DOI:
10.1039/C5DT03947A
(Paper)
Dalton Trans., 2016,
45, 681-689
Ligand noninnocence in FeNO corroles: insights from β-octabromocorrole complexes†
Received
9th October 2015
, Accepted 29th October 2015
First published on 30th November 2015
Abstract
The first FeNO octabromocorroles have been synthesized including four β-octabromo-meso-tris(p-X-phenyl)corrole derivatives Fe[Br8TpXPC](NO) (X = CF3, H, CH3, OCH3) and the β-octabromo-meso-tris(pentafluorophenyl)corrole complex, Fe[Br8TPFPC](NO). The last complex, which proved amenable to single-crystal X-ray structure determination, exhibits the geometry parameters: Fe–N(O) 1.643(8) Å, N–O 1.158(9) Å, and a FeNO angle of 176.4(6)°. The more electron-deficient complexes exhibit increased instability with respect to NO loss and also higher infrared NO stretching frequencies (νNO). Interestingly, DFT calculations and IR marker bands indicate a noninnocent {FeNO}7-(corrole˙2−) formulation for all FeNO corroles, both β-H8 and β-Br8, with essentially the same degree of corrole radical character. Instead, an electron-deficient corrole appears to exert a field effect resulting in reduced Fe-to-NO backdonation, which accounts for both the increased instability with respect to NO loss and the higher νNO's.
Introduction
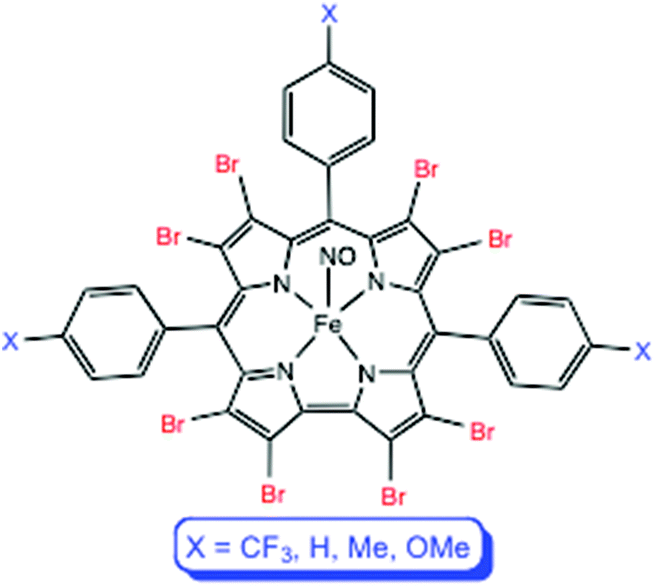
The well-known FeNO corroles have long been thought of as classic {FeNO}6 complexes.1–3 Recently, multiple lines of evidence, including electronic absorption spectroscopy, infrared spectroscopy, and broken-symmetry DFT calculations,4 however, have indicated a noninnocent {FeNO}7-(corrole˙2−) formulation for these complexes.5–9 The question thus arises, to what extent is such a description contingent upon the exact electronic character, i.e., the substitution pattern, of the corrole? Our earlier study focused on FeNO complexes of meso-tris(p-X-phenyl)corrole ligands, i.e., the Fe[TpXPC](NO) series. In this work, we have synthesized and characterized the corresponding β-octabromo complexes, i.e., Fe[Br8TpXPC](NO), where (Fig. 1; X = CF3, H, CH3, OCH3), as well as Fe[Br8TPFPC](NO), where Br8TPFPC is the highly electron-deficient β-octabromo-meso-tris(pentafluorophenyl)corrole ligand. Qualitatively, the stability of the complexes appears to decrease as the corrole ligand becomes increasingly electron-deficient. Thus, it was gratifying that a single crystal X-ray structure could be obtained for Fe[Br8TPFPC](NO), the most electron-deficient complex studied. Electron-poor FeNO corroles also exhibit higher infrared NO stretching frequencies (νNO), which vary over a range of some 40 cm−1. It is tempting to interpret this trend as reflecting a slightly higher degree of {FeNO}6 character of the metal center, along with reduced radical character on the corrole. We shall see, however, that broken-symmetry DFT calculations indicate a different rationale.
 |
| | Fig. 1 Fe[Br8TpXPC](NO) derivatives studied in this work. | |
Results and discussion
Synthesis and chemical stability
The FeNO octabromocorroles were prepared in much the same manner as the corresponding β-H8 complexes, i.e., via iron insertion in pyridine-methanol, followed by addition of aqueous NaNO2. The syntheses were greatly facilitated by the recent improvements in the syntheses of the H3[Br8TpXPC] (X = CF3, H, CH3, OCH3)10,11 and H3[Br8TPFPC] ligands.12,13 The characteristic blood-red color of the FeNO complexes provided a simple visual test of their stability, which, as mentioned above, varied significantly across the compounds studied. Thus, Fe[TpXPC](NO), for X = H, CH3, and OCH3, and Fe[Br8TpXPC](NO), for X = CH3 and OCH3, were all moderately stable in the solid state as well as overnight in dichloromethane solution at room temperature. On the other hand, blood-red dichloromethane and chloroform solutions of Fe[TpCF3PC](NO), Fe[Br8TPC](NO), Fe[TPFPC](NO), Fe[Br8TpCF3PC](NO), and Fe[Br8TPFPC](NO), turned brown in about an hour or so, indicating decomposition. These complexes also decomposed, with NO loss, in the solid state, but only over days. Spectroscopic and electrochemical characterization was therefore carried out on freshly prepared samples, although certain measurements (particularly molar extinction coefficients and elemental analyses) could not be carried out for the more unstable complexes.
Molecular structure of Fe[Br8TPFPC](NO) and general structural considerations14
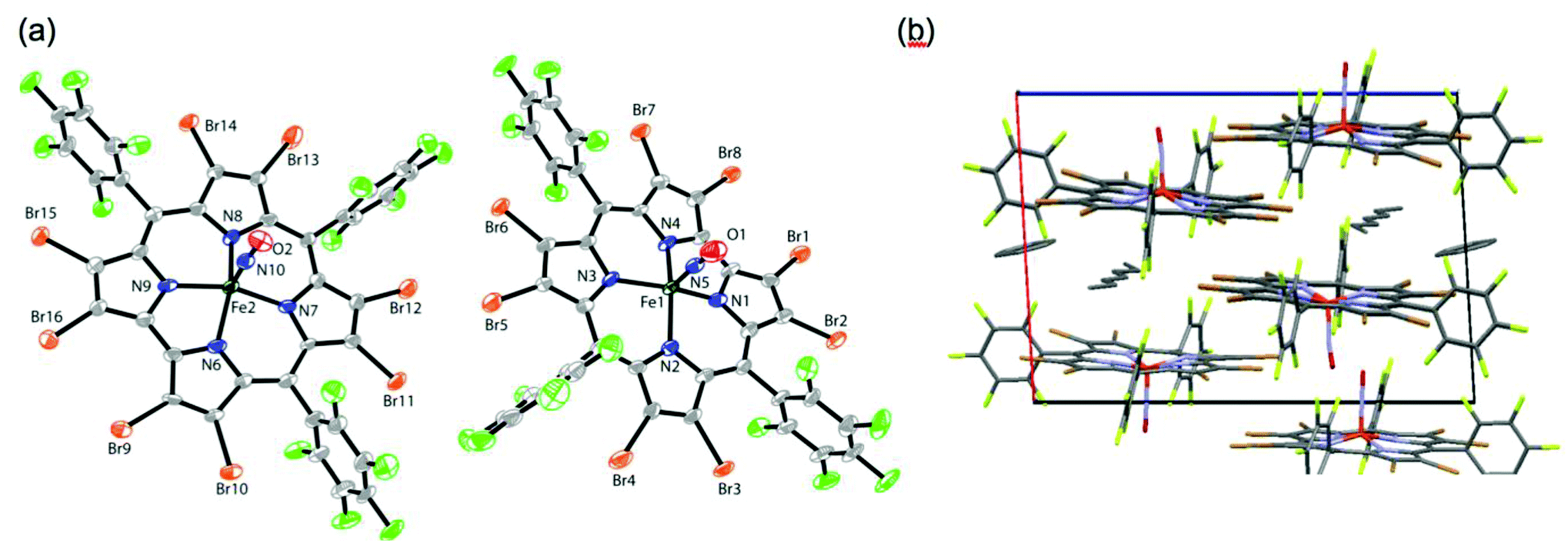
Attempted crystallization and single-crystal X-ray analysis of Fe[Br8TpCH3PC](NO) led, to our surprise, to a rather poor-quality structure for a μ-oxodiiron 10-methoxyisocorrole complex. Fortunately, X-ray quality crystals of Fe[Br8TPFPC](NO) could be obtained by diffusion of benzene into a heptane solution of the complex, providing the first X-ray structure of an FeNO octabromocorrole. The geometrical parameters of the FeNO units (listed under Fig. 2) were found to be nearly identical to those found for {FeNO}6 porphyrin-type complexes,15i.e., Fe–N(O) ∼1.65 Å, N–O ∼1.16 Å, and a nearly linear FeNO angle >170°. We shall see, however, that other probes, notably infrared spectroscopy and broken-symmetry DFT calculations, favor a {FeNO}7-(corrole˙2−) description, for both β-H8 and β-Br8 complexes.16
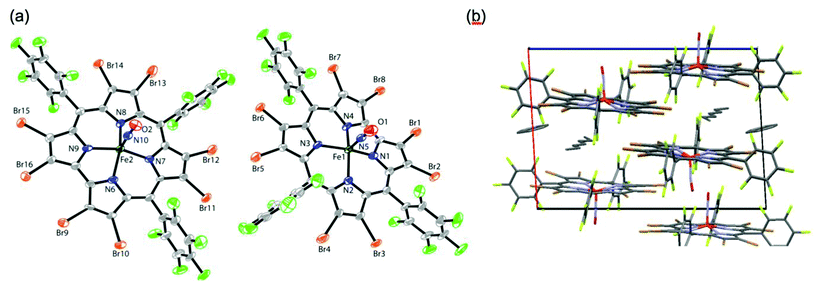 |
| | Fig. 2 X-ray structure of Fe[Br8TPFPC](NO): (a) thermal ellipsoid plot and (b) stick representation of molecular packing. Selected distances (Å) and angles (°) involving the FeNO units: Fe1–N5 1.643(8), N5–O1 1.158(9), Fe1–N5–O1 176.4(6), Fe1–N1 1.900(7), Fe1–N2 1.952(7), Fe1–N3 1.931(7), Fe1–N4 1.928(6); Fe2–N10 1.655(7), N10–O2 1.143(9), Fe2–N10–O2 171.5(7), Fe2–N6 1.919(6), Fe2–N7 1.926(7), Fe2–N8 1.947(6), Fe2–N9 1.915(7). | |
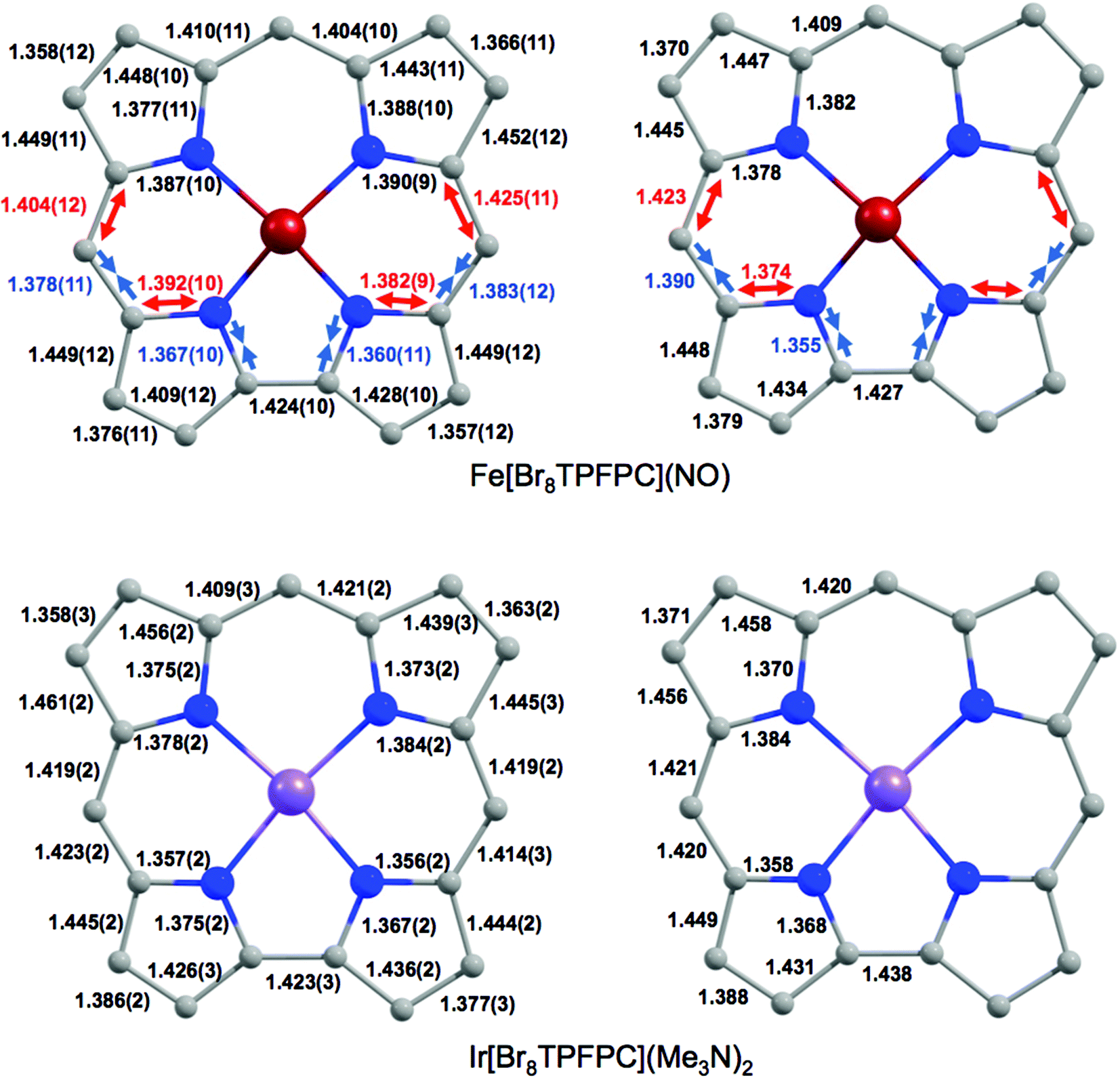
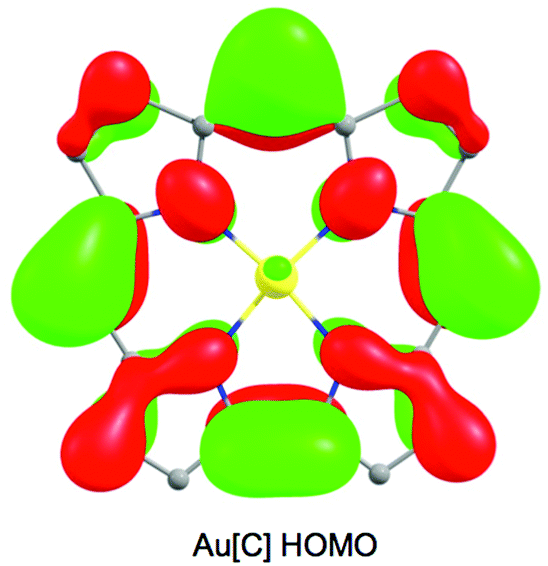
Despite the apparent similarity with {FeNO}6 complexes, careful examination of the skeletal bond distances of Fe[Br8TPFPC](NO) (Fig. 3), as well as those in DFT (B3LYP/STO-TZ2P) optimized structures, does afford evidence of corrole radical character. The bipyrrole part of the corrole macrocycle of Fe[Br8TPFPC](NO) exhibits characteristic bond length alternations that are not seen in an innocent [Br8TPFPC] complex such as Ir[Br8TPFPC](Me3N)2.17 Analogous bond length alternations have also been noted for noninnocent β-H8 complexes such as Fe[TPC](NO) and Fe[TPC]Cl.5 These bond length alternations all correspond to removal of an electron from the corrole HOMO depicted in Fig. 4.
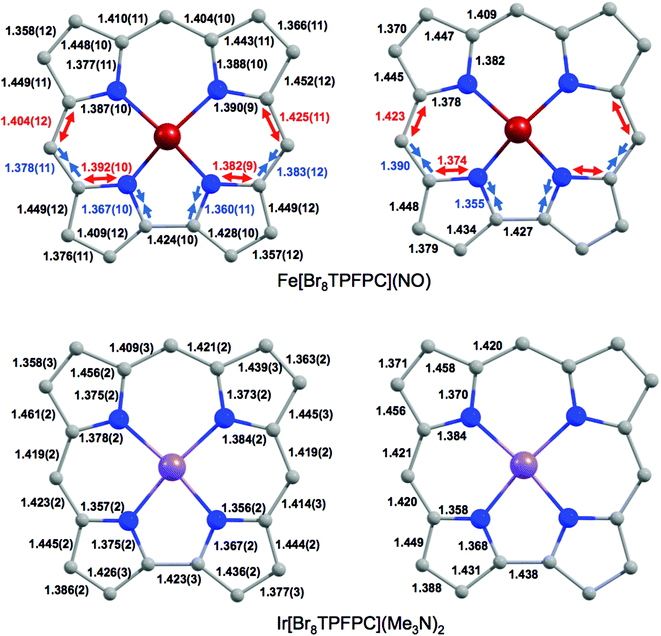 |
| | Fig. 3 Skeletal geometries (Å) of Fe[Br8TPFPC](NO) and Ir[Br8TPFPC](Me3N)2: X-ray (left) and B3LYP/STO-TZ2P (Cs, right) distances. The red and blue arrows indicate ‘longer’ and ‘shorter’ bonds, respectively. | |
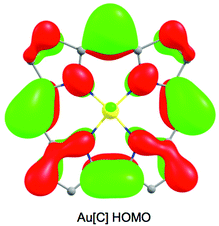 |
| | Fig. 4 The HOMO of Au(III) corrole, a representative innocent metallocorrole. | |
Infrared spectroscopy
The infrared νNO's of the FeNO octabromocorroles examined (1786–1803 cm−1, Table 1) are some 90–100 cm−1 lower than those of genuine {FeNO}6 porphyrins such as Fe(TpivPP)(NO) (1893 cm−1),15 but some 30 cm−1 higher than the values measured for electron-rich FeNO corroles such as Fe[OEC](NO)1 and Fe[TPC](NO)4 (both 1767 cm−1). Based on infrared νNO's, the {FeNO}7-(corrole˙2−) description proposed for β-H8 FeNO triarylcorroles would appear to be applicable to the β-Br8 complexes as well.
Table 1 Soret λmax (nm), IR νNO (cm−1), and redox potentials (V vs. SCE) of FeNO corroles
| Complex |
λ
max
|
ν
NO
|
E
1/2ox
|
E
1/2red
|
| ‘—’: Data unavailable due to compound instability. |
|
FeNO triarylcorroles
|
| Fe[TpOMePC](NO) |
416 |
1761 |
0.83 |
−0.37 |
| Fe[TpMePC](NO) |
400 |
1767 |
0.84 |
−0.36 |
| Fe[TPC](NO) |
390 |
1767 |
0.86 |
−0.33 |
| Fe[TpCO2MePC](NO) |
391 |
1770 |
0.95 |
−0.25 |
| Fe[TpCF3PC](NO) |
385 |
1781 |
0.98 |
−0.22 |
| Fe[TPFPC](NO) |
378 |
1801 |
1.07 |
0.00 |
|
FeNO β-octabromo-meso-triarylcorroles
|
| Fe[Br8TpOMePC](NO) |
394 |
1786 |
1.20 |
0.025 |
| Fe[Br8TpMePC](NO |
395 |
1786 |
1.21 |
0.025 |
| Fe[Br8TPC](NO) |
397 |
1794 |
— |
— |
| Fe[Br8TpCF3PC](NO |
391 |
1803 |
1.33 |
0.200 |
| Fe[Br8TPFPC](NO) |
392 |
1803 |
— |
— |
For β-H8 FeNO corroles, the infrared νNO's vary significantly as a function of the meso substituents, from 1761 cm−1 for Fe[TpOMePC](NO) to 1801 cm−1 for Fe[TPFPC](NO). Intriguingly, the effect of β-octabromination on the νNO varies considerably, from >20 cm−1 for certain Fe[Br8TpXPC](NO) derivatives to only 2 cm−1 for Fe[Br8TPFPC](NO), indicating a leveling-off with increasing electron-deficient character of the corrole ligand (Table 1). Indeed, a value of 1803 cm−1 appears to be the upper limit for the νNO of FeNO corroles.
DFT calculations
Broken-symmetry DFT (B3LYP) calculations played a major role in establishing the noninnocent, radical nature of FeNO corroles.5 An examination of the broken-symmetry spin density profiles (Fig. 5) and Mulliken spin populations (Table 2) now also provide an attractive explanation for the variation of νNO across the complexes studied. Table 2 shows that both the Fe and NO spin populations decrease (in absolute value) with increasing electron-deficient character of the corrole ligand. Because the latter spin density may be viewed as a measure of occupation of the NO π* orbitals, we may conclude that a strongly electron-deficient corrole ligand leads to a lesser degree of backbonding and hence to a stronger NO bond and a higher νNO. Thus, between Fe[TpOMePC](NO) and Fe[Br8TPFPC](NO), the magnitude of the broken-symmetry spin population on the NO decreases by 0.12, which correlates with an experimentally observed upshift of 42 cm−1 in the νNO. Interestingly, there does not appear to be a clear correlation between the amount of broken-symmetry spin density on the macrocycle and its electron-deficient character. Indeed, to a first approximation, all FeNO corroles exhibit the same degree of corrole radical character (as measured by the amount of broken-symmetry spin density on the corrole).
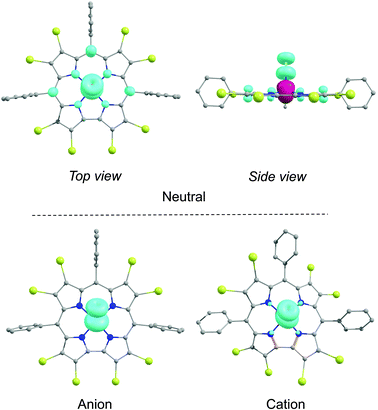 |
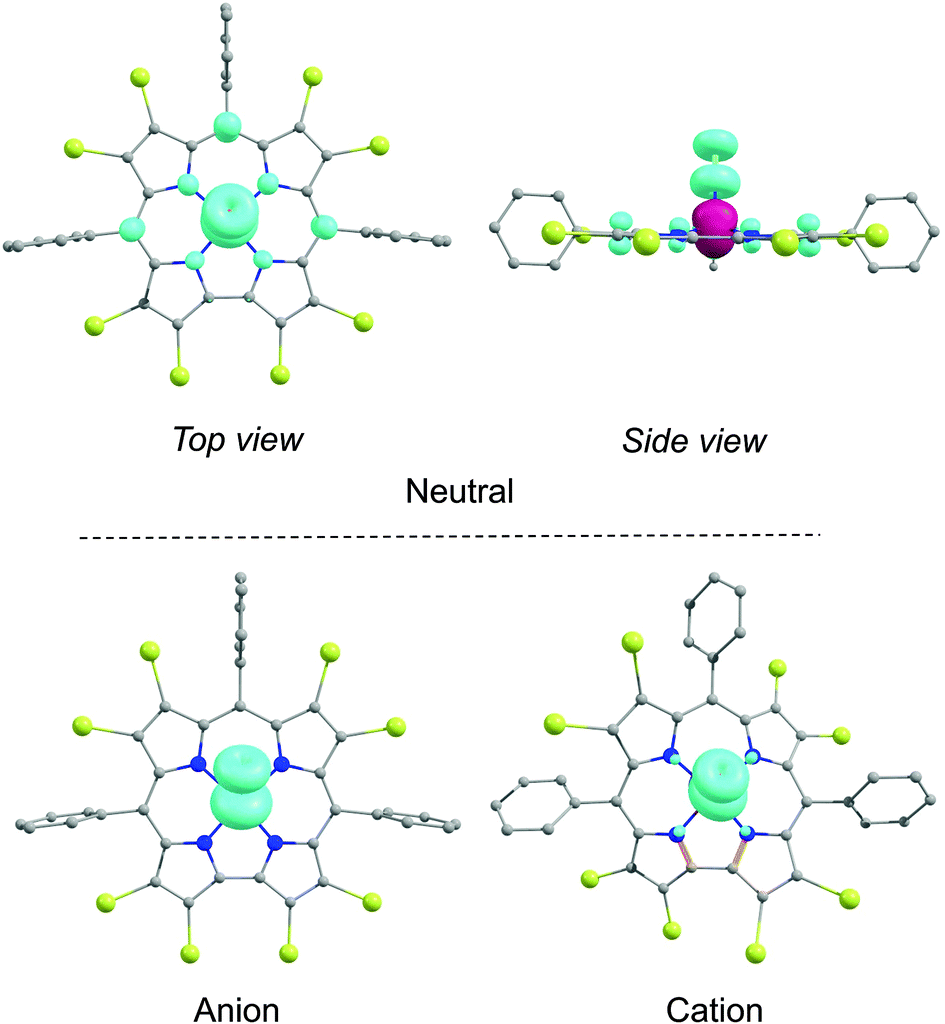
| | Fig. 5 Spin density plots for the neutral, anionic, and cationic states of Fe[Br8TPC](NO). The α and β spin densities are shown in cyan and burgundy, respectively, The contour has been set at 0.009 e Å−3. | |
Table 2 Selected broken-symmetry B3LYP/STO-TZP spin populations
| Ligand |
Fe |
N |
O |
NO |
Corrole |
| TpOMePC |
2.031 |
−0.666 |
−0.488 |
−1.154 |
−0.877 |
| TPC |
2.018 |
−0.659 |
−0.486 |
−1.145 |
−0.873 |
| TPFPC |
1.949 |
−0.615 |
−0.477 |
−1.092 |
−0.857 |
| Br8TpOMePC |
2.013 |
−0.627 |
−0.486 |
−1.113 |
−0.900 |
| Br8TPC |
1.999 |
−0.620 |
−0.484 |
−1.104 |
−0.895 |
| Br8TPFC |
1.918 |
−0.570 |
−0.467 |
−1.036 |
−0.882 |
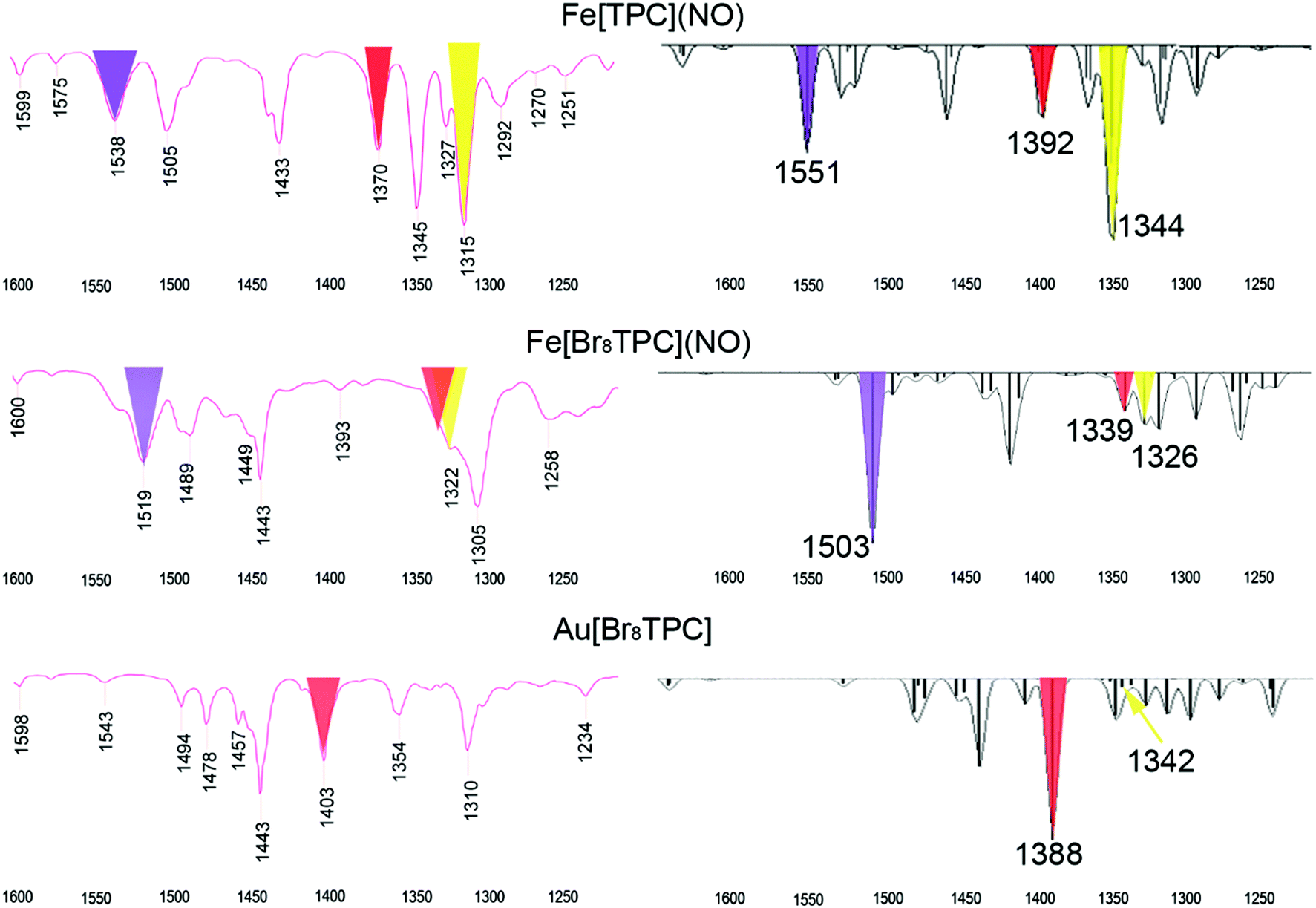
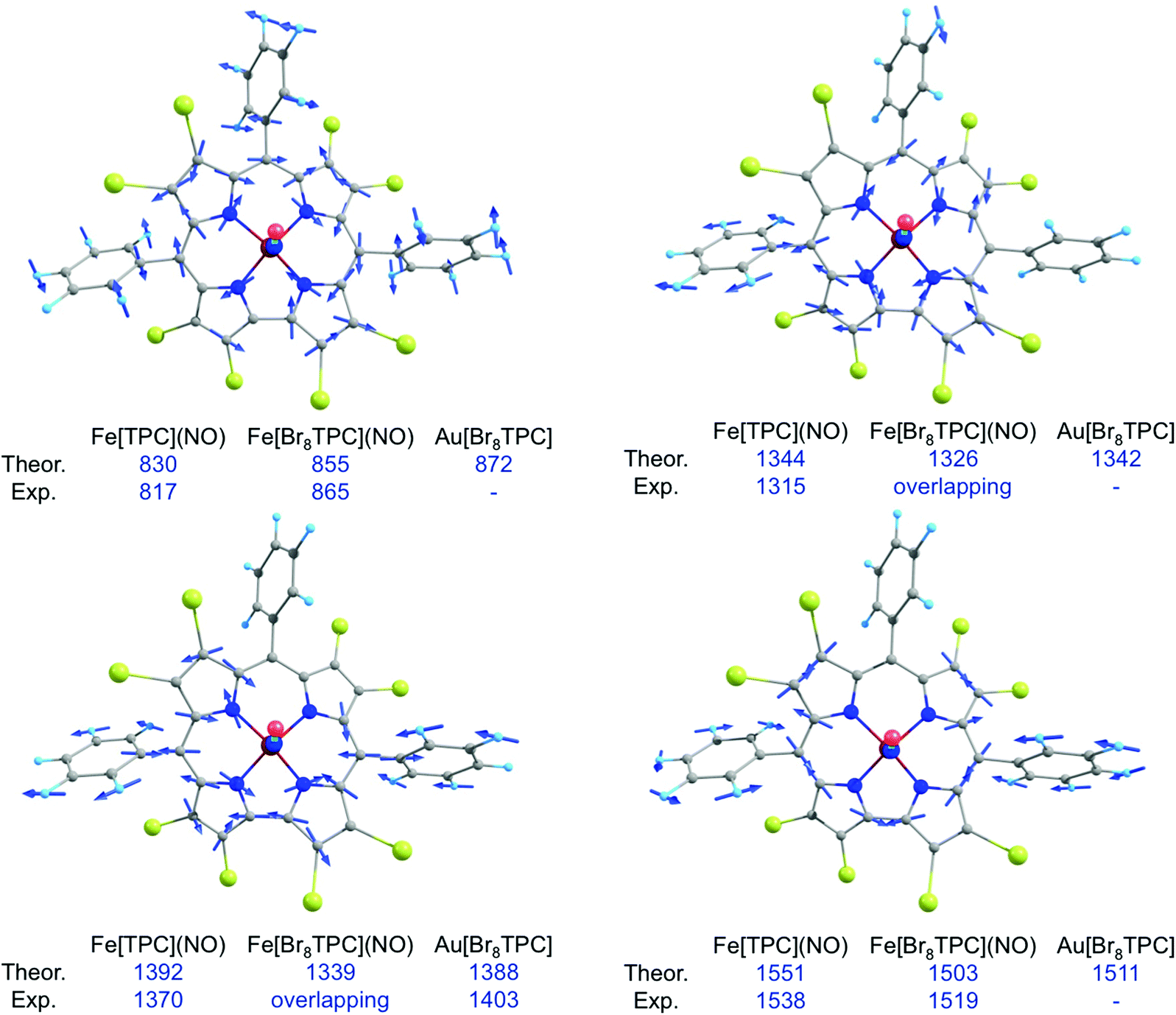
In our earlier study of FeNO meso-triarylcorroles,5 we identified 3–4 IR skeletal modes that are indicative of a noninnocent corrole macrocycle. Using IR spectroscopy and DFT calculations in concert, we have now identified the same modes for FeNO octabromocorroles (Fig. 6 and 7). Fig. 7 depicts the significant shifts in these bands between noninnocent Fe[Br8TPC](NO) and the known, innocent analogue Au[Br8TPC]. These frequency shifts may be qualitatively interpreted in terms of changes in skeletal bond order as a result of removal of an electron from the corrole HOMO depicted in Fig. 4.
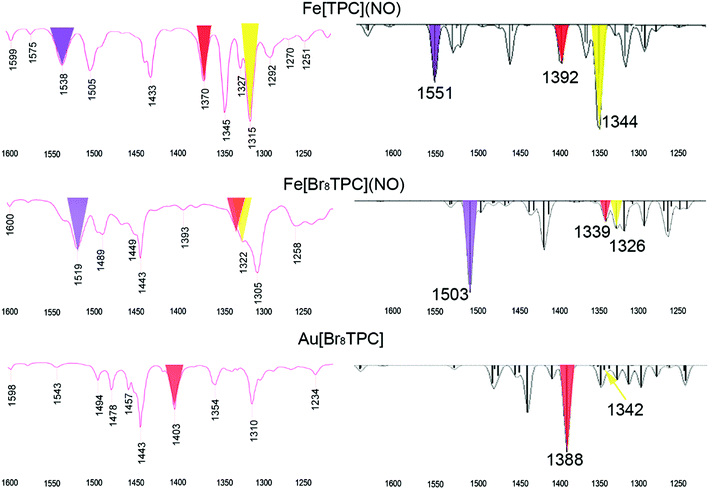 |
| | Fig. 6 Comparison of experimental and broken-symmetry B3LYP/STO-TZP calculated IR spectra for three metallocorroles. | |
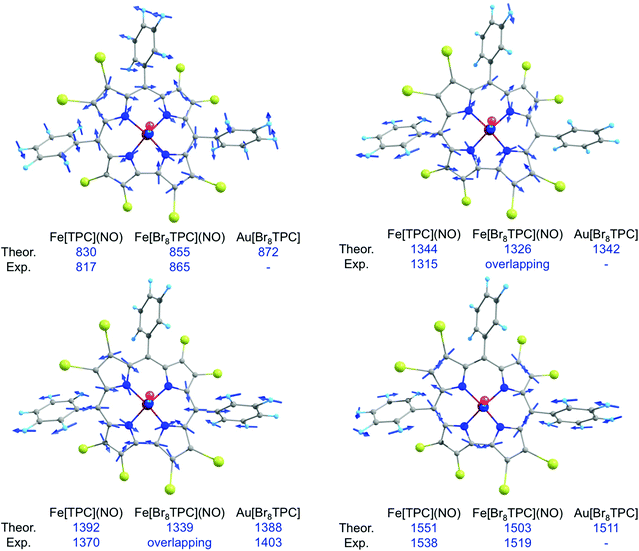 |
| | Fig. 7 Selected IR marker bands, including visual depiction of the eigenvectors and experimental and broken-symmetry B3LYP/STO-TZP frequencies (cm−1). | |
Redox potentials
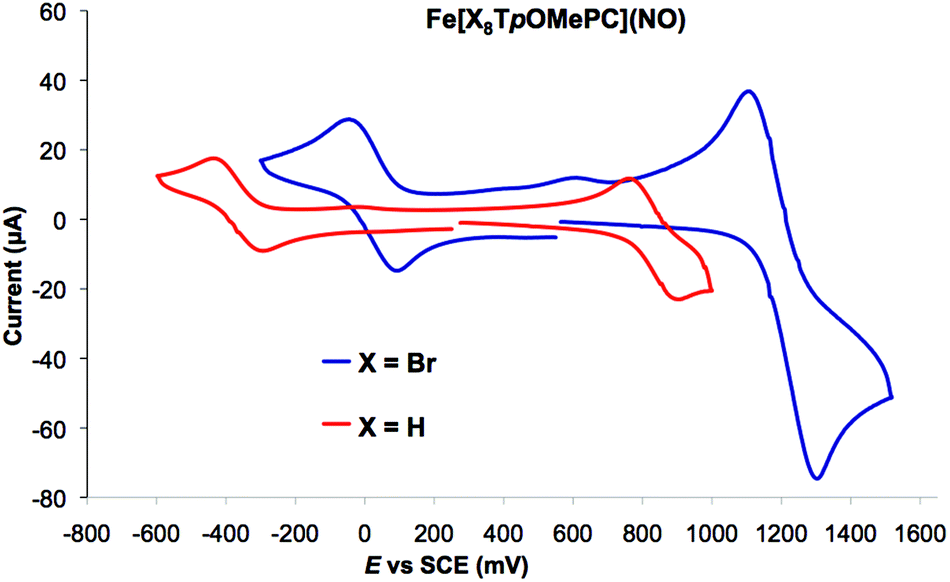
As shown in Table 1, both the meso aryl and β-Br substituents exert a significant effect on the redox potentials of FeNO corroles. Thus, β-octabromination upshifts both the oxidation and reduction potentials by about 400 mV (Fig. 8). As shown in Fig. 5, the calculated spin densities of both the cationic and anionic states of a typical FeNO corrole are almost exclusively localized on the FeNO unit in a manner that is characteristic of a S = 1/2 {FeNO}7 group. The corrole macrocycle in the cationic and anionic states thus may be described as formally monoanionic and trianionic, respectively, a scenario that is analogous to that observed for copper corroles.18–21
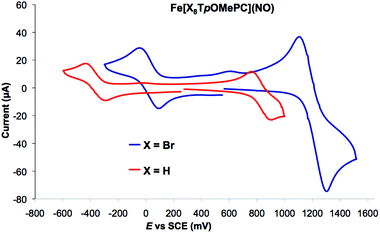 |
| | Fig. 8 Representative cyclic voltammograms of an FeNO triaarylcorrole and its β-Br8 analogue. | |
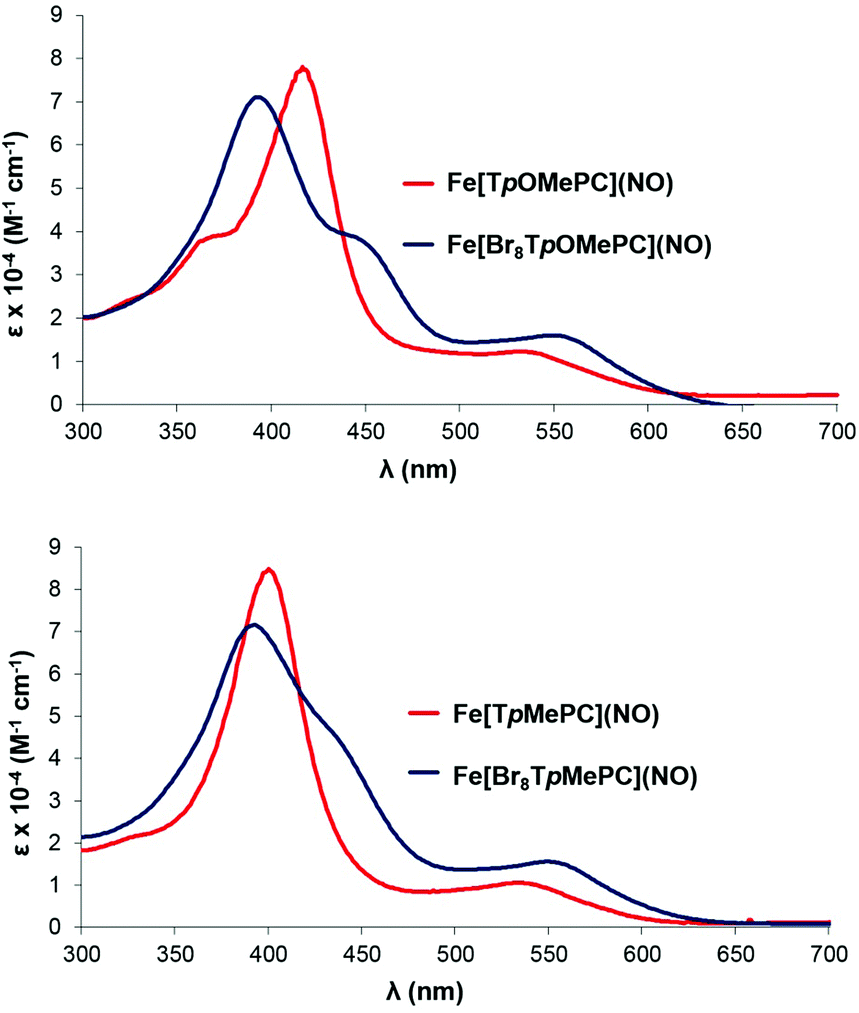
Electronic absorption spectroscopy
The Soret maxima of several but not all families of metallotriarylcorroles redshift markedly in response to increasing electron-donating character of the para substituent on the meso-aryl groups. Over a lengthy series of studies,5,6,18,22–26 we have shown that this effect is specific to noninnocent corroles and the substituent effects arise from aryl-to-corrole charge transfer character in certain transitions in the Soret region. Indeed, the strongly substituent-sensitive Soret maxima of the Fe[TpXPC](NO) series provided the first clue that FeNO corroles are noninnocent, i.e. not {FeNO}6.5 The Fe[Br8TpXPC](NO) series reported herein provides the first example of a series of noninnocent metallotriarylcorroles whose Soret maxima are essentially unaffected by para substituents on the meso-aryl groups (Table 1 and Fig. 9). Based on an examination of the valence MOs of the complexes in question, this unique behavior appears to be clearly attributable to steric inhibition of resonance, i.e., the bulky β-Br groups preclude any significant mesomeric interactions between the aryl groups and the corrole macrocycle in these compounds. This scenario may be contrasted with the Cu[Br8TpXPC] series, where strong saddling leads to enhanced aryl-corrole mesomeric interactions and hence also to strongly substituent-sensitive Soret maxima.18a,d,e
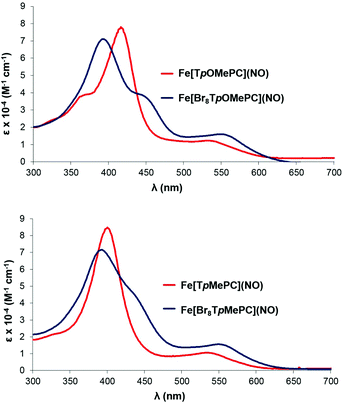 |
| | Fig. 9 Representative electronic absorption spectra. | |
Conclusions
A detailed, multitechnique study of FeNO octabromocorroles has led to considerable deepening of our appreciation of FeNO corroles as strongly noninnocent complexes. Broken-symmetry DFT calculations indicate that the amount of radical character of the corrole macrocycle is essentially constant for all the FeNO corroles studied and does not vary significantly with increasing electron-deficient character of the corrole macrocycle. Instead, an electron-deficient corrole ligand leads to decreased π-backdonation from the iron to the NO, which manifests itself experimentally in a higher NO stretching frequency. The relative instability of the more electron-deficient FeNO corroles with respect to NO loss is also reasonably ascribed to decreased π-backdonation in these complexes.
A combined experimental-DFT approach has also led to the identification of structural and IR spectroscopic signatures for ligand noninnocence in FeNO corroles. Uniquely, the Soret maxima of FeNO octabromocorroles do not shift as a function of para substituents on the meso-aryl groups, as they do for other families of noninnocent metallotriarylcorroles. Steric inhibition of aryl-corrole mesomeric interactions in these sterically crowded complexes appears to be responsible for this unusual phenomenon.
Experimental section
Instrumentation
Ultraviolet–visible (UV–vis) spectra were recorded on an HP 8454 spectrophotometer in CH2Cl2. Cyclic voltammetry was carried out with an EG&G model 263A potentiostat equipped with a three-electrode system: a glassy carbon working electrode, a platinum wire counter electrode, and a saturated calomel reference electrode (SCE). Tetrabutylammonium perchlorate, recrystallized twice from absolute ethanol and dried in a desiccator for at least 2 weeks, was used as the supporting electrolyte. The reference electrode was separated from the bulk solution by a fritted-glass bridge filled with a solvent/supporting electrolyte mixture. All potentials were referenced to the SCE. A scan rate of 100 mV s−1 was used. The anhydrous CH2Cl2 solutions were purged with argon for at least 5 min prior to the electrochemical measurements and an argon blanket was maintained over the solutions during the measurements. 1H NMR spectra (400 MHz) were recorded in CDCl3 (referenced to residual CHCl3 at 7.26 ppm) at 298 K on a Varian Inova 400 spectrometer. High-resolution electrospray-ionization (HR-ESI) mass spectra were recorded on an LTQ Orbitrap XL mass spectrometer. IR spectra were acquired as an average of 32 scans with a 1 cm−1 resolution on a Varian 7000e FT-IR spectrometer.
General synthetic details
All reagents and solvents were obtained from Sigma-Aldrich and used as purchased unless otherwise noted. CHROMASOLV® HPLC grade n-hexane and dichloromethane were used for chromatography. FeCl2·4H2O was obtained from Merck. NaNO2 was obtained from Fluka. Chloroform-d was obtained from Euriso-top. Pyrrole was passed through a short column of basic alumina (Merck, 5 cm height) before use to remove brownish impurities and stored at −20° C. Silica gel 60 (0.04–0.063 mm particle size; 230–400 mesh, Merck) was used for flash chromatography.
Free-base β-octabromo-meso-triarylcorroles H3[Br8TpXPC]11 and H3[Br8TPFPC]12 were prepared via the reductive demetalation of the copper and manganese complexes, respectively, as described earlier. For H3[Br8TpXPC] (X = CH3 and OCH3), iron insertion and nitrosylation could be achieved in good yields via slight modification of a one-pot literature procedure.2a The procedure, however, did not work for H3[Br8TPFPC] and led to relatively poor yields for H3[Br8TPC] and H3[Br8TpCF3PC]. In the latter two cases, the major byproduct could be readily converted to the FeCl corrole,27 and subsequently nitrosylated via another literature procedure. For H3[Br8TPFPC], a two-step procedure via Fe[TPFPC]Cl and subsequent nitrosylation was employed. All of the iron nitrosyl complexes were stored in sealed vials at 4 °C. Because of the limited stability of the compounds, especially with respect to denitrosylation, satisfactory elemental analyses did not prove possible. Full spectroscopic characterization was carried out on all the compounds and, in one case, also single-crystal X-ray structure determination. Detailed procedures for the preparation and purification of the various complexes are as follows.
Synthesis of Fe[Br8TpOMePC](NO)
Free-base β-octabromo-meso-tris(4-methoxyphenyl)corrole (100 mg, 80 μmol) was added to a 50 mL two-necked round-bottomed flask equipped with a magnetic stirrer and a reflux condenser and dissolved in pyridine (4.0 mL) and methanol (8.0 mL). The system was the closed off with rubber septa and vented via an oil bubbler. Argon was bubbled through the solution while it was virgorously stirred for 20 min. Iron(II) chloride tetrahydrate (318 mg, 1.6 mmol) was added to the solution under an argon blanket. The solution was then heated to 69 °C under a slow flow of argon for 1 h after which heating was discontinued and a saturated aqueous solution of NaNO2 (2.0 mL) was added dropwise via syringe, followed by stirring for an additional 30 min. The solution was then placed in an ice bath, quenched with cold water (40 mL), and filtered. The dry, filtered precipitate was washed with CH2Cl2 and the filtrate was rotary-evaporated to dryness. Flash chromatography (silica, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 CH2Cl2:hexane, subsequently 1:1) afforded Fe[Br8TpOCH3PC](NO) as the first, deep red band. Yield: 98 mg (92%). UV-vis (CH2Cl2): λmax (ε × 10−4 (M−1 cm−1)) (nm); 394 (7.1), 549 (1.6). 1H NMR (400 MHz, chloroform-d) δ 7.43 (br s, 4H, 5,15-o), 7.33 (d, J = 8.2 Hz, 2H, 10-o), 7.09 (d, J = 8.4 Hz, 4H, 5,15-m), 7.05 (d, J = 7.3 Hz, 2H, 10-m), 3.98 (s, 6H, 5,15-methoxy), 3.96 (s, 3H, 10-methoxy). MS (HR ESI) m/z 1330.4334 (M−), calcd 1330.4340); IR (thin film): νNO 1786 cm−1.
:2 CH2Cl2:hexane, subsequently 1:1) afforded Fe[Br8TpOCH3PC](NO) as the first, deep red band. Yield: 98 mg (92%). UV-vis (CH2Cl2): λmax (ε × 10−4 (M−1 cm−1)) (nm); 394 (7.1), 549 (1.6). 1H NMR (400 MHz, chloroform-d) δ 7.43 (br s, 4H, 5,15-o), 7.33 (d, J = 8.2 Hz, 2H, 10-o), 7.09 (d, J = 8.4 Hz, 4H, 5,15-m), 7.05 (d, J = 7.3 Hz, 2H, 10-m), 3.98 (s, 6H, 5,15-methoxy), 3.96 (s, 3H, 10-methoxy). MS (HR ESI) m/z 1330.4334 (M−), calcd 1330.4340); IR (thin film): νNO 1786 cm−1.
Synthesis of Fe[Br8TpMePC](NO)
Free-base β-octabromo-meso-tris(4-methylphenyl)corrole (78 mg, 65 μmol) and iron(II) chloride tetrahydrate (259 mg, 1.3 mmol) were used as starting materials and subjected to the same procedure as described above. Flash chromatography (silica, 1:4 CH2Cl2/hexane) afforded Fe[Br8TpCH3PC](NO) as the first, deep red band. Yield: 76 mg (91%). UV-vis (CH2Cl2): λmax (ε × 10−4 (M−1 cm−1)) (nm); 395 (7.2), 551 (1.6). 1H NMR (400 MHz, chloroform-d) δ 7.47–7.29 (m, 5,10,15-aryl, 12H), 2.56 (s, 5,15-methyl, 6H), 2.55 (s, 10-methyl, 3H). MS (HR ESI) m/z 1283.4667 (M−), calcd 1283.4556; IR (thin film): νNO 1786 cm−1.
Synthesis of Fe[Br8TPC](NO)
Free-base β-octabromo-meso-triphenylcorrole (42 mg, 36 μmol) and iron(II) chloride tetrahydrate (144 mg, 0.72 mmol) were subjected to the same procedure as described above. Flash chromatography (silica, 1:4 CH2Cl2:hexane) afforded Fe[Br8TPC](NO) as the first deep red band and also yielded second, darker reddish-brown band. The latter band was collected, evaporated to dryness, and dissolved in 20 mL CH2Cl2. The solution was washed twice with ∼20 mL 7% HCl(aq.) and once with water, dried over MgSO4, and filtered. A saturated aqueous solution of NaNO2 (2.0 mL) was added to the solution, and the solution was stirred for 4 h at room temperature. The solution was washed with water, dried over MgSO4, filtered, and evaporated to dryness. Flash chromatography (silica, 1:4 CH2Cl2:hexane) afforded Fe[Br8TPC](NO) as the first deep red band. Combined yield: 21 mg (47%). UV-vis (CH2Cl2): λmax (ε relative) (nm); 397 (1.0), 553 (0.2). 1H NMR (400 MHz, chloroform-d) δ 7.67–7.39 (m, 15H). MS (HR ESI) m/z 1240.3994 (M−), calcd 1240.4013; IR (thin film): νNO 1794 cm−1.
Synthesis of Fe[Br8TpCF3PC](NO)
Free-base β-octabromo-meso-tris(4-trifluoromethylphenyl)corrole (127 mg, 93 μmol) and iron(II) chloride tetrahydrate (370 mg, 1.86 mmol) were subjected to the same procedure as described above, except that the reaction mixture was heated for 0.5 h instead of 1 h prior to NaNO2 addition. Flash chromatography (silica, 1:4 CH2Cl2:hexane) afforded Fe[Br8TpCF3PC](NO) as the first deep red band. Continued elution with 1:1 CH2Cl2:hexane led to a second, reddish-brown band, which was collected and treated in the same way as described for the second band in the synthesis of Fe[Br8TPC](NO) to yield a second crop of the title compound. Combined yield: 99 mg (74%). UV-vis (CH2Cl2): λmax (ε × 10−4 (M−1 cm−1)) (nm); 392 (7.1), 552 (1.1). 1H NMR (400 MHz, chloroform-d) δ 7.90–7.56 (m, 12H). MS (HR ESI) m/z 1444.3636 (M−), (calcd 1444.3646); IR (thin film): νNO 1803 cm−1.
Synthesis of Fe[Br8TPFPC]Cl
Free-base β-octabromo-meso-tris(pentafluorophenyl)corrole (30 mg, 21 μmol) was added to a 50 mL two-necked round-bottomed flask equipped with a magnetic stirrer and a reflux condenser and dissolved in dry DMF (25 mL). The system was the closed off with rubber septa and vented via an oil bubbler. Argon was bubbled through the solution under vigorous stirring for 20 min. Anhydrous iron(II) chloride (53 mg, 0.42 mmol) was added to the solution under an argon blanket and the solution was heated to reflux. The reaction was monitored by TLC (silica, CH2Cl2:hexane 1:2). After 40 min of refluxing, no starting material could be detected by TLC and heating was discontinued. The solution was cooled to room temperature and the solvent removed via rotary evaporation. The solid material was dissolved in diethyl ether and passed through a short silica column with diethyl ether as eluent. After evaporation of the diethyl ether, the resulting solid was dissolved in CH2Cl2 and washed twice with 2 M HCl(aq.) and once with water. The organic layer was dried over Na2SO4 and evaporated to dryness. Flash chromatography (silica, CH2Cl2) afforded Fe[Br8TPFPC]Cl as a reddish brown band. Yield: 29 mg (91%). UV-vis (CH2Cl2): λmax, nm (logε, M−1 cm−1) 418 (4.66), 563 (0.79).19F NMR (376 MHz, chloroform-d) δ −151.05 (s, 2F), −151.83 (s, 4F), −157.73 (s, 3F), −159.60 (s, 6F). MS (MALDI-TOF) m/z 1480.69 (M − Cl−), calcd 1480.47. MS (HR-ESI) m/z 1515.23 (M−), calcd 1515.92.
Synthesis of Fe[Br8TPFPC](NO)
Chloroiron β-octabromo-meso-tris(pentafluorophenyl)corrole (20 mg, 13 μmol) was dissolved in CH2Cl2 (20 mL) and a saturated aqueous solution of NaNO2 (5.0 mL) was added to the solution. The solution was stirred at room temperature for 6 h, washed with water, dried over Na2SO4, and filtered, and the filtrate was evaporated to dryness. Flash chromatography (silica, 1:2 CH2Cl2:hexane) of the residue afforded Fe[Br8TPFPC](NO) as a deep red band. Yield: 13 mg (68%). X-ray quality crystals were obtained by vapor diffusion of n-heptane into a benzene solution of the title compound within a sealed vial. UV-vis (CH2Cl2): λmax, nm (logε, M−1 cm−1) 395 (5.52), 560 (1.28). 19F NMR (376 MHz, Chloroform-d): δ −137.41 (dd, J = 23.7, 8.1 Hz, 5,15-o, 2F), −138.17 (d, J = 24.0 Hz, 10-o, 1F), −138.76 (d, J = 23.5 Hz, 10-o′, 1F), −138.99 (dd, J = 23.7, 7.9 Hz, 2F, 5,15-o′, 2F), −150.66 to −151.16 (m, 5,10,15-p, 3F), −161.57 to −162.41 (m, 5,10,15-m, 6F). MS (HR-ESI): 1510.26, (Calcd 1510.26). IR (thin film): νNO 1803 cm−1.
Single-crystal X-ray structure determination of Fe[Br8TPFPC](NO)·C7H16·0.5C6H6
A red block of 0.220 × 0.200 × 0.080 mm3 was mounted in the 100(2) K nitrogen cold stream provided by an Oxford Cryostream low temperature apparatus on the goniometer head of a Bruker D85 diffractometer equipped with an ApexII CCD detector, on beamline 11.3.1 at the Advanced Light Source in Berkeley, CA. Diffraction data were collected using synchrotron radiation monochromated with silicon(111) to a wavelength of 0.77490(1) Å. A full sphere of data was collected using 0.3° ω scans. A multi-scan absorption correction was applied using the program SADABS 2014/3. The data consists of 54540 reflections collected, of which 20937 were unique [R(int) = 0.0627] and 13555 were observed [I > 2σ (I)]. The structure was solved by dual-space methods (SHELXT) and refined by full-matrix least-squares on F2 (SHELXL-97) using 1282 parameters and 0 restraints. The hydrogen atoms on carbon atoms were generated geometrically and refined as riding atoms with C–H = 0.95–0.99 Å and Uiso(H) = 1.2 times Ueq(C) for aromatic and CH2 groups and Uiso(H) = 1.5 times Ueq(C) for CH3 groups. The maximum and minimum peaks in the final difference Fourier map were 4.537 and −3.374 e Å−3. Crystal data: C84H19Br16F30Fe2N10O2, MW = 3160.35, triclinic, P1, a = 13.667(3) Å, b = 16.679(3) Å, c = 20.152(4) Å, α = 74.769(3)°, β = 91.4200(10)°, γ = 88.903(3)°, V = 4425.1(15) Å3, T = 100(2) K, Z = 2, R1 [I > 2σ(I)] = 0.0702, wR2 (all data) = 0.2084, GOF (on F2) = 1.056.
Computational details
All calculations were carried out with the ADF 201428 program system, the B3LYP29,30 exchange–correlation functional, and appropriately fine integration grids and tight SCF and geometry optimization criteria. Scalar relativistic effects were taken into account with the ZORA Hamiltonian31 and ZORA STO-TZP basis sets, except for the structures shown in Fig. 3, for which ZORA STO-TZ2P basis sets were employed.32 Grimme's D3 dispersion correction was used throughout.33 All optimized geometries were verified as true minima via numerical frequency calculations.
Acknowledgements
This work was supported by FRINATEK project 231086 of the Research Council of Norway (AG) and the Advanced Light Source, Berkeley, California (CMB, KJG). The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231
References
- E. Vogel, S. Will, A. S. Tilling, L. Neumann, J. Lex, E. Bill, A. X. Trautwein and K. Wieghardt, Angew. Chem., Int. Ed. Engl., 1994, 33, 731–735 CrossRef.
-
(a) C. A. Joseph, M. S. Lee, A. V. Iretski, G. A. Wu and P. C. Ford, Inorg. Chem., 2006, 45, 2075–2082 CrossRef CAS PubMed;
(b) P. Singh, I. Saltsman, A. Mahammed, I. Goldberg, B. Tumanskii and Z. Gross, J. Porphyrins Phthalocyanines, 2012, 16, 663–673 CrossRef CAS;
(c) W. Sinha, N. Deibel, H. Agarwala, A. Garai, D. Schweinfurth, C. S. Purohit, G. K. Lahiri, B. Sarkar and S. Kar, Inorg. Chem., 2014, 53, 1417–1429 CrossRef CAS PubMed.
- J. H. Enemark and R. D. Feltham, Coord. Chem. Rev., 1974, 13, 339–406 CrossRef CAS.
- Reviews on quantum chemical studies on nitrosyls:
(a) A. Ghosh, Acc. Chem. Res., 2005, 38, 943–954 CrossRef CAS PubMed;
(b)
A. Ghosh, K. H. Hopmann and J. Conradie, in Computational Inorganic and Bioinorganic Chemistry, ed. E. I. Solomon, R. A. Scott and R. B. King, John Wiley & Sons Ltd, Chichester, UK, 2009, pp. 389–410 Search PubMed;
(c) L. E. Goodrich, F. Paulat, V. K. K. Praneeth and N. Lehnert, Inorg. Chem., 2010, 49, 6293–6316 CrossRef CAS PubMed.
- H. Vazquez-Lima, H.-K. Norheim, R. Einrem and A. Ghosh, Dalton Trans., 2015, 44, 10146–10151 RSC.
- For earlier studies on ligand noninnocence in iron corroles from our aboratory, see:
(a) E. Steene, T. Wondimagegn and A. Ghosh, J. Phys. Chem. B, 2001, 105, 11406–11413 CrossRef CAS; addition/correction: J. Phys. Chem. B, 2002, 106, 5312–5312.
(b) E. Steene, T. Wondimagegn and A. Ghosh, J. Am. Chem. Soc., 2003, 125, 16300–16309 CrossRef CAS PubMed.
- For other studies of ligand noninnocence in iron corroles, see:
(a) O. Zakharieva, V. Schünemann, M. Gerdan, S. Licoccia, S. Cai, F. A. Walker and A. X. Trautwein, J. Am. Chem. Soc., 2002, 124, 6636–6648 CrossRef CAS PubMed;
(b) Review: F. A. Walker, S. Licoccia and R. Paolesse, J. Inorg. Biochem., 2006, 100, 810–837 CrossRef CAS PubMed.
-
Ab initio CASSCF/CASPT2 studies on FeCl corrole: B. O. Roos, V. Veryazov, J. Conradie, P. R. Taylor and A. Ghosh, J. Phys. Chem., 2008, 112, 14099–14102 CrossRef CAS PubMed.
- A. B. Alemayehu, H. Vazquez-Lima, C. M. Beavers, K. J. Gagnon, J. Bendix and A. Ghosh, Chem. Commun., 2014, 50, 11093–11096 RSC.
- C. Capar, K. E. Thomas and A. Ghosh, J. Porphyrins Phthalocyanines, 2008, 12, 964–967 CrossRef CAS.
- J. Capar, S. Berg, K. E. Thomas, C. M. Beavers, K. J. Gagnon and A. Ghosh, J. Inorg. Biochem. Search PubMed , in press: http://www.sciencedirect.com/science/article/pii/S0162013415300441.
- C. Capar, L.-K. Hansen, J. Conradie and A. Ghosh, J. Porphyrins Phthalocyanines, 2010, 14, 509–512 CrossRef CAS.
- For a crystal structure of H3[Br8TPFPC], see: J. Capar, J. Conradie, C. M. Beavers and A. Ghosh, J. Phys. Chem. A, 2015, 119, 3452–3457 CrossRef CAS PubMed.
- For reviews on corrole coordination chemistry, see:
(a) K. E. Thomas, A. Alemayehu, J. Conradie, C. M. Beavers and A. Ghosh, Acc. Chem. Res., 2012, 45, 1203–1214 CrossRef CAS PubMed;
(b) J. H. Palmer, Struct. Bonding, 2012, 142, 49–89 CrossRef CAS;
(c) H. L. Buckley and J. Arnold, Dalton Trans., 2015, 44, 30–36 RSC.
- M. K. Ellison, C. E. Schulz and W. R. Scheidt, Inorg. Chem., 1999, 38, 100–108 CrossRef CAS.
- For key examples of linear, S = 1/2 {FeNO}7 complexes, see:
(a) K. J. Franz and S. J. Lippard, J. Am. Chem. Soc., 1999, 121, 10504–10512 CrossRef CAS; addition/correction: J. Am. Chem. Soc., 2001, 123, 1266–1266;
(b) For a theoretical study, see: E. Tangen, J. Conradie and A. Ghosh, Inorg. Chem., 2005, 44, 8699–8706 CrossRef CAS PubMed;
(c) J. Conradie, K. H. Hopman and A. Ghosh, J. Phys. Chem. B, 2010, 114, 8517–8524 CrossRef CAS PubMed.
- J. H. Palmer, M. W. Day, A. D. Wilson, L. M. Henling, Z. Gross and H. B. Gray, J. Am. Chem. Soc., 2008, 130, 7786–7787 CrossRef CAS PubMed.
- For selected studies of Cu corroles from our laboratory, see:
(a) I. H. Wasbotten, T. Wondimagegn and A. Ghosh, J. Am. Chem. Soc., 2002, 124, 8104–8116 CrossRef CAS PubMed;
(b) A. B. Alemayehu, E. Gonzalez, L.-K. Hansen and A. Ghosh, Inorg. Chem., 2009, 48, 7794–7799 CrossRef CAS PubMed;
(c) A. B. Alemayehu, L.-K. Hansen and A. Ghosh, Inorg. Chem., 2010, 49, 7608–7610 CrossRef CAS PubMed;
(d) K. E. Thomas, J. Conradie, L.-K. Hansen and A. Ghosh, Eur. J. Inorg. Chem., 2011, 1865–1870 CrossRef CAS;
(e) S. Berg, K. E. Thomas, C. M. Beavers and A. Ghosh, Inorg. Chem., 2012, 51, 9911–9916 CrossRef CAS PubMed.
- For key early papers on Cu corroles, see:
(a) C. Brückner, R. P. Briñas and J. A. K. Bauer, Inorg. Chem., 2003, 42, 4495–4497 CrossRef PubMed;
(b) M. Bröring, F. Brégier, E. C. Tejero, C. Hell and M. C. Holthausen, Angew. Chem., Int. Ed., 2007, 46, 445–448 CrossRef PubMed.
- For spectroelectrochemical studies of Cu corroles, see: Z. Ou, J. Shao, H. Zhao, K. Ohkubo, I. H. Wasbotten, S. Fukuzumi, A. Ghosh and K. M. Kadish, J. Porphyrins Phthalocyanines, 2004, 8, 1236–1247 CrossRef CAS.
- The Fe[Br8TpXPC](NO) complexes appear to exhibit a two-electron oxidation; the origin of this phenomenon remains to be investigated.
- MnCl corroles: A. Ghosh and E. Steene, J. Inorg. Biochem., 2002, 91, 423–436 CrossRef CAS PubMed.
- CrO and MoO corroles: I. Johansen, H.-K. Norheim, S. Larsen, A. B. Alemayehu, J. Conradie and A. Ghosh, J. Porphyrins Phthalocyanines, 2011, 15, 1335–1344 CrossRef CAS.
- TDDFT studies of Cu corroles:
(a) A. B. Alemayehu, J. Conradie and A. Ghosh, Eur. J. Inorg. Chem., 2011, 12, 1857–1864 CrossRef;
(b) A. B. Alemayehu, M. M. Conradie and A. Ghosh, J. Porphyrins Phthalocyanines, 2012, 16, 695–704 CrossRef CAS.
- Ag and Au corroles:
(a) Au: A. B. Alemayehu and A. Ghosh, J. Porphyrins Phthalocyanines, 2011, 15, 106–110 CrossRef CAS;
(b) Ag, Au: K. E. Thomas, A. B. Alemayehu, J. B. Conradie, C. M. Beavers and A. Ghosh, Inorg. Chem., 2011, 50, 12844–12851 CrossRef CAS PubMed . Ag,
Au: K. E. Thomas, H. Vazquez-Lima, Y. Fang, Y. Song, K. J. Gagnon, C. M. Beavers, K. M. Kadish and A. Ghosh, Chem. – Eur. J., 2015, 21, 16839–16847 Search PubMed.
- 5d metallocorroles:
(a) Pt: A. B. Alemayehu, H. Vazquez-Lima, C. M. Beavers, K. J. Gagnon, J. Bendix and A. Ghosh, Chem. Commun., 2014, 50, 11093–11096 RSC;
(b) Os: A. Alemayehu, K. J. Gagnon, J. Terner and A. Ghosh, Angew. Chem., Int. Ed., 2014, 53, 14411–14414 CrossRef CAS PubMed.
- L. Simkhovich, A. Mahammed, I. Goldberg and Z. Gross, Chem. – Eur. J., 2001, 7, 1041–1055 CrossRef CAS.
-
(a) G. te Velde, F. M. Bickelhaupt, S. J. A. van Gisbergen, C. Fonseca Guerra, E. J. Baerends, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS;
(b) C. Fonseca Guerra, J. G. Snijders, G. te Velde and E. J. Baerends, Theor. Chem. Acc., 1998, 99, 391–403 Search PubMed.
- A. D. Becke, Phys. Rev., 1988, A38, 3098–3100 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1998, 37, 785–789 CrossRef.
-
(a) E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef CAS;
(b) E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783–9792 CrossRef CAS;
(c) E. van Lenthe, A. E. Ehlers and E. J. Baerends, J. Chem. Phys., 1999, 110, 8943–8953 CrossRef CAS.
- E. van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1430309. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt03947a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a