
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new metal–organic polymeric system capable of stimuli-responsive controlled release of the drug ibuprofen†
Ana Belén
Lago
*,
Arantxa
Pino-Cuevas
,
Rosa
Carballo
and
Ezequiel M.
Vázquez-López
Departamento de Química Inorgánica, Facultade de Química, Instituto de Investigación Biomédica (IBI)-Universidade de Vigo, 36310 Vigo, Galicia, Spain. E-mail: ablago@uvigo.es
First published on 15th December 2015
Abstract
A series of new zinc compounds has been prepared easily under mild synthetic conditions with the drug ibuprofen incorporated as a ligand to form different coordination polymers. The host materials have a high drug content and exhibit high stability, low cytotoxicity, good biodegradability and high biocompatibility. The ion exchange mechanism involved in the ibuprofen release process and the pH-controlled drug release have been studied.
Introduction
The search for new medical strategies with high therapeutic efficiencies and reduced adverse side-effects remains a challenge for the scientific community. The development of new drug delivery systems (DDS) to generate new solid forms of active pharmaceutical ingredients (APIs) has attracted increased interest in pharmaceutical materials science. A wide range of different architectures have been employed in an effort to optimize drug therapeutics.1 Various solid-phase carriers, such as polymeric resins or metal organic frameworks (MOFs), have emerged as promising platforms for drug delivery systems.2 Many of the drawbacks associated with small molecule drugs can be circumvented through the use of these novel systems.Ion exchange resins are cross-linked polymers that incorporate compounds and these systems have been widely studied in drug delivery research. Numerous ion-exchange resins are commercially available and they allow some control over the rate of drug release.3 The ionic strength and pH at the site of delivery play key roles in the liberation of immobilized drugs from resinates. This class of compounds can be considered as a relevant example in the search for new dosage forms.
Hybrid materials composed of metal ions and organic bridging ligands exhibit promising characteristics as drug carriers, including intrinsic biodegradability as a result of the relatively labile metal–ligand bonds and high drug-loading capacity.4 Moreover, the addition of metal ions to the polymeric system allows changes in the physicochemical and biological properties of the material.5 One particular approach in this field is the direct coupling of the biomolecule as the linker with the metal to prepare MOF compounds.6 The release of the active molecule can be achieved by desorption, anionic exchange or through biodegradation of the material.
An alternative approach consists of the direct coordination of the biomolecule to the metal ion connector but with the coordination polymer built up within a different biodegradable polydentate linker.7 As ion-exchange resins, these coordination polymers will contain two principal parts: a structural unit consisting of a polymeric chain and a functional section, which is the ion-active site group.
An important goal in developing materials with pharmaceutical applications is to design biocompatible carriers with little or zero toxicity. In this sense, in the design of coordination polymers it seems appropriate to include bioactive metal cations such as zinc(II), which is an essential nutrient for life and can also be used as an antibacterial growth agent.8 Moreover, stimuli-responsive controlled-release systems, which undergo physical or chemical changes in response to small changes in the environmental conditions, are highly desired. In comparison to sustained-release systems, these dynamic systems can achieve a site-selective, controlled-release pattern, which improves the therapeutic efficacy.9 However, it remains a challenge to find more convenient ways to achieve a fully controllable drug-release system under normal physiological conditions.10
Herein, we describe a zinc coordination polymer with a fully controlled architecture as a starting material for ibuprofen drug delivery. The choice of ibuprofen as the API is due to its moderate size, poor solubility, its relatively simple molecular geometry, and its major relevance as a typical test molecule in controlled drug delivery experiments.11 Furthermore, in the field of coordination polymer research our group has previously investigated the ability of the ditopic linker ligand bis(4-pyridylthio)methane (SCS) to form polymeric compounds.12
One of the most attractive features of these new compounds is the simplicity of their synthesis and the straightforward loading process, which is achieved in one-pot and in a controlled way. The mechanism involved in the drug delivery process is controlled by anion exchange and this was also studied. These features were of sufficient interest to attract us to study new coordination polymers for applications in materials science and, in particular, as drug delivery systems. The synthesis, characterization, cytotoxicity and release behavior of this system was evaluated. This system could also be regarded as a model for other drugs that contain carboxylic acid groups capable of anionic exchange.
Experimental
Materials and physical measurements
Bis(4-pyridylthio)methane (SCS) was prepared as described previously.13 The other starting materials and solvents were obtained commercially and were used as supplied. Ibuprofen was used as a racemic mixture. Elemental analyses (C, H, N, S) were carried out on a Fisons EA-1108 microanalyser. IR spectra were recorded from KBr discs (4000–400 cm−1) on a Jasco FT/IR-6100 spectrophotometer. 1H-NMR spectra were obtained on a Bruker AMX 400 spectrometer. Mass spectra (ESI+) were recorded on a Hewlett-Packard 5989A spectrometer. UV-Vis spectra were recorded on a Jasco V-670 spectrophotometer. TGA was performed on a SETSYS Evolution Setaram thermogravimetric analyser in a flow of N2 at a heating rate of 10 °C min−1.Synthesis of complexes
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was added to a solution of zinc(II) acetate dihydrate, Zn(CH3CO2)2·2H2O (0.427 mmol) in the same solvent mixture (10 mL). A solution of ibuprofen (Hibu) (0.853 mmol) in the same solvent mixture was added slowly to the above colorless solution. The suspension was stirred and heated under reflux for 8 h. The resulting white precipitate was filtered off, washed with water and ethanol, and dried under vacuum.
:1) was added to a solution of zinc(II) acetate dihydrate, Zn(CH3CO2)2·2H2O (0.427 mmol) in the same solvent mixture (10 mL). A solution of ibuprofen (Hibu) (0.853 mmol) in the same solvent mixture was added slowly to the above colorless solution. The suspension was stirred and heated under reflux for 8 h. The resulting white precipitate was filtered off, washed with water and ethanol, and dried under vacuum.
Yield: 231 mg (80%). Anal. found: C, 61.93; H, 6.24; N, 3.96; S, 8.71; C37H42N2S2O4Zn requires: C, 62.35; H, 5.98; N, 3.96; S, 9.05. 1H-NMR (400 MHz, [D6]-DMSO, 25 °C): δ 0.84–0.86 (d, 12H, C12-H3, C11-H3, ibu); 1.28–1.30 (d, 6H, C3-H3,ibu); 1.77–1.80 (q, 2H, C11-H, ibu), 2.33–2.39 (d, 4H, C10-H2, ibu); 3.47–3.49 (dd, 2H, C2-H2, ibu); 4.99 (s, 2H, C14-H2, SCS); 7.02–7.04 (d, 4H, C5arom, C7arom, ibu), 7.17–7.19 (d, 4H, C6arom, C8arom, ibu), 7.38–7.40 (d, 4H, C15arom, C16arom, SCS), 8.41–8.43 (d, 4H, C17arom, C18arom, SCS). IR (KBr): ν(cm−1) = 2965m, 2927m, 2868m (CH stretching), 1625s (νasym(OCO)), 1595s (ν(CC) + ν(CN)), 1374m, 1362 (νsym(OCO)), 1244m (ω(CH,CH2)), 1063m (ν(CH) + δ(C–H)), 740 m, 717 (ν(CS)). MS-ESI: m/z (%) = 235 (49) |SCS + H|+, 475 (100) |Zn(ibu)2 + H|+, 786 (51) |Zn(ibu)2(SCS)(dmso)|+.
:1) was added to a solution of ZnCl2 (0.427 mmol) in the same solvent mixture (10 mL). A white precipitate formed immediately and the suspension was stirred for 10 min. A solution of Hibu (0.427 mmol) and NaOH (0.475 mmol) in the same solvent mixture (10 mL) was added to the suspension. The suspension was stirred for 16 h. The resulting white precipitate was filtered off, washed with ethanol and water and dried under vacuum. Colorless single crystals were obtained by slow evaporation at r.t. (1 week) of the mother liquor and better quality crystals were obtained by slow diffusion in a test tube of a solution of SCS (0.025 mmol), Hibu (0.025 mmol) and NaOH (0.025 mmol) in ethanol (10 mL) and a solution of ZnCl2 (0.025 mmol) in water (10 mL) at r.t. The powder X-ray diffraction data were compared to those simulated from the crystal structure and showed that the crystal is representative of the bulk material. Data for 1 from the first synthetic procedure:
Yield: 216 mg (95%). Anal. found: C, 53.22; H, 4.77; N, 5.35; S, 11.97; C24H26N2S2O2ClZn requires: C, 53.44; H, 4.86; N, 5.19; S, 11.89. 1H-NMR (400 MHz, [D6]-DMSO, 25 °C): δ 0.84–0.86 (d, 6H, C12-H3, C11-H3, ibu); 1.30–1.32 (d, 3H, C3-H3, ibu); 1.77–1.81 (q, 1H, C11-H, ibu), 2.38–2.40 (d, 2H, C10-H2, ibu); 3.48–3.53 (dd, 1H, C2-H, ibu); 4.99 (s, 2H, C14-H2, SCS); 7.03–7.05 (d, 2H, C5arom, C7arom, ibu), 7.18–7.20 (d, 2H, C6arom, C8arom, ibu), 7.39–7.40 (d, 4H, C15arom, C16arom, SCS), 8.41–8.43 (d, 4H, C17arom, C18arom, SCS). IR (KBr): ν(cm−1) = 2954m, 2912m, 2866m (CH stretching), 1597s (νasym(OCO), ν(CC) + ν(CN)), 1381m (νsym(OCO)), 1233w (ω(CH,CH2)), 1064m (ν(CH) + δ(C–H)), 729m (ν(CS)). MS-ESI: m/z (%) = 235 (100) |SCS + H|+, 475 (40) |Zn(ibu)2 + H|+, 371 (5) |ZnCl2(SCS) + H|+, 786 (9) |Zn(ibu)2(SCS)(dmso)|+.
ZnCl2 (0.418 mmol), Hibu (0.418 mmol), NaOH (0.418 mmol) and ethanol/water (2:1, 20 mL) were added to a high pressure vessel, which was placed in a microwave oven. The power input was limited by temperature Tlim = 90 °C and power Plim = 500 W. For heating up to Tlim a time of 5 min was used and the temperature was maintained for 5 min at 90 °C with a stirring rate of 400 rpm. Under these conditions the pressure reached about 4.0 bar. Cooling was carried out over 15 min (pressurized air). The resulting white precipitate was filtered off, washed with ethanol and water and dried under vacuum.
Colorless single crystals were obtained by slow diffusion in a test tube of a solution of Hibu (0.250 mmol) and NaOH (0.375 mmol) in ethanol (10 mL) and a solution of ZnCl2 (0.250 mmol) in water (10 mL) at r.t. The powder X-ray diffraction data were compared to those simulated from the crystal structure and showed that the crystal was representative of the bulk material.
Yield: 76 mg (71%). Anal. found: C, 61.05; H, 7.56; C26H34O4Zn·2H2O requires: C, 61.24; H, 7.12. 1H-NMR (400 MHz, [D6]-DMSO, 25 °C): δ 0.84–0.86 (d, 6H, C12-H3, C11-H3); 1.28–1.30 (d, 3H, C3-H3); 1.75–1.82 (q, 1H, C11-H), 2.38–2.39 (d, 2H, C10-H2); 3.46–3.51 (dd, 1H, C2-H); 7.02–7.04 (d, 2H, C5arom, C7arom), 7.17–7.19 (d, 2H, C6arom, C8arom); IR (KBr): ν(cm−1) = 2952m, 2921m, 2867m (CH stretching), 1623m (ν(OC)H bonded), 1541vs (νasym(OCO)), 1510s, 1458 (ν(CC) + β(HCH)), 1410s (νsym(OCO)), 1258m (ν(OC) + β(HCH) + τ(HCCC)), 1067m (β(HCC) + β(CCC)).
Yield: 50 mg (63%). Anal. found: C, 35.87; H, 2.62; N, 7.46; S, 17.30; C11H10N2S2Cl2Zn requires: C, 35.64; H, 2.71; N, 7.56; S, 17.30. 1H-NMR (400 MHz, [D6]-DMSO, 25 °C): 5.00 (s, 2H, C14-H2, SCS); 7.40–7.41 (dd, 4H, C15arom, C16arom, SCS), 8.41–8.43 (dd, 4H, C17arom, C18arom, SCS). IR (KBr): ν(cm−1) = 1594s (ν(CC) + ν(CN)), 1214m (ω(CH,CH2)), 1060m (ν(CH) + δ(C–H)), 725s (ν(CS)). MS-ESI: m/z (%) = 235 (100) |SCS + H|+, 567 (3) |ZnCl(SCS)2|+, 705 (40) |Zn2Cl3(SCS)2|.
The labeling scheme for Hibu and SCS are shown in Fig. S1B.†
Cytotoxicity study
The cytotoxicity experiments for SCS, 1, 2, 3 and 4 were carried out with cell line MRC-5 (non-tumor human fibroblast). Cells were cultured in Eagle's Minimum Essential Medium supplemented with 10% FBS (fetal bovine serum) at 37 °C in a humidified atmosphere of 95% of air and 5% CO2.Growth inhibition induced by the compounds was evaluated by a colorimetric method based on the tetrazolium salt MTT ([3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide]), which is reduced by living cells to yield formazan, which is purple. Cells were seeded in 96-well plates at a density of 1 × 104 cells and left to incubate for 24 h. After this time, DMSO solutions of the compounds were added to each well in such a way that the same proportion of DMSO was maintained in each well. Incubation was performed at 37 °C/5% CO2 for 7 days. At the end of the incubation period, 10 μL of MTT solution (5 mg mL−1 in PBS) was added and the solutions were incubated for 4 h at 37 °C/5% CO2. After the addition of 100 μL of SDS (10% in 0.01 M HCl) the solutions were again incubated for 12–14 h. Finally, the cell viability was evaluated by measuring the absorbance at 595 nm using a plate spectrophotometer (Tecan Ultra Evolution). The cell viability was calculated by dividing the absorbance of each well by that of the control wells (cells treated with medium containing 1% DMSO). Each experiment was repeated at least three times. The results of these experiments are shown in Table 1.
| Compound | Concentration (μM) | % Growth inhibition |
|---|---|---|
| SCS | 100 | 32 ± 1 |
| [Zn(ibu)2(SCS)] (1) | 100 | 44 ± 2 |
| [Zn(Cl)(ibu)(SCS)] (2) | 100 | 49 ± 1 |
| [Zn(ibu)(H2O)2] (3) | 100 | 17 ± 3 |
| [ZnCl2(SCS)] (4) | 100 | 39 ± 1 |
Liberation studies and degradation of the compounds
The presence of ibuprofen as a solid or in solution was detected by FTIR, NMR and UV-Vis techniques. The molar ratio of SCS to ibu−, i.e., 1:2 in (1) and 1:1 in (2), is crucial to characterize the solid compounds and also to gain an insight into the liberation and/or degradation processes of the compounds. 1H-NMR and elemental analysis studies were performed at regular intervals in order to shed light on the SCS:ibu− molar ratio, both in the solid state and in solution. UV spectrophotometry was carried out at room temperature on aqueous or PBS solutions in order to confirm the presence of SCS or ibu− in solution. Three absorption bands were observed in the spectrum of ibuprofen in aqueous solution: one intense band at 220 nm and two weak bands at high concentrations at 264 and 272 nm.15 Bands were observed at the same wavelength for a solution of the sodium salt of ibuprofenate and [Zn(ibu)2(H2O)2] (3). The absorption maximum of SCS was observed at 265 nm along with a lower intensity band at 212 nm. These bands experience bathochromic shifts upon protonation (vide infra). The UV-Vis spectra of polymeric compounds 1 and 2 show bands due to the two ligands but the concentration of each analyte could not be determined by this method due to overlap of the bands.
Quantitative NMR (qNMR) was used to quantify the Ibu− and SCS released with D2O as solvent. Solutions with known concentrations in DMSO were used as the inputs for quantification of the ligand concentrations in solution (Tables S6 and S7, ESI†).
Synthesis and characterization
In order to load ibuprofen into coordination polymers in a one-pot synthesis, either an ibuprofen solution or ibuprofen previously deprotonated with NaOH was added to the corresponding metal salt and the organic SCS linker to obtain two mixed-ligand compounds [Zn(ibu)2(SCS)] (1) and [ZnCl(ibu)(SCS)] (2). Moreover, single-ligand compounds [Zn(ibu)2(H2O)2] (3) and [ZnCl2(SCS)] (4) were also isolated (Scheme 1). Our interest in compounds 3 and 4 was to understand fully the potential products that result from the degradation of the polymeric compounds 1 and 2, as well as to ascertain all the possible combined compounds from the building units that participate in the formation of the polymeric ibuprofen systems. Polymeric compounds were isolated as pure phases in excellent yields (80% for compound 1 and 95% for compound 2) from reactions carried out under mild conditions using water and ethanol solvents. The in situ synthesis and loading at room temperature of the drug into the final compounds are two of the advantages of this method over others for the preparation of this kind of drug system. The use of this approach, when compared to conventional drug loading processes, allows the drug to be easily incorporated into the carriers. It is noteworthy that the content of ibuprofen is 35 wt% in 2 and reached 61 wt% in 1. The simplicity and degree of control achieved in the loading process made it easy to determine the drug content in the complexes. The rate of drug loading is therefore constant and is not dependent on the activation processes, pH conditions or temperatures and this represents another advantage of this system compared to drug loading in resins or porous MOF materials.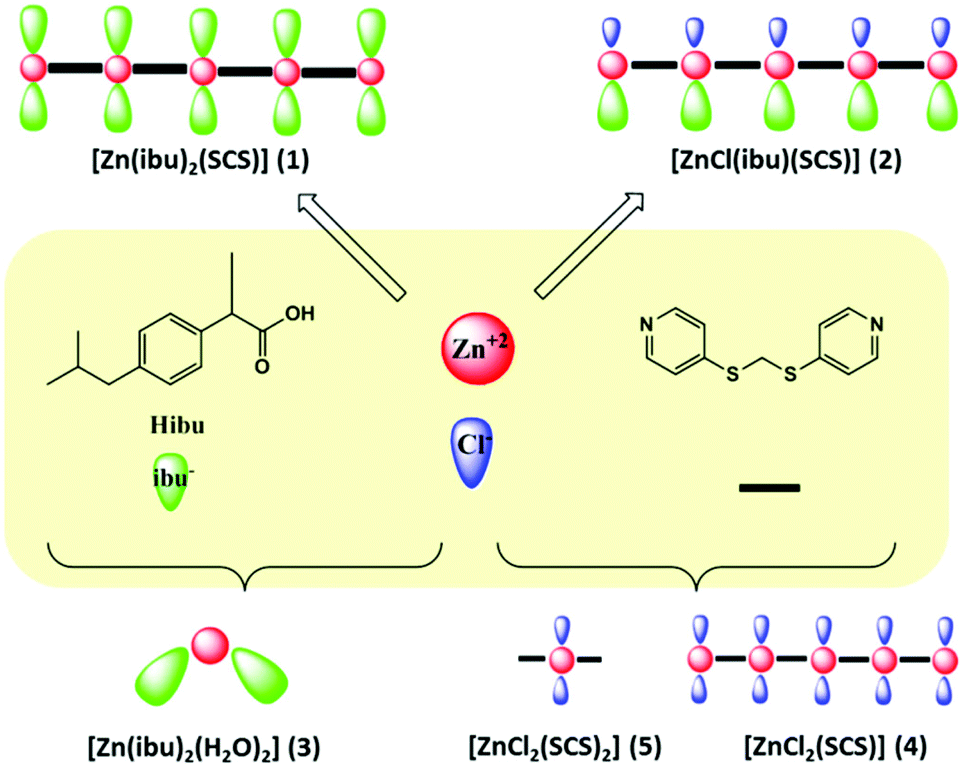 | ||
| Scheme 1 | ||
The compounds are insoluble in water but are soluble in chloroform or dichloromethane. Aqueous stability studies performed on the as-synthesized materials revealed that both complexes (1 and 2) have a relatively high stability against hydrolysis. Indeed, both compounds retain their crystallinity after soaking in water for several weeks at different temperatures up to a maximum of 80 °C, as evidenced by powder X-ray diffraction (PXRD) experiments (Fig. S3 and S4, ESI†). Nevertheless, some level of degradation was observed by UV-Vis spectrophotometry of the remaining aqueous solutions, which showed similar low intensity UV-Vis bands due to the presence of ibuprofen and SCS ligands in both solutions (from 1 and 2) (Fig. S5, ESI†). SEM analysis of the recovered product after treatment in aqueous media did not show any notable change in morphology, although some edge cracks did appear in the crystals (Fig. 1). The thermal stabilities of polymeric compounds 1 and 2 were studied by thermogravimetric analysis. Both compounds were stable up to 250–300 °C, at which point decomposition took place. Thermal analysis of 1 showed decomposition from 250 °C with a step corresponding to a weight loss of 30%, which is consistent with the loss of one ibuprofen molecule (calcd 30%) (Fig. S1A, ESI†).
 | ||
| Fig. 1 SEM images X600 of (a) [Zn(ibu)2(SCS)] (1) and (d) [ZnCl(ibu)(SCS)] (2) with their respective singles crystals (a′ and d′); post-treatment materials; (b) (1) at pH = 5; (e) (2) at pH = 5; (c) product after anionic exchange in PBS solution, (f) (2) in an aqueous environment. | ||
The FT-IR spectra of the compounds contain features corresponding to anionic ibuprofen and the characteristic absorption bands of the SCS ligand.16 The bands at around 2900 cm−1 are assigned to the alkyl moiety of ibuprofen17 and bands at 1600 cm−1 and 1390 cm−1 are assigned to asymmetric and symmetric stretching vibrations of carboxylate groups, respectively. The spectrum of complex 1 contained a band assigned to (νasym(OCO)) at 1625 cm−1 whereas in the spectrum of 2 this band is overlapped with the band due to ν(CC) and ν(CN) of the SCS pyridine groups at 1597 cm−1. In the vibrational spectroscopic study of carboxylate complexes, the difference (Δν) between νasym(CO2−) and νsym(CO2−) bands is often used to draw conclusions about the nature of the carboxylate coordination18 in that the frequency separation for unidentate species usually has the highest value. Compound 2 has a chelating coordinaton mode (crystallography section) and this showed lower separation values (Δνa–s) than 1.
Crystallography
All of the compounds included in this work are crystalline samples and some of them could be isolated as single crystals (Fig. 2). The structures of [Zn(ibu)2(H2O)2] (3), the monomeric [ZnCl2(SCS)2] (5) and polymeric [ZnCl2(SCS)] (4) forms obtained from the synthesis of [ZnCl2(SCS)] (4), the protonated Zn-SCS compound (vide infra) isolated in acidic media [H2-SCS][ZnCl4] (Zn-HSCSH) and the polymer [ZnCl(ibu)(SCS)] (2) were determined by single crystal X-ray diffraction. SEM images revealed a fibrous morphology in 1 whereas compound 2 presented a pillared sheet-like morphology. Examination of single crystals showed the same morphology in the micron range (Fig. 1). | ||
| Fig. 2 View of the structures of 2 (A); 4 (B); 3 (C); 5 (D) and Zn-HSCSH (E) showing their representations in this work. | ||
Significant structural parameters for these structures are listed in Table S2† and crystal structure and refinement data are listed in Table S1.† The coordination environments of zinc atoms are represented in Fig. S2A† along with the atom numbering scheme used. The compounds crystallize in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) except for 3 and 5, which crystallize in the polar C2 and P21 monoclinic groups, respectively.
except for 3 and 5, which crystallize in the polar C2 and P21 monoclinic groups, respectively.
The isolation of these five compounds as single crystals proved to be very helpful to understand the mechanism of the liberation process. It was envisaged that a combination of the linear bidentate N,N-divergent SCS ligand with zinc metallic units would result in the formation of very stable monodimensional neutral chains through coordination bonds.
The zinc centers are arranged in a tetrahedral geometry and are coordinated to different atoms (Cl, N or O) with Zn–O and Zn–N distances of around 2 Å and slightly longer distances for the Zn–Cl interactions (around 2.2 Å). When ibuprofen molecules are present, the metal centers can increase their coordination number due to weaker interactions established with oxygen atoms of the carboxylate groups (2 and 3), which results in an anisobidentate coordination. Thus, the coordination sphere around the zinc metal cations in 3 can be described as 4 + 2 and it consists of four oxygen atoms from water and carboxylate at expected distances and the other oxygen atom of the anisobidentate chelate with longer Zn⋯O distances (around 2.9 Å). In SCS-polymeric compounds the metal centers are coordinated to two nitrogen atoms and two chloride atoms in 4 and to one chloride and one ibuprofen oxygen atom in 2. In compound 2 the zinc is also coordinated to other ibuprofen oxygen atoms with longer bond distances (2.561 Å). The Addison parameter (τ) of 0.94 suggests that the coordination sphere around the zinc atom adopts an almost ideal trigonal bipyramidal geometry, with chloride and oxygen atoms in the apical positions. In Zn-HSCSH the zinc cation is tetracoordinated by chloride ions to form the [ZnCl4]2− anion.
On the basis of these observations, the stability of the coordination environment and the lability of the coordination interactions provide the required flexibility to the coordination environment of the metal center. This characteristic is essential to achieve the anionic exchange and therefore the delivery of ibuprofen. Anionic ibuprofen molecules are pendant ligands in the coordination polymers formed with two coordination sites that can be substituted in 1 and one coordination site in 2. In the polymeric complexes ibuprofenate molecules are the most labile ligands and they can be easily substituted by other anionic ligands.
The flexible twisted SCS molecule has proven to be a suitable ligand that gives rise to polymerization through coordination bonds.12 Each SCS ligand adopts a μ2-bridging mode to connect the zinc centers with Zn⋯Zn distances of 12.695 Å and 12.657 Å and N⋯N distances of 9.399 Å and 9.363 Å in 2 and 4, respectively.
Compounds 2 and 4 can be described as chain-type neutral structures produced by the zinc metal centers and SCS ligands. The anionic ligands can be chloride atoms or ibuprofenate molecules and these are arranged perpendicular to the growth of the chains, with the ligands of the same nature oriented to the same side (2). However, the expected polymerization could be disrupted – as observed in 5 and also in acidic media (vide infra).
Compounds 5 and 4 are the monomeric and the polymeric structures, respectively, obtained in the synthesis of [ZnCl2(SCS)] (4). The less common κN terminal mode of the SCS ligand is found in 5. In accordance with this situation, the structural parameters of the SCS ligand are closer to those of the free ligand with intramolecular N⋯N distances of 8.155 and 9.070 Å.
The values of the C–S–C–S torsion angles are 179.10/84.75° in 4 and 178.34/85.32° in 2, i.e., far from that in the free ligand [73.73(7)°]. These torsion angles in the monodentate terminal ligand (5) are also shorter and have closer values to those in the free molecule; 60.50/62.34° and 76.91/64.29°.
Crystals of Zn-HSCSH obtained in acidic media consist of [ZnCl4]2− anions surrounding the [H2-SCS]2+ cations that result from protonation of the SCS ligand. Isolation of this species provides evidence that the acidic medium can be used to control the degree of polymerization in this system. Protonation of SCS leads to slight changes in the distances and angles related to pyridine rings but appreciable differences in the torsion angle, as observed previously for the analogous structure of [H2-SCS][CuCl4].19 The torsion angles of the C–S–C–S bridge, with values of 174.90/85.73 and 164.67/81.25°, are the largest angles observed in this work and they correspond to the less common TG conformation.20 In accordance with this situation, the intramolecular N⋯N distances are 9.509 and 8.671 Å.
The zinc–ibuprofen compound 3 crystallizes in the chiral non-centrosymmetric space group C2 with the metal atom lying in a 2-fold axis. This compound crystallized with two water molecules and it is a neutral mononuclear complex with two enantiopure ibuprofen ligands acting in a monodentate manner. However, the determination of the absolute structure on a single crystal is not representative of the absolute structure of all crystals in the batch.
Release of ibuprofen anion by ion-exchange
In polymeric compounds 1 and 2 ibuprofenate is the most labile ligand and it can be substituted by other anionic ligands. Both compounds are able to undergo exchange reactions when placed in contact with a solution containing anions that are removed from solution through exchange with the ibuprofen anions coordinated to the polymers.The release of ibuprofen was investigated under simulated physiological conditions in phosphate buffered solution (PBS, pH = 7.4). The release profiles in NaCl solutions were also studied. Compound 1 was soaked in an NaCl solution and a transformation to compound 2 was observed due to the exchange of 30 wt%, which corresponds to the replacement of sodium ibuprofen. In this way, compound 1 exhibits a phase transition to 2 induced by an anionic exchange process. The structural transformation was confirmed by X-ray powder diffraction experiments carried out before and after the treatment (Fig. S6, ESI†). Anion exchange could be detected in the remaining solid powder by determination of the final SCS:ibu− molar ratio (1:1), which corresponds to [ZnCl(ibu)(SCS)] (2). This study was carried out by elemental analysis, NMR and/or IR and techniques (section 4.1, ESI†). The UV-Vis spectra of the resulting crude solution showed the typical bands of the ibu− molecule at high concentrations. Dilution of this solution allowed us to detect the characteristic band at 220 nm (Fig. S6, ESI†).
Two different treatments were performed in order to study the behavior of 2 in sodium chloride solutions; mild conditions involved stirring the mixture for 21 days and harsher conditions involved heating under reflux for 12 hours. Degradation of 2 was observed under both sets of conditions since both ibu− and SCS species were detected in solution (Fig. S7, ESI†). The crystalline integrity of the framework was maintained throughout the process, as evidenced by PXRD and the 1:1 molar ratio (SCS:ibu−) confirmed by NMR. Nevertheless, it was estimated by weight difference that 50% of the initial weight was lost under reflux conditions. Moreover, NaCl was identified in the bulk sample by powder diffraction. A significant decrease in the total percentage of organic elements (C, H, N, S) was also observed in the remaining powders and this loss was more marked after the stirring treatment. This observation indicates the presence of inorganic salts and that the drug release process is accompanied by gradual decomposition of the initial compound 2.
The behavior exhibited by this system could be suitable for the administration of a high dose in an initial stage since the first step has a high exchange capacity and the resulting compound (2), which has a lower drug loading, shows a slower release rate along with decomposition of the material. This level of degradation was not observed in pure water. The possible decomposition of 2 was further evaluated by experiments in PBS solutions with D2O as solvent.
The release of ibuprofen from 1 and 2 was investigated by ligand exchange reactions in simulated physiological PBS buffers at r.t. (Fig. 3). In this medium, the release process for the coordinated drug is triggered by phosphate anions due to the preferential coordination of PO43− anions with zinc cations. In order to determine the release profile, the polymeric samples 1 and 2 were placed in 0.1 M PBS buffer and the drug release was monitored by UV-Vis analysis (Fig. S8 and S9, ESI†). At each time point, aliquots of buffer were removed from the buffer/polymer suspensions and replaced with fresh buffer. The aliquot was then analysed by UV-Vis spectrophotometry. The results showed that the UV intensity of the ibu− (band at 221 nm) was the most intense and it remained practically constant. Compounds were also soaked under continuous stirring followed by solution removal after eight days, when the solvents were separated from the solids and they were completely replaced with fresh PBS buffer solution. The intensity of the ibuprofen UV-band (221 nm) increased significantly after the complete change of the solution. As fresh PBS buffer solution was added the intensity of the UV band of the SCS ligand (at 264 nm) increased until it became more intense than the band related to ibu− (at least six replacements were required). Similar behavior was found for 2, albeit with less pronounced changes in intensity of the UV bands (Fig. S9, ESI†).
 | ||
| Fig. 3 Schematic representation of the anionic exchange process. | ||
The diffraction patterns of the solids remaining after the treatments discussed above show that both systems are crystalline and correspond to a pattern due to the same unidentified structure. SEM images recorded after anionic exchange also showed that the morphology changed completely after the treatment (Fig. 1). A significant absorption band at around 1050 cm−1 due to phosphate was observed in the IR spectrum of the residue after drug release (Fig. S11, ESI†). This suggests the involvement of the phosphate anions in the drug release process through the formation of a compound with Zn2+, SCS and PO43− anions.
Different experiments were carried out in order to shed light on the release mechanism and to quantify the ibuprofen in solution (section 4, ESI†). The decomposition of polymeric carrier 2 was further clarified by NMR studies of the remaining solutions in PBS buffer using D2O as solvent. The use of D2O instead of water allowed an in-depth study by NMR spectroscopy of the mechanism of ibuprofen delivery. Interestingly, a molar ratio of 8:1 (ibu−:SCS) was obtained when the equilibrium was reached and this remained constant during one month of stirring. The ibu− concentration in the solution at this molar ratio was 2.7 × 10−3 M (0.34 × 10−3 M of SCS) and this was stable over time. When the complete replacement of solvent had been achieved the concentration of ibu− was 2.25 × 10−3 M in the first three batches and this started to decrease after the fourth replacement (Table S7, ESI†).
The release of SCS ligand indicated that the drug release process is accompanied by gradual decomposition of the polymeric compounds in the last stage. This process resulted in powders in which the main components were Zn2+ and PO43− anions. This point is also advantageous since degradation of the drug carrier is generally required in clinical applications.
Release of ibuprofen in acidic media
The behavior of 1 and 2 in acidic media was examined. Both compounds were soaked in water with different amounts of 0.1 M HCl (Fig. 4). The pH was measured after 30 minutes and 24 hours of treatment although significant variation in this parameter was not observed. The final values were in the range 4.6–4.9 after the addition of 100 μL of 0.1 M HCl, 4.1–4.5 after 300 μL and around 2.2–2.7 after 1000 μL of 0.1 M HCl. Pristine compounds dissolved completely in more acidic media (pH between 2.2–2.7) with ibuprofen adhering to the walls and floating in the vial due to its insolubility in water. Characterization of the final solid phases after the treatment (pH between 4–5) showed that in all cases the final product corresponded to the original material together with some insoluble ibuprofen (section 5, ESI†). Moreover, under much more acidic conditions (pH around 2) the materials were totally degraded and single crystals of [H2SCS][ZnCl4] were obtained from compounds 1 and 2. | ||
| Fig. 4 Schematic representation of the degradation process in acidic media. | ||
The UV-Vis spectra of the solutions under different pH conditions are shown in Fig. S14 and S15.† Protonation of the SCS ligand induced a bathochromic shift in the band at 265 nm to 296 nm (Fig. S13, ESI†). The addition of acid led to a progressive increase in the intensity of the band at 296 nm and this change was accompanied by a decrease in the absorption bands at 212 and 265 nm. The band at 212 nm was shifted steadily upon protonation to 222 nm. This behavior was observed for both compounds. According to the observed trends, one would expect that in more acidic media the amount of [SCS-H2]2+ species in solution would be higher than in the less acidic media.
The acid dissociation constant (pKa) of ibuprofen (4.91–5.2) means that the vast majority of ibuprofen molecules are present as undissociated ibuprofen under the acidic conditions used and this compound is in the solid state due to its low solubility in water.
The rate of degradation of 1 was further elucidated by NMR studies of the resulting solutions when a similar experiment was performed in D2O rather than water (Table S10, ESI†). The ability to protonate both pyridine groups under these conditions has been studied previously19 and, in this case, the protonation is responsible for the dissolution and degradation of the initial compound. Protonation of SCS led to a significant increase in the solubility of the ligand in water and the NMR signals of the pyridine rings were shifted to lower field (Table S10, ESI†). The amount of [SCS-H2]2+ species in solution increased considerably upon acidification until complete degradation of the initial polymeric compound occurred along with the release of ibuprofen. The [SCS-H2]2+:ibu− molar ratio in solution could be estimated by analyzing the relative intensities of the NMR signals – approximately 2:1 at pH ∼ 5 and approximately 13:1 at pH ∼ 4 – and ibuprofen was not detected in solution in the much more acidic conditions. Moreover, under these conditions single crystals of [H2-SCS][ZnCl4] were isolated from the solutions.
SEM images of the recovered products show evidence for cracking of the crystals, with visible changes in morphology under slightly acidic conditions (Fig. 1) as the roughness of the materials increased with the level of disintegration.
Cytotoxicity
The cytotoxicity levels against normal cells (MRC-5, non-tumor human fibroblasts) of SCS, 1, 2, 3 and 4 were determined. The results of these experiments are included in Table 1. All of the compounds showed low growth inhibition at the maximum concentration assayed (10 ± 3% for 100 μM) and the growth inhibition curve was not calculated. These results suggest that the introduction of high concentrations of SCS and Zn compounds barely changes the cytotoxicity profile of SCS and Zn compounds on MRC-5 cells. The cytotoxicity of the polymeric compounds 1 and 2 are related to the toxicity arising from the interactions between ibuprofen, the SCS ligand and Zn ions of the polymeric compounds with MRC-5 cells.Conclusion
The studies described above provide a new strategy for the development of metal–drug coordination polymers constructed with biomolecular building blocks as potential solid forms of active pharmaceutical ingredients. The nature of these polymeric compounds for the storage and release of the ibuprofen drug molecule was investigated. These systems have high drug contents and high thermal and aqueous stability.The different compositions and coordination environments of the metal centers in the two polymeric carriers (1 and 2) are associated with two distinct drug release stages. The unique properties of the coordination bonds, i.e., thermodynamic stability and lability, play a crucial role in this behavior. The drug release in the initial stage may be regulated by the possibility of anionic exchange in the media (note that 1 is stable in distilled water). In this way, a second stage is reached and this involves polymeric compound 2. Release of ibuprofen in the last stage is a slower process that occurs along with decomposition of the material. An MTT cytotoxicity assay with SCS, 1, 2, 3 and 4 at the maximum concentration tested showed that the new functional materials possess negligible cytotoxicity. These biocompatible polymers appear to be innovative tools with the potential for drug delivery.
Acknowledgements
Financial support from ERDF (EU), MEC (Spain) and the Xunta de Galicia (Spain) (research projects CTQ2010-19386/BQU and 10TMT314002PR) are gratefully acknowledged. A. B. L thanks the Xunta de Galicia for a postdoctoral contract under the Ángeles Alvariño Program.References
- (a) H. Wang, T.-L. Hu, R.-M. Wen, Q. Wang and X.-H. Bu, J. Mater. Chem. B, 2013, 1, 3879–3882 RSC; (b) B. Lou and F. He, New J. Chem., 2013, 37, 309–316 RSC.
- (a) V. Aanad, R. Kandarapu and S. Garg, Drug Discovery Today, 2001, 6, 905–914 CrossRef; (b) P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- X. Guo, R.-K. Chang and M. A. Hussain, J. Pharm. Sci., 2009, 98, 3886–3902 CrossRef CAS PubMed.
- R. C. Huxford, J. D. Roca and W. Lin, Curr. Opin. Chem. Biol., 2010, 14, 262–268 CrossRef CAS PubMed.
- L. A. Belfiore, M. P. M. Curdie and P. K. Das, Polymer, 2001, 42, 9995–10006 CrossRef CAS.
- (a) A. Paloic, A. Puskaric, M. Mazaj, E. Zunkovio, N. Z. Logar and J. Bronic, J. Solid State Chem., 2015, 225, 59–64 CrossRef CAS; (b) S. R. Miller, D. Heurtaux, T. Baati, P. Horcajada, J.-M. Grenèche and C. Serre, Chem. Commun., 2010, 46, 4526–4528 RSC.
- (a) J. An, S. Geib and N. L. Rosi, J. Am. Chem. Soc., 2009, 131, 8376–8377 CrossRef CAS PubMed; (b) N. Motakef-Kazemi, S. A. Shojaosadaati and A. Morsali, Microporous Mesoporous Mater., 2014, 186, 73–79 CrossRef CAS.
- A. S. Prasad, Mol. Med., 2008, 14, 353–357 CAS.
- S. Binauld and M. H. Stenzel, Chem. Commun., 2013, 49, 2082–2102 RSC.
- L. L. Tan, H. Li, Y. C. Qiu, D. X. Chen, X. Wang, R. Y. Pan, Y. Wang, S. X. A. Zhang, B. Wang and Y. W. Yang, Chem. Sci., 2015, 6, 1640–1644 RSC.
- Q. Hu, J. Yu, M. Liu, A. Liu, Z. Dou and Y. Yang, J. Med. Chem., 2014, 57, 5679–5685 CrossRef CAS PubMed.
- (a) A. B. Lago, R. Carballo, S. Rodriguez-Hermida and E. M. Vázquez-López, CrystEngComm, 2013, 15, 1563–1570 RSC; (b) A. B. Lago, R. Carballo, O. Fabelo, N. Fernández-Hermida, F. Lloret and E. M. Vázquez-López, CrystEngComm, 2013, 15, 10550–10562 RSC; (c) R. Carballo, B. Covelo, N. Fernández-Hermida, E. García-Martínez, A. B. Lago and E. M. Vázquez-López, Polyhedron, 2009, 28, 923–932 CrossRef CAS; (d) R. Carballo, B. Covelo, N. Fernández-Hermida, E. García-Martínez, A. B. Lago and E. M. Vázquez-López, Polyhedron, 2008, 27, 3247–3254 CrossRef CAS.
- R. Carballo, B. Covelo, E. García-Martínez, A. B. Lago and E. M. Vázquez-López, Z. Anorg. Allg. Chem., 2007, 633, 780–782 CrossRef CAS.
- (a) B. Tita, M. Stefanescu and D. Tita, Rev. Chim., 2011, 62, 1060–1064 CAS; (b) C. Dendrinou-Samara, G. Tsotsou, L. V. Ekateriniadou, A. H. Kortsaris, C. P. Raptopoulou, A. Terzis, D. A. Kyriakidis and D. P. Kessissoglou, J. Inorg. Biochem., 1998, 71, 171–179 CrossRef CAS PubMed.
- (a) D. S. Wishart, C. Knox, A. C. Guo, S. Shrivastava, M. Hassanali, P. Stothard, Z. Chang and J. Woolsey, Nucleic Acids Res., 2006, 34(Database issue), D668–D672 CrossRef CAS PubMed; (b) A. M. Kumar, A. Swathi, D. Supriya, V. V. L. N. Prasad and P. V. Diwan, Am. J. Pharm. Technol. Res., 2012, 2, 541–547 Search PubMed.
- (a) R. Carballo, B. Covelo, N. Fernández-Hermida, E. García-Martínez, A. B. Lago and E. M. Vázquez-López, Cryst. Growth Des., 2008, 8, 995–1004 CrossRef CAS; (b) R. Carballo, B. Covelo, N. Fernández-Hermida, E. García-Martínez, A. B. Lago and E. M. Vázquez-López, J. Mol. Struct., 2008, 892, 427–432 CrossRef CAS; (c) R. Carballo, B. Covelo, N. Fernández-Hermida, E. García-Martínez, A. B. Lago and E. M. Vázquez-López, Polyhedron, 2007, 633, 780–782 CAS.
- (a) M. L. Vueba, M. E. Pina and L. A. E. Batista de Carvalho, J. Pharm. Sci., 2008, 97, 845–859 CrossRef CAS PubMed; (b) S. Bonora, A. Pisi, S. Ottani, D. Cesini, A. Maris and M. D. Foggia, Vib. Spectrosc., 2014, 73, 45–55 CrossRef CAS.
- (a) A. B. Lago, R. Carballo, S. Rodríguez-Hermida and E. M. Vázquez-López, Cryst. Growth Des., 2014, 14, 3096–3109 CrossRef CAS; (b) K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, J. Wiley & Sons, New York, 4th edn, 1986 Search PubMed.
- A. B. Lago, R. Carballo, E. García-Martínez and E. M. Vázquez López, Cryst. Growth Des., 2011, 11, 59–68 CAS.
- (a) M. Korn, A. Haas and H. Oberhammer, Chem. Ber., 1995, 128, 461–464 CrossRef CAS; (b) E. M. Page, D. A. Rice, K. Aerset, K. Hagen and A. R. J. Genge, J. Phys. Chem. A, 2000, 104, 6672–6676 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1422661–1422665. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt04031k |
| This journal is © The Royal Society of Chemistry 2016 |