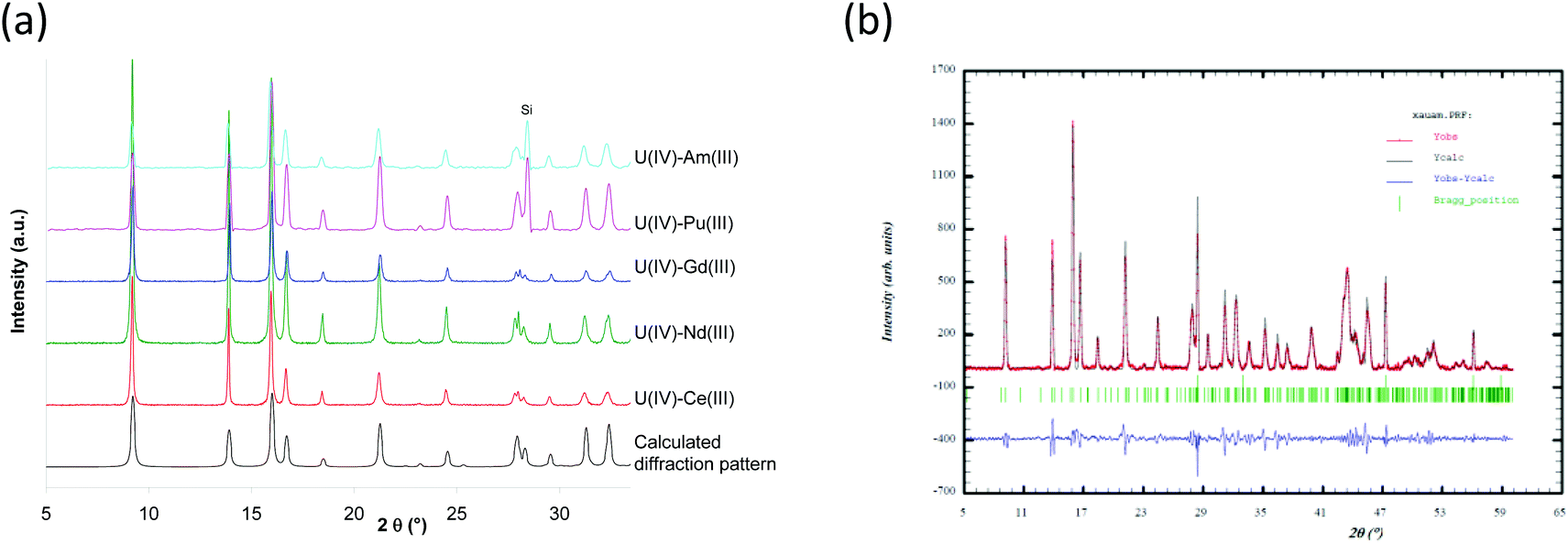 | ||
| Fig. 2 X-ray diffraction patterns of U90(IV)–An/Ln10(III) oxalate co-precipitates compared to the X-ray diffraction pattern calculated from the uranium oxalate hydrate, (NH4)2U2(C2O4)5·nH2O single crystal data;24 (a) example of Fullprof fit results obtained for U(IV)–Am(III) oxalate. Si is used as internal standard. | ||
The diffraction patterns are very similar and characteristic of the hexagonal M2+xUIV2−xAn/LnIIIx(C2O4)5·nH2O compounds.20,24
It is interesting to note that the lattice parameters evolve along the actinide/lanthanide series (Table 2). The decrease in the lattice parameters from Ce(III) to Gd(III) for the hexagonal structure is in agreement with the decreasing ionic radii along the lanthanide series22 (Table 2). Considering the possible effect of alpha radiolysis and the slight difference of the An(III)/(U(IV) + An(III)) ratios (8.8% for Pu and 10.8% for Am), a relevant comparison of the lattice parameters of structures based on those trivalent actinides adjacent on the periodic table is not practicable. XRD analysis shows that mixed oxalate compounds characterized by a similar hexagonal structure are obtained when, starting from (N2H5+,H3O+)2UIV2(C2O4)5·nH2O,18 tetravalent uranium is substituted by a trivalent lanthanide or actinide (An/Ln(III)/(U(IV) + An/Ln(III) ∼ 10%). Considering the lattice parameters determined for the U(IV)–Nd(III) and U(IV)–Am(III) systems, a slight discrepancy is observed from the data previously published.18 This difference is more important for the a parameter, which corresponds to the hexagonal cycles.
| System | a (Å) | c (Å) | AnIII/(AnIII + UIV) (%) | Reference |
|---|---|---|---|---|
| U90(IV)–Ce10(III) | 19.225(2) | 12.745(2) | 9.6 ± 0.9 | This work |
| U90(IV)–Nd10(III) | 19.222(3) | 12.738(2) | 9.7 ± 0.9 | This work |
| U90(IV)–Nd10(III) | 19.205(2) | 12.74(2) | 10 ± 1 | 18 |
| U90(IV)–Gd10(III) | 19.183(2) | 12.707(3) | 10.3 ± 1.0 | This work |
| U90(IV)–Pu10(III) | 19.193(2) | 12.730(2) | 8.8 ± 0.2 | This work |
| U90(IV)–Am10(III) | 19.245(2) | 12.760(2) | 10.8 ± 0.3 | This work |
| U90(IV)–Am10(III) | 19.203(8) | 12.74(1) | 10 ± 1 | 18 |
Water molecules and hydrazinium and oxonium cations located in these hexagonal cavities are very labile and their composition partially depends on the chemical conditions of the precipitation and the storage conditions of the precipitates. Moreover, the decomposition of hydrazinium cations leads to the formation of ammonium cations, which can participate in the charge balance in the hexagonal mixed oxalate and modify the lattice parameters.39
In the U(IV)–An(III)/Ln(III) precipitated solids, hydrazinium cations were quantified in order to study the balance charge mechanisms during the partial substitution of tetravalent uranium by a trivalent lanthanide or actinide. Very similar contents were obtained except for the uranium–americium compound (Table 3). Then a general formula of (N2H5+)1.2(H3O+)1UIV1.8An/LnIII0.2(C2O4)5·nH2O can be suggested. These results show that the balance charge during the partial substitution of tetravalent uranium by a trivalent lanthanide or actinide is ensured by the same mixture of hydrazinium and oxonium cations. Concerning the U(IV)–Am(III) precipitate, radiolysis could be responsible for the partial decrease of hydrazinium contents in the solid phase.
| System | General formula M2+xUIV2−xAn/LnIIIx(C2O4)5·nH2O | ||||
|---|---|---|---|---|---|
| UIV | Ln/AnIII | N2H5+ | H3O+ | H2O | |
| U90(IV)–Ce10(III) | 1.81 | 0.19 | 1.2 | 1.0 | 5.4 |
| U90(IV)–Nd10(III) | 1.81 | 0.19 | 1.2 | 1.0 | 5.3 |
| U90(IV)–Gd10(III) | 1.79 | 0.21 | 1.2 | 1.0 | 6.0 |
| U90(IV)–Pu10(III) | 1.82 | 0.18 | 1.1 | 1.1 | 5.5 |
| U90(IV)–Am10(III) | 1.78 | 0.22 | 0.8 | 1.4 | 5.1 |
Thermogravimetric experiments were carried out on the different mixed U(IV)–An(III)/Ln(III) compounds and led to the determination of the complete general formula of each mixed U(IV)–An(III)/Ln(III) oxalate (Table 3). The different thermogravimetric curves are very close which indicates that very similar mechanisms are implied during thermal decomposition for each mixed U(IV)–An(III)/Ln(III) oxalate (Fig. 3). Qualitatively, the thermal conversion of the different mixed U(IV)–An(III)/Ln(III) oxalates into oxides proceeds through dehydration and hydrazinium cation decomposition steps (between 20 and around 300 °C) followed by the oxalate ligand decomposition step (between 300 and 700 °C) leading to the mixed oxide formation. These decomposition steps are quite similar to those previously reported for the (N2H5+)2UIV2(C2O4)5·nH2O oxalate decomposition.40 Finally, no significant difference is observed in the chemical formula derived from the TGA experiments which confirms also the existence of the same mechanism of oxalic co-precipitation for all U(IV)–An(III)/Ln(III) pairs (An(III) = Pu or Am and Ln(III) = Ce, Nd or Gd).
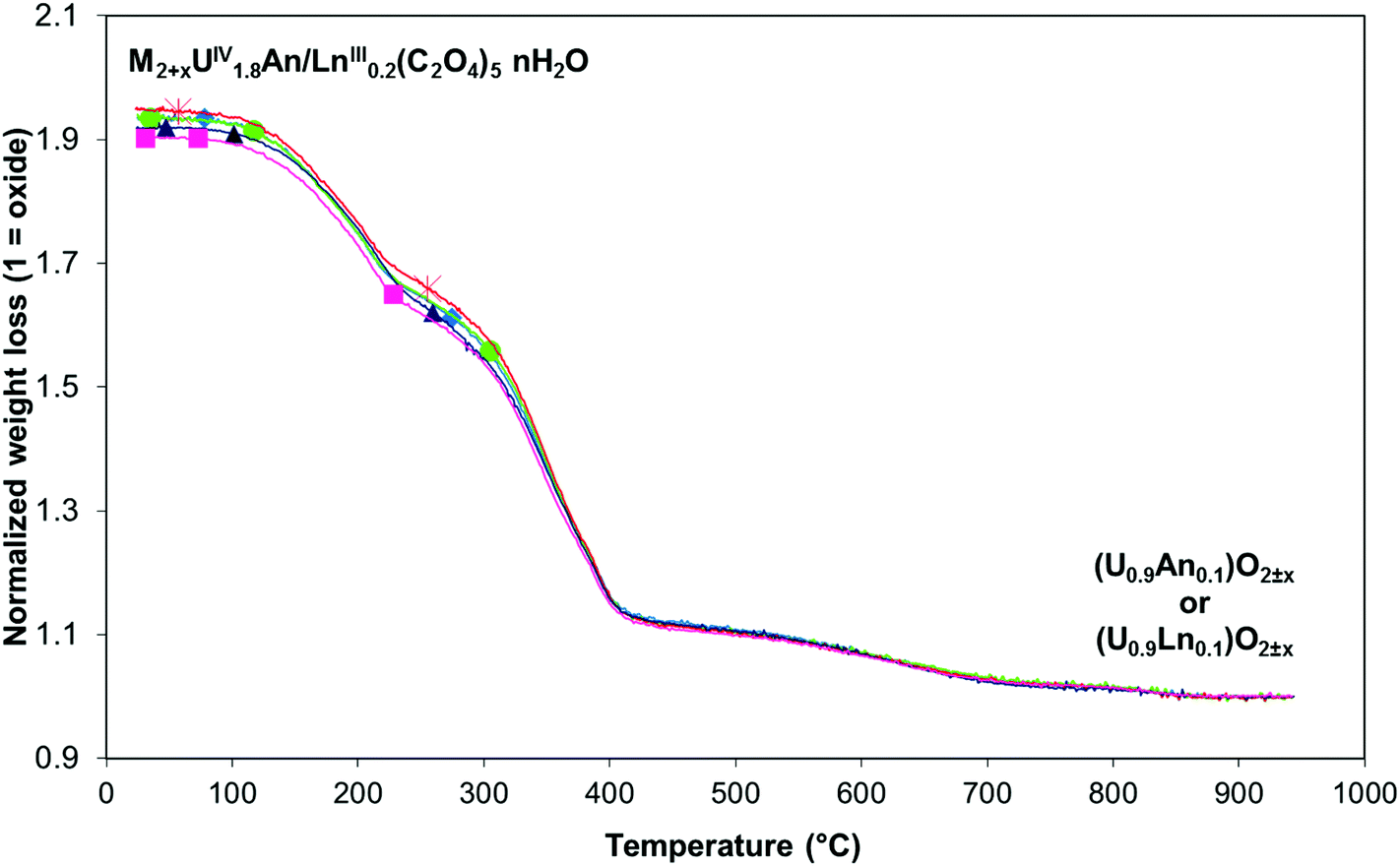 | ||
Fig. 3 TG plot analyses of U(IV)–An(III)/Ln(III) mixed oxalates under Ar: ■ U(IV)–Pu(III); ▲ U(IV)–Am(III); ◆ U(IV)–Ce(III); ● U(IV)–Nd(III);  U(IV)–Gd(III). U(IV)–Gd(III). | ||
The following investigations by EXAFS and XANES experiments aim at unambiguously determining the oxidation states stabilized in the oxalate compounds and precisely investigating the U(IV)–An(III)/Ln(III) oxalate solid solutions.
In fact only the non-substituted U(IV) hexagonal structure has been totally determined.24 For the substituted U(IV)–An(III)/Ln(III) oxalate single crystals only a subcell with as = a/√3, and cs = c/2 is observed and only an average structure in this subcell has been determined for the mixed U(IV)–Pu(III) oxalate (NH4)2.7PuIII0.7UIV1.3(C2O4)5·nH2O.20 Thus, occupation of different sites with different environments for U(IV) and An(III)/Ln(III) atoms in the actual structure cannot be ruled out. In the (NH4+)2UIV2 (C2O4)5·nH2O hexagonal structure, U(IV) is coordinated by 10 oxygens from five bidentate oxalate ligands24 but in Ln(III) and An(III) oxalates the metals are coordinated by six oxygens from three oxalate ligands and three water molecules.41,42 It is therefore essential to ensure that Ln(III) and An(III) have the same environment in the substituted compounds as U(IV).
XANES results
XANES spectra collected at the U LIII edge for (U,An) and (U,Ln) samples are shown in Fig. 4a and b respectively. No difference is observed between the mixed oxalates spectra, demonstrating that the uranium valence state is identical for the five compounds. Furthermore, the unique WL position value (17175.1(5) eV) is very close to the value measured on the UIVO2 reference compound (17175.5(5) eV) unambiguously showing that the oxidation state of the uranium cation is IV in the precipitated solids. Hence, the tetravalent state of uranium is kept during the precipitation step.
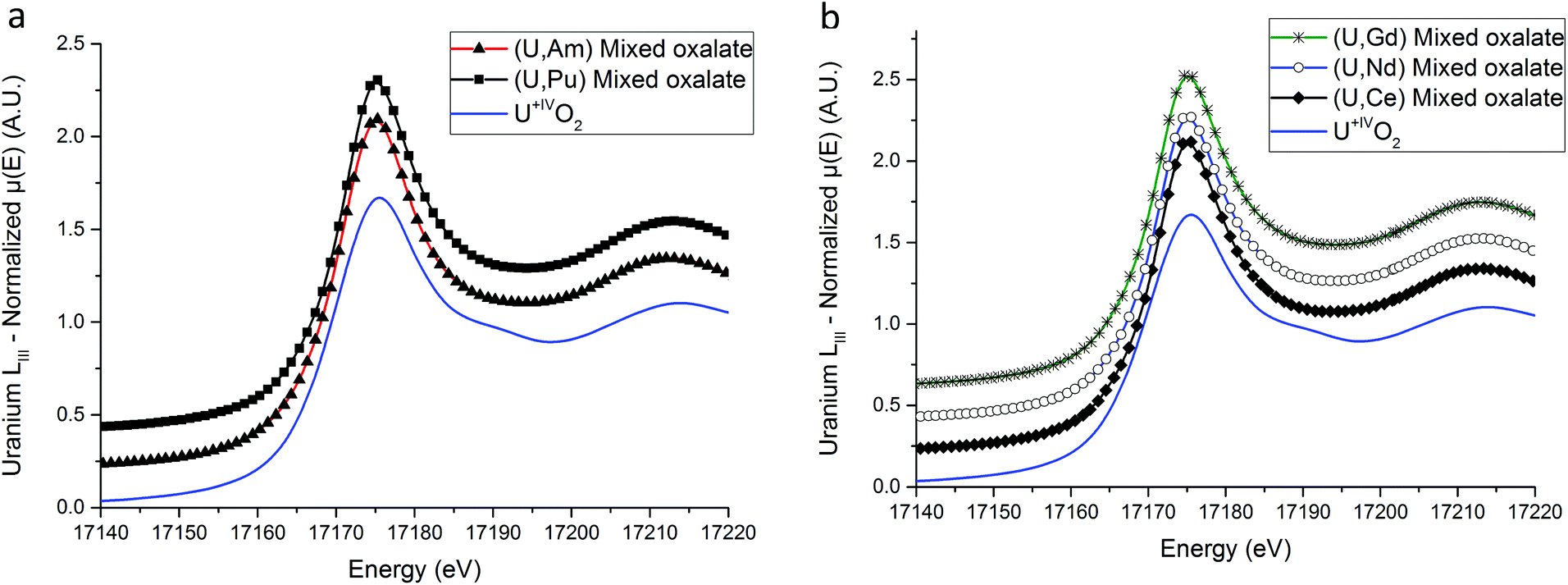 | ||
| Fig. 4 (U,An) (a) and (U,Ln) (b) uranium LIII XANES spectra. | ||
XANES spectra collected at Am LIII and Pu LIII edges are shown in Fig. 5a and b respectively. For the (U,Am) sample, the E0 and WL positions are located at 18513.1(5) eV and 18517.1(5) eV respectively. Both positions are clearly shifted to lower energies compared to AmO2 with 18514.5(5) and 18521.5(5) eV for the E0 and WL positions, respectively. The 4.4(5) eV difference between the WL positions is in very good agreement with the value of 4 eV observed between the WL positions of AmIVO2 and AmIII2O3 by Nishi et al.43 Based on this comparison, the +III state of americium in the (U,Am) mixed oxalate is evidenced. Concerning the (U,Pu) sample, both the E0 (18058.9(5) eV) and the WL (18062.8(5) eV) Pu-LIII XANES positions are slightly below the positions reported for PuIIIF331 which are equal to 18059.3(5) eV and 18063.3(5) eV for the E0 and the WL, respectively. Moreover, similarly to americium, a 4.3(5) eV shift to lower energy compared to the WL maximum reported for PuIVO2 (18067.1(5) eV)33 is observed.
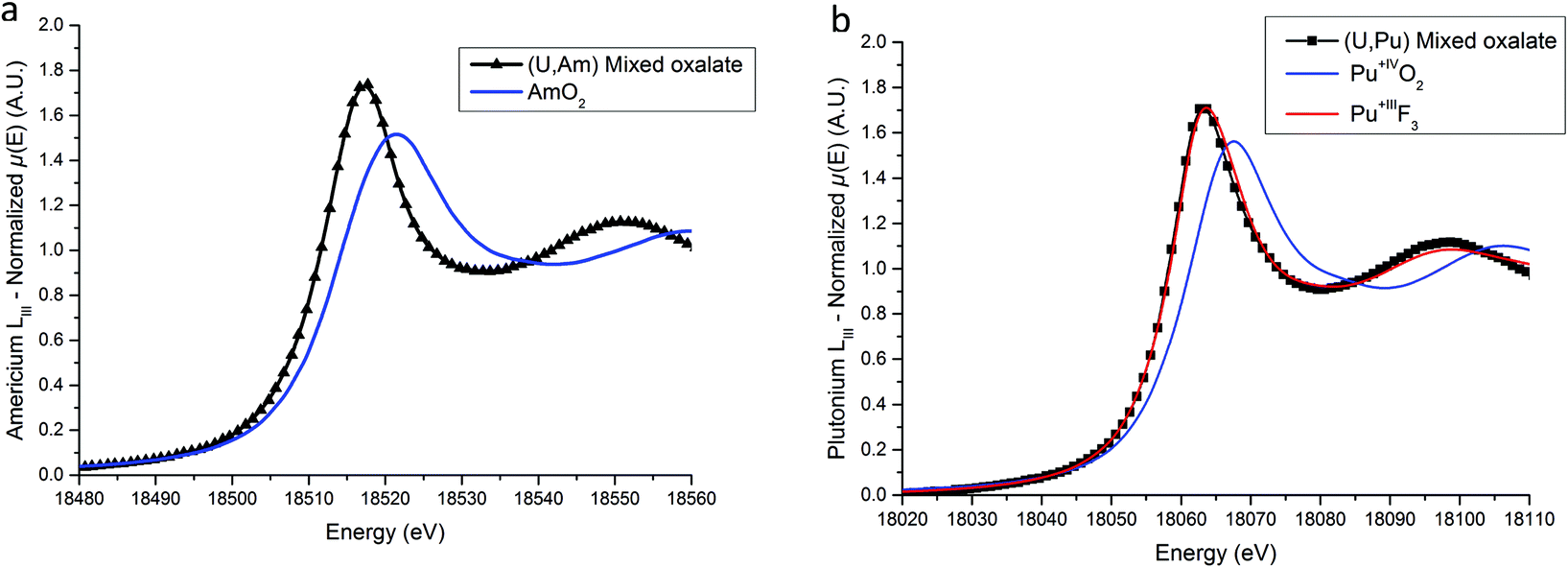 | ||
| Fig. 5 Americium LIII (a) and plutonium LIII (b) XANES spectra compared to reference samples. | ||
Based on these results, we can come to a conclusion on the accuracy of the formulations (UIV,AnIII) and (UIV,LnIII).
EXAFS results
Based on the hexagonal structure resolved by Arab-Chapelet et al.24 a view of a unit cell of mixed oxalates is given in Fig. 6a. Considering the lattice parameter values detailed in Table 2, the distances between actinide cations (a/3) are too large (>6 Å) to be accurately characterized using EXAFS. Despite this fact, the homogeneity at a molecular scale of the mixed oxalates can still be estimated, and in particular, whether the two different valence states of the actinides, confirmed with XANES, lead to the same number of oxalate ligands around U and Am or U and Pu. As shown in Fig. 6b, the uranium LIII-edge EXAFS spectra collected on (U,Am), (U,Pu) oxalates and to (U,Gd) oxalate are very similar. In their Fourier transforms, shown in Fig. 6c, the three intense peaks observed at ∼2, ∼3 and ∼4 Å are due to the three first coordination shells as illustrated by the diagram. Nevertheless, this simplified picture has to be modulated by the fact that the third peak is mostly due to the contribution of multiple-scattering paths as detailed in the materials and methods part.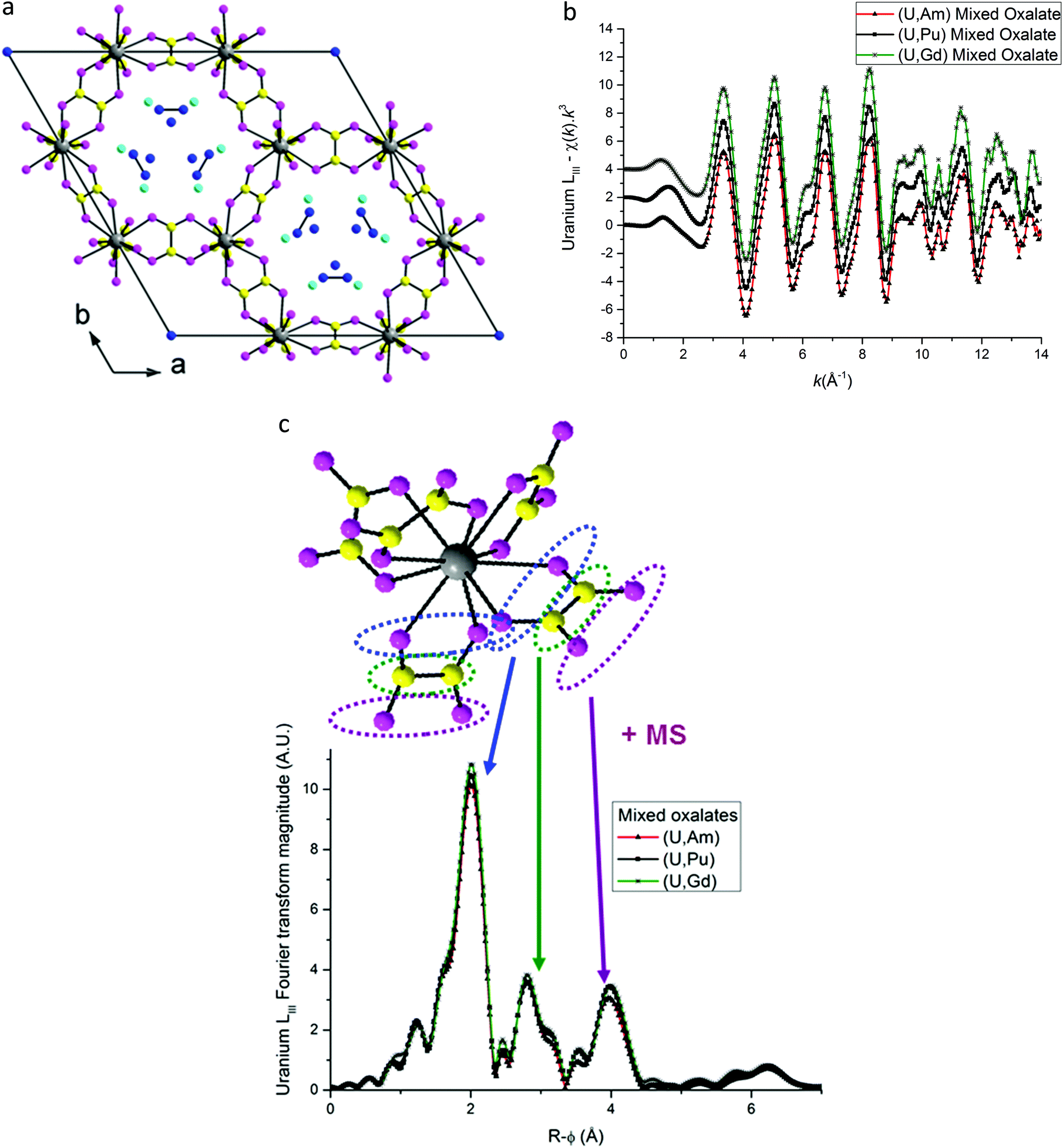 | ||
Fig. 6 (a) Hexagonal unit cell based on resolved (U,Nd) structure.24 ( Actinide/lanthanide cations, Actinide/lanthanide cations,  and and  oxygen and carbon atoms in the C2O4 group, and oxygen and carbon atoms in the C2O4 group, and  monovalent cations and monovalent cations and  oxygen atoms in water molecules). (b) EXAFS data at uranium LIII edge. (c) Fourier transformations of EXAFS shown in (b). oxygen atoms in water molecules). (b) EXAFS data at uranium LIII edge. (c) Fourier transformations of EXAFS shown in (b). | ||
The three FT are almost identical indicating that a similar environment is observed around uranium cations for the three samples.
EXAFS fit results for (U,Am) and (U,Pu) oxalate samples are displayed in Fig. 6–8, and associated metric values are given in Tables 4 and 5. The experimental data are very well reproduced by the fits as illustrated by the low Rfactor values obtained. For each sample, the three coordination shells corresponding to single-scattering paths (O1, C and O2), and coordination numbers equal to ∼10 (by taking into account uncertainties) are systematically obtained for both cations. This result is in very good agreement with the structure including five oxalate ions (C2O4) around U+IV, Pu+III and Am+III cations in the mixed (U0.9Pu0.1) and (U0.9Am0.1) oxalates. The same values of the Debye–Waller factors around both U and Am in the (U,Am) sample compared to values observed in (U,Pu) sample were measured. As the data were collected at 15 K, these similar σ2 values illustrate that a similar structural disorder is observed in the two mixed oxalates.
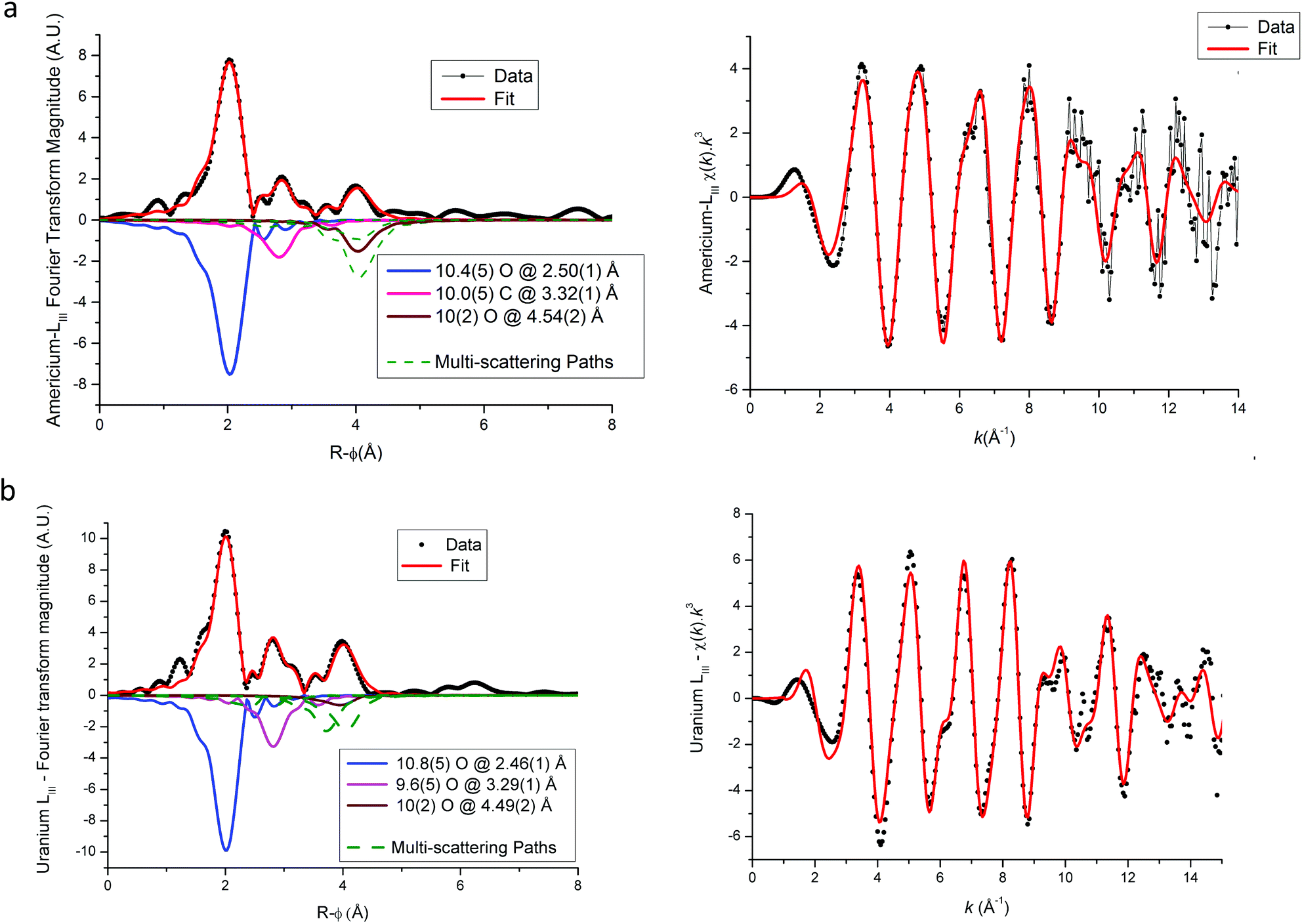 | ||
| Fig. 7 Americium (a) and uranium (b) LIII edge EXAFS and their Fourier transformations for U(IV)–Am(III) mixed oxalate (Experimental data in symbols, global fit in red lines). Individual scattering paths are displayed in negative (full lines for single-scattering paths and dashed green lines for multiple-scattering paths). | ||
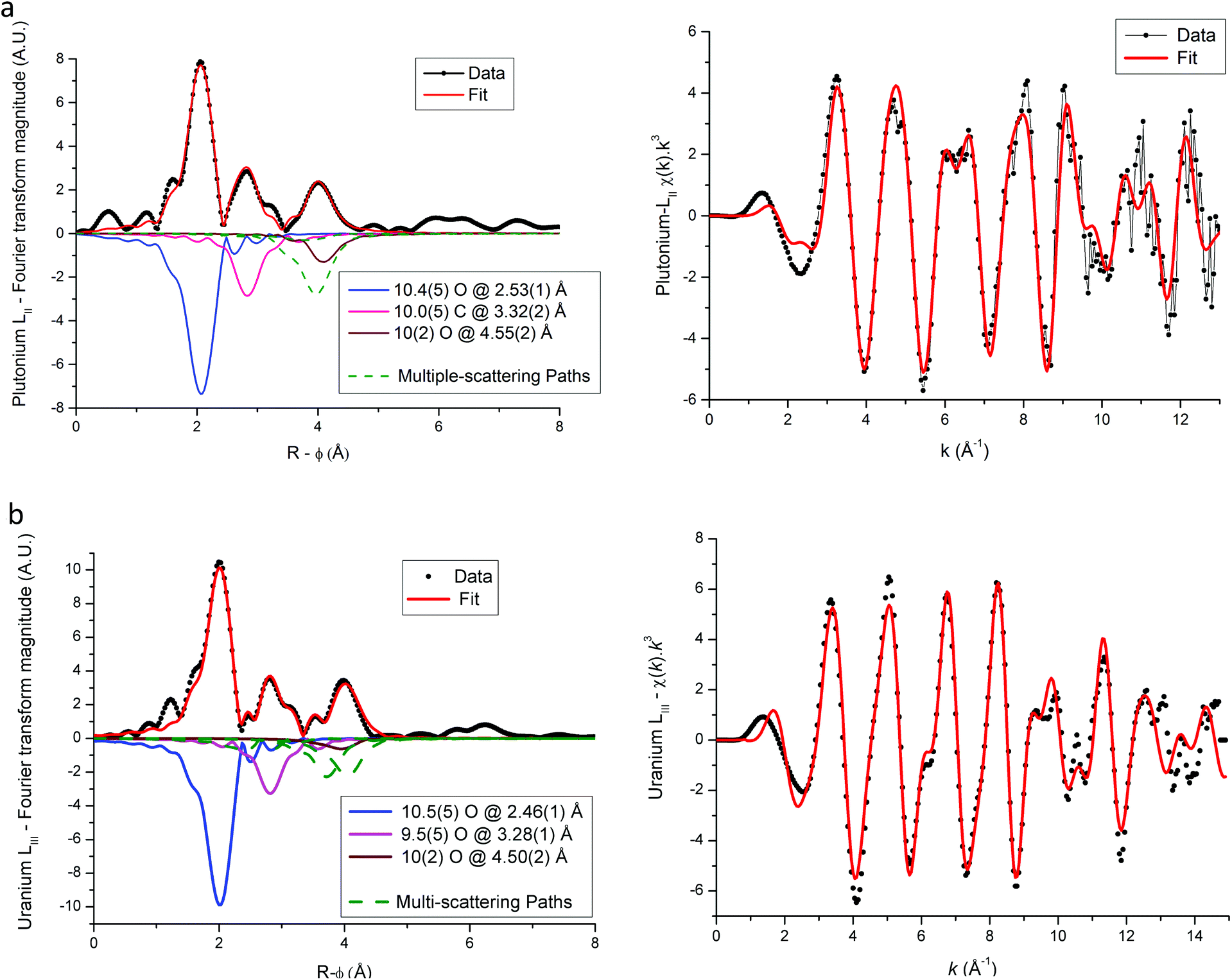 | ||
| Fig. 8 Plutonium LII (a) and uranium LIII (b) edged EXAFS and Fourier transform spectra for U(IV)–Pu(III) mixed oxalate. Individual scattering paths are displayed in negative (full lines for single-scattering paths and dashed green lines for multiple-scattering paths). | ||
| EXAFS | XRD | ||||
|---|---|---|---|---|---|
| Edge | Coordination shell | N | R (Å) | σ 2 (Å2) | R (Å) |
| For multiple-scattering paths, values with a star (*) correspond to the parameters fixed during the fit and are calculated using the correspond single-scattering paths. The XRD corresponds to the values calculated using the cell parameters given in Table 2. | |||||
| U-LIII | O (1) | 10.5(5) | 2.46(1) | 0.005(1) | 2.46 |
| C | 9.2(5) | 3.28(2) | 0.003(1) | 3.28 | |
| O (2) | 10(2) | 4.50(2) | 0.011(2) | 4.52 | |
| C → O(1) | 20* | 3.45(2) | 0.004* | 3.51 | |
| O(2) → C | 20* | 4.58(2) | 0.008* | 4.59 | |
| C → O(1) → C | 10* | 4.51(2) | 0.008* | 4.53 | |
| ΔE0 (eV) | 3.0(5) | ||||
| R factor (k1; k2; k3) | 0.004; 0.006; 0.011 | ||||
| Pu-LII | O (1) | 10.4(5) | 2.53(1) | 0.007(1) | 2.46 |
| C | 10.0(5) | 3.32(1) | 0.004(1) | 3.28 | |
| O (2) | 10(2) | 4.55(2) | 0.004(1) | 4.52 | |
| O(2) → C | 20* | 4.55(2) | 0.004* | 4.59 | |
| C → O(1) → C | 10* | 4.55(2) | 0.005* | 4.53 | |
| ΔE0 (eV) | 3.0(5) | ||||
| R factor (k1; k2; k3) | 0.003; 0.004; 0.008 | ||||
| EXAFS | XRD | ||||
|---|---|---|---|---|---|
| Edge | Coordination shell | N | R (Å) | σ 2 (Å2) | R (Å) |
| For multiple-scattering paths, values with a star (*) correspond to the parameters fixed during the fit and are calculated using the corresponding single-scattering paths. The XRD corresponds to the values calculated using the cell parameters given in Table 2. | |||||
| U–LIII | O (1) | 10.8(5) | 2.46(1) | 0.006(1) | 2.46 |
| C | 9.6(5) | 3.28(2) | 0.005(1) | 3.29 | |
| O (2) | 10(2) | 4.50(2) | 0.012(2) | 4.52 | |
| C → O(1) | 20* | 3.45(2) | 0.004* | 3.51 | |
| O(2) → C | 20* | 4.60(2) | 0.008* | 4.55 | |
| C → O(1) → C | 10* | 4.49(2) | 0.004* | 4.51 | |
| ΔE0 (eV) | 3.1(5) | ||||
| R factor (k1; k2; k3) | 0.010; 0.012; 0.015 | ||||
| Am–LIII | O (1) | 10.4(5) | 2.50(1) | 0.007(1) | 2.46 |
| C | 10.0(5) | 3.32(1) | 0.007(1) | 3.29 | |
| O (2) | 10(2) | 4.54(2) | 0.004(1) | 4.52 | |
| C → O(1) | 20* | 3.54(2) | 0.007* | 3.51 | |
| O(2) → C | 20* | 4.59(2) | 0.006* | 4.55 | |
| C → O(1) → C | 10* | 4.62(2) | 0.006* | 4.51 | |
| ΔE0 (eV) | 0.3(5) | ||||
| R factor (k1; k2; k3) | 0.003; 0.005; 0.012 | ||||
For each compound, the only significant differences observed between the two actinides are systematically shorter distances around uranium than around Pu or Am. These differences could be explained by the difference in ionic radii between U+IV and trivalent Pu and Am, but these values are unknown for such cations that are ten-coordinated. Nevertheless, based on values available for americium(III) and uranium(IV) cations for a coordination number (CN) of 9 (Am+III = 1.162 Å (ref. 23) and U+IV = 1.05 Å (ref. 22)), the Am+III–O bond distance would be significantly longer (about 0.11 Å) than the U+IV–O bond. Similar values would be observed for Pu+III–O and Am+III–O distances as Pu and Am have very similar ionic radii (Am+III = 1.162 Å (ref. 23) and Pu+III = 1.165 Å (ref. 23) for CN = 9). But, as shown in Tables 4 and 5 the differences with U+IV–O determined by EXAFS in this work are only 0.04 Å and 0.07 Å for americium(III) and plutonium(III), respectively.
Conclusion
Mixed (U0.9Pu0.1) and (U0.9Am0.1) oxalates have been synthesized by oxalic co-precipitation in nitric acid medium. The stabilization of the trivalent state for plutonium and the tetravalent one for uranium during the precipitation step has been demonstrated by XANES measurements. Moreover, the EXAFS measurements confirmed that in these mixed oxalates, U+IV, Pu+III and Am+III cations have the same coordination sphere based on five bidentate oxalate ligands. The metal–oxygen distances calculated by the XAS measurements are in agreement with those calculated from the XRD lattice parameters. Thus, these multiscale structural characterizations have confirmed the formation of actinide(IV,III) oxalate solid solutions. Finally, this study confirms that the oxalic co-precipitation process is a convenient route for actinide co-conversion into oxalate solid solutions, which can be used as precursors of actinide oxide solid solutions with a homogeneous actinide distribution at a molecular scale.References
- F. Gauche, Energy Procedia, 2011, 7, 314–316 CrossRef.
- D. Warin, J. Nucl. Sci. Technol., 2007, 44, 410–414 CrossRef CAS.
- F. Lebreton, D. Prieur, D. Horlait, T. Delahaye, A. Jankowiak, C. Léorier, F. Jorion, E. Gavilan and F. Desmoulière, J. Nucl. Mater., 2013, 438, 99–107 CrossRef CAS.
- F. Lebreton, D. Prieur, A. Jankowiak, M. Tribet, C. Léorier, T. Delahaye, L. Donnet and P. Dehaudt, J. Nucl. Mater., 2012, 420, 213–217 CrossRef CAS.
- D. Prieur, A. Jankowiak, T. Delahaye, N. Herlet, P. Dehaudt and P. Blanchart, J. Nucl. Mater., 2011, 414, 503–507 CrossRef.
- T. Delahaye, F. Lebreton, D. Horlait, N. Herlet and P. Dehaudt, J. Nucl. Mater., 2013, 432, 305–312 CrossRef CAS.
- F. Lebreton, D. Horlait and T. Delahaye, Procedia Chem., 2012, 7, 499–504 CrossRef CAS.
- D. Horlait, F. Lebreton, A. Gauthé, M. Caisso, B. Arab-Chapelet, S. Picart and T. Delahaye, J. Nucl. Mater., 2014, 444, 181–185 CrossRef CAS.
- E. Remy, S. Picart, T. Delahaye, I. Jobelin, F. Lebreton, D. Horlait, I. Bisel, P. Blanchart and A. Ayral, J. Nucl. Mater., 2014, 453, 214–219 CrossRef CAS.
- S. Vaudez, R. C. Belin, L. Aufore, P. Sornay and S. Grandjean, J. Nucl. Mater., 2013, 442, 227–234 CrossRef CAS.
- A. K. Chadha, K. D. S. Mudher, I.L. Krishnan and N. C. Jayadevan, Thermochim. Acta, 1991, 191, 323–331 CrossRef CAS.
- K. D. S. Mudher, I. L. Krishnan, A. K. Chadha and V. Venugopal, Thermochim. Acta, 1997, 297, 169–175 CrossRef CAS.
- S. Grandjean, A. Bérès, J. Rousselle and C. Maillard, WO, 2005119699, 2005 Search PubMed.
- B. Chapelet-Arab, S. Grandjean, G. Nowogrocki and F. Abraham, J. Alloys Compd., 2007, 444–445, 387–390 CrossRef.
- S. Grandjean, B. Arab-Chapelet, M. Bertrand, S. Picart, P. Baron, P. Blanc and D. Warin, Proceedings of Global, Paper 9441, Paris, France, 2009 Search PubMed.
- S. Grandjean, B. Arab-Chapelet, A.-C. Robisson, F. Abraham, Ph. Martin, J.-Ph. Dancausse, N. Herlet and C. Léorier, J. Nucl. Mater., 2009, 385, 204–207 CrossRef CAS.
- M. J. Sarsfield, in Reprocessing and Recycling of Spent Nuclear Fuel, ed. R. Taylor, Woodhead publishing series in nuclear energy Nr. 79, 2015, ch. 13 Search PubMed.
- B. Arab-Chapelet, S. Grandjean, G. Nowogrocki and F. Abraham, J. Nucl. Mater., 2008, 373, 259–268 CrossRef CAS.
- D. Horlait, N. Clavier, N. Dacheux, R. Cavalier and R. Podor, Mater. Res. Bull., 2012, 47, 4017–4025 CrossRef CAS.
- C. Tamain, B. Arab-Chapelet, M. Rivenet, F. Abraham, R. Caraballo and S. Grandjean, Inorg. Chem., 2013, 52, 4941–4949 CrossRef CAS PubMed.
- G. T. Seaborg, Radiochim. Acta, 1993, 61, 115–122 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Cryst., 1976, 32, 751–767 CrossRef.
- J. N. Cross, E. M. Villa, S. Wang, J. Diwu, M. J. Polinski and T. E. Albrecht-Schmitt, Inorg. Chem., 2012, 51, 8419–8424 CrossRef CAS PubMed.
- B. Chapelet-Arab, G. Nowogrocki, F. Abraham and S. Grandjean, J. Solid State Chem., 2005, 178, 3046–3054 CrossRef CAS.
- B. Chapelet-Arab, G. Nowogrocki, F. Abraham and S. Grandjean, J. Solid State Chem., 2005, 178, 3055–3065 CrossRef CAS.
- B. Chapelet-Arab, L. Duvieubourg, G. Nowogrocki, F. Abraham and S. Grandjean, J. Solid State Chem., 2006, 179, 4029–4036 CrossRef CAS.
- DIFFRACplus EVA, Bruker AXS, Karlsruhe, Germany, 2007 Search PubMed.
- J. Rodriguez-Carjaval, Abstracts of the satellite on powder diffraction of the XV congress of the IUCr, 127, Toulouse, France, 1990 Search PubMed.
- W. Matz, N. Schell, G. Bernhard, F. Prokert, T. Reich, J. Claußner, W. Oehme, R. Schlenk, S. Dienel, H. Funke, F. Eichhorn, M. Betzl, D. Pröhl, U. Strauch, G. Hüttig, H. Krug, W. Neumann, V. Brendler, P. Reichel, M. A. Denecke and H. Nitsche, J. Synchrotron Radiat., 1999, 6, 1076 CrossRef CAS.
- http: //sixpack.sams-xrays.com/ .
- P. M. Martin, S. Grandjean, C. Valot, G. Carlot, M. Ripert, P. Blanc and C. Hennig, J. Alloys Compd., 2007, 444–445, 410–414 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- R. C. Belin, P. M. Martin, P. J. Valenza and A. C. Scheinost, Inorg. Chem., 2009, 48, 5376–5381 CrossRef CAS PubMed.
- J. J. Rehr and R.C. Albers, Rev. Mod. Phys., 2000, 72, 621–654 CrossRef CAS.
- D. Prieur, P. M. Martin, A. Jankowiak, E. Gavilan, A. C. Scheinost, N. Herlet, P. Dehaudt and P. Blanchart, Inorg. Chem., 2011, 50, 124–137 CrossRef PubMed.
- D. Prieur, P. M. Martin, F. Lebreton, T. Delahaye, A. Jankowiak, J. P. Laval, A. C. Scheinost, P. Dehaudt and P. Blanchart, J. Solid State Chem., 2012, 194, 206 CrossRef CAS.
- J.-F. Vigier, P. M. Martin, L. Martel, D. Prieur, A. C. Scheinost and J. Somers, Inorg. Chem., 2015, 54, 5358–5365 CrossRef CAS PubMed.
- C. J. Mandleberg, K. E. Francis and R. Smith, J. Chem. Soc., 1961, 2464–2468 RSC.
- A. Gil-Martin, B. Arab-Chapelet, M. Rivenet, S. Grandjean, G. Nowogrocki and F. Abraham, Procedia Chem., 2012, 7, 33 CrossRef CAS.
- V. Tyrpekl, J.-F. Vigier, D. Manara, T. Wiss, O. Dieste Blanco and J. Somers, J. Nucl. Mater., 2015, 460, 200–208 CrossRef CAS.
- W. Ollendorff and F. Weigel, Inorg. Nucl. Chem. Lett., 1969, 5, 263–269 CrossRef CAS.
- W. Runde, L. F. Brodnax, A. Bean and B. L. Scott, Inorg. Chem., 2009, 4, 5967–5972 CrossRef PubMed.
- T. Nishi, M. Nakada, C. Suzuki, H. Shibata, A. Itoh, M. Akabori and M. Hirata, J. Nucl. Mater., 2010, 401, 138–142 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |