
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrowave-assisted FLP-catalyzed hydrogenations
S.
Tussing
and
J.
Paradies
*
Institute of Organic Chemistry, University of Paderborn, Warburger Straße 100, D-33098 Paderborn, Germany. E-mail: jan.paradies@uni-paderborn.de
First published on 9th November 2015
Abstract
FLP-catalyzed hydrogenations of 15 substrates were compared using microwave irradiation and conventional heating. The direct comparison revealed that a rate acceleration of up to 2.5 was achieved in the presence of microwaves. This heating method is particularly promising for the hydrogenation of nitrogen-containing heterocycles. Acridine, quinines and especially 1-methyl indole were reduced very efficiently under mild conditions and only 4 bar hydrogen pressure in high yields.
Introduction
The reduction of organic molecules by frustrated Lewis pairs (FLP) using molecular hydrogen (H2) has emerged to be a powerful tool for organic synthesis.1 Electron-deficient as well as electron-rich compounds have been successfully hydrogenated2 and even asymmetric reductions of prochiral substrates were achieved.3 Usually such reactions are conducted in closed reaction systems e.g. thick-walled vials, pressure tubes including NMR-tubes or steel bomb autoclaves. The appropriate reactor must be selected with regard to the required H2-pressure. The closed reaction systems are typically heated in oil baths or with heating jackets above the boiling point of the solvent.4 The same specifications hold true for microwave-reactors including the advantage of the fast equilibration of the temperature and inverted temperature gradients.5 Microwave-assisted transition-metal catalyzed hydrogenations and transfer hydrogenations were reported6 rendering this method attractive for FLP-catalyzed hydrogenations. Interestingly, the application of microwave heating in FLP-catalyzed hydrogenations has not been reported yet.Herein we report the first microwave-assisted FLP-catalyzed hydrogenation of imines, heterocycles, enamines, silyl enol ethers, nitroolefins, malonates and olefins. This study focuses on the comparison of hydrogenations conducted under conventional and microwave heating. Therefore reported catalyst systems (Lewis acids 1–3 and Lewis bases 4–8, Chart 1) and known substrates susceptible to FLP-catalyzed hydrogenation were investigated.
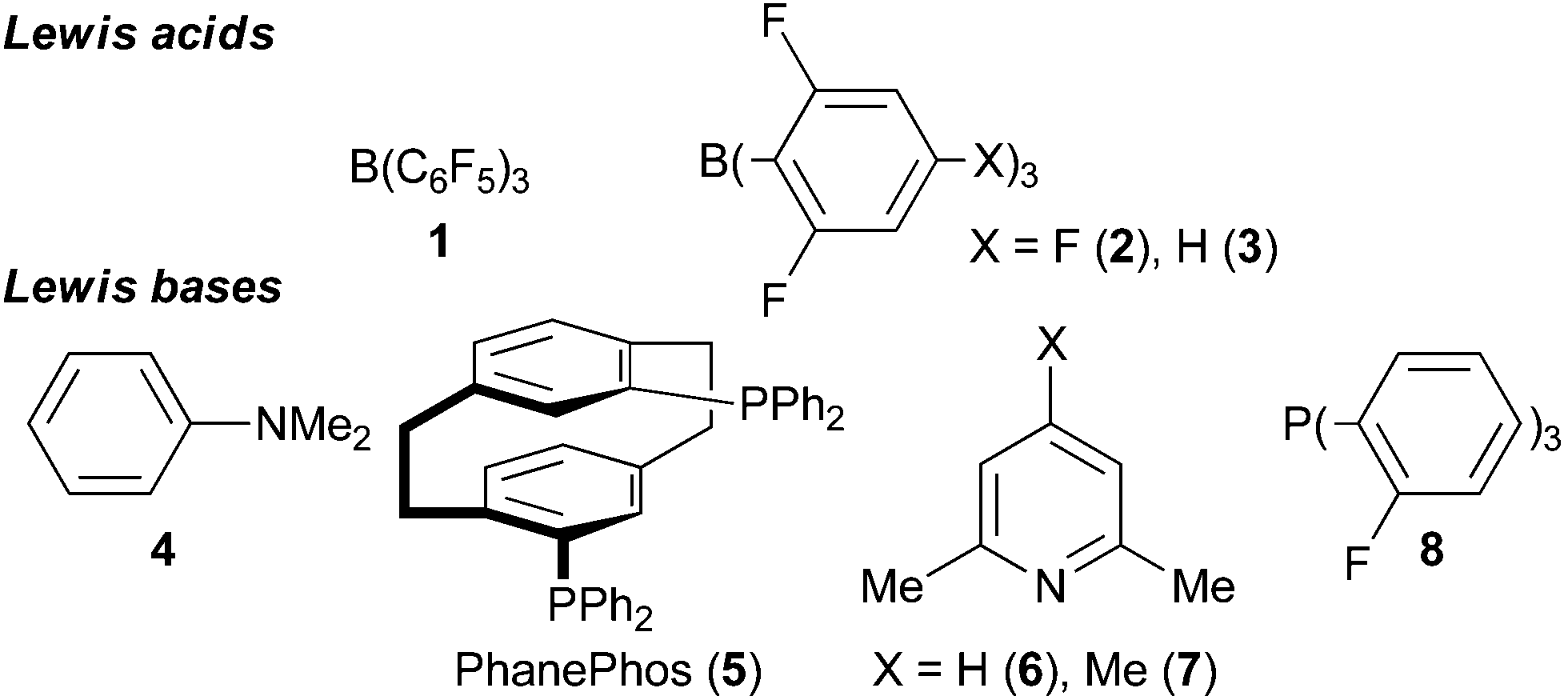 | ||
| Chart 1 Lewis acids and Lewis bases for FLP-mediated hydrogen activation. | ||
Results and discussion
We initiated our studies with the well-established FLP-catalyzed hydrogenation of N-benzylidene-tertbutyl amine1c,7 (9) using conventional and microwave heating. A solution of imine 9 in toluene (0.2 M) and 5 mol% B(C6F5)3 was prepared and split into two identical thick walled glass vessels. The vessels were sealed and pressurized with 4 bar of hydrogen. One sample was immersed into a preheated oil bath and the second sample was introduced into a CEM microwave reactor with a power setting to 150 W. Both reactions were heated to 80 °C for the indicated time (see Table 1).|
|
||
|---|---|---|
| Time [min] | Oil bath | Microwave (150 W) |
| Yield [%] | ||
| 10 | 22 | 60 |
| 15 | 42 | 82 |
| 20 | 47 | 99 |
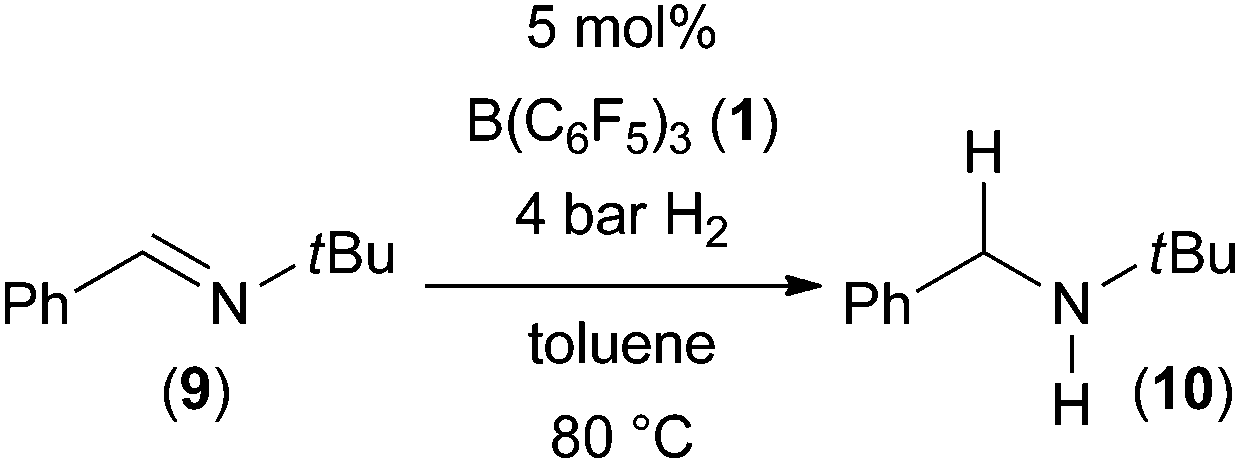
The hydrogenation product 10 was obtained with both heating methods however in very different yields. The reaction using microwave irradiation gave the product in 60% yield after 10 minutes while using conventional heating furnished only 22% yield. The microwave-assisted reaction went to full conversion with prolongation of the reaction time to 20 minutes while the oil bath heated reaction provided 10 in 47% yield. We attribute this significant rate increase to more efficient heating of the reaction by microwave irradiation.
Encouraged by these results we investigated other substrates bearing polarized double bonds in the microwave-assisted reaction. We selected substrates, which are susceptible to FLP-catalyzed hydrogenation under conventional heating and compared them with yields obtained from microwave experiments.
The standard conditions for the hydrogenations using microwave-assisted and conventional heating were benzene as solvent (0.2 M), 4 bar hydrogen and 150 W power-level in CEM Discover microwave reactor with external IR temperature control or a preheated oil-bath respectively.12 For best comparability thermal as well as microwave-assisted hydrogenations were conducted under identical conditions using stock-solutions, identical glassware, temperature, hydrogen pressure and reaction time. The reactions were stopped after 10 to 120 minutes and the yields were determined by 1H NMR with hexamethylbenzene as internal standard. The results are summarized in Table 2. Generally, all substrates underwent FLP-catalyzed hydrogenation under microwave conditions most of them with pronounced rate acceleration. In accord with our observations for imine 9 we found two-fold rate increase for imines 11a and 11b (entries 1 and 2). The imine 11c underwent hydrogenation in comparable yields and diastereoselectivity irrespective of the heating method (entry 3). Particularly, the hydrogenation of nitrogen-containing heterocycles benefits from microwave-assisted heating (entries 4–7). The hydrogenation of acridin (11d), 8-methyl-quinolin (11e) and 2-methyl-quinoline (11f) proceeded in significantly higher yields (72–99%) compared to the reactions with conventional heating (7–68%). Notably, indol 11g was reduced in 57% yield in 40 minutes at 140 °C with only 4 bar of hydrogen pressure under microwave conditions whereas the hydrogenation was not achieved using conventional heating (entry 7). Although yields for the reaction using conventional heating were not reported2e the decrease of the required pressure by almost 100 bar renders the application of microwave-assisted FLP-hydrogenation of indoles as very promising. We also investigated the core hydrogenation of diphenylamine with 20 mol% catalyst loading (11h).4b However, even under forcing conditions only traces of the product could be observed irrespective of the heating techniques (entry 8). The electron-rich double bond in enamines7b and silyl enol ethers3c,9,13 were reduced in good to excellent yields in only 10 to 40 minutes (entries 9–11). Again the microwave-assisted reactions provided the products in shorter reaction times compared to reactions using conventional heating. Densely functionalised malonates10,14 were hydrogenated in quantitative yield in 2 h at 80 °C (entry 12) compared to 12 h at room temperature. However, direct comparison of the reactions using microwave and conventional heating after 40 minutes revealed identical reaction rates using the FLP-catalyst 3/7 for transient hydrogen activation. Such electronically modified FLP-catalysts e.g. consisting of less Lewis-acidic boranes B(C6F3H2)2 (2) and B(C6F2H3)3 (3) or less electron-releasing phosphine 8, display highly reversible H2-activation at room temperature or below according to eqn (1).11
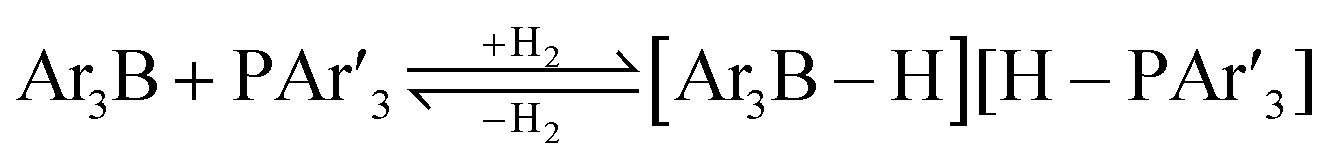 | (1) |
| Entry | Substrate | Catalyst | mol% cat. | Pressure H2 [bar] | Temp. [°C] | Time [min.] | Yield [%] | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| a Conditions: benzene, 150 W, 4 bar hydrogen pressure; 0.2 M. b MW: microwave reactor. c Conv. heating: conventional heating. | |||||||||
| 1 |
|
MWb | 1 | 5 | 4 | 80 | 20 | 12 | |
| Conv. heatingc | 1 | 5 | 4 | 80 | 20 | 6 | |||
| 5 | 5 | 120 | 60 | 99 | 7a | ||||
| 2 |

|
MW | 1 | 10 | 4 | 110 | 40 | 99 | |
| Conv. heating | 1 | 10 | 4 | 110 | 40 | 45 | |||
| 5 | 10 | 120 | 1320 (22 h) | 68 | 3e | ||||
| 3 |
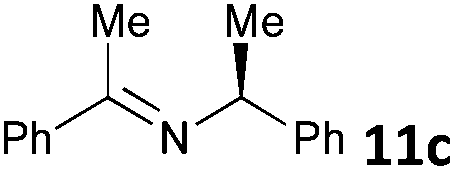
|
MW | 1 | 5 | 4 | 80 | 20 | 33 (32 de) | |
| Conv. heating | 1 | 5 | 4 | 80 | 20 | 27 (32 de) | |||
| 5 | 5 | 80 | 2880 (48 h) | 72 (36 de) | 8 | ||||
| 4 |

|
MW | 1 | 5 | 4 | 80 | 10 | 99 | |
| conv. heating | 1 | 5 | 4 | 80 | 10 | 68 | |||
| 5 | 4 | 25 | 120 | 80 | 4c | ||||
| 5 |
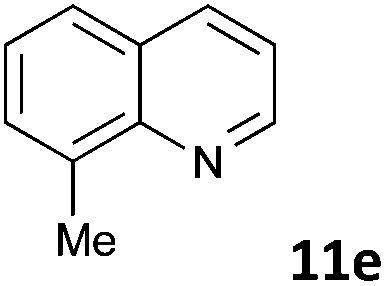
|
MW | 1 | 10 | 4 | 80 | 40 | 99 | |
| Conv. heating | 1 | 10 | 4 | 80 | 40 | 7 | |||
| 10 | 4 | 80 | 360 (6 h) | 88 | 4c | ||||
| 6 |
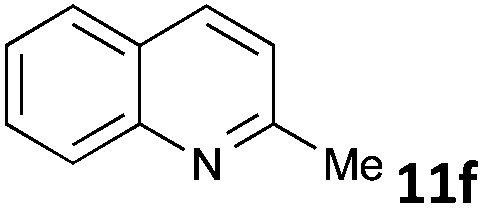
|
MW | 1 | 5 | 4 | 80 | 40 | 75 | |
| Conv. heating | 1 | 5 | 4 | 80 | 40 | 62 | |||
| 5 | 4 | 50 | 960 (16 h) | 74 | 4c | ||||
| 7 |

|
MW | 1 | 5 | 4 | 140 | 40 | 57 | |
| Conv. heating | 1 | 5 | 4 | 140 | 40 | 0 | |||
| 5 | 103 | 80 | 1080 (18 h) | n.a. | 2e | ||||
| 8 |

|
MW | 1 | 20 | 4 | 140 | 120 | 4 | |
| Conv. heating | 1 | 20 | 4 | 140 | 120 | 0 | |||
| 100 | 4 | 110 | 5760 (96 h) | 65 | 4b | ||||
| 9 |
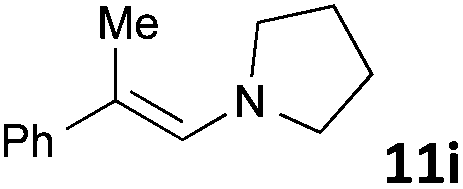
|
MW | 1 | 5 | 4 | 140 | 40 | 66 | |
| Conv. heating | 1 | 5 | 4 | 140 | 40 | 43 | |||
| 10 |
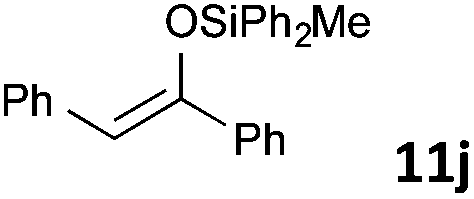
|
MW | 1/4 | 10 | 4 | 80 | 20 | 73 | |
| 20 | 4 | 80 | 10 | 99 | |||||
| Conv. heating | 1/4 | 10 | 4 | 80 | 20 | 76 | |||
| 1/5 | 20 | 5 | 50 | 1440 (24 h) | 78 | 9 | |||
| 11 |
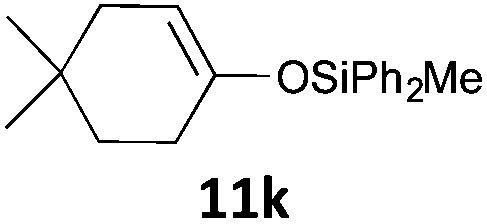
|
MW | 1/4 | 10 | 4 | 80 | 30 | 55 | |
| Conv. heating | 1/4 | 10 | 4 | 80 | 30 | 59 | |||
| 1/5 | 20 | 5 | 50 | 1440 (24 h) | 74 | 9 | |||
| 12 |
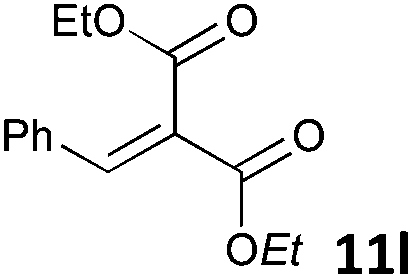
|
MW | 3/7 | 20 | 4 | 80 | 120 | 99 | |
| 20 | 4 | 80 | 40 | 45 | |||||
| Conv. heating | 3/7 | 20 | 4 | 80 | 40 | 42 | |||
| 20 | 4 | 25 | 720 (12 h) | >95 | 10 | ||||
| 13 |

|
MW | 2/6 | 20 | 4 | 50 | 120 | 23 | |
| 20 | 4 | 80 | 120 | 17 | |||||
| Conv. heating | 2/6 | 20 | 4 | 50 | 120 | 59 | |||
| 20 | 5 | 40 | 1440 (24 h) | >95 | 10 | ||||
| 14 |

|
MW | 1/8 | 20 | 4 | 80 | 60 | 16 | |
| 20 | 4 | 110 | 60 | 8 | |||||
| Conv. heating | 1/8 | 20 | 4 | 80 | 60 | 57 | |||
| 20 | 4 | 25 | 1440 (24 h) | 99 | 11 | ||||
According to an almost ergoneutral reaction the temperature increase shifts the equilibrium to the left side resulting in reduced concentrations of the H2-activation products.7c,10,11,15 Consequently, substrates e.g. nitroolefins or olefins (entries 13 and 14), which require the application of highly reversible FLP-catalysts are less susceptible to microwave-assisted hydrogenations and conventional heating provides higher yields. The reduction of nitroolefin 11m was achieved in 60% at 50 °C, but only 32% yield was obtained if the reaction was conducted at 80 °C (entry 13). Similar reactivity was observed for the hydrogenation of the olefin 11n (entry 14). Nevertheless, microwave-assisted FLP-catalyzed hydrogenations are highly valuable for reactions, which require heating leading to a significant rate increase.
Conclusions
In summary we have shown that microwave-assisted heating significantly accelerates FLP-catalyzed hydrogenations rendering this methodology as useful tool in organic synthesis. The yields for the hydrogenation of fifteen common substrates under microwave irradiation and conventional heating were directly compared. Rate increase of 2–2.5 were observed for imines 9, 11a and 11b using only 4 bar of hydrogen pressure. We attribute the reduced reaction times to more efficient heating of the reaction mixture. The hydrogenation of nitrogen-containing heterocycles e.g. acridine (11d), quinolines (11e–f) and 1-methyl indole (11g) proceeds in high yields under mild reaction conditions. Astonishingly, the indole derivative 11g was reduced in 57% yield under only 4 bar hydrogen pressure while the reduction was absent using conventional heating.Experimental
General information
All preparations were carried out in oven-dried glassware under an atmosphere of inert gas (Argon 5.0, Air Liquide) employing both Schlenk line techniques and a Glovebox Systems inert atmosphere glovebox. Microwave assisted reactions were carried out in a CEM Discover microwave system (model 908010) with external IR temperature control in 10 ml reaction vessels equipped with Teflon/rubber crimp caps. [D6]-deuterated benzene and toluene were degassed by 3 freeze–pump–thaw cycles, vacuum transferred from sodium/benzophenone, dispensed into Straus flasks equipped with Young-type Teflon valve stop-cocks and stored over 4 Å molecular sieves. Hydrogen 6.0 (AirLiquide) was purified through a Johnson Matthey Model HIG 35XL gas purifier. Substrates for hydrogenation reactions were purchased from Sigma Aldrich or abcr and were recrystallized or distilled prior to use if necessary. Reagents for borane synthesis were used as received without further purification. B(C6F2H3)3 (3), B(C6F3H2)3 (2) were prepared as described earlier.10General procedure for microwave assisted hydrogenation reactions
In a glovebox, B(C6F5)3 (1) (5–10 mol%), hexamethylbenzene (1.6 mg, 0.01 mmol, 0.1 equiv.) and the corresponding substrate (0.1 mmol, 1.0 equiv.) were dissolved in 0.5 ml of deuterated solvent. The stock solution was divided in three parts, two portions of 0.2 ml were transferred into two 10 ml reaction vessels equipped with magnetic stir bars and were capped with PTFE/butyl rubber crimp caps. The third part of 0.1 ml of the stock solution was used as comparison sample and analysed by 1H NMR spectroscopy. The reaction vessels were attached to an H2/vacuum line via needle, immersed into liquid nitrogen (6 cm), freeze pump thawed for one cycle and charged with H2 at 77 K. The reactions were carried out in two CEM Discover (model 908010) microwave systems using 150 W power at given times and temperatures. The cooled reaction mixtures were separately analysed by 1H NMR spectroscopy. Yield as mean value of both measurements. Non-microwave control samples were completely immersed in a preheated oil bath. For substrates requiring an FLP, a suitable base was added matching the concentration of borane as indicated. For the domino hydrosilylation hydrogenation of enones HSiPh2Me (0.1 mmol, 1.0 equiv.) was added prior to the addition of a suitable base as described earlier.9 Reactions of nitroolefins and malonates required the use of B(2,6-F2-C6H3)3 (3) or B(2,4,6-F3-C6H2)3 (2) (20 mol%).The analytic data of all starting materials and hydrogenation products were reported in the literature. Yields were determined by 1H NMR spectroscopy using hexamethylbenzene as internal standard.
Acknowledgements
The German Science Foundation (DFG) is gratefully acknowledged for financial support.References
- (a) G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124 CrossRef CAS PubMed; (b) G. C. Welch and D. W. Stephan, J. Am. Chem. Soc., 2007, 129, 1880 CrossRef CAS PubMed; (c) P. A. Chase, G. C. Welch, T. Jurca and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 8050 CrossRef CAS PubMed; (d) P. Spies, G. Erker, G. Kehr, K. Bergander, R. Froehlich, S. Grimme and D. W. Stephan, Chem. Commun., 2007, 5072 RSC.
- (a) J. Paradies, Synlett, 2013, 777 CrossRef CAS; (b) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46 CrossRef CAS PubMed; (c) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400 CrossRef CAS PubMed; (d) D. W. Stephan, Acc. Chem. Res., 2015, 48, 306 CrossRef CAS PubMed; (e) D. W. Stephan, S. Greenberg, T. W. Graham, P. Chase, J. J. Hastie, S. J. Geier, J. M. Farrell, C. C. Brown, Z. M. Heiden, G. C. Welch and M. Ullrich, Inorg. Chem., 2011, 50, 12338 CrossRef CAS PubMed; (f) D. W. Stephan, Org. Biomol. Chem., 2008, 6, 1535 RSC; (g) D. W. Stephan, Org. Biomol. Chem., 2012, 10, 5740 RSC; (h) J. Paradies, Angew. Chem., Int. Ed., 2014, 53, 3552 CrossRef CAS PubMed.
- (a) Y. Liu and H. Du, J. Am. Chem. Soc., 2013, 135, 12968 CrossRef CAS PubMed; (b) Y. B. Liu and H. F. Du, J. Am. Chem. Soc., 2013, 135, 6810 CrossRef CAS PubMed; (c) S. Wei and H. Du, J. Am. Chem. Soc., 2014, 136, 12261 CrossRef CAS PubMed; (d) D. J. Chen, Y. T. Wang and J. Klankermayer, Angew. Chem., Int. Ed., 2010, 49, 9475 CrossRef CAS PubMed; (e) D. J. Chen and J. Klankermayer, Chem. Commun., 2008, 2130 RSC; (f) G. Ghattas, D. J. Chen, F. F. Pan and J. Klankermayer, Dalton Trans., 2012, 41, 9026 RSC.
- (a) Y. Segawa and D. W. Stephan, Chem. Commun., 2012, 48, 11963 RSC; (b) T. Mahdi, Z. M. Heiden, S. Grimme and D. W. Stephan, J. Am. Chem. Soc., 2012, 134, 4088 CrossRef CAS PubMed; (c) S. J. Geier, P. A. Chase and D. W. Stephan, Chem. Commun., 2010, 46, 4884 RSC.
- (a) C. O. Kappe, A. Stadler and D. Dallinger, Microwaves in Organic and Medicinal Chemistry, Wiley-VCH, Weinheim, 2nd edn, 2012 Search PubMed; (b) C. O. Kappe, Chem. Soc. Rev., 2008, 37, 1127 RSC; (c) C. O. Kappe, B. U. Pieber and D. Dallinger, Angew. Chem., Int. Ed., 2013, 52, 1088 CrossRef CAS PubMed.
- (a) M. J. Gracia, J. M. Campelo, E. Losada, R. Luque, J. M. Marinas and A. A. Romero, Org. Biomol. Chem., 2009, 7, 4821 RSC; (b) T. Kato, S.-I. Matsuoka and M. Suzuki, Chem. Commun., 2015, 51, 13906 RSC; (c) Z. Lu and W. Zhang, Chin. J. Chem. Eng., 2014, 22, 227 CrossRef CAS; (d) N. R. Guha, D. Bhattacherjee and P. Das, Tetrahedron Lett., 2014, 55, 2912 CrossRef CAS; (e) R. Liu, Y. Wang, H. Cheng, Y. Yu, F. Zhao and M. Arai, J. Mol. Catal. A: Chem., 2013, 366, 315 CrossRef CAS; (f) T. Marimuthu and H. B. Friedrich, ChemCatChem, 2012, 4, 2090 CrossRef CAS; (g) C. Schmoeger, A. Stolle, W. Bonrath and B. Ondruschka, Curr. Org. Chem., 2011, 15, 151 CrossRef CAS; (h) C. Schmoeger, T. Gallert, A. Stolle, B. Ondruschka and W. Bonrath, Chem. Eng. Technol., 2011, 34, 445 CrossRef CAS; (i) T. Gallert, M. Hahn, M. Sellin, C. Schmoeger, A. Stolle, B. Ondruschka, T. F. Keller and K. D. Jandt, ChemSusChem, 2011, 4, 1654 CrossRef CAS PubMed; (j) G. Cravotto, L. Orio, E. C. Gaudino, K. Martina, D. Tavor and A. Wolfson, ChemSusChem, 2011, 4, 1130 CrossRef CAS PubMed; (k) Y. M. Ma, X. Zhou, X. Y. Wei and Z. M. Zong, Energy Sources, Part A, 2010, 32, 1201 CrossRef CAS; (l) A. M. R. Galletti, C. Antonetti, A. M. Venezia and G. Giambastiani, Appl. Catal., A, 2010, 386, 124 CrossRef CAS; (m) M. J. Gracia, J. M. Campelo, E. Losada, R. Luque, J. M. Marinas and A. A. Romero, Org. Biomol. Chem., 2009, 7, 4821 RSC; (n) L. Piras, E. Genesio, C. Ghiron and M. Taddei, Synlett, 2008, 1125 CAS; (o) G. S. Vanier, Synlett, 2007, 131 CrossRef CAS; (p) E. M. Gordon, D. C. Gaba, K. A. Jebber and D. M. Zacharias, Organometallics, 1993, 12, 5020 CrossRef CAS.
- (a) P. A. Chase, T. Jurca and D. W. Stephan, Chem. Commun., 2008, 1701 RSC; (b) P. Spies, S. Schwendemann, S. Lange, G. Kehr, R. Fröhlich and G. Erker, Angew. Chem., Int. Ed., 2008, 47, 7543 CrossRef CAS PubMed; (c) S. Tussing, L. Greb, S. Tamke, B. Schirmer, C. Muhle-Goll, B. Luy and J. Paradies, Chem. – Eur. J., 2015, 21, 8056 CrossRef CAS PubMed; (d) T. A. Rokob, A. Hamza, A. Stirling and I. Papai, J. Am. Chem. Soc., 2009, 131, 2029 CrossRef CAS PubMed.
- Z. M. Heiden and D. W. Stephan, Chem. Commun., 2011, 47, 5729 RSC.
- L. Greb, P. Ona-Burgos, A. Kubas, F. C. Falk, F. Breher, K. Fink and J. Paradies, Dalton Trans., 2012, 41, 9056 RSC.
- L. Greb, C. G. Daniliuc, K. Bergander and J. Paradies, Angew. Chem., Int. Ed., 2013, 52, 5876 CrossRef CAS PubMed.
- L. Greb, S. Tussing, B. Schirmer, P. Ona-Burgos, K. Kaupmees, M. Lokov, I. Leito, S. Grimme and J. Paradies, Chem. Sci., 2013, 4, 2788 RSC.
- It is known that external IR temperature control in microwave-assisted reactions is not fully reliable and internal temperature probes should be used. However, such an experimental setup is not available for pressure reactions in the microwave.
- H. D. Wang, R. Fröhlich, G. Kehr and G. Erker, Chem. Commun., 2008, 5966 RSC.
- J. A. Nicasio, S. Steinberg, B. Ines and M. Alcarazo, Chem. – Eur. J., 2013, 19, 11016 CrossRef CAS PubMed.
- L. Greb, P. Ona-Burgos, B. Schirmer, S. Grimme, D. W. Stephan and J. Paradies, Angew. Chem., 2012, 124, 10311 CrossRef.
| This journal is © The Royal Society of Chemistry 2016 |