
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Platinum phosphinothiolato hydride complexes: synthesis, structure and evaluation as tin-free hydroformylation catalysts†
Julio
Real
*a,
Esther
Prat-Gil
a,
Montserrat
Pagès-Barenys
a,
Alfonso
Polo
b,
Joan F.
Piniella
c and
Ángel
Álvarez-Larena
c
aDepartament de Química, Universitat Autònoma de Barcelona, 08193 Bellaterra, Barcelona, Spain. E-mail: juli.real@uab.cat
bDepartament de Química, Universitat de Girona. Campus de Montilivi, 17071 Girona, Spain
cServei de Difracció de Raigs-X, Universitat Autònoma de Barcelona, 08193 Bellaterra, Barcelona, Spain
First published on 3rd February 2016
Abstract
Ligand 2-diphenylphosphinothiophenol (Hsarp) reacted with Pt(PPh3)4 to yield trans-[PtH(sarp)(PPh3)], which undergoes fast exchange with free PPh3 on the NMR time scale and very slowly and reversibly formed some cis-[PtH(sarp)(PPh3)] over time in solution (11%, 24 h). Reaction of trans-[PtH(sarp)(PPh3)] with Hsarp in boiling toluene gave cis- and trans-[Pt(sarp)2]; the cis isomer being more stable. These complexes were characterized by 1H and 31P NMR and also analyzed by XRD in the case of trans-[PtH(sarp)(PPh3)], trans-[Pt(sarp)2], and cis-[Pt(sarp)2]. trans-[PtH(sarp)(PPh3)] was evaluated as a preformed, tin-free hydroformylation catalyst on styrene and found active at 100 °C, at pressures over 75 bar, yielding phenylpropanal (regioselectivities up to 83% in 2-phenylpropanal), with total conversions to aldehydes up to 100% at styrene/platinum ratios from 400/1 to 1000/1 and minimal hydrogenation products.
Introduction
Phosphinothiolato ligands behave as soft–soft chelates with very different donors in terms of size, electronic properties and Brønsted basicity. This difference makes phosphinothiolato-species unique heterotopic ligands. Transition metal complexes of sulfur–phosphorus ligands have been studied for their potential applications.1 The areas of interest include carbonylation reactions,2S-alkylation/S-dealkylation reactions,3 carbothiolations,4 and copolymerizations.5 These complexes are relevant in hydrodesulfurization (HDS),6 in the modeling of sulfur-containing metalloproteins7 and in imaging and radiotherapy.8Olefin hydroformylation is a topic of continuing interest,9 including alternate metal catalysts (non-rhodium), which have been reviewed recently.10 The substitution of rhodium as the metal of choice for hydroformylation is not expected, but in some exceptional cases a third row metal (Ir) has been reported to outperform a second row metal (Rh) in catalytic carbonylation (see reports on the Cativa process).11
As part of a project on the synthesis of new amino- and phosphino-thiolates of group 10 metals, and on the analysis of the ligand-based stereoelectronic effects that are determinant in their properties and potential applications,2b,12 we now report on platinum 2-diphenylphosphinothiophenolato complexes, their synthesis and structural features and the first tests as homogeneous, tin halide free hydroformylation catalysts.13
Results and discussion
Synthetic aspects
Ligand Hsarp (2-diphenylphosphinothiophenol) reacted with Pt(PPh3)4 to yield modest amounts of 1 (30%) when used stoichiometrically (Scheme 1). When Hsarp was used in larger amounts the yield increased accordingly (up to 83%); this is interpreted as a need to displace three moles of PPh3 from the sphere of coordination of platinum(0) to maximize the amount of platinum hydride formed before the addition of Et2O to precipitate 1 and to keep PPh3 in solution (Scheme 1).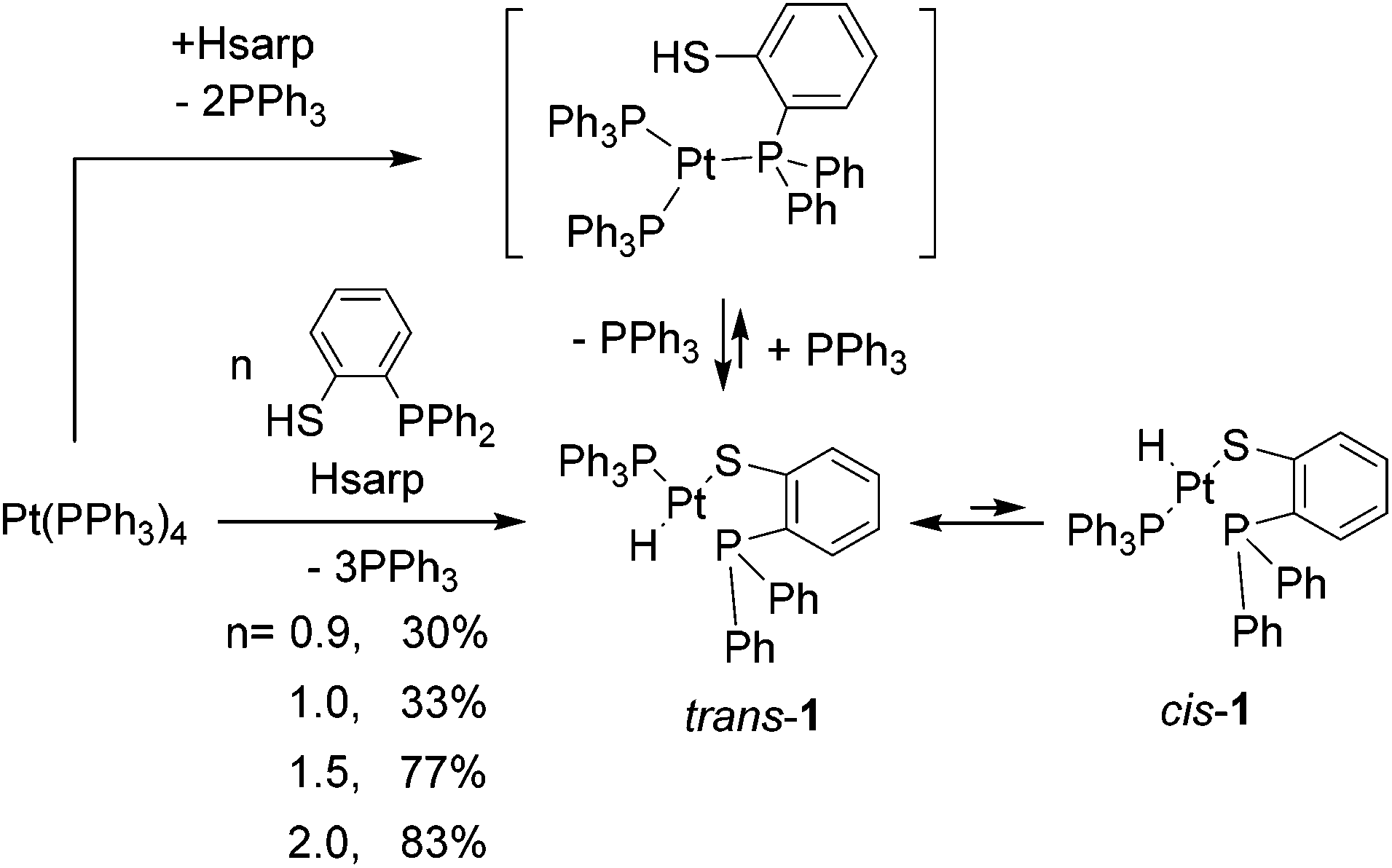 | ||
| Scheme 1 Synthesis of [PtH(sarp)PPh3], n is the Hsarp/Pt ratio used. | ||
The yield of 1 is only dependent on the excess of Hsarp, and is independent of the temperature or the reaction time, within reasonable limits. Crude 1 contains basically trans-1 with small amounts of free PPh3 (ca. 0.5–1%). Crude 1 could be further purified by column chromatography on silica or by slow recrystallization to obtain pure trans-1. Solutions of trans-1 are stable to decomposition but evolve to an equilibrium mixture of trans-1 (89%) and cis-1 (11%) isomers, which is complete in 24 h.
Upon heating Hsarp with Pt(PPh3)4 or trans-1 in toluene at 110 °C, the platinum hydride was observed to decrease in concentration with time, until all the hydride turned into cis-[Pt(sarp)2] (cis-2). A complete reaction takes 28 h; if the reaction is stopped before completion, the corresponding amounts of 1 and 2 can be recovered without much loss, which demonstrates the remarkable thermal stability of hydride 1. We envision this reaction as two step: the fast substitution of PPh3 by Hsarp and the slow reaction of platinum hydride with –SH, to give hydrogen (Scheme 2). Furthermore, in a similar addition involving HSCH2CH2PPh2 (dppet), this has been found to be the rate-determining step, according to conceptual DFT calculations.14
 | ||
| Scheme 2 | ||
The bischelate obtained in this way may contain small amounts of trans-[Pt(sarp)2] which can be converted to cis-[Pt(sarp)2] by further heating at 110 °C in toluene. The greater thermodynamic stability of the cis isomer is predicted by the greater trans influence of phosphorous over sulfur (or by the so called antisymbiosis rule)15 while steric hindrance would favour the trans isomer. In the case of palladium, we have shown that trans and cis bischelates [Pd(sarp)2] interconvert in solution and the equilibrium can be displaced towards the former or the latter by the use of polar or non-polar crystallization solvents respectively.16 In the case of platinum this procedure proved impractical, as trans-2 does not form in sufficient amounts in any solvent tested in reasonable reaction times. Bischelate cis-2 is also the identifiable platinum product of the thermal decomposition of 1 in boiling toluene.
The bulkier ligand, 2-diphenylphosphino-6-trimethylsililthiophenol (Hsarp′), reacted in the same way as Hsarp to yield the corresponding hydride [PtH(sarp′)(PPh3)] (3) (Scheme 3). For comparison purposes, the ligand EtSCH2CH2SH was also prepared and used in the synthesis of [PtH(EtSCH2CH2S-κ2S,S)(PPh3)], which was made from Pt(PPh3)4 in the same way as 1 and 3, but by using a stoichiometric amount of the ligand.17 At room temperature, fast exchange was not observed in the NMR of 4 and isomerization was not detected (Scheme 3).
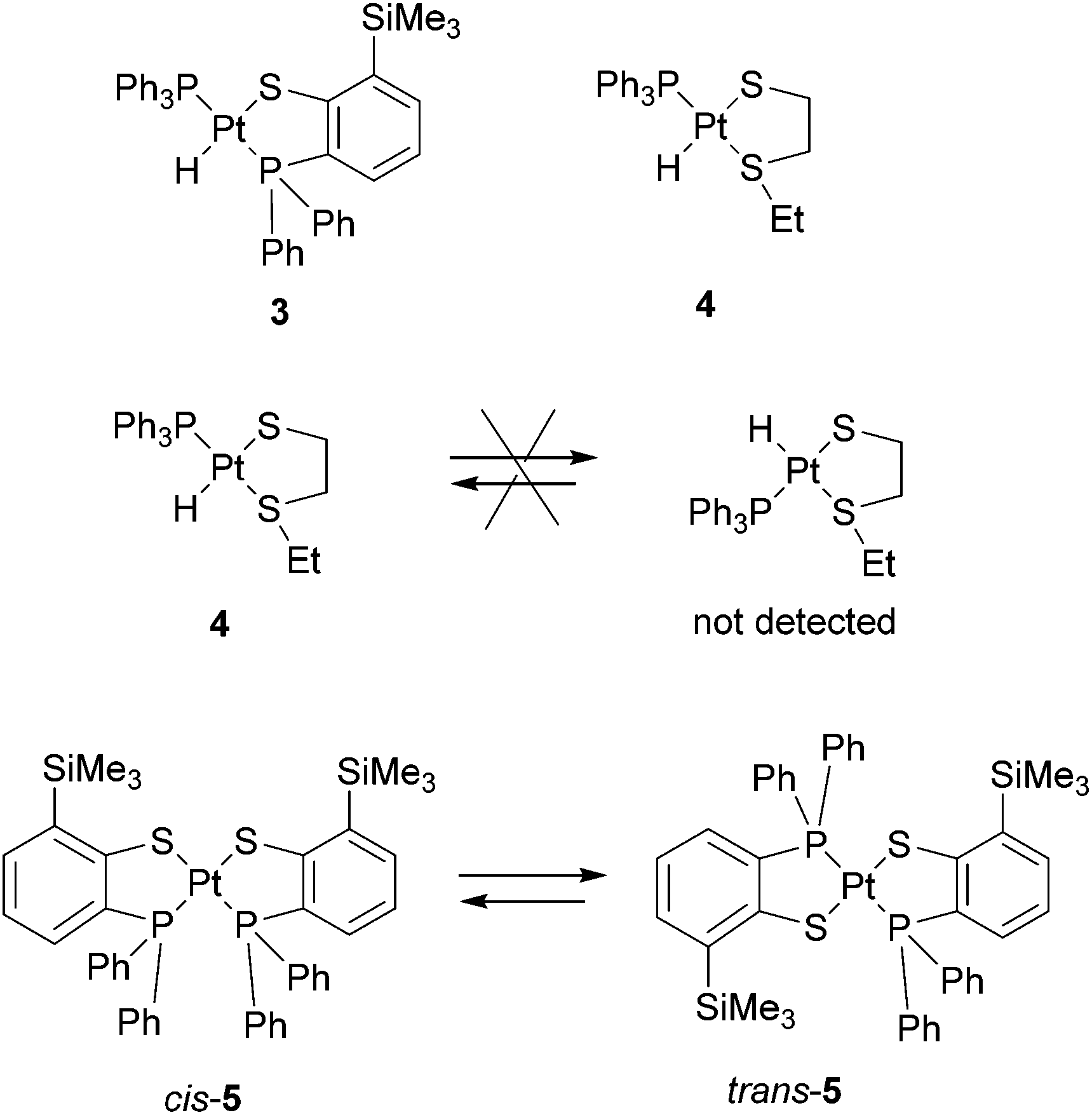 | ||
| Scheme 3 | ||
The reaction of Hsarp′ with Pt(PPh3)4 in boiling toluene gave the bischelates 5, with the difference compared to 2, that a longer time (48 h) was needed for the bulkier ligand and the product was a mixture of cis and trans isomers, containing a slight excess of the latter (Scheme 3). Clearly, in the case of sarp′ the trans isomer becomes more stable for steric reasons.
NMR spectroscopy
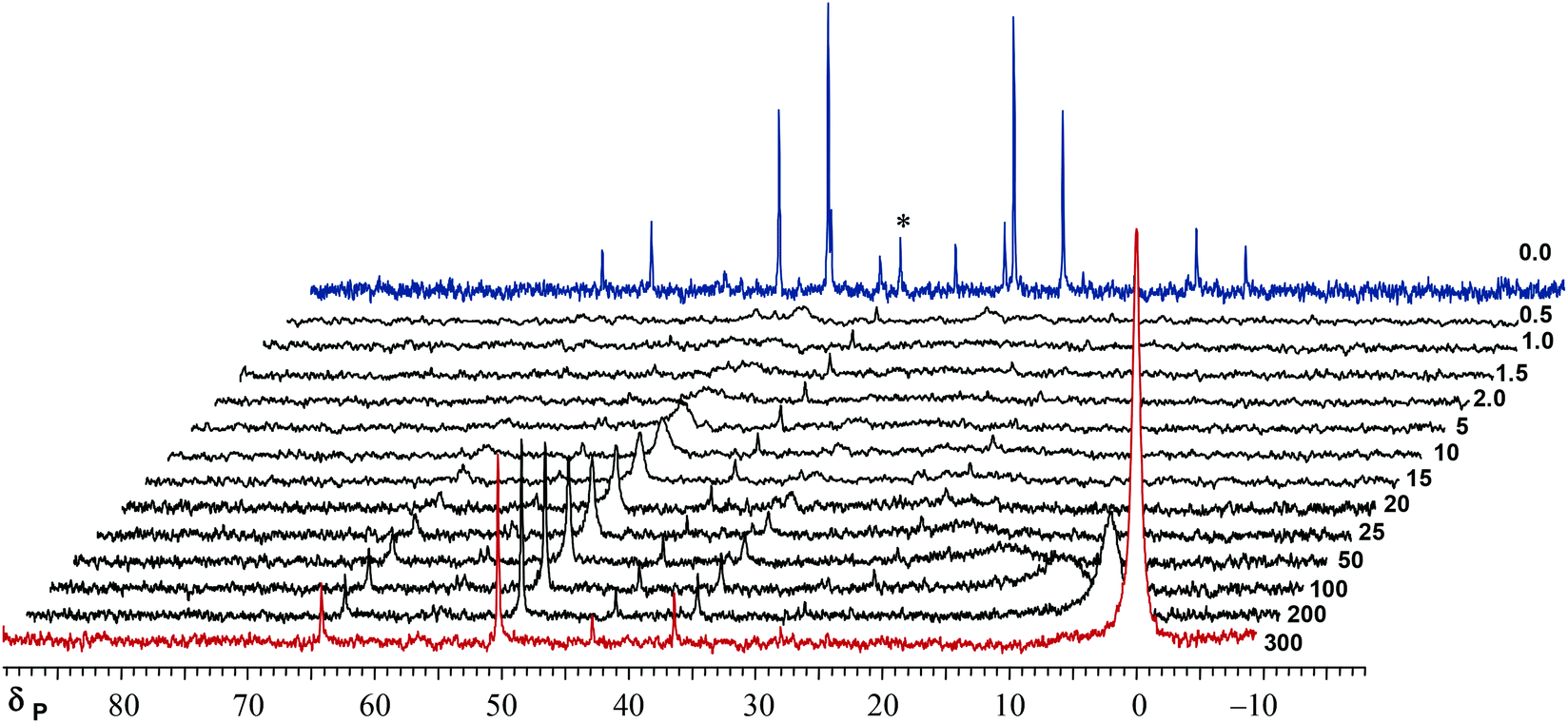
The hydride region of the 1H NMR spectra of crude 1 consisted of a doublet with 195Pt satellites and the 31P NMR showed just noise at room temperature. This was attributed to the exchange phenomena, so the NMR were taken at low temperature (down to −60 °C) but the slow exchange limit was not achieved in this way. Purification of 1 by chromatography or recrystallization gave samples free of PPh3 which exhibited non-exchanging, narrow lined 1H and 31P NMR spectra. The obvious conclusion is that exchange takes place between 1 and free PPh3 and this is a bimolecular reaction that cannot proceed in the absence of PPh3. The long accumulation of 31P NMR spectra on samples of crude 1 at room temperature showed the NMR of Fig. 1, which consists of the very broad signals of trans-1, which exchanges with free PPh3, and very small amounts of cis-1 and cis-2 (narrow lines). | ||
| Fig. 1 Long time accumulation 31P{1H} NMR (101 MHz, CD2Cl2, rt) spectrum of crude 1 (22 K transients), the broad signals correspond to trans-1 (the major product, red marks) and the narrow ones to cis-1 (green marks), the broadness is caused by exchange of trans-1 with trace amounts of free PPh3 (estimated at ca. 0.5%); 195Pt (33.8% abundance) satellites are observable. The signal at δ 43 corresponds to a small amount of cis-[Pt(sarp)2]. | ||
Relevant NMR data are collected in Tables 1 and 2. Hydride signals are well shifted to low frequencies into negative delta values. Hydride coupling to platinum (1JPtH) is typically very large and it is observed in all cases. Coupling to the sarp phosphorus (2JPH, PA) is also observed in fast exchange and no exchange conditions, while coupling to the PPh3 phosphorus (2JPH, PB) is only observed in the absence of free PPh3, that is, in the absence of exchange. In this case the spectra have been obtained from samples that contained minute amounts of free PPh3 (see Tables 1 and 2). Complex 4 showed no such exchange with added PPh3 at room temperature, so that, we conclude that the exchange is directly related to the trans effect of the phosphorus of the sarp ligand.
| Complex | δ(H–Pt) | J(H–Pt) | J(H–PA) | J(H–PB) |
|---|---|---|---|---|
| a Chemical shifts on the δ scale, coupling constants in Hz. PA values are assigned to the phosphinothiolato ligand and PB values to PPh3. All spectra were recorded in CD2Cl2 at rt (298 K), signals have 195Pt satellites of correct intensity for mononuclear complexes at natural abundance. | ||||
| trans-1 | –7.61 dd | 1022 | 8.1 | 19.8 |
| cis-1 | –4.77 dd | 950 | 186 | 9.9 |
| trans-1 + PPh3 | –7.57 d | 1023 | 8.0 | — |
| trans-3 + PPh3 | –7.08 d | 1151 | 6.6 | — |
| 4 | –10.63 d | 1206 | — | 20.0 |
| Complex | δ PA | J(PA–Pt) | δ PB | J(PB–Pt) | J(PA–PB) |
|---|---|---|---|---|---|
| a Chemical shifts on the δ scale, coupling constants in Hz. PA values are assigned to the phosphinothiolato ligands and PB values to PPh3. All spectra were recorded in CD2Cl2 at rt (298 K), signals have 195Pt satellites of correct intensity for mononuclear complexes at natural abundance. | |||||
| trans-1 | 50.5 d | 2819 | 31.9 | 2915 | 391.2 |
| cis-1 | 56.6 d | 1922 | 24.3 | 3187 | 10.1 |
| trans-1 + PPh3 | 50.5 s | 2801 | — | — | — |
| trans-3 + PPh3 | 48.2 s | 2990 | — | — | — |
| 4 | 23.7 | 3215 | |||
| cis-2 | 42.8 s | 2912 | |||
| trans-2 | 50.8 s | 2645 | |||
| cis-5 | 41.9 s | 2867 | |||
| trans-5 | 48.9 s | 2629 | |||
31P NMR mirrors in most ways the results observed in 1H NMR. In the absence of free PPh3, the resonances of 1 are narrow and all δ and J can be assigned. In the presence of PPh3 (the fast exchange limit) only the chelate ligand phosphorus is observed, including its coupling to platinum (JPtP, PA) but not to PPh3 phosphorus (PB).
The effect of free PPh3 on the NMR of trans-1 was better understood by recording the 31P NMR spectra of a sample of trans-1 after adding known amounts of free PPh3 in an NMR titration-like experiment (Fig. 2). The addition of just 0.5 mol% PPh3 to pure trans-1 caused immediate collapse of the 31P signals; at 5% the signal corresponding to PA started to rise (broad), at about 50% the signal of PA (with 195Pt satellites) was narrower and the signal of free PPh3 started to rise from the noise (broad), at 300% the exchange was fast enough for the PPh3 to be quite narrow, but clearly displaced down-field from the known chemical shift of PPh3. To see cis-1 in the NMR spectrum, solutions of trans-1 were allowed to age. After 24 h equilibrium was reached at room temperature; the equilibrium mixture in dichloromethane solution contained both trans-1 (89%) and cis-1 (11%), but crystallization from CH2Cl2/Et2O yielded only trans-1. The thermodynamic stability of the trans-P,P isomer is in agreement with the high trans influence of hydride, in contrast with the low trans influence of chloride which gives cis-P,P-[PtCl(dppet-κ2P,S)PPh3].7a
 | ||
| Fig. 2 The addition of free PPh3 to a sample of pure trans-1 (uppermost trace) caused a dramatic change in its 31P{1H} NMR spectrum (101 MHz, CD2Cl2, rt), the added amounts are indicated by the traces in mol% of added PPh3. A small amount of cis-[Pt(sarp)2] was added for reference (*). | ||
Coupling constants were useful in the characterization of these compounds.18 The 2JPH coupling constant in cis-1 is much larger (186 Hz) than any other JPH because this is the only case in which the hydride ligand is trans to phosphorus (Table 1). In the case of phosphorus to phosphorus, 2JPP is much larger in trans-1 (391 Hz) than in cis-1 (10.1 Hz). Coupling to platinum is strong both for 1H and 31P, but much larger 1JPtP coupling constants are expected for cis-P,P isomers than for trans-P,P isomers, this criterion has been followed to assign the signals of trans-2 and cis-2 (Table 2), which was supported by the XRD studies (vide infra). However, coupling of the sarp phosphorus to platinum 1JPtP is comparatively small in cis-1 (Table 2), this is most probably caused by the strong trans influence of the hydride ligand. There are numerous precedents of this effect: in cis-[PtH2(PEt3)2], 1JPtP = 1984 Hz, while in trans-[PtH2(PEt3)2], 1JPtP = 2764 Hz; and in cis-[PtH2(PMe3)2], 1JPtP = 1875 Hz, while in trans-[PtH2(PMe3)2], 1JPtP = 2594 Hz.19a,b Complexes cis-[PtR2L2] (R = alkyl,19c aryl;19d L = PPh3, PMe3, PEt3) also feature comparatively small 1JPtP.
The behavior of 1 in solution is summarized in Scheme 4. The first reaction represents the fast exchange we observe in the NMR. Platinum(II) will form a more stable trigonal bipyramidal (tbp) intermediate with the bulky phosphorus ligands in equatorial (eq) positions and with the sarp chelate bonding in an axial–equatorial fashion. To form cis-1, the tbp intermediate must rearrange to a less favourable intermediate (in Scheme 4 this is depicted by L in an axial position), and this is presumably the reason why trans/cis isomerisation is such a slow process for 1.
 | ||
| Scheme 4 Proposed mechanism of ligand exchange. On the NMR time scale exchange with free L is fast for trans-1; conversely, the ligand rearrangement (Hax to Heq and Leq to Lax) that gives rise to cis-1 is understandably slow. | ||
In the exchange process of trans-1 we believe that sarp remains coordinated in the Sax–Peq mode, supported by the fact that the chemical shift of PA is the same in the fast exchange limit and the no exchange (slow exchange) limit. The chemical shift of 31P is strongly affected by the formation of cycles and any opening of the chelate should affect the δP of PA. The coupling of PA to platinum is also retained in the fast exchange limit (Fig. 2).
XRD crystal structures
Slow crystallization of 1 gave yellow crystals of trans-1 exclusively; crystals of minor isomer cis-1 could not be obtained from normal laboratory solvents. Crystallization of 2 in CH2Cl2/Et2O gave mainly chunky yellow crystals of cis-2·CH2Cl2, which is isostructural with the palladium complex cis-[Pd(sarp)2]·CH2Cl2.16 However, careful inspection revealed the presence of a few smaller crystals of a more intense colour and an apparently different habit, which upon XRD analysis proved to be trans-2.The structures exhibit slightly distorted square planar platinum(II) coordination. In the case of trans-1 (Fig. 3) it is worth noting the relatively long Pt–S distance (Table 3), which is ascribed to the strong trans influence of hydride. The chelate angle of sarp (Table 3) is always about 87°, as it is a rigid ligand. This angle makes sarp a well suited ligand both for cis coordination in sqp structures and for Sax–Peq coordination in tbp intermediates, which are stabilized by the nature of sarp in terms of the bite angle and size of the coordinating groups (see above, Scheme 4).
 | ||
| Fig. 3 ORTEP plots of trans-1, in the projection of the coordination plane parallel to the paper (above); the hydride ligand takes the position directly trans to the sulfur to complete the square-planar coordination of platinum(II); in the perpendicular projection (below) the phenyl rings of –PPh2 have been omitted for clarity (except for the ipso carbons). | ||
| trans-1 | trans-2 | cis-2·CH2Cl2 | |||
|---|---|---|---|---|---|
| a Chelate angle. b Open angle, platinum is at the inversion center, S′ is related to S by –x, –y, –z. c Chelate angle, S1 and P1 belong to one ligand, and S2 and P2 belong to the other. | |||||
| Pt–S | 2.357(3) | Pt–S | 2.315(2) | Pt–S1 | 2.313(5) |
| Pt–P1 | 2.243(3) | Pt–P | 2.293(2) | Pt–S2 | 2.326(4) |
| Pt–P2 | 2.278(3) | Pt–P1 | 2.263(4) | ||
| Pt–P2 | 2.249(4) | ||||
| S–Pt–P1a | 86.9(1) | S–Pt–Pa | 86.96(7) | S1–Pt–P1c | 87.7(2) |
| S–Pt–P2 | 98.6(1) | S′–Pt–Pb | 93.04(7) | S2–Pt–P2c | 87.4(2) |
| P1–Pt–P2 | 171.6(1) | Pt–S–C2 | 105.7(2) | S1–Pt–S2 | 84.7(2) |
| Pt–S–C1 | 104.6(3) | Pt–P–C1 | 107.5(2) | P1–Pt–P2 | 100.3(2) |
| Pt–P1–C2 | 109.1(3) | S1–Pt–P2 | 172.0(2) | ||
| S2–Pt–P1 | 171.7(2) | ||||
The Pt–P2 distance in trans-1, corresponding to PPh3 is somewhat longer than the Pt–P1 distance corresponding to sarp (Fig. 3). The P–Pt–P angle is deformed (ca. 172°) from linearity in trans-1 and cis-2, compared to symmetric trans-2 (Fig. 4, center of symmetry at Pt), owing to steric reasons. In trans-1, the hydride ligand has not been located but it must be placed trans to sulfur (NMR, IR), thus the PPh3 is somewhat displaced towards the smaller ligand. In the structures of 2 the sarp ligands are symmetrically disposed only in the trans isomer (Fig. 4), while in the cis isomer the P–Pt–P angle increases to 100.3(2)° and the S–Pt–S angle decreases to 84.7(2)° owing to the repulsion of the –PPh2 groups. The crystal structure of cis-1 could not be determined by XRD, but considering the structures of trans-1, trans-2 and cis-2, together with the spectroscopic data, leaves little doubt about its structure.
 | ||
| Fig. 4 ORTEP plots of trans-[Pt(sarp)2], the platinum lies at a crystallographic inversion center, thus P′ and S′ are related to P and S by –x, –y, –z. | ||
Catalytic hydroformylation
Complex trans-1 exhibits desirable characteristics for a homogeneous catalyst: it undergoes fast ligand exchange, it contains a hydride ligand susceptible to olefin insertion reactions and also triaryl phosphines which usually favor small molecule activation, and addition and elimination processes associated with homogeneous catalysis. However, ligand sarp is a sulfur donor and these ligands are often associated with catalyst deactivation rather than catalyst activation.20 The thiophilicity of late transition metals, coupled with the ability of thiolates to give one, two or even three electron pairs in terminal or bridged compounds respectively,21 may block catalytic activity in homogeneous systems, but this should not be the case here because of the presence of good competing ligands.Nevertheless, trans-1 was tested as a hydroformylation catalyst on styrene in the absence of tin, chloride or any other extra ligands. The first results were disappointing: no activity was detected with syngas pressures below 75 bar. At 75 bar and higher pressures the results were positive (Table 4), using styrene/platinum ratios of 400/1 conversions were essentially complete in 24 h, with very low hydrogenation to ethylbenzene and a regioselectivity towards branched aldehyde of ca. 70%. Lowering the styrene/platinum ratio it can be seen that average TON numbers are about 400 and TOF numbers around 18 h−1 (Table 4, entries 3 and 4). Selectivity increased somewhat upon increasing the hydrogen partial pressure (Table 4, entry 4). Complex cis-2 was identified in the reaction mixtures as one of the platinum products formed after the reaction was over (after all, it is the thermal decomposition product of trans-1). However, testing showed that cis-2 was not responsible for the previous results, as it turned out to perform very poorly, with residual activity and low selectivity (Table 4, entry 5). Instead of preformed trans-1, the mixture of Pt(PPh3)4 plus Hsarp was also tested with the above olefins, and found inactive; one reason for this could be the inherent presence of excess PPh3.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
| Catalyst | [S]/[Pt] | P T | t (h) | C (%) | TONe | R (%) | H (%) |
|---|---|---|---|---|---|---|---|
| a Conditions: 19 mmol styrene (S) in 20 mL toluene at 100 °C. b Total pressure (bar), H2/CO = 1. c Reaction time. d Conversion of styrene to products. e Average turnover number. f Regioselectivity in iso-aldehyde. g Hydrogenation product (ethylbenzene). h In this experiment the pressure ratio H2/CO = 1.3. | |||||||
| trans-1 | 400 | 75 | 24 | 100 | 400 | 70 | 0.4 |
| trans-1 | 400 | 125 | 97 | 98 | 400 | 76 | 0.6 |
| trans-1 | 900 | 125 | 24 | 48 | 432 | 66 | 1.2 |
|
trans-1h |
1000 | 100 | 24 | 46 | 460 | 84 | 0.9 |
| cis-2 | 400 | 75 | 24 | 5 | 20 | 58 | 0.2 |
We have drawn (Scheme 5) a working mechanism that could rationalize the activity of complex 1 (before more data available), which also considers the special characteristics of ligand sarp. The insertion of the olefin into the Pt–H bond gives rise to an alkyl complex, which can be linear or branched, thus determining the regioselectivity of the reaction. It should be noted that for the branched alkyl, η3-C8H9 coordination is possible, thus favoring both activity towards styrene and regioselectivity towards a branched product. It is very difficult to ascertain if L = PPh3 or CO at this point (besides, the presence of CO in the coordination sphere of platinum does not even seem necessary until later). Insertion of CO causes the formation of the acyl complex which must react with hydrogen to yield the aldehyde and to regenerate the starting platinum species. For the activation of hydrogen by transition metal complexes, the first identified mechanism was oxidative addition, which would yield a platinum(IV), d6, species (central path in Scheme 5, in blue). After the elimination of the aldehyde, the cycle would be complete. However, given the nature of sarp (containing a proton-basic moiety not present in other ligands) we suggest that a different form of H2 activation can be considered (the lower path in Scheme 5, in green), with no need for oxidation of Pt(II) to Pt(IV) in a reducing medium. This would be a heterolytic cleavage across the Pt–S bond, or hydogenolysis-type activation. A similar activation has been described for rhodium and iridium arylthiolates.22 Moreover, the activation of H2 to Pt–H and H3O+ (i.e. platinum mediated disproportionation of H2 to H− and H+) has been implicitly proposed in the context of the platinum catalyzed hydroformylation of styrene.23 In this mechanism the elimination of aldehyde gives a platinum(0) species that was already invoked in the synthetic part of this paper, and which we assume to readily react to form a platinum hydride and follow the cycle.
 | ||
| Scheme 5 Mechanistic proposal for hydroformylation with trans-1 as the catalyst. | ||
Experimental
Reactions were performed under nitrogen using standard techniques.24 The isolated products are fairly air stable, and hydride complexes were kept refrigerated. Solvents were dried, distilled under nitrogen, kept over molecular sieves and deaerated prior to use.25 Tetrakis(triphenylphosphine)platinum(0) was prepared by established procedures.26 The synthesis of the ligands Hsarp and Hsarp′ have been described.27 NMR data are given on the δ scale and referenced in the usual manner. Catalysis experiments were performed in a stainless steel Autoclave Engineers EZE seal reactor of a standard vase (ca. 80 mL) and cover design, mechanical stirring (magnetic drive), heating and temperature control through a thermocouple, and sample port. GC analyses were performed using capillary columns HP-5, 0.25 mm diameter × 30 m length, using FID and MS detectors. Elemental analyses (C, H and S) were performed with a Carlo Erba CHNS EA-1108. The platinum content was determined by repeated cycles of mineralization of a weighed sample with conc. HNO3 in a heated crucible (fume hood), followed by ignition in an electric furnace at temperatures over 900 °C (open crucible, 24 h), this gave platinum metal that was weighed as such.X-ray diffraction structure determination
Data were collected using Mo Kα radiation. For compounds trans-1 and cis-2·CH2Cl2 an Enraf-Nonius CAD4 diffractometer (2θmax = 50°) was used; for compound trans-2, a Kuma KM4CCD (2θmax = 56°) was used. Empirical absorption corrections were applied. Structures were solved by direct methods (SHELXS-86) and refined by full-matrix least-squares methods on F2 for all reflections (SHELXL-97). Non-hydrogen atoms were refined anisotropically. Hydrogen atoms bonded to carbon atoms were placed in calculated positions with isotropic displacement parameters fixed at 1.2 times the Ueq of the corresponding carbon atoms. In trans-1, the hydrogen bonded to the platinum atom was not localized in the difference Fourier map, and it has not been included in the refinement. Crystal data and further refinement details are presented as ESI.† Compound cis-2·CH2CL2 is a solvate and some decay (caused by solvent loss) was observed during data collection; this may be the reason why in this structure high residual peaks (5.43 and −4.43 e Å−3) were found in the final difference density map, near to the Pt atom (1.05 Å and 1.12 Å respectively).Hydroformylation experiments
The reactor was conditioned by vacuum and syngas replenishment cycles. The reaction mixture was injected into the reactor through an inlet valve (followed by some fresh solvent in order to clean the valve) at room temperature and the pressure was set to about 30 bar. The temperature was raised to working temperature using the temperature regulator and then the pressure was adjusted to working pressure through the gas supply system. The mechanical stirring was initiated (400 s−1) and this was considered time zero. The reaction mixture consisted of, typically, the alkene (in the case of styrene, 2.00 g, 0.0192 mol), complex 1 (0.0364 g, 4.75 × 10−5 mol; this value depends on the entry in Table 4) and toluene (20 mL total solvent). The organic products were analyzed by GC and NMR. Blank experiments under the conditions of Table 4 run without complex 1 added or with PPh3 added instead of complex 1 (0.02–0.20 g) gave no conversion. Out of the reactor all solutions were homogeneous and yellow in color with no black platinum or other precipitates. After some time, the formation of some crystals was generally observed; they were identified as cis-2.Conclusions
Ligands Hsarp and Hsarp′ readily reacted with Pt(PPh3)4 to give hydride complexes trans-[PtH(sarp)(PPh3)]. The yield of 1 depends mainly on the stoichiometry employed, an excess of Hsarp being necessary for good yields. This does not seem to be a kinetic effect as yields are not influenced by the reaction time or temperature. This dependence is ascribed to an equilibrium in the chelate-assisted oxidative addition of ArS–H to Pt(0). The crude also contains very small amounts of cis-[PtH(sarp)(PPh3)], cis-[Pt(sarp)2)] and PPh3. Under these conditions the trans-P,P hydride undergoes very fast exchange of coordinated PPh3 with free PPh3 and has very broad signals in the NMR, while the cis-P,P hydride exhibits narrow signals at the same temperature, which means that it does not undergo fast PPh3 exchange. These hydrides are remarkably stable towards oxygen and moisture, which allowed purification by column chromatography. Pure trans-1 does not show any broadness in the NMR; the addition of free PPh3 shows that the exchange of PPh3 is bimolecular. Only trans-1 is isolated as solid crystals upon recrystallisation, fresh solutions show the signals of trans-1 only, but with time the cis isomer becomes apparent. Equilibrium was reached in 24 h and the solution contained 89% trans-1 and 11% cis-1. Fast exchange and trans/cis equilibrium was not observed in other non-sarp containing complexes such as 4. The reaction of Pt(PPh3)4 or 1 with excess Hsarp (at 110 C, 24 h) yields bischelates [Pt(sarp)2], the cis isomer being more stable thermodynamically. In 31P NMR, cis-2 shows a signal at δ 42.80 with 195Pt satellites (1JPtP = 2912 Hz) and trans-2 shows a signal at δ 50.80 (1JPtP = 2645 Hz) which is in agreement with XRD results and with the general observation that direct Pt–P coupling constants are larger for cis-P,P isomers.Hydride complex trans-1 was found worth evaluating as a hydroformylation catalyst but was found inactive at low pressures. At higher pressures, however, it was found active on styrene. The activity of trans-1 is comparable to platinum/diphosphine/SnCl2 systems published recently,28 but its selectivity is different. This difference in selectivity would support trans-1 as a new catalytic system.
Acknowledgements
This work was financially supported by the Dirección General de Enseñanza Superior e Investigación Científica (DGESIC) of Spain though project BQU2002-04070-C02.Notes and references
- (a) J. R. Dilworth and N. Wheatley, Coord. Chem. Rev., 2000, 199, 89–158 CrossRef CAS; (b) D. K. Dutta and B. Deb, Coord. Chem. Rev., 2011, 255, 1686–1712 CrossRef CAS.
- (a) P. W. N. M. van Leeuwen, Homogeneous Catalysis: Understanding the Art, Kluwer AP, Dordrecht (The Netherlands), 2004, p. 122 and references therein Search PubMed; (b) N. Brugat, A. Polo, A. Álvarez-Larena, J. F. Piniella and J. Real, Inorg. Chem., 1999, 38, 4829–4837 CrossRef CAS PubMed.
- (a) J. S. Kim, J. H. Reibenspies and M. Y. Darensbourg, J. Am. Chem. Soc., 1996, 118, 4115–4423 CrossRef CAS; (b) C. A. Grapperhaus, K. B. Venna and M. S. Mashuta, Inorg. Chem., 2007, 46, 8044–8050 CrossRef CAS PubMed.
- H. Kuniyasu, F. Yamashita, T. Hirai, J.-H. Ye, S.-I. Fujiwara and N. Kambe, Organometallics, 2006, 25, 566–570 CrossRef CAS.
- L.-P. He, M. Hong, B.-X. Li, J.-Y. Liu and Y.-S. Li, Polymer, 2010, 51, 4336–4339 CrossRef CAS.
- (a) R. J. Angelici, Organometallics, 2001, 20, 1259–1275 CrossRef CAS; (b) D. A. Vicic and W. D. Jones, J. Am. Chem. Soc., 1999, 121, 7606–7617 CrossRef CAS.
- (a) P. V. Rao, S. Bhaduri, J. Jiang, D. Hong and R. H. Holm, J. Am. Chem. Soc., 2005, 127, 1933–1945 CrossRef CAS PubMed; (b) S. Ye, F. Neese, A. Ozarowski, D. Smirnov, J. Krzystek, J. Telser, J.-H. Liao, C.-H. Hung, W.-C. Chu, Y.-F. Tsai, R.-C. Wang, K.-Y. Chen and H.-F. Hsu, Inorg. Chem., 2010, 49, 977–988 CrossRef CAS PubMed; (c) T.-W. Chiou and W.-F. Liaw, Inorg. Chem., 2008, 47, 7908–7913 CrossRef CAS PubMed; (d) C.-M. L. Lee, Y.-L. Chuang, C.-Y. Chiang, G.-H. Lee and W.-F. Liaw, Inorg. Chem., 2006, 45, 10895–10904 CrossRef CAS PubMed; (e) T. Ohkubo, H. Sakashita, T. Sakuma, M. Kainosho, M. Sekiguchi and K. Morikawa, J. Am. Chem. Soc., 1994, 116, 6035–6036 CrossRef CAS; (f) L. C. Myers, M. P. Terranova, A. E. Ferentz, G. Wagner and G. L. Verdine, Science, 1993, 261, 1164–1167 CAS.
- N. Salvarese, N. Morellato, A. Venzo, F. Refosco, A. Dolmella and C. Bolzati, Inorg. Chem., 2013, 52, 6365–6377 CrossRef CAS PubMed.
- (a) Rhodium Catalyzed Hydroformylation, ed. P. W. N. M. van Leeuwen and C. Claver, Kluwer, Dordrecht (The Netherlands), 2000 Search PubMed; (b) R. Franke, D. Selent and A. Borner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed; (c) E. V. Gusevskaya, J. Jiménez-Pinto and A. Börner, ChemCatChem, 2014, 6, 382–411 CrossRef CAS.
- (a) J. Pospech, I. Fleischer, R. Franke, S. Buchholz and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 2852–2872 CrossRef CAS PubMed. See also: (b) C. Kubis, W. Baumann, E. Barsch, D. Selent, M. Sawall, R. Ludwig, K. Neymeyr, D. Hess, R. Franke and A. Börner, ACS Catal., 2014, 4, 2097–2108 CrossRef CAS.
- (a) A. Haynes, P. M. Maitlis, G. E. Morris, G. J. Sunley, H. Adams, P. W. Badger, C. M. Bowers, D. B. Cook, P. I. E. Elliott, T. Ghaffar, H. Green, T. R. Griffin, M. Payne, J. M. Pearson, M. J. Taylor, P. W. Vickers and R. J. Watt, J. Am. Chem. Soc., 2004, 126, 2847–2861 CrossRef CAS PubMed, and references therein. (b) G. J. Sunley and D. J. Watson, Catal. Today, 2000, 58, 293–307 CrossRef CAS; (c) J. H. Jones, Platinum Met. Rev., 2000, 44, 94–105 CAS; (d) J. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Sausalito CA, 2010, p. 749. ISBN 978-1-891389-53-5 Search PubMed.
- (a) J. Duran, A. Polo, J. Real, J. Benet-Buchholz, A. Poater and M. Solà, Eur. J. Inorg. Chem., 2003, 4147–4151 CrossRef CAS; (b) J. Duran, N. Brugat, A. Polo, J. Real, X. Fontrodona and J. Benet-Buchholz, Organometallics, 2003, 22, 3432–3438 CrossRef CAS; (c) N. Brugat, J. Duran, A. Polo, J. Real, A. Álvarez-Larena and J. F. Piniella, Tetrahedron: Asymmetry, 2002, 13, 569–577 CrossRef CAS.
- L. Jánosi, T. Kégl and L. Kollár, J. Organomet. Chem., 2008, 693, 1127–1135 CrossRef.
- J. Duran, A. Polo, J. Real, J. Benet-Buchholz, M. Solà and A. Poater, ChemistryOpen, 2016 DOI:10.1002/open.201500136.
- R. G. Pearson, Inorg. Chem., 1973, 12, 712–713 CrossRef CAS.
- (a) J. Real, E. Prat, A. Polo, A. Alvarez-Larena and J. F. Piniella, Inorg. Chem. Commun., 2000, 3, 221 CrossRef CAS; (b) D. Canseco-González, S. Gómez-Benítez, S. Hernández-Ortega, R. A. Toscano and D. Morales-Morales, J. Organomet. Chem., 2003, 679, 101–109 CrossRef.
- T. B. Rauchfuss and D. M. Roundhill, J. Am. Chem. Soc., 1975, 97, 3386–3392 CrossRef CAS.
- (a) P. E. Garrou, Chem. Rev., 1981, 81, 229 CrossRef CAS; (b) O. Kühl, Phosphorus-31 NMR Spectroscopy, Springer, 2008, ISBN 978-3-540-79117-1 Search PubMed; (c) Prosphorus-31 NMR Spectroscopy in Stereochemical Analysis, ed. J. G. Verkade and L. D. Quin, VCH, 1987, ISBN 0-89573-149-5 Search PubMed; (d) Phosphorus-31 NMR, Principles and Applications, ed. D. G. Gorenstein, Academic Press, 1984, ISBN 0-12-291750-2 Search PubMed; (e) S. O. Grim, R. L. Keitler and W. McFarlane, Inorg. Chem., 1967, 6, 1133–1137 CrossRef CAS.
- (a) R. S. Paonesa and W. C. Trogler, J. Am. Chem. Soc., 1982, 104, 1138–1040 CrossRef; (b) R. S. Paonesa and W. C. Trogler, Inorg. Chem., 1983, 22, 1038–1048 CrossRef; (c) R. H. Reamey and G. M. Whitesides, J. Am. Chem. Soc., 1984, 106, 81–85 CrossRef CAS; (d) P. S. Pregosin and H. Rügger, Inorg. Chim. Acta, 1984, 86, 55–60 CrossRef CAS. See also: (e) O. J. Scherer and H. Jungman, J. Organomet. Chem., 1981, 208, 153–159 CrossRef CAS; (f) P. S. Pregosin, R. Favez, R. Roulet, T. Boschi, R. A. Michelin and R. Pos, Inorg. Chim. Acta, 1980, 45, L7–L9 CrossRef CAS.
- Poisoning by sulfur is common in heterogeneous, supported metal catalysts that contain highly dispersed metal atoms, clusters, or larger “rafts” or crystallites with no ligands, thus coordinatively unsaturated, and tend to abstract sulfur from sulfur containing impurities in the gas flow and form layers of metal sulfide that block the reactive sites. See, for instance: B. C. Gates, Catalytic Chemistry, Wiley, New York, 1992, p. 352. ISBN 0-471-55914-8 Search PubMed.
- A. Polo and J. Real, in Houben-Weyl Science of Synthesis, ed. N. Kambe, Thieme, New York, 2008, vol. 39, p. 437, ISBN 978-1-58890-530-7 Search PubMed.
- (a) Y. Ohki, M. Sakamoto and K. Tatsumi, J. Am. Chem. Soc., 2008, 130, 11610–11611 CrossRef CAS PubMed; (b) M. Sakamoto, Y. Ohki, G. Kehr, G. Erker and K. Tatsumi, J. Organomet. Chem., 2009, 694, 2820–2824 CrossRef CAS.
- M. Gottardo, A. Scarso, S. Paganelli and G. Strukul, Adv. Synth. Catal., 2010, 352, 2251–2262 CrossRef CAS . See page 2260, Scheme 4.
- D. F. Shriver and M. A. Drezdzon, The Manipulation of Air Sensitive Compounds, Wiley-Interscience, New York, 2nd edn, 1986, ISBN 0-471-86773-X Search PubMed.
- D. D. Perrin, W. L. F. Armarego and D. R. Perrin, Purification of Laboratory Chemicals, Pergamon Press, Oxford, 3rd edn, 1988, ISBN 0-08-034714-2 Search PubMed.
- R. Ugo, F. Cariati and G. LaMonica, Inorg. Synth., 1968, 11, 105 CrossRef CAS.
- (a) E. Block, G. Ofori-Okai and J. Zubieta, J. Am. Chem. Soc., 1989, 111, 2327 CrossRef CAS; (b) E. Block, V. Eswarakrishnan, M. Gernon, G. Ofori-Okai, C. Saha, K. Tang and J. Zubieta, J. Am. Chem. Soc., 1989, 111, 658 CrossRef CAS.
- P. Pongrácz, L. Kollár and L. T. Mika, Green Chem., 2016 10.1039/c5gc01778e , in press.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, IR spectra and crystal data. CCDC 1431425–1431427. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt04107d |
| This journal is © The Royal Society of Chemistry 2016 |