
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Scandium(III) complexes of monophosphorus acid DOTA analogues: a thermodynamic and radiolabelling study with 44Sc from cyclotron and from a 44Ti/44Sc generator†
R.
Kerdjoudj
a,
M.
Pniok
b,
C.
Alliot
cd,
V.
Kubíček
b,
J.
Havlíčková
b,
F.
Rösch
e,
P.
Hermann
*b and
S.
Huclier-Markai
*ac
aLaboratoire Subatech, UMR 6457, Ecole des Mines de Nantes/CNRS/IN2P3/Université de Nantes, 4 Rue A. Kastler, BP 20722, F-44307 Nantes Cedex 3, France. E-mail: sandrine.huclier@subatech.in2p3.fr; Fax: +33-251858452; Tel: +33-251858537
bDepartment of Inorganic Chemistry, Faculty of Science, Universita Karlova, Hlavova 2030, CZ-12843 Prague 2, Czech Republic. E-mail: petrh@natur.cuni.cz; Fax: +420-221951253; Tel: +420-221951263
cARRONAX GIP, 1 rue Arronax, F-44817 Nantes Cedex, France
dCRCNA, Inserm/CNRS/Université de Nantes, 8 quai Moncousu, 44007 Nantes Cedex 1, France
eInstitute of Nuclear Chemistry, Johannes-Gutenberg-University of Mainz, Fritz-Strassmann-Weg 2, D-55128 Mainz, Germany
First published on 30th November 2015
Abstract
The complexation ability of DOTA analogs bearing one methylenephosphonic (DO3AP) or methylenephosphinic (DO3APPrA and DO3APABn) acid pendant arm toward scandium was evaluated. Stability constants of their scandium(III) complexes were determined by potentiometry combined with 45Sc NMR spectroscopy. The stability constants of the monophosphinate analogues are somewhat lower than that of the Sc–DOTA complex. The phosphorus acid moiety interacts with trivalent scandium even in very acidic solutions forming out-of-cage complexes; the strong affinity of the phosphonate group to Sc(III) precludes stability constant determination of the Sc–DO3AP complex. These results were compared with those obtained by the free-ion selective radiotracer extraction (FISRE) method which is suitable for trace concentrations. FISRE underestimated the stability constants but their relative order was preserved. Nonetheless, as this method is experimentally simple, it is suitable for a quick relative comparison of stability constant values under trace concentrations. Radiolabelling of the ligands with 44Sc was performed using the radioisotope from two sources, a 44Ti/44Sc generator and 44mSc/44Sc from a cyclotron. The best radiolabelling conditions for the ligands were pH = 4, 70 °C and 20 min which were, however, not superior to those of the parent DOTA. Nonetheless, in vitro behaviour of the Sc(III) complexes in the presence of hydroxyapatite and rat serum showed sufficient stability of 44Sc complexes of these ligands for in vivo applications. PET images and ex vivo biodistribution of the 44Sc–DO3AP complex performed on healthy Wistar male rats showed no specific bone uptake and rapid clearance through urine.
Introduction
On the way to personalized medicine, nuclear medicine offers both diagnostic tools and therapeutic drugs utilizing various radioisotopes. Until recently, most radiopharmaceuticals were designed to be used solely for either diagnostics or therapeutics. Radionuclides currently used for imaging, such as 68Ga or 111In, are different from those used for therapy, such as 90Y or 177Lu. One radionuclide would be used to image individual patient disease states and evaluate their receptor expression, metabolic rate, clearance and handling, and a second radionuclide would be used for therapy. Problems with this approach arose due to differences in the chemistry of the radionuclides themselves which have been shown to affect the overall pharmacology, in particular accumulation of the radiopharmaceuticals at target and non-target sites, resulting in an over- or underestimation of the dose to critical tissues and the dose being outside the optimum range of efficacy. In contrast, using the same metal to perform both diagnosis and therapy would result in a better determination of the absorbed dose and would give a better indication of the therapeutic activity necessary to administer the dose. Such a radiopharmaceutical pair utilizing diagnostic and therapeutic radioisotopes is called “theranostics”.1Among the radionuclides available, there is significant interest in the therapeutic radioisotope 47Sc (β−, τ1/2 3.35 d, Eβ 0.143 (68%) and 0.204 MeV (32%); γ, Eγ 159.4 keV, 68%) as it matches with the positron-emitting 44Sc (β+, τ1/2 3.97 h, Eβ 0.63 MeV, 94.3%) or 43Sc (β+, τ1/2 3.89 h, Eβ 0.344 MeV (17.2%) and 0.508 MeV (70.9%)) and, thus, they form an ideal theranostic pair. The potential of 47Sc for nuclear medicine has been already investigated.2–4 The possibility of 47Sc production by neutron irradiation of 47Ti and consecutive solid-phase extraction chromatography has been evaluated.5 Very recently, the feasibility of photonuclear production of 47Sc from 48Ca or 47Ca/47Sc generators has been studied.5,6 Due to its dominant positron emission, 44Sc is very suitable for PET imaging. Its half-life perfectly matches the pharmacokinetics of small molecules or (oligo)peptides. In addition to two collinear γ rays produced by annihilation, 44Sc offers a third γ ray suitable for three-photon coincidence imaging which may further increase resolution of the current PET imaging.7 In addition, the radioisotope can be produced together with its long-lived isomeric excited nucleus, 44mSc (γ, τ1/2 2.44 d, 98.8%, Eγ 270.9 keV), decaying to 44Sc with soft γ emission. The half-life of 44mSc matches the in vivo pharmacokinetics of antibodies and, due to its low-energy transition (recoil energy only 0.89 eV), it can serve as an in vivo generator of 44Sc as the daughter 44Sc stays inside the chelator after the decay of the parent 44mSc nucleus.8 The 44mSc/47Sc theranostic pair with both radionuclides having similar half-lives is very suitable for radiopharmaceuticals utilizing antibodies or their fragments and, thus, they form a unique and very promising theranostic pair for cancer treatments. Utilizations of the Sc-based theranostic pairs can be spread from antibody radioimmunotherapy (the 44mSc/47Sc pair) to treatments with labeled oligopeptides or small molecules (the 44Sc/47Sc pair).
44Sc can be produced by a generator employing 44Ti as a long-lived parent radioisotope.9,10 The radioisotope can be also produced in most medical cyclotrons designed for 18F production.11 The ARRONAX cyclotron produces the 44mSc/44Sc pair from an enriched 44CaCO3 target via the deuteron production route12 which seems to be a promising route to obtain non-carrier-added (NCA) 44Sc with an optimized 44mSc/44Sc ratio. Other production routes allow a better 44mSc/44Sc ratio by bombarding a 45Sc target13–15 leading to a carrier-added product and it is a major drawback for the production of radiopharmaceuticals.
Metallic radioisotopes utilized in nuclear medicine must be tightly bound in a complex to avoid non-specific deposition in tissues. These complexes must exhibit a high thermodynamic stability, a high selectivity for a particular metal ion, a fast complexation of the metallic radioisotopes, kinetic inertness as well as an ability to be conjugated to a biological vector molecule (bifunctional ligands). The design of new radiopharmaceuticals is a viable multidisciplinary field involving physics, chemistry, biology and medicine.16–22
Scandium is a cousin of lanthanides but with some differences. Sc(III) is smaller than Ln(III) (thus, being harder and with a higher preference for hard oxygen donor ligands) and it prefers donor numbers from six to eight. However, the chemistry of trivalent scandium is much less developed than that of trivalent lanthanides.23 Mostly, multidentate ligands already used in Gd(III)-based MRI contrast agents as well as for radiolanthanides, i.e. derivatives of DTPA (DTPA = diethylenetriamine-N,N,N′,N′′,N′′-pentaacetic acid) or DOTA (DOTA = 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid), are the first choice to bind the ion. It has been shown that DOTA derivatives are suitable ligands for scandium radioisotopes.24 Their oligopeptide,10,25–29 antibody30,31 or other32,33 conjugates have been investigated for complexation of the scandium radionuclides.
Recently, we have investigated the chemistry of Sc(III)–DTPA and Sc(III)–DOTA complexes in detail.34 The study confirms that DOTA is a very suitable chelator for trivalent scandium. Efficient radiolabeling of DOTA with 44m/44Sc requires elevated temperatures (>70 °C).8 For oligopeptides, such a high temperature is not a critical parameter. However, antibodies or their fragments need a much lower labelling temperature (mostly below 37 °C) to preserve their immunoreactivity. Slow formation kinetics of DOTA-like chelators remains an important obstacle limiting their use in some radiopharmaceuticals. Therefore, ligands permitting formation of complexes at much lower temperatures to label antibody conjugates are sought.
Scandium(III) is a “harder” metal ion than trivalent lanthanides and oxygen atoms in derivatives of phosphoric acid have a “harder” character than those in the carboxylate group. Thus, phosphonic (R–PO3H2) or phosphinic (R2PO2H) acid pendants may alter ligand behaviour in the desired direction.35 Such DOTA derivatives form thermodynamically stable and kinetically inert complexes with somewhat enhanced complexation kinetics.36–38 Their complexes/conjugates are stable in vivo and show good pharmacokinetic properties due to their high hydrophilicity.39–41 Trivalent gallium is a similar very hard metal ion and phosphinic acid derivatives of NOTA (NOTA = 1,4,7-triazacyclononane-1,4,7-triacetic acid) showed much faster labeling with 68Ga as compared to the parent ligand.42,43
Therefore, monophosphorus acid DOTA analogs, DO3AP, DO3APABn and DO3APPrA (Fig. 1) were considered as better ligands than DOTA. Solution investigations of their complexes were complemented by radiochemical studies with non-carrier-added (NCA) 44Sc from two sources, a 44Ti/44Sc generator and from an ARRONAX cyclotron, and by in vivo/in vitro evaluation of the radiolabelled complexes.
 | ||
| Fig. 1 Structures of ligands discussed in this work. | ||
Experimental
General
The phosphorus acid DOTA analogues, DO3APPrA, DO3AP and DO3APABn (prepared as hydrates according to elemental analysis) were synthesized according to the literature.37,44,45 DOTA (reagent grade) was obtained from Macrocyclics. Analytical grade (conc. aq. HCl, conc. aq. ammonia, MeOH, EtOH, HNO3, ammonium acetate) or pure reagent grade (all other chemicals) reagents were obtained commercially and were used as received unless otherwise specified. Deionized water (18.2 MΩ cm−1; Milli-Q, Millipore) was used throughout the work. Solid-phase extraction resin based on DGA (N,N,N′,N′-tetra(n-octyl)diglycolamide; Triskem), cation AG1-X8 (200–400 mesh, Cl−-form; Bio-Rad), anion AG50W-X8 (200–400 mesh, H+-form; Sigma) exchangers, and Chelex-100 (imino-diacetate functionalized resin; Sigma) were purchased. The concentrations of stable scandium together with potential metals present in the solutions were analysed by ICP-AES (ThermoFischer ICAP 6500 DUO ICP-AES). The wavelengths for each element were selected in order to give the best relative emissions and to prevent interferences. To calibrate the ICP-AES, commercially available single- and multi-element standards (∼10 ppm, SCP Science) were used in dilute nitric acid (1% w/w). The acidic solutions of the radioisotope batches were collected and were diluted in aq. HNO3 (1% w/w) prior to the analysis.Production of 44Sc
Potentiometry
Potentiometric titrations were performed by a general method described elsewhere:34,36 25 °C, I = 0.1 M (NMe4)Cl, starting volume 5 mL (ligands) or 1 mL (complexes), ligand concentration 0.004 M, ligand![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Sc molar ratio 1:0.95, PHM 240 pH-meter, 2 mL ABU901 autoburette and GKB2401B combined electrode (glass/Ag–AgCl, all Radiometer), pKw = 13.81. Electrode calibration by strong acid–strong base titration was performed before each titration to give a pair calibration/titration which was used for calculations of the constants; the procedure gives a reliable pH even at pH below 2 and above 12. To obtain very acidic protonation constants of DO3APPrA and DO3APABn which were necessary for the correct determination of Sc(III) complex stability constants, the ligands were titrated in the pH range 1.4–11.8 with electrode calibration performed in the pH range 1.5–11.8 (∼50 points per titration, four titrations per sample). As complexation equilibria in Sc(III)–ligand systems were slow (time to reach full equilibrium was six days; checked by NMR), the titrations were carried out by the out-of-cell method;34,36 pH range 1.4–5.5, 23–25 points per titration, two parallel titrations. To help with the determination of the first protonation constant of the complexes and enable correct fitting of all equilibrium data for the Sc(III)–DO3APABn and –DO3APPrA systems, the acid–base titrations of the pre-formed complexes were performed.46 Stock solutions of the ligands (0.02 M) and ScCl3 (0.05 M) were mixed in ampoules (L:Sc molar ratio 1:0.95, cL = 0.006 M in the final samples) and a stock solution of (NMe4)OH was added (98% necessary for complete neutralization of the added ligand amount). The ampoule was flame-sealed, heated at 90 °C in an oven for 24 h to ensure complete complexation of the Sc(III) ion, cooled to room temperature and opened. The solution was pipetted into a titration vessel, water and excess of HCl/(NMe4)Cl solution were added (to ensure acidic starting pH and 0.1 M ionic strength), and the solution (starting volume 5 mL) was immediately titrated by (NMe4)OH stock solution (pH range 1.7–12, ∼40 points per titration, 4 parallel titrations, the complex concentration ∼0.003 M). Mathematical treatment of all equilibrium data (together with the NMR titration data, see below) was done with the OPIUM program package.47 The constants are concentration constants and were calculated as overall protonation/stability constants βhlm defined generally as βhlm = [HhLM]/([H]h·[L]·[M]); charges are omitted. The calculated overall protonation/stability constants (with standard deviations given directly by the program) are presented in the ESI (Tables S1 and S2†). Throughout the text, pH means −log[H+].
:Sc molar ratio 1:0.95, PHM 240 pH-meter, 2 mL ABU901 autoburette and GKB2401B combined electrode (glass/Ag–AgCl, all Radiometer), pKw = 13.81. Electrode calibration by strong acid–strong base titration was performed before each titration to give a pair calibration/titration which was used for calculations of the constants; the procedure gives a reliable pH even at pH below 2 and above 12. To obtain very acidic protonation constants of DO3APPrA and DO3APABn which were necessary for the correct determination of Sc(III) complex stability constants, the ligands were titrated in the pH range 1.4–11.8 with electrode calibration performed in the pH range 1.5–11.8 (∼50 points per titration, four titrations per sample). As complexation equilibria in Sc(III)–ligand systems were slow (time to reach full equilibrium was six days; checked by NMR), the titrations were carried out by the out-of-cell method;34,36 pH range 1.4–5.5, 23–25 points per titration, two parallel titrations. To help with the determination of the first protonation constant of the complexes and enable correct fitting of all equilibrium data for the Sc(III)–DO3APABn and –DO3APPrA systems, the acid–base titrations of the pre-formed complexes were performed.46 Stock solutions of the ligands (0.02 M) and ScCl3 (0.05 M) were mixed in ampoules (L:Sc molar ratio 1:0.95, cL = 0.006 M in the final samples) and a stock solution of (NMe4)OH was added (98% necessary for complete neutralization of the added ligand amount). The ampoule was flame-sealed, heated at 90 °C in an oven for 24 h to ensure complete complexation of the Sc(III) ion, cooled to room temperature and opened. The solution was pipetted into a titration vessel, water and excess of HCl/(NMe4)Cl solution were added (to ensure acidic starting pH and 0.1 M ionic strength), and the solution (starting volume 5 mL) was immediately titrated by (NMe4)OH stock solution (pH range 1.7–12, ∼40 points per titration, 4 parallel titrations, the complex concentration ∼0.003 M). Mathematical treatment of all equilibrium data (together with the NMR titration data, see below) was done with the OPIUM program package.47 The constants are concentration constants and were calculated as overall protonation/stability constants βhlm defined generally as βhlm = [HhLM]/([H]h·[L]·[M]); charges are omitted. The calculated overall protonation/stability constants (with standard deviations given directly by the program) are presented in the ESI (Tables S1 and S2†). Throughout the text, pH means −log[H+].
NMR measurements
The 45Sc (97.15 MHz) and 31P{1H} (161.91 MHz) NMR spectra were recorded on Varian UNITY Inova 400. All experiments, unless stated otherwise, were carried out in 5 mm NMR tubes at 25.0 °C. The 31P NMR chemical shifts were referenced to 85% aq. H3PO4 (δP = 0.0 ppm). The 45Sc NMR chemical shifts were measured with respect to 0.1 M Sc(ClO4)3 in 1 M aq. HClO4 (δSc = 0.0 ppm), 0.01 M [Sc(ox)4]5− (δSc = 8.31 ppm)34 or 0.01 M [Sc(tmu)6](ClO4)3 in CH3NO2 (tmu = N,N,N′,N′-tetramethylurea; δSc = −53.3 ppm)48 as external standards in the insert tube. The equation pD = pH + 0.4 (pH is a standard pH-meter reading) was used for D2O solutions.Samples for NMR titrations were prepared similarly to the out-of-cell titration as given above and analogously to the previous paper.34 Stock solutions of the ligands, ScCl3, HCl, KCl (if necessary), and water were mixed into tubes (final cL = 0.004 M, L:Sc molar ratio 1:0.95, V0 = 1 mL; I = 0.1 M (H,K)Cl, with no control of the ionic strength at pH < 1.1) to obtain pH in the range 0.5–1.3 and the tubes were tightly closed (12–15 titration points); the equilibration time was seven days at room temperature. Species abundance in each tube (i.e. the titration point) was determined by 45Sc NMR measurements. Two parallel titrations were carried out. To determine the equilibration time, other samples were prepared as above at three pHs, the samples were left at room temperature and 45Sc NMR spectral changes with time were followed.
Samples for equilibrium studies with the Sc(III)–DO3AP system were prepared by mixing stock solutions of DO3AP, ScCl3, 17% or 5% aq. HCl, 1.1 M or 0.2 M HCl, 0.2 M KCl (if necessary), and water into tubes (conditions as above). The solutions were left to equilibrate for at least seven days at room temperature and pH values were determined as above. Job's method was employed to determine stoichiometry of the out-of-cage species in the Sc(III)–DO3AP system. Each sample was prepared in a vial by mixing 0.1 M aq. ScCl3 and 0.1 M aq. DO3AP stock solutions with 1 M aq. HCl (final volume 1 mL, pH 0.22, cSc = 4–20 mM, cSc+L = 40 mM, 7 points, equilibration time seven days). To determine protonation constants of [Sc(DO3AP)]2− isomeric species, a pre-prepared complex was dissolved in H2O in a 10 mm NMR tube and the desired pH was adjusted with aq. HCl or aq. (NMe4)OH (cScL ∼ 0.01 M, pH range 4.15–7.67, 26 points). The pH was determined by a freshly calibrated (three buffers) combined pH electrode fitting the NMR tube.
To characterize the [Sc(DO3APPrA)]2−, [Sc(DO3APABn)]− and [Sc(DO3AP)]2− complexes, their solutions were prepared by dissolving the appropriate ligand (0.05 mmol), ScCl3·6H2O (0.055 mmol) in water (1 mL) and adjusting the solution pH to ∼7 with solid Li2CO3. Then, the solutions were stirred in sealed vials at 90 °C for 2 h. Excess of Sc(III) precipitated as white Sc(III)–carbonate/hydroxide and was filtered off with a syringe filter and the filtrate was evaporated to dryness. The solid was dissolved in D2O (0.5 mL) to obtain a solution with the complex concentration ∼0.1 M.
Free-ion selective radiotracer extraction (FISRE) method
An aliquot of the Chelex-100 chelating resin was mixed with bulk aq. solution containing 45Sc to give a final concentration of the isotope 10−5 M. To prevent the potential interference of nitrate ions on the distribution behaviour of the metallic content, aliquots of standard solutions were transferred into a dry Teflon beaker pre-cleaned with ultrapure aq. HNO3 (2% w/w). Then, they were evaporated to dryness and recovered in a suitable solution three times. The ligand stock solution was added to reach a final ligand concentration ranging from 10−7 to 10−3 M. All measurements were performed at ionic strength I = 0.1 M NaCl. The pH of the suspension was adjusted to pH = 2.6; 2.5 and 2.3 according to the ligand (DO3AP, DO3APPrA or DO3APABn, respectively). These pH values were estimated by taking into account stability constant values (logKScL) obtained from the equilibrium data. As the distribution coefficients were calculated as a function of dried resin mass, humidity content was determined by drying five samples of each pre-conditioned resin in an oven at 105 °C. The final distribution coefficients were calculated on the basis of the arithmetic averages of replicate analyses (at least triplicate analysis). The resin concentration (in g mL−1) was chosen for each batch to minimize the global uncertainty of the partition coefficient. The reproducibility of logKd was better than 5%.
The resulting suspension was equilibrated for 24 h and the pH was re-adjusted, if necessary. In preliminary experiments, the sufficient equilibration time was found to be six days for all the systems. Solid and liquid phases were separated by sedimentation. Aliquots (1 mL) of the supernatant were taken for ICP-AES analysis. Scandium concentrations were determined and experimental Kd values were plotted as a function of the total ligand concentration in the solutions. These dependences were used for stability constant determinations. To check the potential presence of protonated complexes, other sets of experiments were performed at fixed ligand concentrations (10−3 M) in the pH range 2–7 utilizing the same protocol as described above.
Radiolabelling with 44Sc
In vitro and in vivo experiments
The in vitro challenge studies (stability in serum, hydroxoapatite adsorption) were performed by standard procedures and details are given in the ESI.†Animal studies were carried out in accordance with the guidelines of the French law on Animal Studies. Biodistribution of the 44Sc–DO3AP complex was followed in four healthy Wistar rats injected intravenously with 44Sc–DO3AP solution (100 μL, 1 MBq). The animals were imaged at selected time intervals, then sacrificed and dissected. The rats were sacrificed at 30 min and 1 h, two rats per time point. Organs were collected, weighed, counted on a gamma counter, and the percentage of injected dose per gram of tissue (% ID per g) was calculated. PET images were acquired using a Siemens Inveon micro PET/CT instrument.
Results and discussion
Characterization of the Sc(III) complexes in solution
Structures of the in-cage complexes in solution were determined by 1H, 31P{1H} and 45Sc NMR measurements. In all cases, 1H NMR spectra were very complicated due to the non-equivalence of all protons in a rigid structure. The 31P{1H} NMR spectra showed the presence of two signals (Fig. 2). On the basis of a number of data for lanthanide(III) complexes of DOTA-like ligands, such an observation can be explained by the presence of two isomeric complexes having square antiprismatic (SA) and twisted square antiprismatic (TSA) arrangements. These isomers have been observed for lanthanide(III) complexes of the title ligands.44,45 It is surprising as only one isomer, SA, was observed in solution for the [Sc(DOTA)]− complex,50 although both isomers have been found in the solid state.34,50 It was observed that the 31P NMR chemical shift and relative abundance of the TSA/SA isomers of the Sc(III)–DO3AP complex is changed in the pH range 4–7. Such behaviour has been observed for lanthanide(III) complexes of DO3AP44 and is connected with protonation of the coordinated phosphonate group. Thus, 31P{1H} NMR spectra were recorded over the pH range (Fig. S4†) and used for calculation of protonation constants for each complex isomer. The results, logKa 5.33 and 5.53, correspond well to the potentiometric data (see below) and the values are similar to those found for the Ln(III) analogues.44 The isomer ratio did not change with pH for the complexes of the phosphinate ligands.
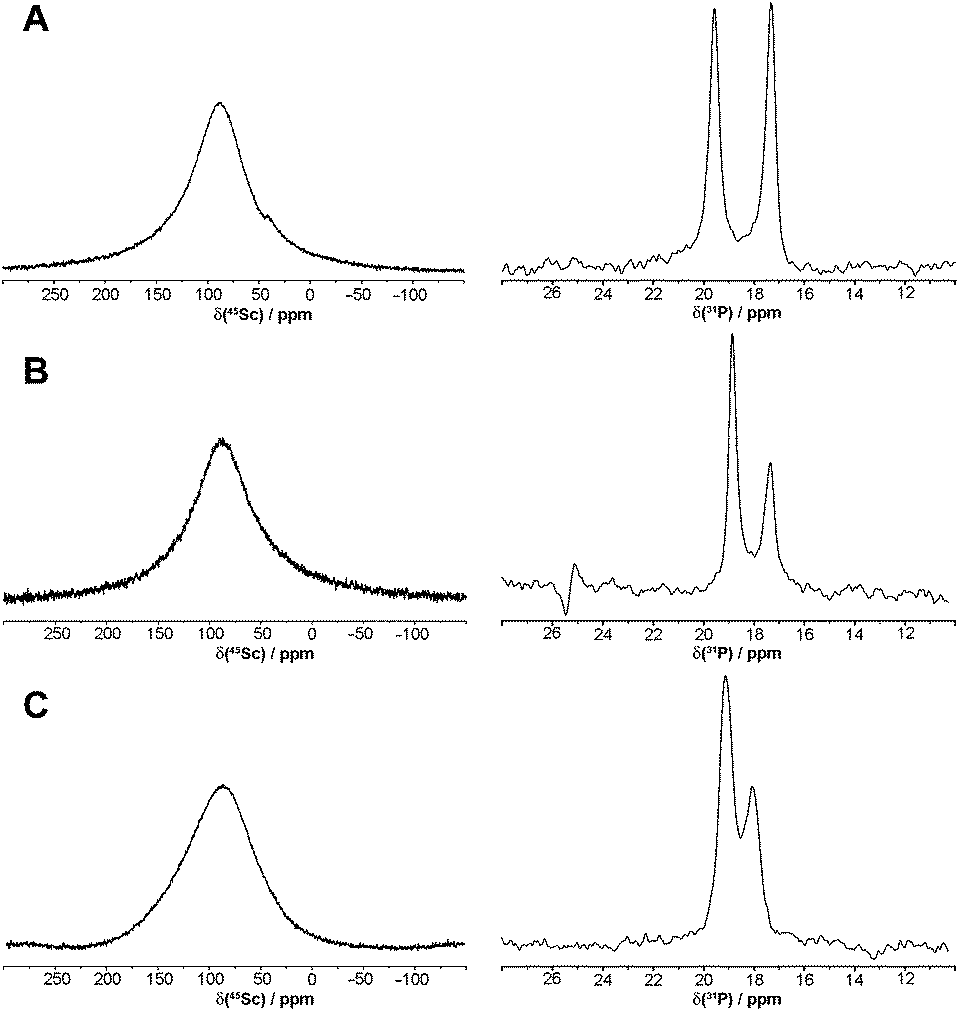 | ||
| Fig. 2 45Sc and 31P{1H} NMR spectra of [Sc(DO3AP)]2− (A), [Sc(DO3APPrA)]2− (B) and [Sc(DO3APABn)]− (C) in D2O (0.1 M, pD = 7.1). | ||
The 45Sc NMR spectra (Fig. 2) recorded in 0.1 M solutions of pre-prepared complexes showed very broad peaks 87 ppm (ϖ1/2 ∼ 5200 Hz), 87 ppm (ϖ1/2 ∼ 5700 Hz) and 90 ppm (ϖ1/2 ∼ 7200 Hz) for the [Sc(DO3AP)]2−, [Sc(DO3APPrA)]2− and [Sc(DO3APABn)]− complexes, respectively. The values are similar to those observed for [Sc(DOTA)]− (δSc ∼ 100 ppm, ϖ1/2 ∼ 4300 Hz)34 also confirming the formation of the in-cage complex for all ligands.
Equilibrium studies
For the Sc(III)–DO3APABn and –DO3APPrA systems, it was found that equilibrium at pH > 1.8 (i.e. formation of in-cage complexes, see below) is established during several days and the rate of complexation is faster with a higher pH (Fig. S1†). Equilibria in very acidic solutions (formation of out-of-cage complexes) were established almost immediately. Similarly to the Sc(III)–DOTA system, the Sc(III)-aqua complex disappeared above pH ∼ 1.8. However, some complex formation started even at pH 0–0.5 (Fig. S2†) as indicated by a broad 45Sc NMR peak (∼35 ppm). This chemical shift suggests a purely oxygen coordination sphere. It can be facilitated by the presence of a phosphinic acid group able to bind metal ions even in very acidic solutions (see also below). Therefore, the species can be assigned as out-of-cage complexes where the Sc(III) ion is coordinated by oxygen atom(s) from one or more pendant arms of the ligands. Once the in-cage complexes (the Sc(III) ion is sandwiched between N4 and O4 planes as in the [Sc(DOTA)]− complex50) are formed, the 45Sc NMR signal of the out-of-cage complexes starts to broaden and decreases in intensity and, finally, no 45Sc NMR signal is observed (due to a low concentration of the complexes in these titration experiments). In parallel, two broad closely located 31P{1H} NMR signals assignable to the Sc(III)-bound phosphinate group in the isomeric in-cage complexes start to be observed. To quantify the abundance of the Sc(III)-aqua ion for stability constant determination (see below), [Sc(tmu)6]3+ had to be used as the secondary standard48 as other standards34 overlapped with the signal of the out-of-cage species.The behaviour of the Sc(III)–DO3AP system was analogous to that observed for the Sc(III)–DO3APABn and –DO3APPrA systems only at pH above ∼3.8; no 45Sc and two 31P{1H} NMR peaks were observed confirming the formation of the in-cage complex (Fig. 3). From potentiometric titrations in this pH range, only the constant corresponding to protonation of the coordinated phosphonate group in the in-cage complex, logKa 5.29, could be determined (Table S2†). However, unexpected results were obtained for the Sc(III)–DO3AP system in more acidic solutions. Here, other peaks appeared in both NMR spectra: δSc ∼ 23 ppm, and δP −8.3 ppm (Fig. 3). The 45Sc NMR signal of the Sc(III)-aqua complex starts to be observable below pH ∼ 0.8. Surprisingly, the signal at δSc ∼ 23 ppm was present even in extremely acidic solutions (up to 1:1 aq. HCl). Most probably, the NMR signal can be assigned to an out-of-cage complex species where at least the protonated phosphonate group (and, possibly, the acetate group(s) as well) is bound to the scandium(III) ion. Such a coordination mode of the phosphonate group has been observed with metal ions (e.g. trivalent lanthanides) even under highly acidic conditions.51,52 The overall stoichiometry of the Sc(III)–DO3AP species present in the acidic solutions can be roughly estimated as Sc:L = 1:2 from a Job-like plot of NMR signal intensities (Fig. S3†) and, overall, the speciation is rather complicated. Unfortunately, such solution behaviour (i.e. not determinable correct Sc/DO3AP stoichiometry as well as the number of protons in the species) precluded determination of the Sc(III)–DO3AP complex stability constants as the system is too complex to be modeled by equilibrium constant calculations.
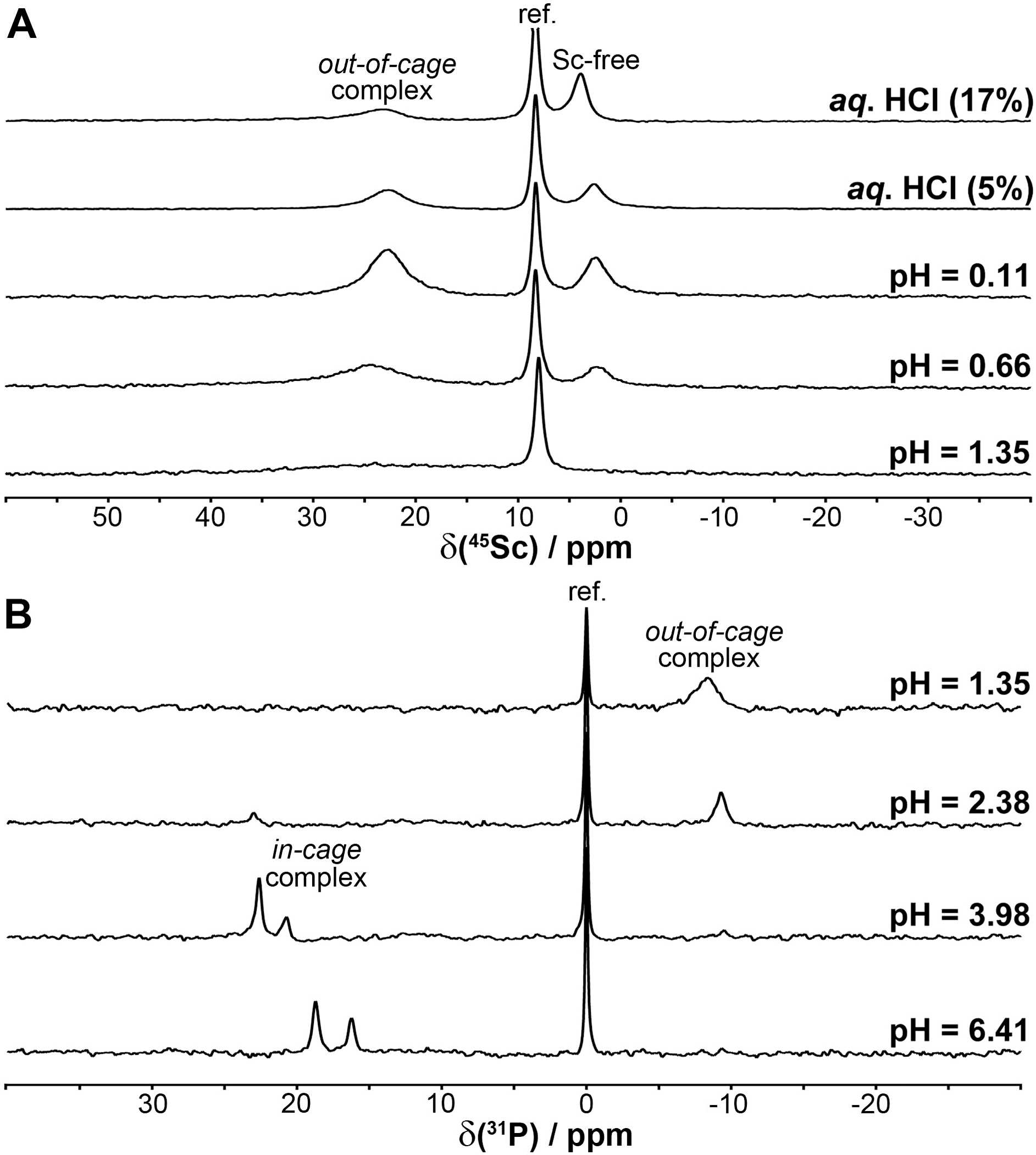 | ||
| Fig. 3 45Sc (A) and 31P{1H} (B) NMR spectra of equilibrated solutions prepared by mixing ScCl3 and DO3AP (cSc = cL = 0.004 M). The given pH values are those of the equilibrated solutions. | ||
The stability constants were determined by a combination of three techniques, 45Sc NMR, out-of-cell and direct (in-cell) potentiometry (for more detailed discussion, see the ESI†). The stability constants are presented in Table 1 and the experimentally determined overall stability constants are given in Table S2.† The corresponding distribution diagrams are shown in Fig. 4. The Sc(III) complexes of DO3APABn and DO3APPrA are more stable than their lanthanide(III) complexes (logKLuL 24.0 and 25.5, respectively)37 but less stable than the [Sc(DOTA)]− complex (logKScL 30.79).34 The difference in stability constants between the lutetium(III) and scandium(III) complexes is similar for all ligands. The first Sc(III) complex protonation constants, corresponding to the protonation of amino or carboxylate groups in the phosphorus side chain, are almost identical to those of lutetium(III) complexes.37 The next two protons are attached to the ring nitrogen atoms with the formation of out-of-cage complexes where the scandium(III) ion is bound only to pendant arm oxygen atom(s). Such a chemical model has already been suggested for lanthanide(III)–DO3AP systems36 and such out-of-cage species diprotonated on ring amines have been observed for lanthanide(III) complexes in the solid state as well as in solution.41,53 The very high abundance of the triprotonated species (Fig. 4) can be caused by a strong preference of trivalent scandium for hard phosphinate oxygen donors in the out-of-cage species.
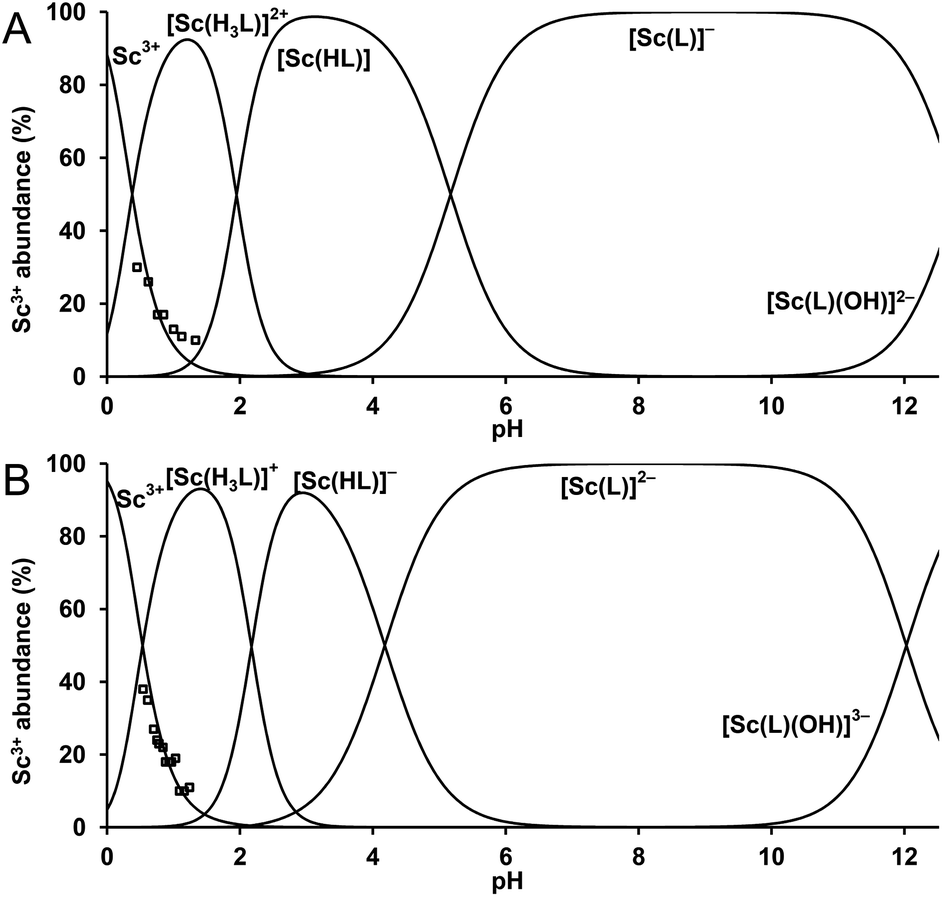 | ||
| Fig. 4 Distribution diagram for the Sc3+–DO3APABn system (A) and the Sc3+–DO3APPrA system (B). cSc = cL = 0.004 M. The abundance of free Sc3+ ion (□) was determined by 45Sc NMR spectroscopy. | ||
| Equilibriuma | DO3APABn | DO3APPrAb |
DO3APc |
DOTA | DTPA |
|---|---|---|---|---|---|
|
a Charges are omitted for clarity.
b The most acidic protonation constants of DO3APPrA were re-determined: logK5 2.94 and logK6 1.54 (Table S1).
c Other constants could not be determined (see text).
|
|||||
| L + Sc ⇌ [Sc(L)] | 27.03 | 28.31 | 30.79 | 27.43 | |
| [Sc(L)] + H ⇌ [Sc(HL)] | 5.17 | 4.18 | 5.29 | 1.0 | 1.36 |
| [Sc(HL)] + 2H ⇌ [Sc(H3L)] | 3.96 | 4.35 | |||
| [Sc(OH)(L)] + H ⇌ [Sc(L)] + H2O | 12.83 | 12.03 | 12.44 | ||
As stability constants of the Sc(III)–DO3AP complex cannot be determined by the above method, a competition method with trivalent ytterbium (as the metal ion with a very high stability constant with DO3AP, logKYbL ∼ 28.5)36 as well as transchelation with DTPA were tested. In both cases, the determination failed due to problems with too slow kinetics of the transmetallation or transchelation.
Determination of stability constants under trace concentrations
Another technique, the Free Ion Selective Radiotracer Extraction (FISRE) method54 was employed to determine stability constants in trace (micromolar) concentrations of the ligands. The technique uses a chelating ion exchanger (here, Chelex-100 containing imino-diacetate groups was used) as a competing “ligand”. The chelating resin competes with the ligands for the Sc3+ ion and the speciation is determined by solid/liquid separation. Recently, the method has been used for the determination of thermodynamic stability of metallo-radiopharmaceuticals containing Y(III).55 The technique has been also used to estimate stability constants of scandium(III) complexes with EDTA, DTPA, NOTA and DOTA.24Adsorption of the free Sc3+-aqua ion on imino-diacetate chelating groups 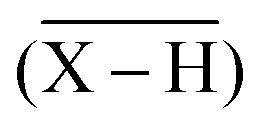 can be described by the following equilibrium (eqn (1)):
can be described by the following equilibrium (eqn (1)):
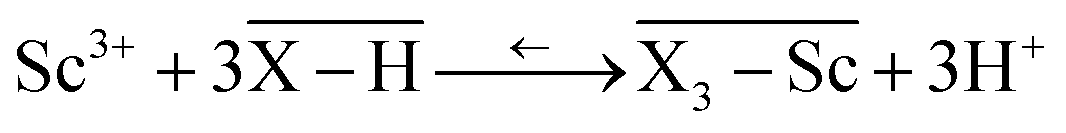 | (1) |
The overlined species refer to the species present on the resin (adsorbed species). The electroneutrality of each phase is required. Stability constants could be estimated by fitting dependence of the distribution coefficient (Kd) of Sc(III) between the resin and the supernatant on the total ligand concentration in the supernatant. If both the ligand and the exchange resin are used in a large excess in comparison with the initial Sc concentration, Kd values could be expressed as eqn (2):
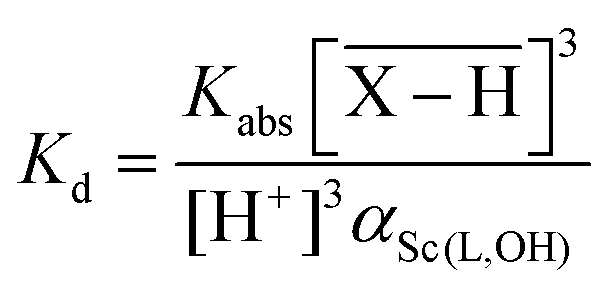 | (2) |
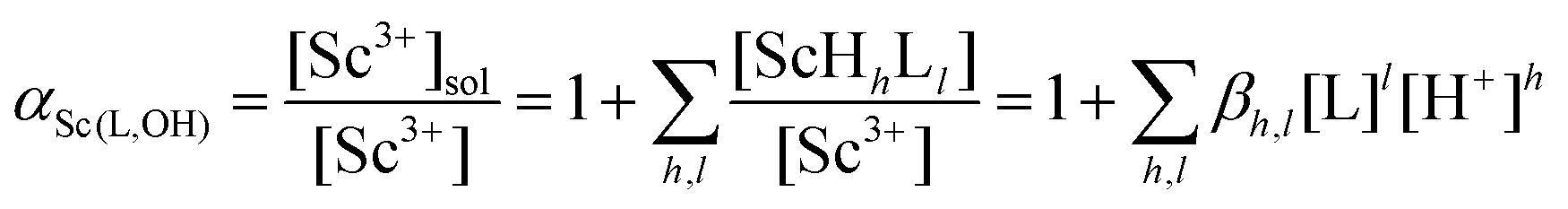 | (3) |
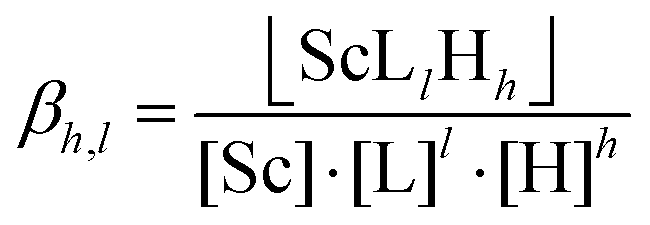 | (4) |
As αSc(L,OH) is a function of the ligand protonation constants and stability constants of the corresponding Sc(III) complexes, the stability constants could be calculated by fitting of the Kd values determined at various ligand excesses and at various solution pH values according to eqn (2). For the full set of equations and more explicit explanation of the method, see the ESI.†
Transchelation kinetics of macrocyclic ligands is generally slow. Therefore, the equilibration time was at least six days. The experimental data are depicted in Fig. 5 and S5.† To obtain a good fit of the data obtained for various pH values, not only [Sc(L)] but also [Sc(HL)] species had to be included in all systems (for a comparison of different models, see Fig. S5†). The results are summarized in Table 2. The values obtained by the FISRE method are in a reasonable qualitative agreement with the values obtained by potentiometry/NMR (Table 1) if errors naturally accompanying utilization of trace concentrations and very high absolute values of the constants are taken into account.
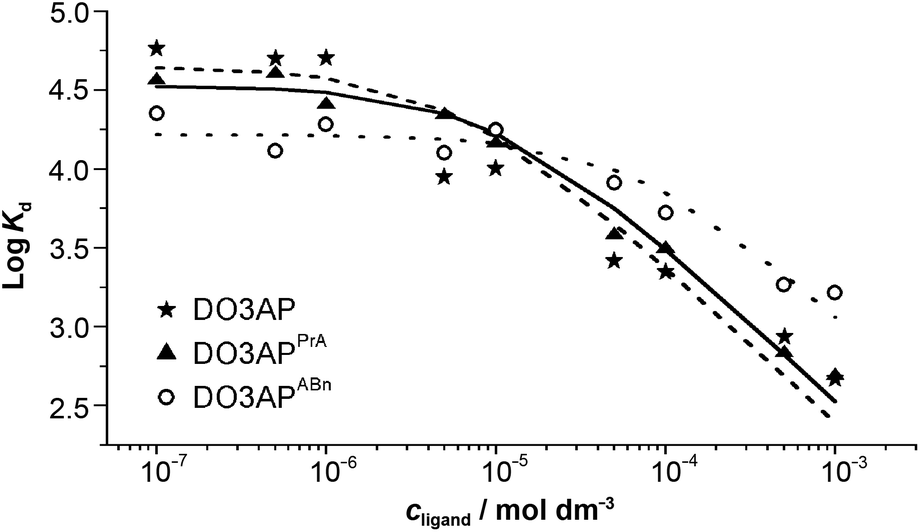 | ||
| Fig. 5 The Sc(III)–ligand isotherms obtained by the FISRE method: DO3AP (full line); DO3APPrA (dashed line) and DO3APABn (dotted line). The lines correspond to the fitting as explained in the ESI.† Solution pH and concentration of the Chelex resin were adjusted for each batch to minimize the global uncertainty of the partition coefficient (I = 0.1 M NaCl). | ||
K) of scandium(III)–DO3AP, –DO3APABn and –DO3APPrA complexes as determined by the FISRE method
The results show that the method can be used for fast screening of complex stabilities as it works with significantly lower amounts of compounds in comparison with common techniques such as potentiometry or NMR. Further, it could also be used for studying “problematic” metal ions (e.g. trivalent or tetravalent metal ions) or ligands. It can be used for a qualitative evaluation of metal ion–ligand interactions at the trace level. In addition, the FISRE method is more easy to carry out, faster and operationally cheaper than the “standard” methods (here, the stability constant of the [Sc(DO3AP)]2– complex cannot be determined by the conventional methods) and gives results which can be used for evaluation of the complexation ability of new ligands toward metal ions intended to be utilized in radiopharmaceuticals.
Labelling of the title ligands with generator- (44Ti/44Sc) and cyclotron-produced (44m/44Sc) radioscandium
The labelling efficiency of the title ligands was tested at different solution pHs, temperatures and ligand concentrations (or more correctly, at different radiometal-to-ligand molar ratios). The labelling was done with no-carrier-added (NCA) radioscandium from two sources: 44Sc eluated from a 44Ti/44Sc generator (finally obtained in 0.25 M aq. ammonium acetate buffer, pH 4)9,10 and a mixture of 44mSc and 44Sc radioisotopes produced by an accelerator (finally obtained in 0.1 M aq. HCl)12 as both sources can be considered for the future regular production of 44Sc. The radioscandium from each source differs in the specific activity commonly obtained and/or in the cold metal ion impurity content. The labelling results are summarized in Table 3 and Fig. 6 and 7.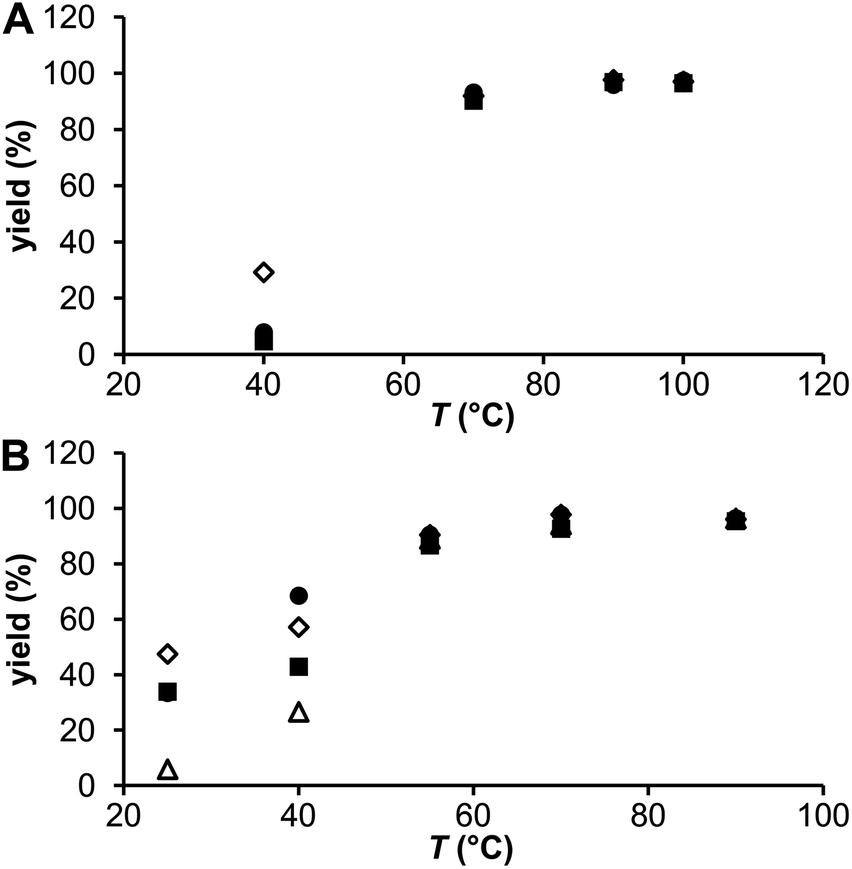 | ||
| Fig. 6 Radiolabeling of DOTA (●) and its monophosphorus acid analogues (◊ DO3AP, ■ DO3APABn and △ DO3APPrA) at different temperatures (pH 4.0, labelling time 30 min): (A) generator 44Sc, n(ligand) = 20 nmol; n(Sc) = 4.5 × 10−5 to 1 × 10−4 nmol; A(44Sc) = 3 kBq. (B) Cyclotron 44mSc/44Sc, n(ligand) = 0.2 nmol; n(Sc) = 1.5 × 10−6 nmol; A(44Sc) = 3 kBq. | ||
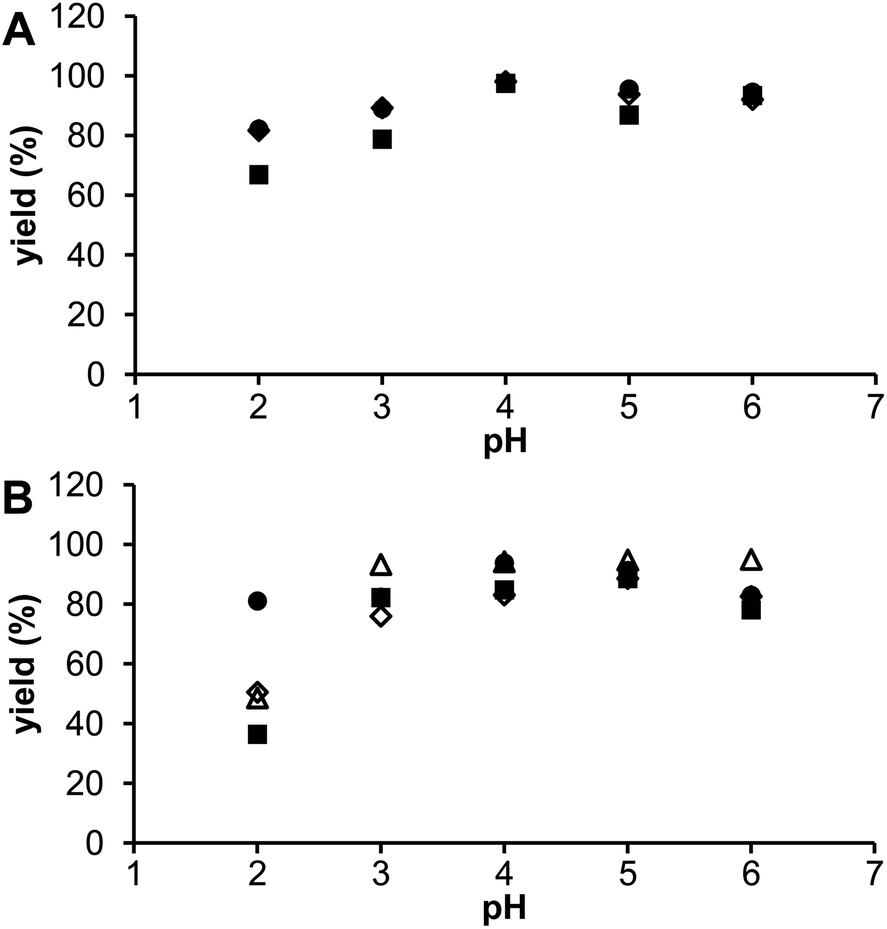 | ||
| Fig. 7 Radiolabelling of DOTA (●) and its monophosphorus acid analogues (◊ DO3AP, ■ DO3APABn and △ DO3APPrA) at different pH values (t = 70 °C, labeling time 30 min): (A) generator 44Sc, n(ligand) = 20 nmol; n(Sc) = 4.5 × 10−5 to 1 × 10−4 nmol; A(44Sc) = 3 kBq. (B) Cyclotron 44mSc/44Sc, n(ligand) = 0.2 nmol; n(Sc) = 1.5 × 10−6 nmol; A(44Sc) = 3 kBq. | ||
:ligand ratios. Experimental conditions: t = 70 °C, 30 min, pH = 4. All yields are given within the experimental uncertainties of the cyclone device of ±5%
| Radioisotope source | n ligand (nmol) | Ligand | |||
|---|---|---|---|---|---|
| DOTA | DO3AP | DO3APPrA | DO3APABn | ||
| Generator 44Sc | 1 | 10.1 | 10.0 | 8.6 | 1.9 |
| 3 | 96.5 | 91.6 | 88.5 | 4.3 | |
| 5 | 96.7 | 93.8 | 95.7 | 20.8 | |
| 10 | 96.8 | 95.3 | 97.0 | 91.1 | |
| 20 | 97.3 | 95.7 | 97.1 | 94.7 | |
| 25 | 97.1 | 96.6 | 96.5 | 95.1 | |
| 30 | 97.6 | 97.6 | 97.7 | 96.6 | |
| Cyclotron 44mSc/44Sc | 0.02 | 31.1 | 36.1 | 28.4 | 32.5 |
| 0.07 | 97.8 | 97.4 | 88.3 | 90.6 | |
| 0.12 | 98.9 | 98.8 | 94.2 | 92.7 | |
| 0.17 | 99.4 | 98.8 | 98.9 | 94.7 | |
| 0.24 | 99.6 | 99.1 | 99.1 | 96.4 | |
As the excess of a chelator over a metal radioisotope determines the accessible specific activity, the influence of the chelator/radioscandium molar ratio on the radiochemical yield was tested under conditions previously successfully used for DOTA derivatives: labelling time 30 min, pH 4 and temperature 70 °C. Results are presented in Table 3. With generator 44Sc, overall radiolabelling yields for 44Sc–DOTA and 44Sc–DO3AP were quite similar ranging from 10% to 95% if 1 or 5 nmol of the ligand, respectively, were added. In contrast, the radiolabeling yield for 44Sc–DO3APABn was much lower (approx. 25% if <10 nmol of DO3APABn was used) and 20 nmol of the ligand was required to reach 95% radiolabeling. With the cyclotron 44m/44Sc, the amount of the ligands required to reach the same radiolabelling yields was significantly lower, although the same general trend was observed. Thus, 0.2 nmol of the ligands is enough for the cyclotron 44m/44Sc to obtain a minimum of 95% radiolabeling versus more than 5 nmol of the ligands for the 44Sc from the 44Ti/44Sc generator. The differences between the radioscandium from the two sources could be explained by the different contents of cold metallic impurities competing with the ligands for the scandium radioisotope.
Specific activity (SA), a measure of the radioactivity per unit mass of the compound, is one of the major criteria for radiopharmaceuticals and should be as high as possible. Specific activities of the radioscandium from each source and for each ligand were calculated and the values are summarized in Table S3.† The calculated specific activity of the cyclotron 44m/44Sc is always higher than 10 MBq nmol−1 (4 h after end of beam). However for the generator 44Sc, the specific activity was estimated to be max. ∼2 MBq nmol−1 (for DOTA; 4 h after the end of elution).
The influence of temperature and solution pH was tested under conditions suggested by the experiments above: 20 nmol and 0.2 nmol of the ligands for the generator 44Sc and the cyclotron 44m/44Sc, respectively, and at a reaction time of 30 min. The results are plotted in Fig. 6. At 40 °C, the generator 44Sc revealed 30% labeling for DO3AP whereas 5–8% yield was achieved for the other ligands. The yields increased with temperature as expected, to more than 90% and 95% at 70 °C and 90 °C, respectively, but with no difference among the ligands. Surprisingly at low temperatures, the labelling yield with DOTA was significantly lower than that with DO3AP. On the other hand with the cyclotron 44m/44Sc, even if concentrations of the ligands were much lower compared to those for the generator 44Sc, higher radiolabeling yields, mainly at lower temperatures, were obtained. At 40 °C (a temperature still suitable for antibody labelling), radiolabelling yields >40% (44Sc–DO3APABn), >55% (44Sc–DO3AP) and ∼70% (44Sc–DOTA) were observed; the values for DOTA agreed well with those previously published.8 Complete binding (>95%) was achieved at 70 °C for all ligands. Thus, replacement of an acetate group on the DOTA skeleton does not lead to noticeably improved radiolabelling at lower temperatures if compared with the parent DOTA. Monophosphinate ligands were labelled worse than DOTA or DO3AP at lower temperatures.
The influence of solution pH was investigated in the range 2–6 and results are shown in Fig. 7. For radioscandium from both sources, the best labelling was observed at pH 4–5 with some decrease at a higher pH, probably due to the formation of colloidal scandium(III)–hydroxide species, as found in the previous studies.9,10 When the solution pH was reduced to 2, labelling efficiency significantly decreased for all the ligands but more for the phosphorus acid DOTA analogues. It is in agreement with the equilibrium data (see above) as monophosphorus acid DOTA analogs form the out-of-cage complexes with higher abundances than DOTA does. So, the optimal radiolabeling yields were achieved using facile conditions – pH 4, reaction time 30 min, and incubation temperature 70 °C.
Unlike for Ln(III) ions,37–40 labelling with 44Sc was not improved. As discussed above, the formation of metal ion complexes of DOTA-like ligands is a two-step process,41,53i.e. formation of the out-of-cage complex is followed by proton removal from, and metal ion transfer into, the ligand cavity as a rate-determining step. Both steps should be optimized to improve labelling efficiency. Here, the basicity of ring nitrogen atoms is similar in all investigated ligands leading to the similar labelling efficiency. In addition, small Sc(III) ions may not fit well into the cavity of the title ligands. A somewhat better labelling with DO3AP might be connected with the rather hard character of the phosphonate group leading to a good interaction with small and hard Sc(III) ions. The effect of the right combination of cavity size, basicity of ring amines and suitable properties of pendant donor atoms for efficient formation of the out-of-cage complex has been shown for phosphinic acid NOTA analogues. Very small trivalent gallium perfectly fits the small cavity of NOTA-like ligands, hard phosphinates are very selective for hard Ga(III) ions and they also decrease the ring amine basicity. It all leads to a very efficient labelling of these ligands with 68Ga.42
In vitro and in vivo studies
Even the labelling studies with the monophosphorus acid DOTA analogs did not show improved properties; in vitro/in vivo properties of their 44m/44Sc complexes were evaluated. The serum stability can be seen as a benchmark of the behaviour of compounds in an extracellular environment and provides information on possible pathways by which the radiopharmaceutical in question can be demetallated.49 It is well known that phosphonate-containing compounds may have a high affinity to bone and sorption on hydroxoapatite (HA), as an in vitro model material of bone, was also investigated. Challenging studies against either hydroxyapatite, or rat serum were monitored as a function of time and are presented in Fig. S6.† It was observed that 44Sc–DO3AP, 44Sc–DO3APABn and 44Sc–DO3APPrA complexes were stable over several hours in serum, similarly to 44Sc–DOTA. It is also in accordance with previous studies on radiolanthanide complexes of DO3APABn and DO3APPrA.39,40 Therefore, ligands with a DOTA-like structure seem to be good chelators for Sc(III) forming stable complexes. Negligible sorption of all complexes, even that of 44Sc–DO3AP, on HA shows that no bone uptake should be expected after intravenous applications.To better prove that 44Sc–DO3AP (as the best ligand in the investigated series) has no uptake on bone, its biodistribution was investigated in healthy rats and the results are shown in Fig. 8 and Table S4.† These data show no specific uptake and rapid clearance through urine. The PET image confirmed this observation. Thus, the 44Sc remains in the chelate in vivo and the 44Sc–DO3AP complex is not adsorbed on bone.
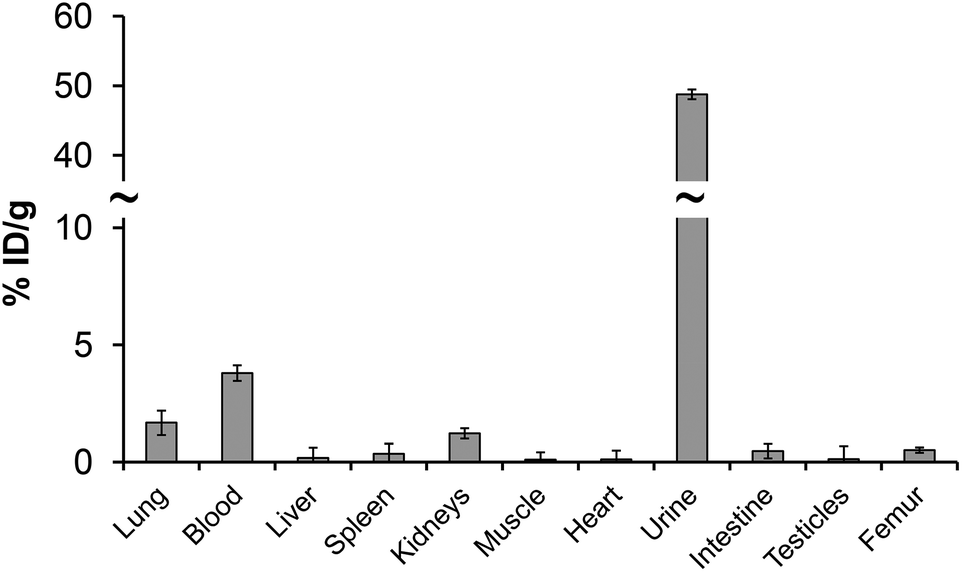 | ||
| Fig. 8 Biodistribution of 44Sc–DO3AP in healthy Wistar rats (n = 4, 1 h p.i.). | ||
Conclusions
The DOTA derivatives bearing one methylphosphonic/phosphinic acid pendant arm, DO3AP, DO3APPrA and DO3APABn, were evaluated as chelators for trivalent scandium. As expected, stability constants with Sc(III) are several orders of magnitude higher than those for trivalent lanthanides. Thermodynamic stability constants were also examined by the FISRE method. The method gave qualitatively similar results but it has an advantage over the common method that, as it is based on trace amounts of metal ions, it can be used for several radiometal ions where conventional methods for stability constant determination can be hardly implemented. Such metal ions of potential radiopharmaceutical interest include e.g. easily hydrolyzing metal ions as Zr(IV), Bi(III), Ac(III) or Th(IV). In addition, experimental conditions of the method are more close to the real ones used for the preparation of radiopharmaceuticals.The labelling efficiency of DOTA and its analogues was, for the first time, investigated on radioscandium from two sources, generator- or cyclotron-produced 44Sc. Chelator excess over radioscandium necessary for efficient labelling was higher for the generator 44Sc. The difference might be attributed to various amounts of cold metal impurities in radioscandium from each source as cold metal ions compete with radiometals during radiolabelling.56 The best labelling conditions for DO3AP, DO3APABn and DO3APPrA were the same as for DOTA. The phosphonate ligand, DO3AP, showed somewhat better labelling efficiency at low temperatures and it is a hint for possible future ligand designs. The specific activity after labelling was higher for the cyclotron produced 44m/44Sc (∼10 MBq nmol−1) as compared to that for the generator produced 44Sc (∼2 MBq nmol−1). In vitro stability of the Sc(III) complexes is very high as expected for complexes of macrocyclic ligands. No specific uptake and a rapid urine clearance of 44Sc–DO3AP were observed in healthy rats. Slow formation kinetics remains the main challenge in the design of chelators for scandium and lanthanide radioisotopes.
Acknowledgements
We thank the French–Czech travel grant (Barrande PHC 21934B) and PICS-CNRS 5196 (to R. K., C. A., and S. H.), the Grant Agency of the Czech Republic (13-08336S; to M. P., J. H., V. K., and P. H.) for financial support. The work was done in the frame of TD1004 COST Action. The ARRONAX cyclotron is a project promoted by the Regional Council of Pays de la Loire financed by local authorities, the French government and the European Union. This work was also supported by grants from the Pays de la Loire Council “Nucléaire pour la Santé” (NucSan) and from the French National Agency for Research called “Investissements d'Avenir” no. ANR-11-LABX-0018-01.References
- F. Rösch and R. P. Baum, Dalton Trans., 2011, 40, 6104–6111 RSC.
- K. L. Kolsky, V. Joshi, L. F. Mausner and S. C. Srivastava, Appl. Radiat. Isot., 1998, 49, 1541–1549 CrossRef CAS PubMed.
- M. Polosak, A. Kasperek, S. Krajewski and A. Bilewicz, J. Radioanal. Nucl. Chem., 2013, 295, 1867–1872 CrossRef CAS PubMed.
- C. Müller, M. Bunka, S. Haller, U. Köster, V. Groehn, P. Bernhardt, N. van der Meulen, A. Türler and R. Schibli, J. Nucl. Med., 2014, 55, 1658–1664 CrossRef PubMed.
- M. Mamtimin, F. Harmon and V. N. Starovoitova, Appl. Radiat. Isot., 2015, 102, 1–4 CrossRef CAS PubMed.
- J. T. Harris and V. N. Starovoitova, Appl. Radiat. Isot., 2015, 97, 188–192 CrossRef PubMed.
- C. Grignon, J. Barbet, M. Bardiès, T. Carlier, J. F. Chatal, O. Couturier, J. P. Cussonneau, A. Faivre, L. Ferrer, S. Girault, T. Haruyama, P. Le Ray, L. Luquin, S. Lupone, V. Métivier, E. Morteau, N. Servagent and D. Thers, Nucl. Instrum. Methods Phys. Res., Sect. A, 2007, 571, 142–145 CrossRef CAS.
- S. Huclier-Markai, R. Kerdjoudj, C. Alliot, A. C. Bonraisin, N. Michel, F. Haddad and J. Barbet, Nucl. Med. Biol., 2014, 41, e36–e43 CrossRef CAS PubMed.
- F. Rösch, Curr. Radiopharm., 2012, 5, 187–201 CrossRef.
- (a) M. Pruszyński, N. S. Loktionova, D. V. Filosofov and F. Rösch, Appl. Radiat. Isot., 2010, 68, 1636–1641 CrossRef PubMed; (b) D. V. Filosofov, N. S. Loktionova and F. Rösch, Radiochim. Acta, 2010, 98, 149–156 CrossRef CAS.
- S. Krajewski, I. Cydzik, K. Abbas, A. Bulgheroni, F. Simonelli, U. Holzwarth and A. Bilewicz, Radiochim. Acta, 2013, 101, 333–338 CrossRef CAS.
- C. Alliot, R. Kerdjoudj, N. Michel, F. Haddad and S. Huclier-Markai, Nucl. Med. Biol., 2015, 42, 524–529 CrossRef CAS PubMed.
- J. Luo, R. Liu, L. Jiang, Z. Liu, G. Sun and S. Ge, Radiochim. Acta, 2013, 101, 607–612 CAS.
- M. Bostan and S. M. Qaim, Phys. Rev. C: Nucl. Phys., 1994, 49, 266–271 CrossRef CAS PubMed.
- S. Bailey, Phys. Rev. C: Nucl. Phys., 1961, 123, 579–582 CAS.
- T. J. Wadas, E. H. Wong, G. R. Weisman and C. J. Anderson, Chem. Rev., 2010, 110, 2858–2902 CrossRef CAS PubMed.
- B. M. Zeglis and J. S. Lewis, Dalton Trans., 2011, 40, 6168–6195 RSC.
- M. D. Bartholomä, Inorg. Chim. Acta, 2012, 389, 36–51 CrossRef.
- V. Carroll, D. W. Demoin, T. J. Hoffman and S. S. Jurisson, Radiochim. Acta, 2012, 100, 653–667 CrossRef CAS PubMed.
- C. S. Cutler, H. M. Hennkens, N. Sisay, S. Huclier-Markai and S. S. Jurisson, Chem. Rev., 2013, 113, 858–883 CrossRef CAS PubMed.
- C. F. Ramogida and C. Orvig, Chem. Commun., 2013, 49, 4720–4739 RSC.
- E. Price and C. Orvig, Chem. Soc. Rev., 2014, 43, 260–290 RSC.
- (a) G. A. Melson and R. W. Stotz, Coord. Chem. Rev., 1971, 7, 133–160 CrossRef CAS; (b) S. A. Cotton, Polyhedron, 1999, 18, 1691–1715 CrossRef CAS; (c) P. R. Meehan, D. R. Aris and G. R. Willey, Coord. Chem. Rev., 1999, 181, 121–145 CrossRef CAS.
- S. Huclier-Markai, A. Sabatie, S. Ribet, V. Kubíček, M. Paris, C. Vidaud, P. Hermann and C. S. Cutler, Radiochim. Acta, 2011, 99, 653–662 CrossRef CAS.
- S. Krajewski, I. Cydzik, K. Abbas, A. Bulgheroni, F. Simonelli, U. Holzwarth and A. Bilewicz, Radiochim. Acta, 2013, 101, 333–338 CrossRef CAS.
- A. Majkowska-Pilip and A. Bilewicz, J. Inorg. Biochem., 2011, 105, 313–320 CrossRef CAS PubMed.
- M. Pruszyński, A. Majkowska-Pilip, N. S. Loktionova, E. Eppard and F. Rösch, Appl. Radiat. Isot., 2012, 70, 974–979 CrossRef PubMed.
- E. Koumarianou, N. S. Loktionova, M. Fellner, F. Rösch, O. Thews, D. Pawlak, S. C. Archimandritis and R. Mikolajczak, Appl. Radiat. Isot., 2012, 70, 2669–2676 CrossRef CAS PubMed.
- R. Hernandez, H. F. Valdovinos, Y. Yang, R. Chakravarty, H. Hong, T. E. Barnhart and W. Cai, Mol. Pharmaceutics, 2014, 11, 2954–2961 CrossRef CAS PubMed.
- L. Moghaddam-Banaem, A. R. Jalilian, M. R. Pourjavid, E. Radfar, A. Bahrami-Samani, K. Yavari, M. Mazidi and M. Ghannadi-Maragheh, Radiochim. Acta, 2012, 100, 215–221 CrossRef CAS.
- R. Chakravarty, S. Goel, H. F. Valdovinos, R. Hernandez, H. Hong, R. J. Nickles and W. Cai, Bioconjugate Chem., 2014, 25, 2197–2204 CrossRef CAS PubMed.
- S. Eigner, D. R. Beckford-Vera, M. Fellner, N. S. Loktionova, M. Piel, O. Lebeda, F. Rösch, T. L. Roß and K. Eigner-Henke, Mol. Imaging Biol., 2013, 15, 79–86 CrossRef PubMed.
- C. Müller, M. Bunka, J. Reber, C. Fischer, K. Zhernosekov, A. Türler and R. Schibli, J. Nucl. Med., 2013, 54, 2168–2174 CrossRef PubMed.
- M. Pniok, V. Kubíček, J. Havlíčková, J. Kotek, A. Sabatie-Gogová, J. Plutnar, S. Huclier-Markai and P. Hermann, Chem. – Eur. J., 2014, 20, 7944–7955 CrossRef CAS PubMed.
- I. Lukeš, J. Kotek, P. Vojtíšek and P. Hermann, Coord. Chem. Rev., 2001, 216&217, 287–312 Search PubMed.
- P. Táborský, P. Lubal, J. Havel, J. Kotek, P. Hermann and I. Lukeš, Collect. Czech. Chem. Commun., 2005, 70, 1909–1942 CrossRef.
- M. Försterová, I. Svobodová, P. Lubal, P. Táborský, J. Kotek, P. Hermann and I. Lukeš, Dalton Trans., 2007, 535–549 RSC.
- P. Táborský, I. Svobodová, P. Lubal, Z. Hnatejko, S. Lis and P. Hermann, Polyhedron, 2007, 26, 4119–4130 CrossRef.
- M. Försterová, M. Petřík, A. Lázničková, M. Lázníček, P. Hermann, I. Lukeš and F. Melichar, Appl. Radiat. Isot., 2009, 67, 21–29 CrossRef PubMed.
- S. Lacerda, F. Marques, P. Campello, L. Gano, V. Kubíček, P. Hermann and I. Santos, J. Labelled Compd. Radiopharm., 2010, 53, 36–43 CrossRef CAS.
- J. Šimeček, P. Hermann, J. Havlíčková, E. Herdtweck, T. G. Kapp, N. Engelbogen, H. Kessler, H.-J. Wester and J. Notni, Chem. – Eur. J., 2013, 19, 7748–7757 CrossRef PubMed.
- J. Notni, P. Hermann, J. Havlíčková, J. Kotek, V. Kubíček, J. Plutnar, N. S. Loktionova, P. J. Riss, F. Rösch and I. Lukeš, Chem. – Eur. J., 2010, 16, 7174–7185 CrossRef CAS PubMed.
- J. Notni, J. Šimeček and H.-J. Wester, ChemMedChem, 2014, 9, 1107–1115 CrossRef CAS PubMed.
- J. Rudovský, P. Cígler, J. Kotek, P. Hermann, P. Vojtíšek, I. Lukeš, J. A. Peters, L. V. Elst and R. N. Muller, Chem. – Eur. J., 2005, 11, 2375–2384 CrossRef PubMed.
- J. Rudovský, J. Kotek, P. Hermann, I. Lukeš, V. Mainero and S. Aime, Org. Biomol. Chem., 2005, 3, 112–117 Search PubMed.
- V. Kubíček, J. Havlíčková, J. Kotek, G. Tircsó, P. Hermann, É. Tóth and I. Lukeš, Inorg. Chem., 2010, 49, 10960–10969 CrossRef PubMed.
- M. Kývala and I. Lukeš, Chemometrics '95, in Abstract book, Pardubice, Czech Republic, 1995, p. 63. Full version of the OPIUM program package is available at http://www.natur.cuni.cz/~kyvala/opium.html Search PubMed.
- G. A. Kirakosyan, V. P. Tarasov and Yu. A. Buslaev, Magn. Reson. Chem., 1989, 27, 103–111 CrossRef CAS.
- W. C. Cole, S. J. DeNardo, C. F. Meares, M. J. McCall, G. L. DeNardo, A. L. Epstein, H. A. Obrien and M. K. Moi, J. Nucl. Med., 1987, 28, 83–90 CAS.
- F. Benetollo, G. Bombieri, L. Calabi, S. Aime and M. Botta, Inorg. Chem., 2003, 42, 148–157 CrossRef CAS PubMed.
- K. L. Nash, J. Alloys Compd., 1997, 249, 33–40 CrossRef CAS.
- Z. Piskula, I. Svobodová, P. Lubal, S. Lis, Z. Hnatejko and P. Hermann, Inorg. Chim. Acta, 2007, 360, 3748–3755 CrossRef CAS.
- (a) S. L. Wu and W. Horrocks Jr., Inorg. Chem., 1995, 34, 3724–3732 CrossRef CAS; (b) A. Stenson, A. L. Thompson and D. Parker, Dalton Trans., 2006, 3291–3293 RSC; (c) P. Vojtíšek and J. Rohovec, Collect. Czech. Chem. Commun., 2006, 71, 264–278 CrossRef; (d) P. Vojtíšek, J. Rohovec and J. Klimentová, Eur. J. Inorg. Chem., 2008, 3948–3956 CrossRef.
- J. Shubert, J. Phys. Chem., 1948, 62, 340–343 CrossRef.
- (a) D. Jurkin, F. J. Gildehaus and B. Wierczinski, Anal. Chem., 2007, 79, 9420–9426 CrossRef CAS PubMed; (b) D. Jurkin and B. Wierczinski, J. Radioanal. Nucl. Chem., 2008, 277, 91–96 CrossRef CAS; (c) D. Jurkin, F. J. Gildehaus and B. Wierczinski, J. Labelled Compd. Radiopharm., 2009, 52, 33–40 CrossRef CAS.
- W. A. P. Breeman, M. de Jong, T. J. Visser, J. L. Erion and E. P. Krenning, Eur. J. Nucl. Med., 2003, 30, 917–920 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details on potentiometry, FISRE and NMR measurements, figures with NMR and titration data, FISRE isotherms, experimental details and figures for challenge studies, table of specific activities of labelled ligands. See DOI: 10.1039/c5dt04084a |
| This journal is © The Royal Society of Chemistry 2016 |