
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Contributions of inner and outer coordination sphere bonding in determining the strength of substituted phenolic pyrazoles as copper extractants†
Mary R.
Healy
a,
James W.
Roebuck
a,
Euan D.
Doidge
a,
Lucy C.
Emeleus
a,
Philip J.
Bailey
a,
John
Campbell
b,
Adam J.
Fischmann
b,
Jason B.
Love
a,
Carole A.
Morrison
*a,
Thomas
Sassi
b,
David J.
White
a and
Peter A.
Tasker
*a
aEaStCHEM School of Chemistry, University of Edinburgh, Edinburgh, EH9 EJJ, UK. E-mail: c.morrison@ed.ac.uk; p.a.tasker@ed.ac.uk
bCytec Industries, 1937 West Main St, Stamford, CT 06902, USA
First published on 14th January 2016
Abstract
Alkyl-substituted phenolic pyrazoles such as 4-methyl-2-[5-(n-octyl)-1H-pyrazol-3-yl]phenol (L2H) are shown to function as Cu-extractants, having similar strength and selectivity over Fe(III) to 5-nonylsalicylaldoxime which is a component of the commercially used ACORGA® solvent extraction reagents. Substitution in the phenol ring of the new extractants has a major effect on their strength, e.g. 2-nitro-4-methyl-6-[5-(2,4,4-trimethylpentyl)-1H-pyrazol-3-yl]phenol (L4H) which has a nitro group ortho to the phenolic hydroxyl group unit and has an extraction distribution coefficient for Cu nearly three orders of magnitude higher than its unsubstituted analogue 4-methyl-6-[5-(2,4,4-trimethylpentyl)-1H-pyrazol-3-yl]phenol (L8H). X-ray structure determinations and density functional theory (DFT) calculations confirm that inter-ligand hydrogen bonding between the pyrazole NH group and the phenolate oxygen atom stabilise the Cu-complexes, giving pseudomacrocyclic structures. Electron-accepting groups ortho to the phenol oxygen atoms buttress the inter-ligand H-bonding, enhancing extractant strength but the effectiveness of this is very dependent on steric factors. The correlation between the calculated energies of formation of copper complexes in the gas phase and the observed strength of comparably substituted reagents in solvent extraction experiments is remarkable. Analysis of the energies of formation suggests that big differences in strength of extractants arise principally from a combination of the effects of the substituents on the ease of deprotonation of the proligands and, for the ortho-substituted ligands, their propensity to buttress inter-ligand hydrogen bonding.
Introduction
Some of the earliest reports of the use of metal complexes of phenolic pyrazoles with the generic structure I (Scheme 1) concern their use in biomimetic investigations.1 They have also been shown to have anti-bacterial activity2 and oxorhenium(V) and dioxomolybdenum(VI) complexes have been used as catalysts for epoxidation reactions.3,4 The proximity of the oxygen and the two nitrogen donor atoms in the proligands and their ability to lose protons from both the phenol and pyrazole moieties to become dianionic bridging ligands favours the formation of polynuclear complexes. Clusters containing up to 21 metal atoms have been characterised by X-ray crystallography5 and in several cases their magnetic properties have been studied in detail.6–8 Six different modes of coordination of the ligand have been identified in solid state structures (Fig. S1, ESI†). | ||
| Scheme 1 Pseudomacrocyclic structures of copper complexes formed by phenolic pyrazoles I and phenolic oximes II. | ||
In this paper we focus on the solution chemistry of phenolic pyrazoles and in particular their ability to function as copper solvent extractants in the pH-dependent equilibrium shown in eqn (1). Some preliminary results have been reported in patents.9 Comparisons are made with the structurally related phenolic oximes (II, Scheme 1) which are used in hydrometallurgical processes to recover approximately 25% of the world's copper,10,11 and have large alkyl substituents to ensure solubility in hydrocarbon solvents.
| Cu2+ + 2HLorg ⇌ [CuL2]org + 2H+ | (1) |
A feature of particular interest in this equilibrium is the extent to which the formation of hydrogen bonds between the pyrazole N–H group and the oxygen atoms of the phenol favour complex formation. The 14-membered pseudomacrocyclic structure shown in Scheme 1 is very similar to that found in the commercial phenolic oxime reagents. Introduction of a substituent ortho to the phenol group in these extractants has been shown to have a major effect on the strength of copper extraction, with substituents which can function as hydrogen bond acceptors buttressing the intermolecular hydrogen-bond from the oximic proton, contributing to a significant increase in the stability of the copper complex and hence the extraction strength.12
In this paper we examine whether similar trends result from 2-substitution (ortho to the OH group, X in Fig. 1) in phenolic pyrazoles. A comparison with the effects of 4-bromo substitution (Y = Br in Fig. 1) was undertaken to establish whether the resulting changes in electronic properties, which influence the ease of deprotonation of the extractants and the strength of binding of the resulting N2O22− donor set in the inner coordination sphere, or whether the changes induced in the hydrogen bonding in the outer coordination sphere are more important in determining overall extractant strength.
 | ||
| Fig. 1 Hydrogen-bonding in proligand monomers and dimers and in copper(II) complexes of substituted phenolic pyrazoles. Outer sphere interactions in the complex are shown by dashed lines. | ||
The reagents L1H–L9H in Fig. 2 that carry hydrophobic Y or Z substituents have been used in solvent extraction experiments. Analogues, L10H–L16H with smaller or no Y or Z substituents have been used in computational work in order to reduce the numbers of conformers with very similar energies; L10H and L12H were also used in X-ray crystal structure analysis.
 | ||
| Fig. 2 Proligands L1H–L16H reported in this work. L1H–L9H were used in the solvent extraction experiments, L10H–L16H in the computational study, and L10H and L12H in the X-ray diffraction study. | ||
Results and discussion
The proligands L1H–L9H and L12H were prepared from the appropriate acetophenones (1–6) by conversion to the diketones (7–18) and subsequent reaction with hydrazine. Details are provided in the ESI.† The unsubstituted proligand L10H was also synthesised by the method of Maib and Jermanowska13via the reaction of chromone and thiosemicarbazide followed by treatment with formic acid.Solvent extraction
The methods employed for carrying out extractions and for analysing results are provided in the ESI.† L1H–L3H, which lack substituents on the phenol unit, were used to compare the performance of the new phenolic pyrazoles as copper extractants with phenolic oximes of the type used in commercial leach/solvent extraction/electrowin operations.10,11 The aqueous solutions employed for loading the dichloromethane (DCM) solutions of the extractants had CuSO4, Fe2(SO4)3, and H2SO4 concentrations (Table 1) similar to those used in commercial feed solutions in copper recovery.14 After separation the Cu-loaded DCM solutions were stripped with an aqueous solution which had a composition similar to those of the “spent electrolytes” used in industry (see ESI†). The data presented in Table 1 demonstrate that the new phenolic pyrazoles have similar strength and selectivity over iron(III) to 5-nonylsalicylaldoxime (ACORGA P50 oxime solvent extraction reagent), one of the active components used in industrial formulations.10,11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ‡
‡
| Extractant (substituent Z, Fig. 2) | (i) Cu content (g L−1) after loading from 5.00 g L−1 feed | (ii) Cu content (g L−1) after stripping with H2SO4(aq) (125 g L−1, containing 5.00 g L−1 Cu) | (iii) Cu and Fe content (g L−1) after loading from a mixed Cu/Fe (5.00/5.00 g L−1) feed | |
|---|---|---|---|---|
| Cu | Fe | |||
| a The stripping experiment was not undertaken for [Cu(L3)2]. | ||||
| L1H (t-Bu) | 1.10 | 0.005 | 1.1 | 0.005 |
| L2H (n-C8H17) | 1.30 | 0.005 | 1.4 | 0.001 |
| L3H (C2H5OC(O)) | 1.10 | 0.003 | —a | —a |
| 5-Nonyl-salicylaldoxime (ACORGA P50 oxime) | 1.44 | 0.196 | 1.4 | <1 |
As the data presented in Table 1 indicate that phenolic pyrazoles could meet the fundamental requirements of commercial Cu-recovery operations, a more detailed study of the effects of substituents on extraction strength was undertaken. The pH-dependence of copper-uptake from aqueous sulphate solutions by chloroform solutions of L4H–L8H is given in Fig. 3 together with pH0.5 values, i.e. the equilibrium pH at which 50% of the available extractant is loaded as [Cu(L)2]. In accordance with the results shown in Table 1, the pyrazole extractant L8H having no substituent ortho to the phenol group is a very slightly weaker extractant than the analogous oxime extractant, 2-hydroxy-5-tert-octylbenzaldehyde oxime, which is of the type used in commercial copper recovery plants10,12 and shows a pH0.5 value of 1.7 under the same conditions as those for Fig. 3.6
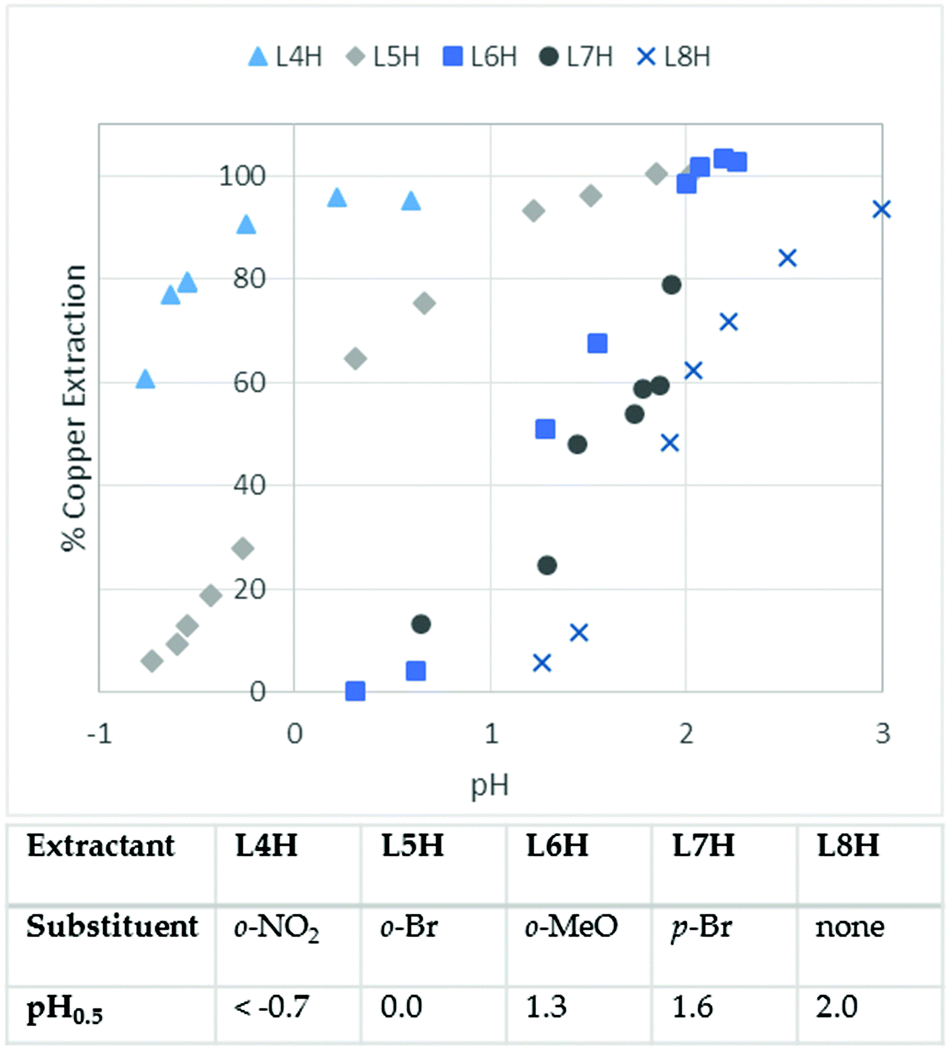 | ||
| Fig. 3 The pH dependence of Cu-loading by 0.005 M chloroform solutions of L4H–L8H from equal volumes of 0.005 M aqueous solution of CuSO4 and the pH0.5 values of the extractants; 100% Cu-loading is based on the formation of a CuL2 complex. | ||
The effect of the introduction of substituents ortho to the phenolic hydroxyl group on the strength of copper extraction is even greater than that found12 in the oximes, increasing the distribution ratio by nearly three orders of magnitude for the nitro-derivative L4H, based on its pH0.5 value being 2.7 units lower than the unsubstituted extractant L8H. Understanding the origins of such changes in extraction strength and its dependence on the nature and location of substituents is important in designing extractants to meet the requirements of particular metal recovery processes. Whilst the introduction of electron withdrawing groups ortho or para to the phenolic OH group will facilitate deprotonation of the extractant to generate the coordinating form and favour complex formation, it will also reduce the basicity of the N2O22− donor set, thereby diminishing the strength of the interactions with the copper cation. H-bond acceptors ortho to the phenolic OH group (X in Fig. 1) could buttress inter-ligand hydrogen bonding, thus favouring the formation of copper complexes, whilst the same group para to the OH group cannot buttress the H-bonding but will produce similar electronic effects. In order to evaluate the relative importance of these effects, DFT calculations were carried out on L10H–L16H. These are lower mass homologues of the extractants L4H–L9H and do not carry the large solubilising alkyl groups that can adopt many similar energy conformations, which would have complicated the task of locating the global minimum optimised structures. Details of the methods used in the computational work are provided in the ESI.†
The energy-minimised structure of [Cu(L12)2] is compared with its single crystal X-ray diffraction structure in Scheme 1. Agreement between the calculated and experimental geometries of the coordination spheres is good, taking into account that there are strong intermolecular Cu⋯O contacts in the solid state structure which lead to the oxygen donor atoms being chemically non-equivalent.
Bromine substitution in [Cu(L12)2] noticeably shortens the Cu–O bond in both the experimentally and computationally determined structures. This can be attributed to the buttressing interaction, labelled ‘a’ in Fig. 4, weakening the other H-bond, labelled ‘b’, which in turn increases electron density on the oxygen atom, and thus strengthening the Cu–O bond.
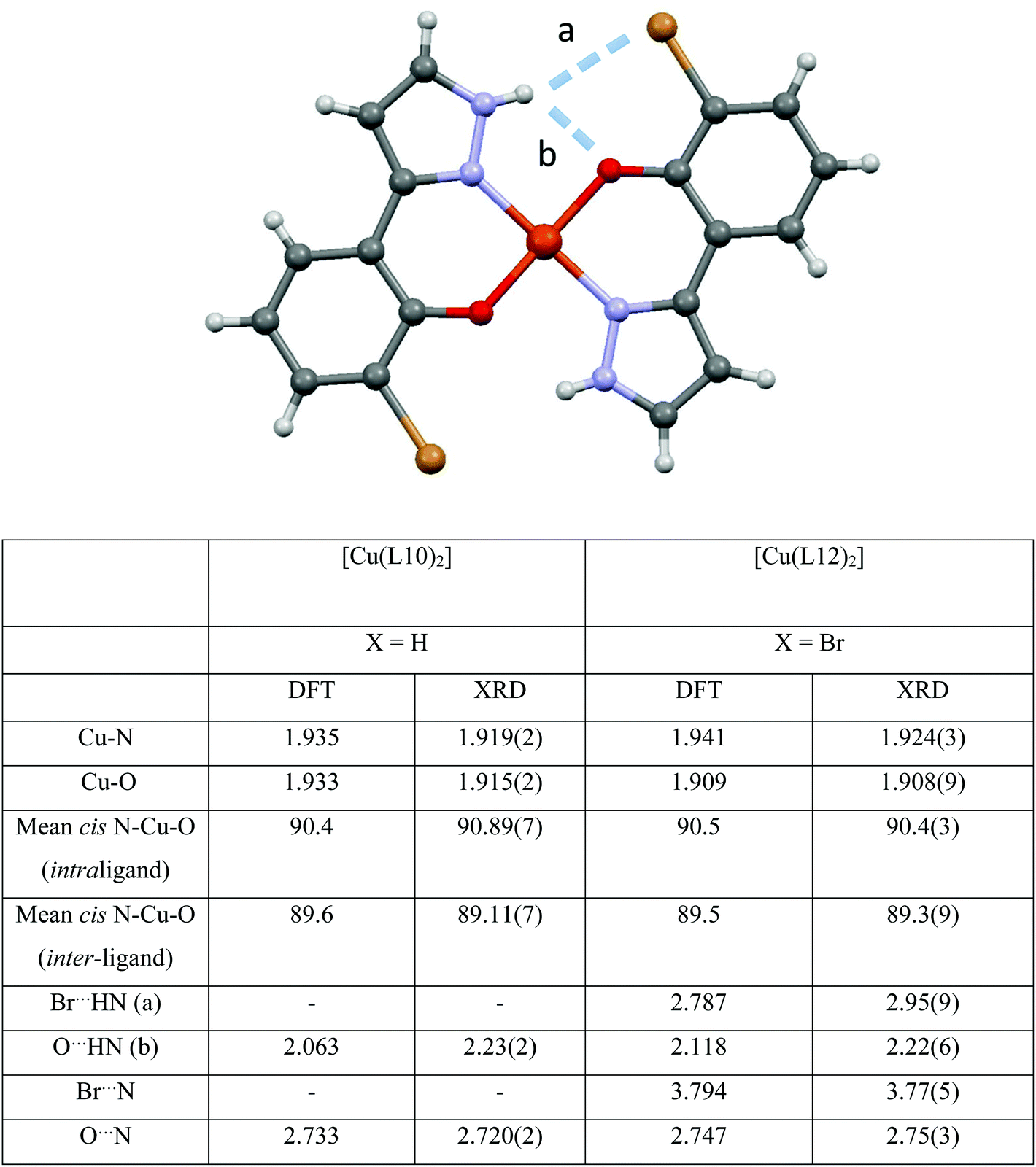 | ||
| Fig. 4 The X-ray crystal structure of [Cu(L12)2]. The bond lengths/Å and angles/° in the inner coordination sphere are compared with those in the calculated structure and those in [Cu(L10)2] LAPXUW,15 together with some outer sphere contact distances. | ||
The introduction of the bromine substituent also results in a small lengthening of the Cu–N bond and the NH⋯O contact distances compared to the non-substituted [Cu(L10)2] complex, which is consistent with the bromine atom forming a weak bonding interaction with the pyrazole N–H group.
There are subtle but important differences between the inter-ligand H-bonding which is displayed by the copper complexes of the phenolic pyrazoles and their phenolic oxime analogues, shown in Fig. 4. The rigidity of the pyrazole ring and its internal bond angles (111.0(2)–105.7(2)° in [Cu(L12)2]) constrain the pyrazole N–H group to point further away from the phenolate oxygen atom than the oxime O–H donor, with the N–H⋯O contact distance (labelled ‘b’ in Fig. 4) measuring 2.25(5) Å compared with O–H⋯O 1.918(3) Å in the analogous oxime, bis(3-bromo-5-t-butylsalicylaldimato)copper(II), ABAXEI.12 The greater flexibility of the oxime group also allows its OH group to make a closer contact with the buttressing bromine substituent than the NH group in [Cu(L12)2]), with O⋯Br and N⋯Br being 3.666(6) and 3.769(3) Å respectively.
Whilst the N–H⋯Br contacts (labelled ‘a’ in Fig. 4) are too long to be described as conventional hydrogen bonds, they are associated with weak bonding interactions as judged by NBO calculations (see below). Based on the structural evidence it therefore appears that the buttressing contribution made by single atom electronegative substituents (X in Fig. 1) that act to stabilise the copper complexes will be smaller in the pyrazole extractants, L1H–L16H, than in their oxime analogues.
Free energies of complex formation were calculated in accordance with eqn (2), using the energy-minimised forms of the proligands (L10H–L16H) and their copper complexes (see Table 2). Note the data presented here do not include solvation effects. The justification for this stems from the fact that the formation energies calculated from eqn (2) rely on calculating differences in the absolute energies for the component species. The aqueous species (H3O+ and Cu2+), are identical in all extractions, leaving only the solvation energies of the preorganised LH dimers and CuL2 complexes in the organic phase for further consideration. The differences in structure between these two species are small, and so consequently the differences in their corresponding solvation energies in chloroform will not be large. As a test that this reasoning is sound, we repeated the calculations for L10H, L12H and L15H using the implicit PCM solvent model.16 The results of this analysis are reported in the ESI.† Absolute formation energies were affected (notably due to the large solvation energy of the Cu2+ cation), but crucially the relative ordering of formation energies for the three complexes remained unchanged. Since it is the relative ordering, rather than the absolute energies, which is needed for comparison against the experimental data, the modelling approach adopted is sound.
| Substituent | Proligand | ΔGdeprotcf. L10H kJ mol−1 | ΔGformcf. L10H kJ mol−1 | ΔGbindcf. L10H kJ mol−1 | ΔUdimercf. L10H kJ mol−1 |
|---|---|---|---|---|---|
| H | L10H | 0.0 | 0.0 | 0.0 | 0.0 |
| o-NO2 | L11H | −130.3 | −70.7 | 59.6 | −34.8 |
| o-Br | L12H | −56.7 | −22.9 | 33.8 | −9.3 |
| o-MeO | L13H | 4.9 | −19.9 | −24.8 | −11.1 |
| p-NO2 | L14H | −144.8 | −9.6 | 135.2 | 8.0 |
| p-Br | L15H | −46.5 | −0.5 | 45.9 | 2.0 |
| p-MeO | L16H | 11.7 | 5.2 | −6.5 | −1.5 |
The dependence of the calculated energies of formation (ΔGform column 3, Table 2) on the nature of substituents in the ortho- and para-position in the phenyl group correlates well with the experimentally determined strength of the extractants, which decrease in the order o-NO2 > o-Br > o-MeO > p-Br > H. The origins of these variations were analysed by breaking down the complex-formation reaction into two steps: (i) deprotonation of the proligand monomer (eqn (3)) and (ii) binding of the resulting anion to the Cu2+ ion (eqn (4)).
| Cu2+ + 2LH + 2H2O ⇌ [CuL2] + 2H3O+ | (2) |
| 2LH + 2H2O ⇌ 2L− + 2H3O+ | (3) |
| Cu2+ + 2L− ⇌ [CuL2] | (4) |
Fig. 5 is a pictorial representation of the deprotonation and binding free energies from Table 2 and helps to demonstrate that the more favourable deprotonation energies resulting from the introduction of electron withdrawing bromo- and nitro-substituents are only partially offset by less favourable binding energies of the more weakly basic donor sets to the Cu2+ ion. As a consequence, formation of copper complexes of the proligands which carry these substituents in either ortho- or para-positions (L11H, L12H, L14H and L16H) is predicted to be more favourable than formation of the complex of the unsubstituted proligand L10H. This trend is mirrored by the observed enhanced copper extraction strength shown by the bromo- and nitro-substituted reagents L4H, L5H and L7H in the solvent extraction experiments (see Fig. 3).
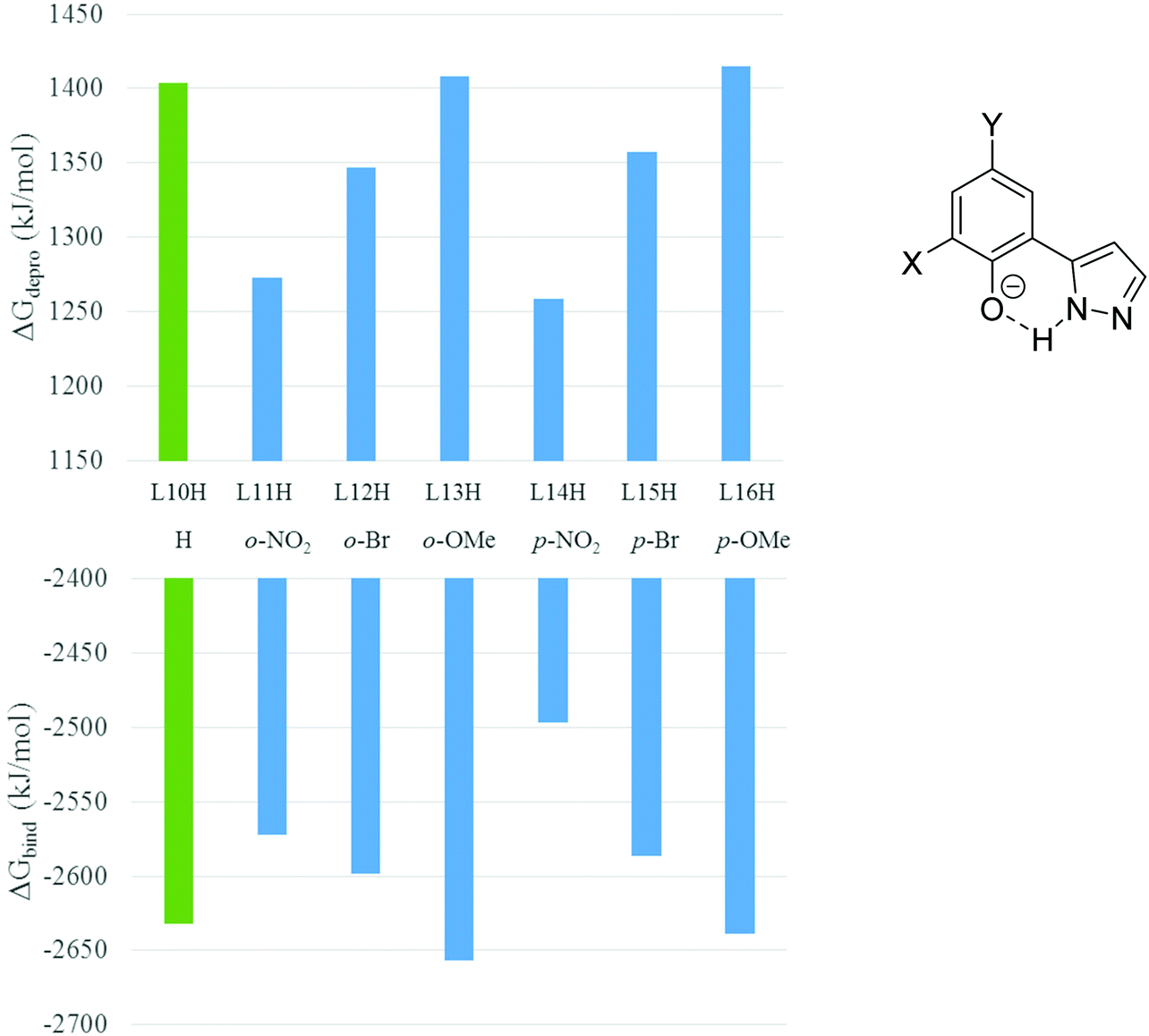 | ||
| Fig. 5 Gibbs free energies of deprotonation of two proligand molecules (ΔGdepro, eqn (3)), as shown in the upper chart, and those of binding of two ligand anions to Cu2+ (ΔGbind, eqn (4)), lower chart, together with the structure of the most stable tautomer of the ligand anions used in calculations. | ||
The more favourable complex formation energy for the ortho-bromo-substituted L12H compared with its para-isomer, L15H, arises from a combination of the deprotonation energy for L12H and the binding energy of the resulting anion both being more favourable (see Table 2). The ability of the ortho-substituent to form a buttressing H-bond with the pyrazole N–H group contributes to the latter.
As might be expected, the introduction of electron-donating substituents such as the ortho- and para-methoxy groups in L13H and L16H, disfavours deprotonation. This effect is compensated in both cases by increased binding energies (see column 5, Table 2) and is predicted to lead to formation of copper complexes that are slightly more favourable than for the unsubstituted L10H.
The natural bond orbital method NBO617 was used to evaluate the relative strengths of the inner sphere Cu–O and Cu–N bonds in complexes of L10–L16 (Table 3). The Cu–N bond strength shows only a small dependence on the nature and position of substitution on the phenol unit. Larger variations are observed for the Cu–O bond, with a stronger interaction observed in complexes of the ortho-substituted ligands [L11]−, [L12]− and [L13]−. This is consistent with the substituent forming a bonding interaction with the pyrazole N–H groups (see Fig. 4) which weakens its interaction with the phenolate oxygen atom thus rendering the pyrazole N atom a better electron donor. The data in Table 3 suggest that this effect is much smaller in the complex of [L13]− for which the strengthening of the Cu–O bond arises principally from the electron-donating properties of the methoxy group.
17 calculations
| Ligand substituent | L10 | L11 | L12 | L13 | L14 | L15 | L16 |
|---|---|---|---|---|---|---|---|
| none | o-NO2 | o-Br | o-MeO | p-NO2 | p-Br | p-MeO | |
| [CuL2] complexes | |||||||
| Cu–N | 212 | 207 | 200 | 204 | 211 | 209 | 212 |
| Cu–O | 221 | 234 | 228 | 238 | 203 | 216 | 208 |
| NH⋯O | 16 | 12 | 13 | 15 | 14 | 15 | 19 |
| NH⋯X | — | 37 | 11 | 2 | — | — | — |
| [LH]2 dimers | L10H | L11H | L12H | L13H | L14H | L15H | L16H |
|---|---|---|---|---|---|---|---|
| none | o-NO2 | o-Br | o-MeO | p-NO2 | p-Br | p-MeO | |
| NH⋯O | 40 | 25 | 37 | 41 | 35 | 39 | 41 |
| NH⋯X | — | 38 | 5 | 1 | — | — | — |
The strength of the buttressing interaction NH⋯X in the copper complexes depends greatly on steric factors. The nitro group, when utilising one of its oxygen atoms as an H-bond acceptor, forms a sterically-favourable six-membered ring, accounting for the much stronger bonding interaction (37 kJ mol−1) with the pyrazole N–H group than those shown by bromo and methoxy substituents (just 11 and 2 kJ mol−1, respectively).
Similar trends are observed in the proligand dimers [LH]2, the structures of which are shown in the top right in Fig. 1. Formation of these dimers is favourable for all the proligands L10H–L16H (see ESI Table S4†), but particularly so for those containing a buttressing NO2, Br or MeO group ortho to the phenol group, i.e. L11H, L12H and L13H. For the o-nitro-substituted proligand [L11H]2, the buttressing H-bond (NH⋯X in Table 3) is stronger than the H-bond to the phenolic oxygen atom, NH⋯O in Table 3, (38 cf. 25 kJ mol−1, respectively). In contrast, for the ortho-bromo and methoxy substituted proligand dimers, [L12H]2 and [L13H]2, in which the H-bond accepting atom is constrained to be well separated from the N–H group, the buttressing interaction is very weak. As a consequence it is largely the inter-ligand NH⋯O interaction which favours dimer formation, with energies comparable to those found for the para-substituted analogues [L15H]2 and [L16H]2.
Only in the case of the ortho nitro-substituted proligand L8H can the buttressing interactions in the Cu-complexes and the proligand dimers be described by a conventional hydrogen bonding model. The much weaker interactions found in the bromo and methoxy analogues L12H and L13H are associated with a small electrostatic contribution which arises from the electronegativity of the ortho substituent.
Conclusions
The very good correlation between the calculated energies of formation of the copper complexes in the gas phase and the observed relative strengths of comparably substituted reagents in solvent extraction experiments may at first sight be surprising. The rationale for it, however, is sound. It arises for the reported extraction equilibria because the solvation/desolvation energies in the aqueous phase are identical, involving only H+aq and Cu2+aq in every case. Consequently only the differences in the solvation energies of the proligands and their copper complexes in the organic phase (chloroform), arising from variations of the X and Y substituents, will influence the correlation between calculated formation energies and the observed strength of extraction. Variations in the nature of X and Y are likely to lead to only minor differences in the energies of solvation by chloroform because these substituents are small in comparison with the solubilising 5-(2,4,4-trimethylpentyl)-group used in the extractants, L4H–L9H. Also the structures of the preorganised proligand dimers [(LH)2] are similar to those of their copper complexes [Cu(L)2] and consequently the differences in their solvation energies will not be large. Under these circumstances, as demonstrated above, computational methods can be very helpful in providing an insight into the origins of variation strength of extractants and in principle could be used to guide the design of new extractants of the same chemical type. We have also reported similar excellent correlations between energies of formation of metal assemblies in the gas phase and the observed strength of extraction in very different classes of process, for example the recovery of their chloridometalates as in eqn (5).18–20| yLorg + yH+aq + [MClx]aqy− ⇌ [(LH)yMClx]org | (5) |
An analysis of the energies of formation of the copper complexes of the phenolic pyrazoles, as summarised in Table 2, suggests that big differences in strength of the extractants arise principally from a combination of the effects of the substituents on the ease of deprotonation of the proligands and, for the ortho-substituted ligands, their propensity to buttress inter-ligand hydrogen bonding.
The pyrazole N–H donor is less accessible to ortho buttressing groups than the oxime O–H group in the structurally related phenolic oxime reagents and only in the case of the o-nitro derivatives is a strong, conventional inter-ligand hydrogen bond formed. This extractant (L4H) is stronger than its oxime analogue,12 whilst the unsubstituted (L5H) is slightly weaker. The comparable strength of the pyrazoles suggests that they might function as alternatives to the oximes in commercial operations in which the latter are used.
Acknowledgements
We thank the EPSRC, Cytec Industries and Anglo American for the funding of PhD studentships for MRH and JWR, Zeneca for PDRA funding (DJW), the EaStCHEM Research Computing Facility for access to software and the Edinburgh Computer and Data Facilities (ECDF) for access to hardware.References
- E. W. Ainscough, A. M. Brodie, J. E. Plowman, K. L. Brown, A. W. Addison and A. R. Gainsford, Inorg. Chem., 1980, 19, 3655–3663 CrossRef CAS.
- R. B. Palkar and H. E. Master, Indian J. Heterocycl. Chem., 1996, 1999, 315–318 Search PubMed.
- J. A. Schachner, P. Traar, C. Sala, M. Melcher, B. N. Harum, A. F. Sax, M. Volpe, F. Belaj and N. C. Mösch-Zanetti, Inorg. Chem., 2012, 51, 7642–7649 CrossRef CAS PubMed.
- P. Traar, J. A. Schachner, L. Steiner, A. Sachse, M. Volpe and N. C. Mösch-Zanetti, Inorg. Chem., 2011, 50, 1983–1990 CrossRef CAS PubMed.
- E. D. Doidge, J. W. Roebuck, M. R. Healy and P. A. Tasker, Coord. Chem. Rev., 2015, 288, 98–117 CrossRef CAS.
- C.-M. Liu, D.-Q. Zhang and D.-B. Zhu, Inorg. Chem., 2009, 48, 4980–4987 CrossRef CAS PubMed.
- M. Viciano-Chumillas, S. Tanase, O. Roubeau, S. J. Teat, L. J. de Jongh and J. Reedijk, Eur. J. Inorg. Chem., 2010, 2010, 947–951 CrossRef.
- Y.-L. Bai, J. Tao, W. Wernsdorfer, O. Sato, R.-B. Huang and L.-S. Zheng, J. Am. Chem. Soc., 2006, 128, 16428–16429 CrossRef CAS PubMed.
- J. Campbell, R. M. Swart, L. Emeleus and S. Owens, Cytec Technology Corp., US Pat., US8088810B2, 2012 Search PubMed.
- J. Szymanowski, Hydroxyoximes and Copper Hydrometallurgy, CRC Press, Inc., 1993 Search PubMed.
- A. M. Wilson, P. J. Bailey, P. A. Tasker, J. R. Turkington, R. A. Grant and J. B. Love, Chem. Soc. Rev., 2014, 43, 123–134 RSC.
- R. S. Forgan, B. D. Roach, P. A. Wood, F. J. White, J. Campbell, D. K. Henderson, E. Kamenetzky, F. E. McAllister, S. Parsons, E. Pidcock, P. Richardson, R. M. Swart and P. A. Tasker, Inorg. Chem., 2011, 50, 4515–4522 CrossRef CAS PubMed.
- P. Maib and Z. Jermanowska, Pol. J. Chem., 1987, 61, 111–122 CAS.
- K. C. Sole and O. Tinker, Copper Cobalt Africa, 8th Southern African Base Metals Conference Livingstone, Zambia, Editon edn., 2015.
- P. A. Tasker, L. Emeleus, S. Parsons and D. Messenger, CCDC, Editon edn., 2005.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- J. E. D. Glendening, K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis and F. Weinhold, NBO 6.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2013 Search PubMed.
- R. J. Ellis, J. Chartres, D. K. Henderson, R. Cabot, P. R. Richardson, F. J. White, M. Schröder, J. R. Turkington, P. A. Tasker and K. C. Sole, Chem. – Eur. J., 2012, 18, 7715–7728 CrossRef CAS PubMed.
- J. R. Turkington, V. Cocalia, K. Kendall, C. A. Morrison, P. Richardson, T. Sassi, P. A. Tasker, P. J. Bailey and K. C. Sole, Inorg. Chem., 2012, 51, 12805–12819 CrossRef CAS PubMed.
- I. Carson, K. J. MacRuary, E. D. Doidge, R. J. Ellis, R. A. Grant, R. J. Gordon, J. B. Love, C. A. Morrison, G. S. Nichol, P. A. Tasker and A. M. Wilson, Inorg. Chem., 2015, 54, 8685–8692 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1430501 and 1430499. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt04055h |
| ‡ In commercial operations, “spent electrolyte” obtained from electrowinning the copper (CuSO4 + H2O = Cu + H2SO4) is used to strip copper from the loaded organic phase (CuL2org + H2SO4 = CuSO4 + 2LHorg), generating fresh electrolyte and regenerating the extractant which is reused.10,11 In this work the sulphuric acid concentration in the strip solution (125 g L−1) was lower than that in a typical spent electrolyte,14 ensuring that both the phenolic oxime and the phenolic pyrazole were not fully stripped, allowing a judgement to be made on their relative ease of stripping. |
| This journal is © The Royal Society of Chemistry 2016 |