
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sedaxicenes: potential new antifungal ferrocene-based agents?†
R.
Rubbiani
*,
O.
Blacque
and
G.
Gasser
 *
*
Department of Chemistry, University of Zurich, Winterthurerstrasse 190, 8057 Zurich, Switzerland. E-mail: riccardo.rubbiani@chem.uzh.ch; gilles.gasser@chem.uzh.ch; Fax: +41-44-635-68-02; Tel: +41-44-635-46-86, +41-44-635-46-30
First published on 1st March 2016
Abstract
Fungal infections are a group of diseases spread all over the world with an extremely high morbidity. Worryingly, although several pathogenic fungi were found to develop resistance towards traditional therapy, research towards the discovery of novel antimycotic agents is very limited. Considering the promising results obtained with the ferrocene-based drug candidates Ferroquine and Ferrocifen as antimalarial and anticancer drug candidates, respectively, we envisaged derivatizing the organic scaffold of a new broad-spectrum fungicide, namely sedaxane, with a ferrocenyl moiety in order to obtain new metal-based antifungal agents. The new ferrocenyl sedaxane derivatives called herein Sedaxicenes (1a, 2 and 3) were characterized using different analytical techniques and the structures were confirmed by X-ray crystallography. As expected for antimycotic agents, 1a, 2 and 3 were found to have a low or even no toxicity towards human cells (IC50 > 100 μM). Interestingly, while the parent drug did not display any mycotoxicity (EC50 > 100 μM), complex 1a was found to have some antifungal activity with an IC50 value of 43 μM under the same experimental conditions. In order to investigate the possible redox-mediated mode of action of 1a, we synthesized the ruthenocene analogue of 1a, namely 1b. Ruthenocene is known to have a completely different electrochemical behaviour from ferrocene although both the compounds are isostructural. As anticipated, complex 1a was found to induce an increase of the reactive oxygen species level in S. cerevisiae, contrary to its 1b analogue and to the parent compound sedaxane.
Introduction
Fungal infections are provoked by tiny parasites present in water, soil, plants, invertebrates and mammals called fungi.1 Fungi belong to a broad family of eukaryotes which display features at the crossroads between mammalian cells and bacteria.2–4 Like eukaryotes, they possess the genetic material located in the nucleus, mitochondria, an endoplasmic reticulum and a digestion vacuole system. Similar to bacteria, they are a spore-producing microorganism.2–4 Fungi also have a characteristic cell wall responsible for nutrient trafficking with the host or the environment and with a protective function.5Despite their limited mortality (<10 million per year), fungi displayed a very alarming morbidity.6 For example, the lifetime incidence of candidiasis (fungal infections provoked by the Candida yeast family) in women reaches 75% and the recent data showed that 25% of the whole world population is affected by Tinea pedis (a mold responsible for the “ringworm of the foot”).6 This represents several hundred millions of infections per year, a number comparable to those of serious pathologies like malaria (data of the World Health Organization, WHO, updated December 2014). Notably, new diseases linked to new human life conditions (e.g. diabetes, obesity, cancer) enable pathogenic fungi to cause severe complications. Fungal infections are therefore involved in the worsening of other diseases, impacting the economic system of different nations. In the worst cases, they lead to human death.7 Surprisingly, conventional therapies against fungi rely on a very limited number of drugs. These therapeutic agents were discovered in the last century and just 7 of them are nowadays in the list of WHO – Essential Medicine (data of October 2013).8 Moreover, antifungal research is stagnant, especially if compared to other pathologies. In the last few decades, just a few new antifungal agents have been unveiled and they were mainly based on structural modifications of the already discovered active drugs. Worryingly, there have recently been an increasing number of reports showing that different pathological fungal strains are developing resistance against conventional therapy.9 This alarming observation is a strong incentive for the discovery of novel compounds or strategies to fight this clearly underestimated problem.
Research on the medicinal potential of metal complexes has witnessed a very rapid growth in the last few decades. Platinum- and gadolinium-based compounds are nowadays heavily used as anticancer and imaging agents, respectively. Other metal complexes like Ferroquine (containing iron) or KP1339 (containing ruthenium) are in clinical trials.10 The reason behind this “success story” is that metal complexes present very favourable features, which make them, in some cases, superior to organic compounds. For example, metal complexes display an enhanced stereochemistry and reactivity compared with organic molecules and can have a large repertoire of physico-chemical properties (lipophilicity, redox potential, etc.).10,11 Surprisingly, there are only a limited number of studies on the activity of metal complexes [Co(II), Ni(II), Cu(II), Mn(I), Pd(II) Bi(III) complexes made a variety of ligands (e.g. azole moieties, thiosemicarbazones, carboxamides, indoles)] against fungal strains, hence leaving this very field pretty much unexplored.12 Due to the encouraging results obtained by many research groups on the biological potential of metallocenes,12,13 we recently embarked on a project to design novel metal-based antifungal agents. The synthesis, characterisation and biological evaluation of three new organometallic complexes based on a known fungicide are herein reported.
Results and discussion
Drug design
Recently, a potent broad-spectrum fungicide, namely sedaxane (ISO common name, CAS no. 874967-67-6, see Scheme 1 for chemical structure), was approved as a pesticide.15 Sedaxane efficiently inhibits the fungal enzyme succinate dehydrogenase involved in the fungal oxidative phosphoryalation.15 Of utmost interest is the fact that sedaxane displayed low toxicity and good clearance, with the majority of the drug excreted within 24 h (in mice models). Moreover, no oral, inhaling or dermal toxicity was observed for concentrations below 5 g per kg per day.14 Interestingly, there are no studies on the possible application of sedaxane to fight human fungal infections.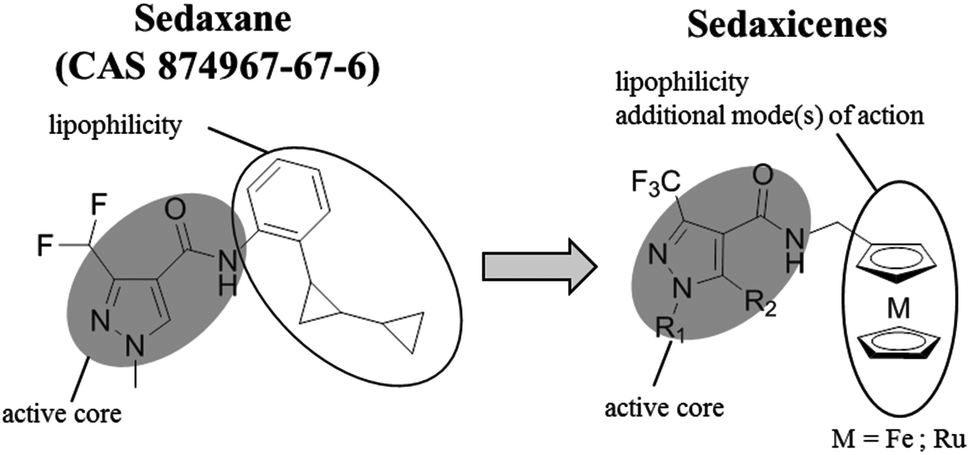 | ||
| Scheme 1 Structures of sedaxane and sedaxicenes. The role of each unit in the molecules is described. | ||
Motivated by the work of several research groups which derivatized a known drug with a ferrocenyl moiety to obtain a synergistic increase in activity (e.g. Ferrocifen, Ferroquine, ferrocenyl praziquantel, ferrocenyl platensimycin, etc.),10,11 we envisaged coupling the favourable therapeutic profile of sedaxane with the potential of the ferrocenyl group (see Scheme 1).12,13
In this work, the core of sedaxane was modified at the bicycloprop-2-yl group, replacing it with a methylferrocene moiety. This replacement was assumed to not influence the overall bioactivity, which is hypothesized to be carried out by the pyrazole carboxamide group by interacting with the iron cluster of the active site of the enzyme.14 Additionally, the presence of the ferrocenyl group was anticipated to afford a potential synergistic mode(s) of action due to the presence of a redox moiety. It was, for example, demonstrated by Biot and co-workers that the presence of the ferrocenyl moiety in Ferroquine was allowing for the formation of ˙OH. Since chloroquine cannot produce ˙OH, this radical generation was assumed to be responsible for the activity of Ferroquine on chloroquine-resistant parasite strains.12,13 To further confirm the contribution of redox activity to the overall antifungal profile of our ferrocenyl derivatives, we synthesized a ruthenocenyl sedaxane analogue (1b, see Scheme 1). Ruthenocene is isostructural to ferrocene but has a significantly different electrochemical behaviour. This difference was shown in the past to play an important role in the formation or not of ROS by Biot and co-workers (e.g. the ruthenocene analogue of Ferroquine, namely Ruthenoquine was not producing ROS while Ferroquine was).13 Of note is that different alkylic moieties were also inserted into the pyrazole core to have a small structure activity relationship (SAR) study.
Synthetic procedures
The synthesis of the metallocenyl analogues of sedaxane, that we have named sedaxicenes, was performed according to the established protocols (see Scheme 2).16 The products 1a, 1b, 2 and 3 were all obtained in moderate yields (23–58%). The new complexes were characterized by 1H, 13C and 19F NMR spectroscopy as well as ESI-MS and X-ray diffraction studies, and their purity was assessed by elemental analysis (<0.5% from the calculated value).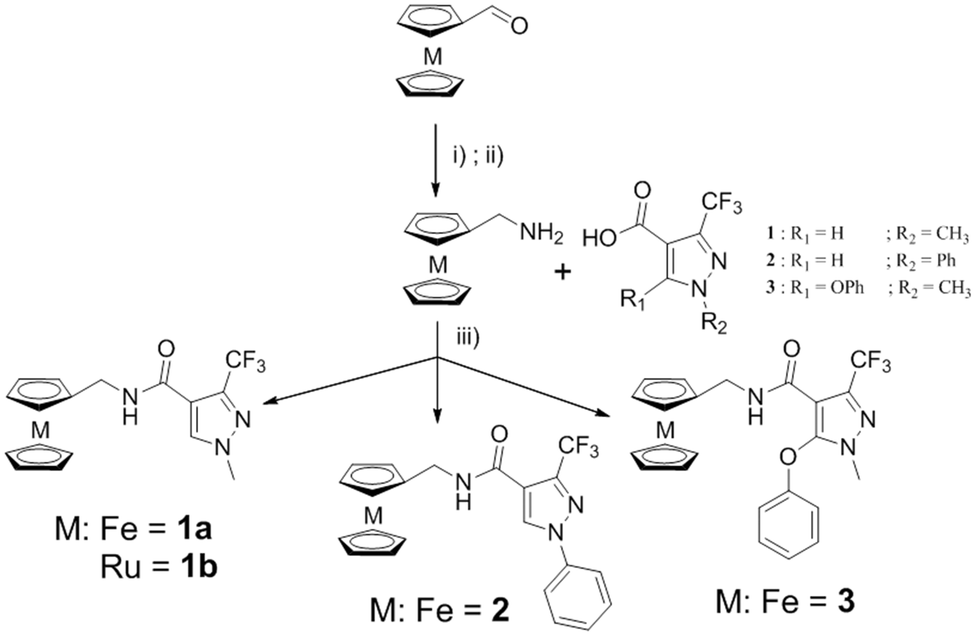 | ||
| Scheme 2 Synthesis of the new antifungal ferrocenyl drug candidates; (i) NH2OH in EtOH under reflux for 4 h; (ii) LiAlH4 in THF under reflux for 3 h, (iii) DIPEA, HATU in DMF at room temperature for 24 h. | ||
The X-ray diffraction studies were performed on suitable single crystals which were obtained after slow evaporation of a concentrated solution of the complex in chloroform at 25 °C over a few days. The exemplar perspective view of the molecular structure of complex 1a is shown in Fig. 1 (see the ESI† for the molecular structures of 2 and 3). Specific features that confirmed the presence of the products were the disappearance of the acidic proton signal between 12.5 and 13.0 ppm in the 1H NMR spectra, the up-field shift of the carbonyl carbon from ∼190 ppm to ∼160 ppm in the 13C NMR spectra, the presence of a single signal in the 19F NMR spectra and the M+ peak in the mass spectra.
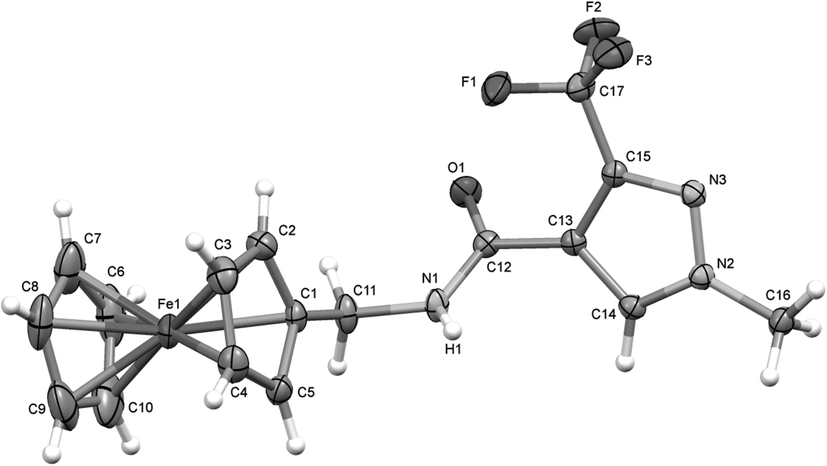 | ||
| Fig. 1 X-ray crystal structure of 1a with selective atomic numbering scheme. Thermal ellipsoids are drawn at the 30% probability level. | ||
Host cytotoxicity
With the steady increase of mycoses, antifungal agents are required to be often administered both topically and systemically, and to lack therefore host toxicity. In order to investigate the impact of our sedaxicenes on the host organism, 1a, 2 and 3 were tested for their cytotoxicity against human cells in vitro. For these experiments, two cells lines were chosen, namely human fibroblast (MRC-5) and retinal pigment epithelial (RPE1 hTert), representing both superficial and internal cells, respectively. The influence of the metal complexes on the cellular proliferation was aimed to provide important information on the efficacy of the sedaxicenes as topical as well as systemic drug candidates. Sedaxane was used as a reference for comparative purposes and Clotrimazole as a positive control (see Table 1). As expected, sedaxane did not affect the cell viability. Complexes 1b, 2 and 3 showed a very mild toxicity, in the mid-micromolar range (towards both the cell lines for 2 and just towards MRC-5 for 1b and towards RPE for 3). Remarkably, complex 1a did not display any activity towards both cell lines. Interestingly, Clotrimazole was found to have a higher cytotoxicity than all sedaxicenes, with IC50 values in the low micromolar range.| Compound | Human cell lines | S. Cerevisiae | |
|---|---|---|---|
| MRC-5 | RPE | (OD = 0.02) | |
| Clotrimazole | 35.8 ± 3.5 | 13.3 ± 0.6 | 0.66 ± 0.26 |
| Sedaxane | >100 | >100 | >100 |
| Complex 1a | >100 | >100 | 43.3 ± 6.2 |
| Complex 1b | 41.0 ± 5.3 | >100 | >100 |
| Complex 2 | 48.9 ± 3.1 | 50.5 ± 7.9 | 98.6 ± 1.7 |
| Complex 3 | >100 | 67.8 ± 9.4 | 91.9 ± 3.6 |
Antifungal activity
Once established that the sedaxicenes had no or only minor toxicity towards human cell lines, the antifungal profile of these metal complexes was evaluated. Saccharomyces cerevisiae (SC) was chosen as a valid fungus model for preliminary biological evaluation.17 The wild type colony of SC was cultured and diluted to reach the beginning of the growing phase just before treatment. SC was then seeded at different concentrations on the agar terrain poured into a 12-well plate and pre-treated with different concentrations of the target sedaxicenes. The plates were then incubated for 24 h at 30 °C and the colony formation was monitored via an Alpha Digitec camera with white light excitation. Sedaxane was used in the study for comparative purposes and Clotrimazole as a positive control.Complexes 1a, 2 and 3 were found to reduce the colony formation of SC, displaying moderate IC50 values in the mid-high micromolar range. Complex 1a, bearing a less hindered pyrazole ring in comparison with 2 and 3, was the most active throughout the series with an EC50 value of 43 μM (see Table 1 and Fig. 2 for an exemplar depicted experiment). Interestingly, sedaxane as well as 1b did not display any activity up to the highest concentration used in this study (100 μM).
 | ||
| Fig. 2 SC growth inhibition upon treatment with increasing concentrations of sedaxane and complex 1a (exemplary figures concerning 1b, 2, 3 and Clotrimazole are reported in the ESI†); experiment performed at different SC growing indexes (OD = 0.002 and 0.02, respectively). | ||
Reactive oxygen species evaluation
The drug design of the sedaxicenes was based on the insertion of a ferrocenyl moiety into the known broad-spectrum fungicide sedaxane. The rationale behind this approach is that the redox iron centre could improve the therapeutic profile of sedaxane with (an) additional mode(s) of action. Complex 1a displayed the best antimycotic profile of all compounds tested in this study. In order to assess the possible redox activity of complex 1a, we investigated the rise of the reactive oxygen species (ROS) level in fungal culture upon treatment with this compound and compared the data with sedaxane (as parent drug) and 1b (as redox inactive 1a analogue). For this purpose, the cell-permeable dye 2′,7′-dichlorofluorescein di-acetate (H2DCF-DA), which turns to be highly fluorescent upon oxidation in the presence of ROS, was used, as previously reported by our group for a cytotoxic Au(III) complex.18 Hence, SC culture was treated for 16 h with 100 μM of complexes 1a and 1b. The medium was then replaced with fresh YPD (not containing the metal compounds) treated with H2DCF-DA. The SC culture was further incubated for 1 h, allowing for the internalization of the dye before the ROS level was quantified. Strikingly, the ROS level in SC treated with complex 1a displayed an increase of 33% in comparison with the untreated control (see Fig. 3). This effect was not observed in the case of 1b. SC treated with sedaxane showed a similar ROS level than the basal value, suggesting that the ferrocenyl moiety is playing indeed a pivotal role to achieve a valuable fungicidal profile.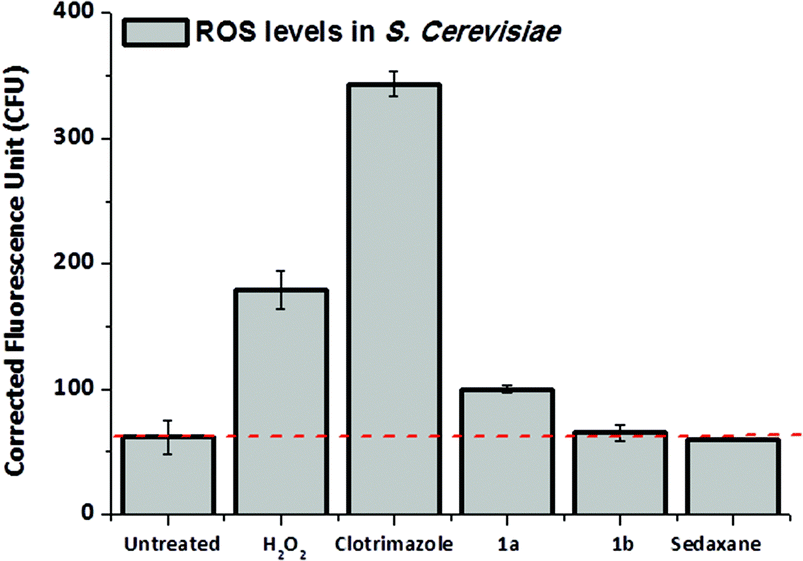 | ||
| Fig. 3 ROS level determination in SC upon treatment with complex 1a and 1b; sedaxane (100 μM) was used for comparative purposes, H2O2 (10 μM) and Clotrimazole (5 μM) as a positive control. | ||
Experimental
General
Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane:acetone, 7:2:1, Rf = 0.70). The fractions were collected and the solvent was removed in vacuo to obtain yellow powder.
:hexane:acetone, 7:2:1, Rf = 0.70). The fractions were collected and the solvent was removed in vacuo to obtain yellow powder.
Ruthenocenyl oxyme was then dissolved in dry 5 mL of THF and reduced to methylaminoferrocene in the presence of LiAlH4 (190 mg, 5 mmol) under reflux conditions for 6 h under a N2 atmosphere. The mixture was cooled down and hydrolyzed with 20 mL of bidistilled water. Methylaminoruthenocene was then extracted with diethylether. The combined organic layers were then removed under reduced pressure.
3-(Trifluoromethyl)-1-methyl-1H-pyrazole-4-carboxylic acid (0.50 mmol) was dissolved in dry DMF (2 mL) under a N2 atmosphere. This solution was added to a 1 mL solution of N,N′-diisopropylethylamine (DIPEA, 1.5 mmol) and 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) (1.5 mmol) in dry DMF. The new solution was then stirred for 20 min at room temperature. Methylaminoruthenocene dissolved in 10 mL of dry DMF was then added to the mixture which was then stirred in the dark overnight. The solvent was evaporated using a high-vacuum pump. The reddish oil was then purified by column chromatography (silica, ethylacetate:hexane, 8:2, Rf = 0.75). The fractions were collected and the solvent was removed in vacuo to obtain a brownish powder. Yield 58%. 1H NMR (500 MHz), CDCl3, δ (ppm) 7.91 (s, 1H, ArH), 6.10 (s, 1H, NH), 4.59 (m, 2H, RcH) 4.50 (m, 5H, RcH), 4.49 (m, 2H, RcH), 4.13 (d, 2H, CH2), 3.93 (s, 3H, CH3); 13C NMR (500 MHz), CDCl3, δ (ppm) 160.6, 136.3, 122.7, 120.6, 117.9, 89.0, 71.2, 70.8, 70.5, 40.4, 39.4; 19F NMR (300 MHz), CDCl3, δ (ppm) −55.8; MS (ESI+): m/z 438.1 [M + H]+, 896.1 [2M + Na]+; elemental analysis (%) for [C17H16F3RuN3O]: calc. C: 46.79, H: 3.70, N: 9.63; found C: 46.83, H: 3.73, N: 9.90.
Crystallographic data collections and structure refinements
Single-crystal X-ray diffraction data were collected at 183(1) K on an Agilent Technologies SuperNova diffractometer for 1a–2 and on an Agilent Technologies Xcalibur Ruby area-detector diffractometer for 3 using a single wavelength Enhance X-ray source with Mo Kα radiation (λ = 0.71073 Å) for 1a and 3 or with Cu Kα radiation (λ = 1.54184 Å) for 2 from a micro-focus X-ray source, and an Oxford Instrument's Cryojet XL cooler. The selected suitable single crystal was mounted using polybutene oil on a flexible loop fixed on a goniometer head and immediately transferred to the diffractometer. Pre-experiment, data collection, data reduction and analytical absorption correction19 were performed with the program suite CrysAlisPro.19 Using Olex2,19 the structures were solved with the ShelXS9719 structure solution program using direct methods and refined with the SHELXL201419 program package by full-matrix least-squares minimization on F2. PLATON19 was used to check the result of the X-ray analyses. All non-H atoms were anisotropically refined. All hydrogen positions were calculated after each cycle of refinement using a riding model with C–H = 0.93 Å and Uiso(H) = 1.2Ueq(C) for aromatic H atoms, with C–H = 0.97 Å and Uiso(H) = 1.2Ueq(C) for methylene H atoms, and with C–H = 0.96 Å and Uiso(H) = 1.5Ueq(C) for methyl H atoms, except for the amido H atoms which were freely refined. For more details about the data collection and refinements parameters, see the Crystallographic Information files (ESI†). Crystal data for C17H16F3FeN3O (1a) (M = 391.18 g mol−1): orthorhombic, space group Pbca (no. 61), a = 10.3579(2) Å, b = 8.72054(14) Å, c = 36.8998(7) Å, V = 3333.04(10) Å3, Z = 8, T = 183(1) K, μ(MoKα) = 0.946 mm−1, Dcalc = 1.559 g cm−3, 22551 reflections measured (4.416° ≤ 2Θ ≤ 50.692°), 3034 unique (Rint = 0.0293, Rsigma = 0.0172) which were used in all calculations. The final R1 was 0.0380 (I > 2σ(I)) and wR2 was 0.0929 (all data). Crystal data for C22H18F3FeN3O·CHCl3 (2) (M = 572.61 g mol−1): monoclinic, space group I2/a (no. 15), a = 10.6241(2) Å, b = 15.1983(4) Å, c = 29.6784(7) Å, β = 90.352(2)°, V = 4792.03(19) Å3, Z = 8, T = 183(1) K, μ(CuKα) = 8.534 mm−1, Dcalc = 1.587 g cm−3, 16614 reflections measured (5.956° ≤ 2Θ ≤ 136.496°), 4393 unique (Rint = 0.0432, Rsigma = 0.0284) which were used in all calculations. The final R1 was 0.0370 (I > 2σ(I)) and wR2 was 0.0994 (all data). Crystal data for C23H20F3FeN3O2 (3) (M = 483.27 g mol−1): triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 9.4458(3) Å, b = 10.3520(4) Å, c = 11.0470(4) Å, α = 96.727(3)°, β = 102.651(3)°, γ = 100.958(3)°, V = 1020.46(6) Å3, Z = 2, T = 183(1) K, μ(MoKα) = 0.793 mm−1, Dcalc = 1.573 g cm−3, 24870 reflections measured (4.534° ≤ 2Θ ≤ 61.016°), 6226 unique (Rint = 0.0381, Rsigma = 0.0276) which were used in all calculations. The final R1 was 0.0344 (I > 2σ(I)) and wR2 was 0.0941 (all data). CCDC 1433288 (for 1), 1433289 (for 2), and 1433290 (for 3) contain the supplementary crystallographic data for this paper.
(no. 2), a = 9.4458(3) Å, b = 10.3520(4) Å, c = 11.0470(4) Å, α = 96.727(3)°, β = 102.651(3)°, γ = 100.958(3)°, V = 1020.46(6) Å3, Z = 2, T = 183(1) K, μ(MoKα) = 0.793 mm−1, Dcalc = 1.573 g cm−3, 24870 reflections measured (4.534° ≤ 2Θ ≤ 61.016°), 6226 unique (Rint = 0.0381, Rsigma = 0.0276) which were used in all calculations. The final R1 was 0.0344 (I > 2σ(I)) and wR2 was 0.0941 (all data). CCDC 1433288 (for 1), 1433289 (for 2), and 1433290 (for 3) contain the supplementary crystallographic data for this paper.
Biological evaluation
Conclusions
The invasiveness and aggressiveness of pathogenic fungi are too often underestimated or even ignored and have not been reflected in an adequate new drug output – the majority of the benchmark products were discovered in the last century! Worryingly, pathogenic fungi have started to develop drug tolerance, defeating an increasing number of therapies. Because of the incidence, resistance and health risk of fungi-related diseases, the discovery of novel drugs or strategies to fight fungal infections is highly necessary. In this article, we coupled a new effective fungicide, namely sedaxane, with ferrocenyl and ruthenocenyl moieties. This strategy has already been successfully employed in medicinal chemistry to increase the therapeutic potential of the accepted drugs. More specifically, we synthesised four new organometallic compounds (complexes 1a, 1b, 2 and 3). The complexes were fully characterized by 1H, 13C and 19F NMR spectroscopy, ESI-MS, elemental analysis (deviation < 0.5%) and X-ray crystallography (for the ferrocene derivatives 1a, 2 and 3). The four complexes were found to have a very low or even no toxicity towards human immortalized fibroblast or epithelial cells. This result is favourable in relation to topical as well as systemic applications. Complex 1a, bearing a methyl group on the pyrazole core, was found to have an antifungal toxicity in the micromolar range contrary to 2 and 3, which bear a phenyl or a phenoxy group on the pyrazole core, respectively. This fact suggests that structural modifications, which hinder the pyrazole ring of the compound, affect drastically the overall activity. Strikingly, investigation of the ROS level in SC upon treatment with 1a and its isostructural but not redox-active ruthenocene analogue 1b evidenced an increase of these noxious species for 1a but not for 1b. Moreover, it has to be noted that the parent drug sedaxane was found to not have any fungicidal profile and to not induce ROS formation under the same experimental conditions. It can therefore be assumed that the increase in mycotoxicity depends on the presence of the ferrocenyl moiety and its redox activity. Altogether these data represent a promising starting point for the development of new potent metal-based antifungal agents.Acknowledgements
This work was financed by the Swiss National Science Foundation (Professorships PP00P2_133568 and PP00P2_157545 to G. G.), the University of Zurich (G. G.), the Stiftung für wissenschaftliche Forschung of the University of Zurich (G. G.), the Novartis Jubilee Foundation (G. G. and R. R.), the Forschungskredit of the University of Zurich (R. R.) and the UBS Promedica Stiftung (G. G. and R. R.). The authors thank Syngenta AG for providing a sedaxane sample for the comparative biological experiments as well as Dr Murat Aykut and PD Dr Stefano Ferrari for helpful discussions.References
- P. N. Lipke and R. Ovalle, J. Bacteriol., 1998, 180, 3735–3740 Search PubMed; D. A. Enoch, H. A. Ludlam and N. M. Brown, J. Med. Microbiol., 2006, 55, 809–818 CrossRef CAS PubMed.
- S. M. Bowman and S. J. Free, BioEssays, 2006, 28, 799–808 CrossRef PubMed.
- A. Caridi, Antonie van Leeuwenhoek, 2006, 89, 417–422 CrossRef PubMed.
- E. Barreto-Bergter, M. R. Pinto and M. L. Rodrigues, An. Acad. Bras. Cienc., 2004, 76, 67–84 CAS.
- D. Gozalbo, M. V. Elorza, R. Sanjuran, A. Marcilla, E. Valentin and R. Sentandreu, Pharmacol. Ther., 1993, 60, 337–345 CrossRef CAS PubMed; M. Debono and R. S. Gordee, Annu. Rev. Microbiol., 1994, 48, 471–497 CrossRef PubMed.
- M. P. English and J. Turvey, Br. Med. J., 1968, 4, 228–230 CrossRef CAS PubMed; S. Perea, M. J. Ramos, M. Garau, A. Gonzales, A. R. Norviega and A. Del Palacio, J. Clin. Microbiol., 2000, 38, 3226–3230 Search PubMed; B. Havlickova, V. A. Czaika and M. Friedrich, Mycoses, 2008, 51, 2–15 CrossRef PubMed; R. P. Hobson, J. Hosp. Infect., 2003, 55, 159–168 CrossRef PubMed.
- D. A. Enoch, H. A. Ludlam and N. M. Brown, J. Med. Microbiol., 2006, 55, 809–818 CrossRef CAS PubMed; J. Menzin, J. L. Meyers, M. Friedman, J. R. Korn, J. R. Perfect, A. A. Langston, R. P. Danna and G. Papadopoulos, Am. J. Infect. Control, 2011, 39, 15–20 CrossRef PubMed.
- M. A. Pfaller, Am. J. Med., 2012, 125, S3–S13 CrossRef CAS PubMed; C. M. Martel, J. E. Parker, O. Bader, M. Weig, U. Gross, A. G. S. Warrilow, D. E. Kelly and S. L. Kelly, Antimicrob. Agents Chemoter., 2010, 54, 3578–3583 CrossRef PubMed; H. Hof, Drug Resist. Updates, 2008, 11, 25–31 CrossRef PubMed.
- T. Ohyama, S. Miyakoshi and F. Isono, Antimicrob. Agents. Chemoter., 2004, 48, 319–322 CrossRef CAS PubMed.
- G. Gasser and N. Metzler-Nolte, Curr. Opin. Chem. Biol., 2012, 16, 84–91 CrossRef CAS PubMed; G. Jaouen, A. Vessieres and S. Top, Chem. Soc. Rev., 2015, 44, 8802–8817 RSC; C. H. Huang and A. Tsourkas, Curr. Top. Med. Chem., 2013, 13, 411–421 CrossRef PubMed; M. Patra, G. Gasser, M. Wenzel, K. Merz, J. E. Bandow and N. Metzler-Nolte, Organometallics, 2010, 29, 4312–4319 CrossRef.
- C. G. Hartinger, N. Metzler-Nolte and P. J. Dyson, Organometallics, 2012, 31, 5677–5685 CrossRef CAS; U. Schatzschneider and N. Metzler-Nolte, Angew. Chem., Int. Ed., 2006, 118, 1534–1537 CrossRef; M. Dorr and E. Meggers, Curr. Opin. Chem. Biol., 2014, 19, 76–81 CrossRef PubMed; I. Ott, Coord. Chem. Rev., 2009, 253, 1670–1681 CrossRef; N. P. E. Barry and P. J. Sadler, Chem. Commun., 2013, 49, 5106–5131 RSC.
- Y. Wang, P. Pigeon, S. Top, M. J. McGlinchey and G. Jaouen, Angew. Chem., Int. Ed., 2015, 54, 10230–10233 CrossRef CAS PubMed; A. Nguyen, A. Vessieres, E. A. Hillard, S. Top, P. Pigeon and G. Jaouen, Chimia, 2007, 61, 716–724 CrossRef; F. Dubar, T. J. Egan, B. Pradines, D. Kuter, K. K. Ncokazi, D. Forge, J. F. Paul, C. Pierrot, H. Kalamou, J. Khalife, E. Buisine, C. Rogier, H. Vezin, I. Forfar, C. Slomianny, X. Trivelli, S. Kapishnikov, L. Leiserowitz, D. Dive and C. Biot, ACS Chem. Biol., 2011, 6, 275–287 CrossRef PubMed; M. Barends, A. Jaidee, N. Khaohirun, P. Singhasivanon and F. Nosten, Malar. J., 2007, 6, 81 CrossRef PubMed; M. Patra, K. Ingram, V. Pierroz, S. Ferrari, B. Spingler, J. Keiser and G. Gasser, J. Med. Chem., 2012, 55, 8790–8798 CrossRef PubMed.
- F. Dubar, C. Slomianny, J. Khalife, D. Dive, H. Kalamou, Y. Guerardel, P. Grellier and C. Biot, Angew. Chem., Int. Ed., 2013, 52, 7690–7693 CrossRef CAS PubMed; F. Dubar, T. J. Egan, B. Pradines, D. Kuter, K. K. Ncokazi, D. Forge, J.-F. Paul, C. Pierroz, H. Kalamou, J. Khalife, E. Buisine, C. Rogier, H. Vezin, I. Forfar, C. Slomianny, X. Trivelli, S. Kapishnikov, L. Leiserowitz, D. Dive and C. Biot, ACS Chem. Biol., 2011, 6, 275–287 CrossRef PubMed; G. Gasser, Chimia, 2015, 69, 442–446 CrossRef PubMed; J. Hess, J. Keiser and G. Gasser, Future Med. Chem., 2015, 7, 821–830 CrossRef PubMed; Y. C. Ong, V. L. Blair, L. Kedzierski and P. C. Andrews, Dalton Trans., 2014, 43, 12904–12916 RSC; M. Patra, M. Wenzel, P. Prochnow, V. Pierroz, G. Gasser, J. E. Bandow and N. Metzler-Nolte, Chem. Sci., 2015, 6, 214–224 RSC; M. Patra and G. Gasser, ChemBioChem, 2012, 13, 1232–1252 CrossRef PubMed.
- C. Biot, N. Francois, L. Maciejewski, J. Brocard and D. Poulain, Bioorg. Med. Chem. Lett., 2001, 10, 839–841 CrossRef CAS; A. Garoufis, S. K. Hadikakou and N. Hadjiliadis, Coord. Chem. Rev., 2009, 253, 1384–1397 CrossRef; N. V. Loginova, T. V. Koval'chuk, N. P. Osipovich, G. I. Polozov, V. L. Sorokin, A. A. Chernyaskaya and O. I. Shadyro, Polyhedron, 2008, 27, 985–991 CrossRef; M. B. Halli, R. S. Malipatil, R. B. Sumathi and K. Shivakumar, Pharm. Lett., 2013, 5, 182–188 Search PubMed; M. Kaya, E. Demir and H. Bekci, J. Enzyme Inhib. Med. Chem., 2013, 28, 885–893 CrossRef PubMed; E. Pahontu, V. Fala, A. Gulea, D. Poirier, V. Tapcov and T. Rosu, Molecules, 2013, 18, 8812–8836 CrossRef PubMed; E. Rodriguez-Fernandes, J. L. Manzano, J. J. Benito, R. Hermosa, E. Monte and J. J. Criado, J. Inorg. Biochem., 2005, 99, 1558–1572 CrossRef PubMed; V. S. Pore, M. A. Jagtap, S. G. Agalave, A. K. Pandey, M. I. Siddiqi, V. Kumar and P. K. Shukla, MedChemComm, 2012, 3, 484–488 RSC; J. Li, T. Xi, B. Yan, Y. Guan, M. Yang, J. Song and H. Ma, New J. Chem., 2015 10.1039/c5nj01845e; P. V. Simpson, C. Nagel, H. Bruhn and U. Schatzschneider, Organometallics, 2015, 34, 3809–3815 CrossRef.
- R. Zeun, G. Scalliet and M. Oostendorp, Pest Manage. Sci., 2013, 69, 527–534 CrossRef CAS PubMed; M. Yoshida, P. V. Shah and D. McGregor, J. Med. Plants Res., 2012, 769–839 Search PubMed; W. Donovan, Sedaxane – Food and Agriculture Organization report, 2012, 1827–1917 CrossRef PubMed; H. Walter, H. Tobler, D. Gribkov and C. Corsi, Chimia, 2015, 69, 425–434 CrossRef PubMed.
- M. Patra, J. Hess, S. Konatsching, B. Spingler and G. Gasser, Organometallics, 2013, 32, 6098–6105 CrossRef CAS; P. D. Beer and D. K. Smith, Dalton Trans., 1998, 417–423 RSC; A. Baramee, A. Coppin, M. Mortuaire, L. Pelinski, S. Tomavo and J. Brocard, Bioorg. Med. Chem., 2006, 14, 1294–1302 CrossRef PubMed; D. V. Muratov, A. S. Romanov and A. R. Kudinov, Russ. Chem. Bull., 2014, 63, 2485–2492 CrossRef.
- M. E. Cardenas, M. C. Cruz, M. Del Poeta, N. Chung, J. R. Perfect and J. Heitman, Clin. Microbiol. Rev., 1999, 12, 583–611 CAS; H. Karathia, E. Vilaprinyo, A. Sorribas and R. Alves, PLoS ONE, 2011, 6, e16015 Search PubMed.
- R. Rubbiani, T. N. Zehnder, C. Mari, O. Blacque, K. Venkatesan and G. Gasser, ChemMedChem, 2014, 9, 2781–2790 CrossRef CAS PubMed; M. Zhang, J. Shi and L. Jiang, Electron. J. Biotechnol., 2015, 18, 202–209 CrossRef; A. Leonidova, V. Pierroz, R. Rubbiani, Y. Lan, A. G. Schmitz, A. Kaech, R. K. O. Sigel, S. Ferrari and G. Gasser, Chem. Sci., 2014, 5, 4044–4056 RSC.
- Agilent Technologies (formerly Oxford Diffraction), Yarnton, Oxfordshire, England, 2012; R. C. Clark and J. S. Reid, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1995, 51, 887 CrossRef CAS; CrysAlisPro, Version 1.171.36.20, Agilent Technologies, Yarnton, Oxfordshire, England, 2012 CrossRef; O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. J. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef; G. M. Sheldrick, Acta Crystallogr., 2008, A64, 112 CrossRef PubMed; G. M. Sheldrick, Acta Crystallogr., 2015, A71, 3 CrossRef PubMed; A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Analytical data and crystal representation of 1a, 2 and 3 (Fig. S1–S5); crystal data refinement and bond/angle calculations for complex 1 (Tables S1–S7); crystal data refinement and bond/angle calculations for complex 2 (Tables S9–S118); crystal data refinement and bond/angle calculations for complex 3 (Tables S19–S22); general crystal experiment settings and an example of antifungal toxicity in a 12-well plate (Fig. S6). CCDC 1433288–1433290. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt04231c |
| This journal is © The Royal Society of Chemistry 2016 |