
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pentamethylcyclopentadienyl-rhodium and iridium complexes containing (N^N and N^O) bound chloroquine analogue ligands: synthesis, characterization and antimalarial properties†
Erik
Ekengard‡
a,
Kamlesh
Kumar‡
a,
Thibault
Fogeron
a,
Carmen
de Kock
b,
Peter J.
Smith
b,
Matti
Haukka
c,
Magda
Monari
d and
Ebbe
Nordlander
*a
aInorganic Chemistry Research Group, Chemical Physics, Center for Chemistry and Chemical Engineering, Lund University, Box 124, SE-221 00 Lund, Sweden. E-mail: Ebbe.Nordlander@chemphys.lu.se
bDivision of Pharmacology, Department of Medicine, University of Cape Town Medical School, Observatory 7925, South Africa
cDepartment of Chemistry, University of Jyväskylä, P.O. Box-35, Jyväskylä, FI-40014, Finland
dDipartimento di Chimica “G. Ciamician”, Alma Mater Studiorum Università di Bologna, Via Selmi 2, 40126 Bologna, Italy
First published on 12th January 2016
Abstract
The synthesis and characterization of twenty new pentamethylcyclopentadienyl-rhodium and iridium complexes containing N^N and N^O-chelating chloroquine analogue ligands are described. The in vitro antimalarial activity of the new ligands as well as the complexes was evaluated against the chloroquine sensitive (CQS) NF54 and the chloroquine resistant (CQR) Dd2 strains of Plasmodium falciparum. The antimalarial activity was found to be good to moderate; although all complexes are less active than artesunate, some of the ligands and complexes showed better activity than chloroquine (CQ). In particular, rhodium complexes were found to be considerably more active than iridium complexes against the CQS NF54 strain. Salicylaldimine Schiff base ligands having electron-withdrawing groups (F, Cl, Br, I and NO2) in para position of the salicyl moiety and their rhodium complexes showed good antiplasmodial activity against both the CQS-NF54 and the CQR-Dd2 strains. The crystal structures of (η5-pentamethylcyclopentadienyl){N1-(7-chloroquinolin-4-yl)-N2-(pyridin-2-ylmethyl)ethane-1,2-diamine)} chlororhodium(III) chloride and (η5-pentamethylcyclopentadienyl){(4-chloro-2-(((2-((7-chloroquinolin-4-yl)amino)ethyl)imino)methyl)phenolate)}chlororhodium(III) chloride are reported. The crystallization of the amino-pyridyl complex (η5-pentamethylcyclopentadienyl){(N1-(7-chloroquinolin-4-yl)-N2-(pyridin-2-ylmethyl)ethane-1,2-diamine)}chloroiridium(III) chloride in acetone resulted in the formation of the imino-pyridyl derivative (η5-pentamethylcyclopentadienyl){(N1-(7-chloroquinolin-4-yl)-N2-(pyridin-2-ylmethylene)ethane-1,2-diamine)}chloroiridium(III) chloride, the crystal structure of which is also reported.
Introduction
Malaria is a parasitic disease and constitutes a serious societal problem in many countries in the tropical and sub-tropical regions of Africa, Asia and Latin America. There are around 200 million cases of malaria each year, and malaria leads to more than half a million deaths every year worldwide.1 The causative agents for malaria are five Plasmodium species, viz. P. falciparum, P. vivax, P. ovale, P. malariae and P. knowlesi. Out of these five species, P. falciparum is the most lethal and responsible for most of the deaths from malaria.2Quinoline-based drugs, in particular chloroquine (CQ, structure a, Fig. 1), have been widely used for the treatment of malaria,3 but resistance to chloroquine and other antimalarial agents has become a major obstacle in the efforts to control malaria.4 It has been postulated that 4-aminoquinoline-based antimalarial agents bind with haematin, which is formed by the degradation of hemoglobin in the food vacuole of the parasite and is toxic to the parasite, and prevent detoxification of haematin. This process inhibits the formation of haemozoin (β-haematin; malaria pigment) in the food vacuole of the parasite, thus causing a build-up of haematin that eventually leads to the death of parasites by haematin poisoning.5–12 Therefore, inhibition of haemozoin formation remains an excellent target for new antimalarial drug discovery.
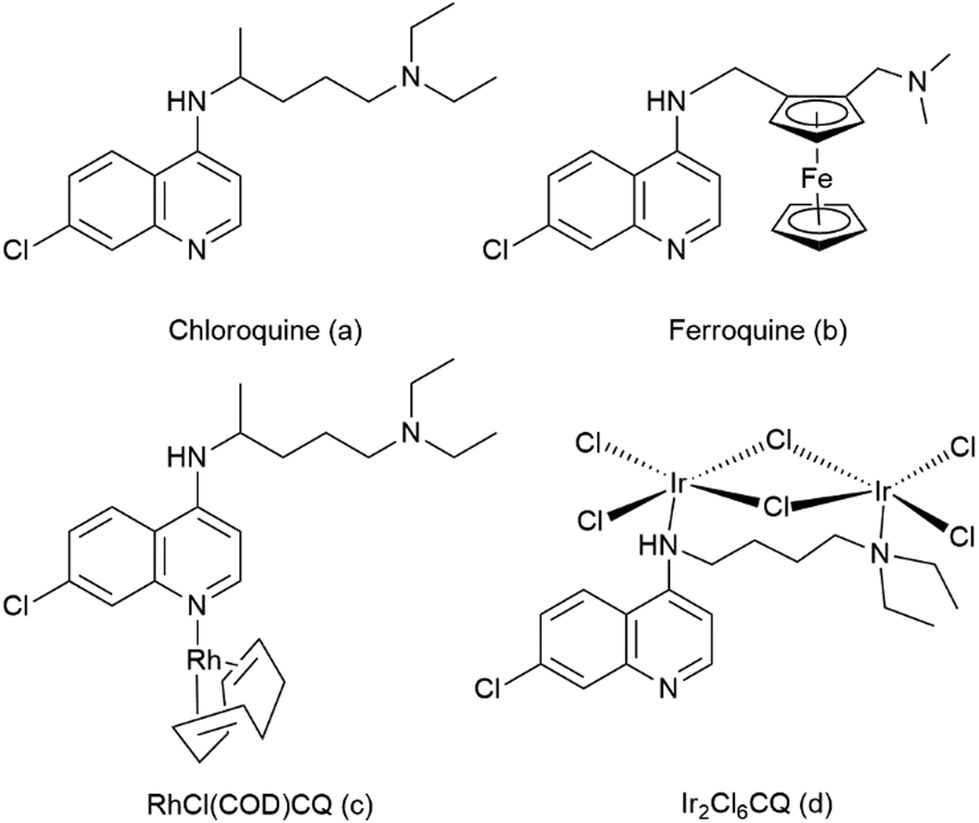 | ||
| Fig. 1 Structures of chloroquine, ferroquine, RhCl(COD)CQ and Ir2Cl6CQ. | ||
After the discovery of cisplatin (cis-[(NH3)2PtCl2]), an anticancer agent commonly used for the treatment of testicular and ovarian cancer,13,14 in the 1960s a wide variety of metal complexes have been synthesized and tested for a number of medical purposes. During the last three decades there has been increased interest in expanding the pharmaceutical potential of coordination compounds, especially organometallic complexes. This has been achieved by the incorporation of organometallic moieties into a large number of bioactive compounds. The focus of this research has mainly been the development of organometallic anticancer compounds, but other classes of bioactive organometallics, e.g. antibacterial and antimalarial, have also been investigated.15,16
A large number of 7-chloro-4-aminoquinoline derivatives have been evaluated in the search for chloroquine analogues that overcome the widespread chloroquine resistance developed by malaria parasites. In 1997, Biot et al. synthesized ferroquine (FQ, structure b, Fig. 1), a chloroquine analogue with a ferrocene moiety in the side chain that shows no cross resistance with chloroquine.17–19 Ferroquine has entered phase IIb clinical trials in association with artesunate.
Other attempts to create metal-containing chloroquine derivatives that overcome chloroquine resistance include ruthenocene compounds and half sandwich compounds of chromium and rhenium, as well as ruthenium, rhodium, iridium and gold coordination complexes.5 Sanchez-Delgado et al. have shown that RuII coordination complexes with CQ enhance the antimalarial activity against resistant parasite strains as compared to free chloroquine.20,21 Hence, there is sustained interest to synthesize new metal conjugates of chloroquine with enhanced antimalarial activity. We have previously investigated ruthenium- and osmium cymene complexes with N^O- and N^N-coordinating chloroquine analogue ligands.22,23 The initial results were promising and indicated that Ru cymene complexes of the N^O-coordinating ligand were more active than the free ligand, while further studies and expanding the ligand scope indicated lowered anti-malarial activity in vitro on coordinating the Ru cymene moiety to both the N^N- and N^O-coordinating ligands. Furthermore, we observed that the heavier osmium congeners of a subset of these complexes exhibited a further reduction in anti-plasmodial activity.22,23
The square planar Rh(I) complex [RhCl(COD)CQ] (structure c, Fig. 1) has been found to exhibit antimalarial activity similar to chloroquine diphosphate (CQDP)20 while the iridium chloroquine conjugate, [Ir2Cl6(CQ)] (structure d, Fig. 1) shows only moderate activity against P. bergei in vitro.24 The rhodium(III) and iridium(III) pentamethylcyclopentadienyl (Cp*) moieties are isoelectric to the ruthenium(II) and osmium(II) arene moieties and have similar coordination chemistry. The Cp* complexes of Rh and Ir are generally more stable than the corresponding cyclopentadienyl (Cp) complexes, a fact that is normally attributed to a combination of steric shielding of the metal center and the greater electron density in the Cp* ring compared to unsubstituted Cp. The large lipophilic Cp* also ought to increase the overall lipophilicity of the potential anti-malarial complex, something that is generally believed to be beneficial to overcome chloroquine resistance.25–27 Smith and coworkers have recently presented a number of Rh and Ir Cp* complexes with anti-malarial properties, based on both quinoline containing ligands and ligands not bearing a quinoline moiety.28–32 In general, these complexes were found to be less active than the corresponding ruthenium-arene complexes, but in several cases it is evident that the RhCp* moiety is preferable over the Ru-arene moiety for anti-plasmodial activity.
We have thus decided to investigate the anti-malarial properties of Rh- and Ir-Cp* complexes of the same family of chloroquine-mimicking ligands used in the studies with ruthenium and osmium. In this study, we have synthesized and characterized twenty new rhodium and iridium-Cp* complexes containing (N^N and N^O)-bound chloroquine analogue ligands. These complexes have been examined against the chloroquine sensitive (CQS) NF54 and the chloroquine resistant (CQR) Dd2 strains of P. falciparum.
Results and discussion
Synthesis and characterization
The ligands, L1, L2 and HL3–10 were synthesized according to a recently reported procedure.23 Reaction of L1 or L2 with the chloro-bridged dimeric rhodium complex [Cp*RhCl2]2 or the structurally analogous iridium complex [Cp*IrCl2]2 in 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio in dichloromethane at room temperature gave the complexes [RhCp*(L1)Cl]Cl (1a), [IrCp*(L1)Cl]Cl (1b), [RhCp*(L2)Cl]Cl (2a) and [IrCp*(L2)Cl]Cl (2b) in good yields. The rhodium complexes 3a–10a and their iridium analogues 3b–10b were synthesized by deprotonating the ligands HL3–10 with triethylamine followed by reaction with [Cp*MCl2]2 (M = Rh and Ir) (Scheme 1). All complexes were found to be air-stable yellow/orange-colored solids that exhibit good solubility in polar solvents.
:1 molar ratio in dichloromethane at room temperature gave the complexes [RhCp*(L1)Cl]Cl (1a), [IrCp*(L1)Cl]Cl (1b), [RhCp*(L2)Cl]Cl (2a) and [IrCp*(L2)Cl]Cl (2b) in good yields. The rhodium complexes 3a–10a and their iridium analogues 3b–10b were synthesized by deprotonating the ligands HL3–10 with triethylamine followed by reaction with [Cp*MCl2]2 (M = Rh and Ir) (Scheme 1). All complexes were found to be air-stable yellow/orange-colored solids that exhibit good solubility in polar solvents.
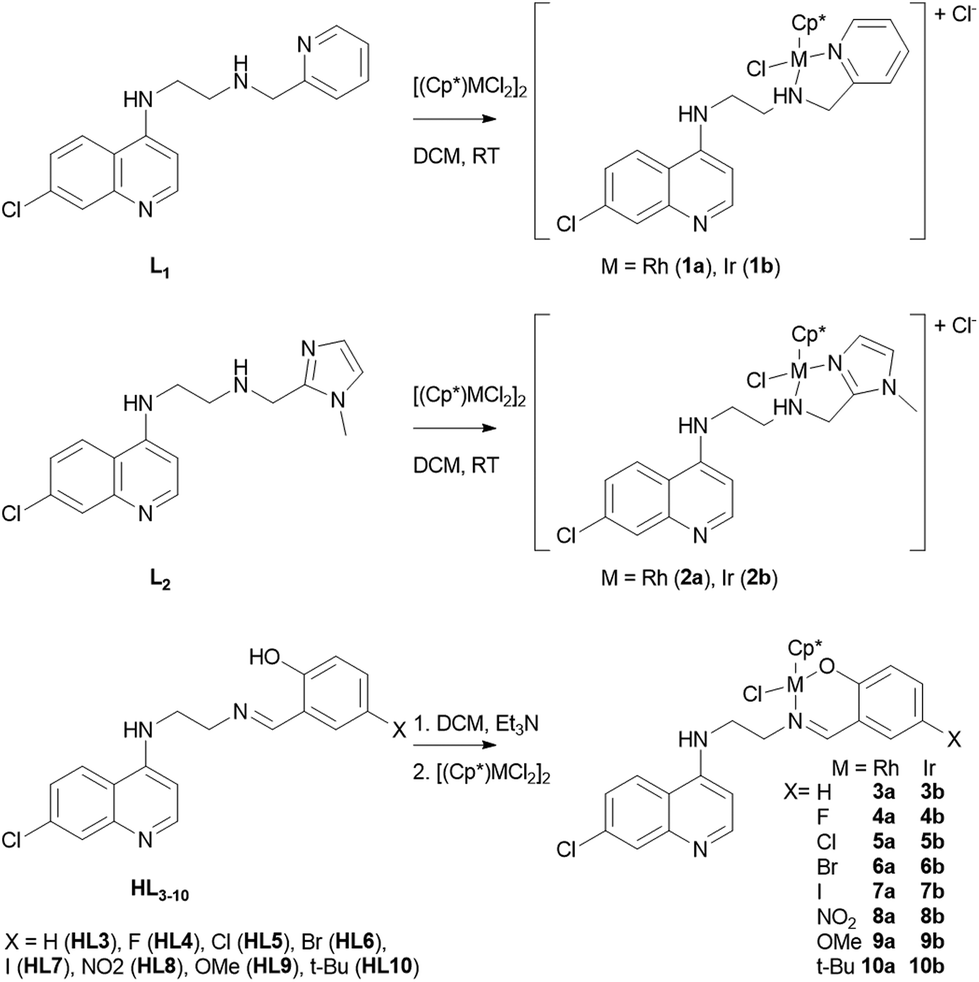 | ||
| Scheme 1 Synthesis of rhodium (1a–10a) and iridium (1b–10b) complexes. | ||
All complexes have been fully characterized by infrared, 1H- and 13C-NMR spectroscopy and mass spectrometry. Furthermore, the molecular structures of complexes 1a and 5a have been authenticated by single crystal X-ray diffraction analysis. Efforts to grow single crystals of complex 1b in acetone gave X-ray quality crystals, but not of 1b but rather the corresponding imino-iridium(III) complex 11 (Scheme 2), in which the ligand has been oxidized to form the imine analogue of L1 (vide infra). The 1H spectra of the complexes show the expected differences relative to the uncoordinated ligands; i.e. a downfield shift of ligand protons close to the coordinating sites and diastereotopic splitting of the methylene (CH2) protons. The diastereotopic splitting can be ascribed to the coordination of the ligand with the metal in a bidentate coordination mode, inducing chirality at the metal center.22
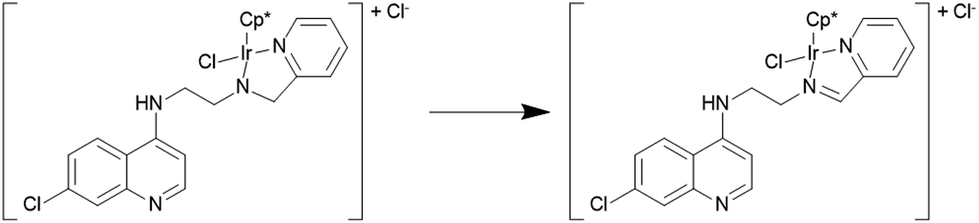 | ||
| Scheme 2 Spontaneous ligand dehydrogenation of 1b to 11. | ||
The most abundant peaks in the ESI mass spectra of complexes 1a and 1b occur at m/z 585 and 675, respectively, which are attributed to the [M]+ peak for each complex. While the iridium complex 2b showed the most abundant peak at m/z 642 ([M − HCl]+), the analogous rhodium complex 2a showed the most abundant peak at m/z 276 which is attributed to [M − HCl]2+. These results suggest cleavage of the M–Cl bond and loss of a chloride ligand for both 2a and 2b. The complexes 3a–10a and 3b–10b all showed [M + 1]+ molecular peaks, but all except 6a and 9a showed [M − Cl]+ as the most abundant peak, indicating that complexes based on salicylaldimine ligands, like the imidazole in L2, are prone to lose a chloride.
Molecular structures of complexes 1a, 5a and 11
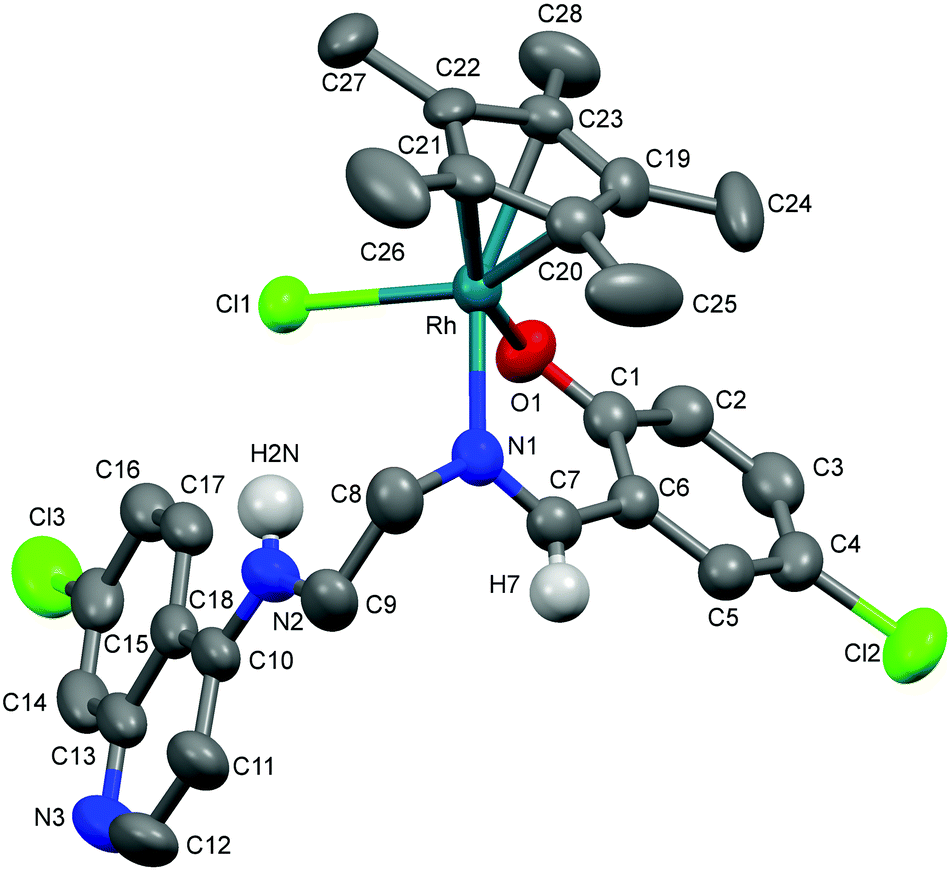
The molecular structures of complexes 1a, 5a and 11 have been determined by X-ray crystallography. Single crystals suitable for X-ray crystallographic analyses were obtained by layering hexane on a dichloromethane solution of 1a kept at low temperature (−20 °C) and by slow evaporation of a dichloromethane/hexane solution of 5a at room temperature. When an acetone solution of complex 1b was allowed to slowly evaporate at low temperature (−20 °C), single crystals suitable for X-ray crystallography were obtained. However, as already mentioned, it was found that the crystal structure showed the structure of an imino-iridium(III) complex, 11 (Scheme 2), as a result of oxidation of the ligand in the corresponding amino-iridium(III) complex 1b. Relevant crystallographic data and structure refinement parameters are compiled in Table 1. Selected bond lengths and bond angles are given in Table 2 and molecular structures with numbering schemes are shown in Fig. 2–4, for 1a, 5a, and 11, respectively. The three complexes adopt the expected piano stool geometry, with all bond lengths and angles around the metal centers similar to reported structures of RhCp* and IrCp* complexes with amino-pyridyl, salicylaldimine-based and imino-pyridyl ligands.33–36 The distances of the Rh atom from the Cp* centroid are 1.779 and 1.816 Å in 1a and 5a, respectively. For 1a, the Rh–Cl, Rh–Namine and Rh–Npy bond distances are 2.4203(6), 2.1729(19) and 2.1089(19) Å, respectively, and for 5a the Rh–Cl, Rh–N and Rh–O bond distances are 2.4212(8), 2.098(3) and 2.080(2) Å, respectively.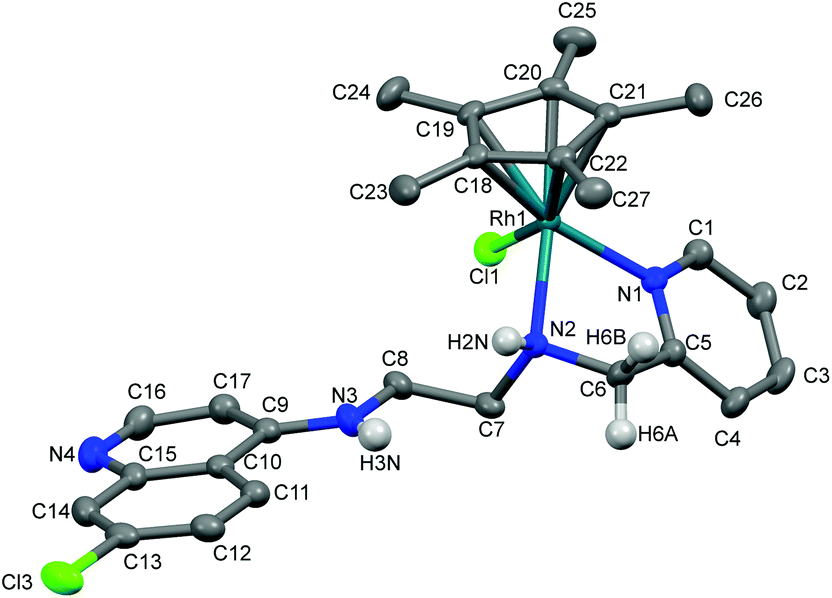 | ||
| Fig. 2 Crystal structure of 1a with atom numbering schemes (thermal ellipsoids drawn at 50% probability level). Solvent of crystallization, counter anion (Cl−) and most of the hydrogen atoms have been omitted for clarity (hydrogen atoms attached to C6, N2 and N3 are shown). | ||
 | ||
| Fig. 3 Crystal structure of complex 5a with atom numbering schemes (thermal ellipsoids drawn at 50% probability level). Solvent of crystallization, hydrogen atoms and disorder at the pentamethylcyclopentadienyl ring have been omitted for clarity (hydrogen atoms attached to N2 and C7 are shown). | ||
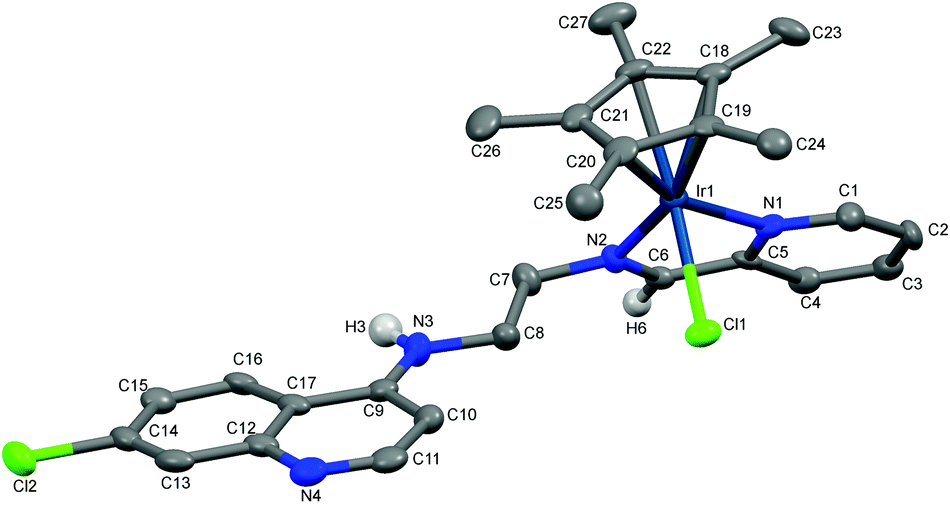 | ||
| Fig. 4 Crystal structure of 11 with atom numbering schemes (thermal ellipsoids drawn at 50% probability level). Solvent of crystallization, counter anion (Cl−) and hydrogen atoms have been omitted for clarity (hydrogen atoms attached to C6 and N3 are shown). | ||
| Complex | 1a | 5a | 11 |
|---|---|---|---|
| a R 1 = ∑||Fo| − |Fc||/∑|Fo|. b wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2 where w = 1/[σ2(Fo2) + (aP)2 + bP] where P = (Fo2 + Fc2)/3. | |||
| Empirical formula | C28.50H35Cl6N4Rh | C28H29Cl3N3ORh·CH2Cl2 | C27H36Cl3IrN4O3 |
| Formula weight | 749.21 | 717.73 | 763.15 |
| Temperature (K) | 170(2) | 293 | 120(2) |
| Wavelength (Å) | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | Triclinic | Monoclinic | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
P |
| Unit cell dimensions | |||
| a (Å) | 11.6470(6) | 12.3489(2) | 11.5226(10) |
| b (Å) | 12.1871(5) | 13.4149(2) | 11.8000(9) |
| c (Å) | 13.4363(5) | 18.5804(3) | 12.6405(8) |
| α (°) | 76.108(3) | 90.00 | 92.389(6) |
| β (°) | 89.564(4) | 99.424(1) | 98.090(6) |
| γ (°) | 61.569(5) | 90.00 | 118.661(8) |
| Volume (Å3) | 1615.33(14) | 3036.47(8) | 1481.3(2) |
| Z | 2 | 4 | 2 |
| Density (calculated, Mg m−3) | 1.540 | 1.570 | 1.711 |
| Absorption coefficient (mm−1) | 1.051 | 1.031 | 4.813 |
| F(000) | 762 | 1456 | 756 |
| Crystal size (mm3) | 0.432 × 0.167 × 0.115 | 0.30 × 0.25 × 0.10 | 0.932 × 0.047 × 0.023 |
| Theta range for data collection | 3.148 to 30.747° | 1.672 to 28.454° | 2.850 to 26.499° |
| Index ranges | −16 ≤ h ≤ 16 | −16 ≤ h ≤ 16 | −13 ≤ h ≤ 14 |
| −17 ≤ k ≤ 17 | −17 ≤ k ≤ 17 | −14 ≤ k ≤ 14 | |
| −19 ≤ l ≤ 19 | −24 ≤ l ≤ 24 | −15 ≤ l ≤ 13 | |
| Reflections collected | 18611 |
55008 |
10684 |
| Independent reflections | 10031 [R(int) = 0.0229] |
7557 [R(int) = 0.0515] | 6081 [R(int) = 0.0458] |
| Completeness to theta = 25.242° | 99.8% | 100.0% | 99.5% |
| Absorption correction | Analytical | Multi-scan | Gaussian |
| Max. and min. transmission | 0.913 and 0.752 | 0.725 and 0.920 | 0.898 and 0.762 |
| Refinement method | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 10031/14/366 |
7557/5/430 | 6081/0/348 |
| Goodness-of-fit on F2 | 1.036 | 0.974 | 0.966 |
| Final R indices [I > 2sigma(I)] | R 1 = 0.0409, wR2b = 0.0974 | R 1 = 0.0368, wR2b = 0.0824 | R 1 = 0.0395, wR2b = 0.0603 |
| R indices (all data) | R 1 = 0.0468, wR2 = 0.1030 | R 1 = 0.0634, wR2 = 0.0971 | R 1 = 0.0607, wR2 = 0.0649 |
| Largest diff. peak and hole (e Å−3) | 1.461 and −1.571 | 0.514 and −0.551 | 1.190 and −0.885 |
| Complex 1a | Complex 5a | Complex 11 |
|---|---|---|
| Rh1–N1 = 2.1089(19) | Rh1–O1 = 2.080(2) | Ir1–N1 = 2.093(4) |
| Rh1–N2 = 2.1729(19) | Rh1–N1 = 2.098(3) | Ir1–N2 = 2.104(4) |
| Rh1–Cl1 = 2.4203(6) | Rh1–Cl1 = 2.4212 (8) | Ir1–Cl1 = 2.4051(13) |
| Rh1–C18 = 2.160(2) | Rh1–C19 = 2.178(8) | Ir1–C18 = 2.187(5) |
| Rh1–C19 = 2.175(2) | Rh1–C20 = 2.128(7) | Ir1–C19 = 2.167(5) |
| Rh1–C20 = 2.148(2) | Rh1–C21 = 2.143(8) | Ir1–C20 = 2.188(4) |
| Rh1–C21 = 2.162(2) | Rh1–C22 = 2.250(7) | Ir1–C21 = 2.181(5) |
| Rh1–C22 = 2.143(2) | Rh1–C23 = 2.202(9) | Ir1–C22 = 2.158(5) |
| Rh1–C5(Cp*) (avg) = 2.157 | Rh1–C5(Cp*) (avg) = 2.180 | Ir1–C5(Cp*) (avg) = 2.176 |
| Rh1–centroid (Cp*) = 1.779 | Rh1–centroid (Cp*) = 1.816 | Ir1–centroid (Cp*) = 1.802 |
| N1–C1 = 1.352(3) | O1–C1 = 1.303(4) | N1–C1 = 1.323(6) |
| N1–C5 = 1.346(3) | N1–C5 = 1.366(6) | |
| C5–C6 = 1.501(3) | C6–C7 = 1.455(4) | C5–C6 = 1.431(7) |
| C6–N2 = 1.494(3) | C7–N1 = 1.280(4) | C6–N2 = 1.296(6) |
| N2–C7 = 1.489(3) | N1–C8 = 1.486(4) | N2–C7 = 1.447(6) |
| N1–Rh1–N2 = 77.52(7) | O1–Rh–N1 = 83.20(9) | N1–Ir1–N2 = 76.11(15) |
| N1–Rh1–Cl1 = 85.28(5) | Cl1–Rh–N1 = 90.06 (7) | N1–Ir1–Cl1 = 85.16(12) |
| N2–Rh1–Cl1 = 95.90(5) | Cl1–Rh–O1 = 88.84 (6) | N2–Ir1–Cl1 = 88.74(12) |
| Rh1–N1–C5 = 115.80(15) | Rh1–O1–C1 = 118.6(2) | Ir–N1–C5 = 115.7(3) |
| N1–C5–C6 = 114.2(2) | O1–C1–C6 = 124.2(3) | N1–C5–C6 = 114.1(4) |
| C5–C6–N2 = 109.10 (18) | C1–C6–C7 = 120.8(3) | C5–C6–N2 = 117.4(4) |
| C6–C7–N1 = 125.2(3) | ||
| Rh1–N2–C6 = 104.88(13) | Rh–N1–C7 = 121.8(2) | Ir1–N2–C6 = 116.4(3) |
| Rh1–N2–C7 = 117.74(15) | Rh–N1–C8 = 120.5(2) | Ir1–N2–C7 = 125.2(3) |
| C6–N2–C7 = 110.27(18) | C7–N1–C8 = 117.4(3) | C6–N2–C7 = 118.5(4) |
As discussed above, coordination of the bidentate quinoline derivatives renders the complexes chiral at the metal (in addition to the stereogenic centers at N2 and N3 for 1a, at N2 for 5a and at N3 for 11), but the complexes crystallize as racemic mixtures of enantiomers, and the NMR spectra of the complexes indicate that no diastereomers are present in solution.
The structure of 11 closely resembles that of 1a, exhibiting a distorted piano-stool geometry around the iridium center; an η5-Cp* group occupies three facial positions of an ideal metal-octahedral environment, and the chelating pyridine-imine ligand and a terminal chloride complete the octahedral coordination sphere. The crystal structure reveals that the Ir1–N1–C5–C6–N2 metallacycle is essentially planar, and coplanar with the plane of the pyridine ring, while in the structure of 1a the corresponding metallacycle is in a classic envelope conformation with N2 in the endo position. The Ir–Cl, Ir–Nimine, Ir–Npy, Ir–centroid(Cp*), and Ir–C5(Cp*) bond distances are similar to the distances in complex 1a and these bond distances are comparable to the values reported for (η5-Cp*)Ir(III)-complexes in the literature.36 However, the major structural differences observed between complexes 1a and 11 are the bond angles and bond distances around the C6 and N2 atoms. The C6–N2 bond length in complex 11 is 1.296(6) Å, which is considerably smaller than the corresponding distance in complex 1a, 1.494(3) Å, indicative of an imine rather than an amine functionality. Also, the bond angles around both C6 and N2 in complex 11 are ∼120°. The short N–C distance and the angles around C6 and N2 unambiguously indicate the sp2 nature of the C6 carbon in complex 11 and a double bond between C6 and N2. However, 1b can still be assumed to be the amino complex indicated in Scheme 1 as the combined spectroscopic data agree very well with this formulation. There is very good agreement between the 1H- and 13C-NMR spectra of 1a and 1b, suggesting an un-oxidized state of L1 in 1b. Additionally, there is no trace of an imine C–H signal in the 1H-NMR spectrum, or of an imine N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretch in the IR-spectrum of 1b. Thus the oxidation of complex 1b to 11 must be assumed to have occurred during the crystallization. Metal-assisted dehydrogenation of coordinated amines to form imines is a well-known phenomenon, especially for Ru(II) and Os(II) complexes.37 However, this is a relatively rare phenomenon for iridium complexes, and only a few examples of amino iridium pentamethylcyclopentadienyl complexes undergoing ligand dehydrogenation to form the corresponding imine complex are known. Jerphagnon et al.38 observed that the ligand in the metallacycle formed from [IrCp*Cl2]2 and N-methylbenzylamine slowly oxidizes to the imine N-benzylidenemethylamine during catalytic experiments or on standing in CDCl3. Similarly, Barloy et al.39 obtained significant amounts of the oxidized pyrroline complex from the reaction between (2R,5R)-2,5-diphenylpyrrolidine and [IrCp*Cl2]2 even when the reaction was performed under anaerobic conditions, and only the oxidized product was obtained when the reaction was run under air.
C stretch in the IR-spectrum of 1b. Thus the oxidation of complex 1b to 11 must be assumed to have occurred during the crystallization. Metal-assisted dehydrogenation of coordinated amines to form imines is a well-known phenomenon, especially for Ru(II) and Os(II) complexes.37 However, this is a relatively rare phenomenon for iridium complexes, and only a few examples of amino iridium pentamethylcyclopentadienyl complexes undergoing ligand dehydrogenation to form the corresponding imine complex are known. Jerphagnon et al.38 observed that the ligand in the metallacycle formed from [IrCp*Cl2]2 and N-methylbenzylamine slowly oxidizes to the imine N-benzylidenemethylamine during catalytic experiments or on standing in CDCl3. Similarly, Barloy et al.39 obtained significant amounts of the oxidized pyrroline complex from the reaction between (2R,5R)-2,5-diphenylpyrrolidine and [IrCp*Cl2]2 even when the reaction was performed under anaerobic conditions, and only the oxidized product was obtained when the reaction was run under air.
Assessment of anti-malarial activity in vitro
The anti-malarial activity of all rhodium (1a–10a) and iridium (1b–10b) complexes has been evaluated against the chloroquine-sensitive (CQS) NF54 and the chloroquine-resistant (CQR) Dd2 strains of Plasmodium falciparum. Chloroquine and artesunate have been used as reference drugs in this study and the antiplasmodial activity was determined in vitro using the parasite lactate dehydrogenase assay. The results are given in Fig. 5 and 6, and Table 3. The anti-malarial properties of all ligands and p-cymene-ruthenium complexes analogous to the complexes presented in this work, and p-cymene-osmium complexes of ligands L1, L3, L4, L5 and L7, have been reported previously.22,23 All ligands display good to moderate activity against both the CQS-NF54 and CQR-Dd2 strains. In particular, the Schiff base ligands HL3–HL10 exhibit higher antimalarial activity than the amine ligands L1 and L2 and in some cases, the Schiff base ligands even showed better activity than chloroquine. However, the coordination of a ruthenium arene moiety was found to be detrimental for anti-malarial activity, and the osmium arene complexes exhibited a further decrease in activity.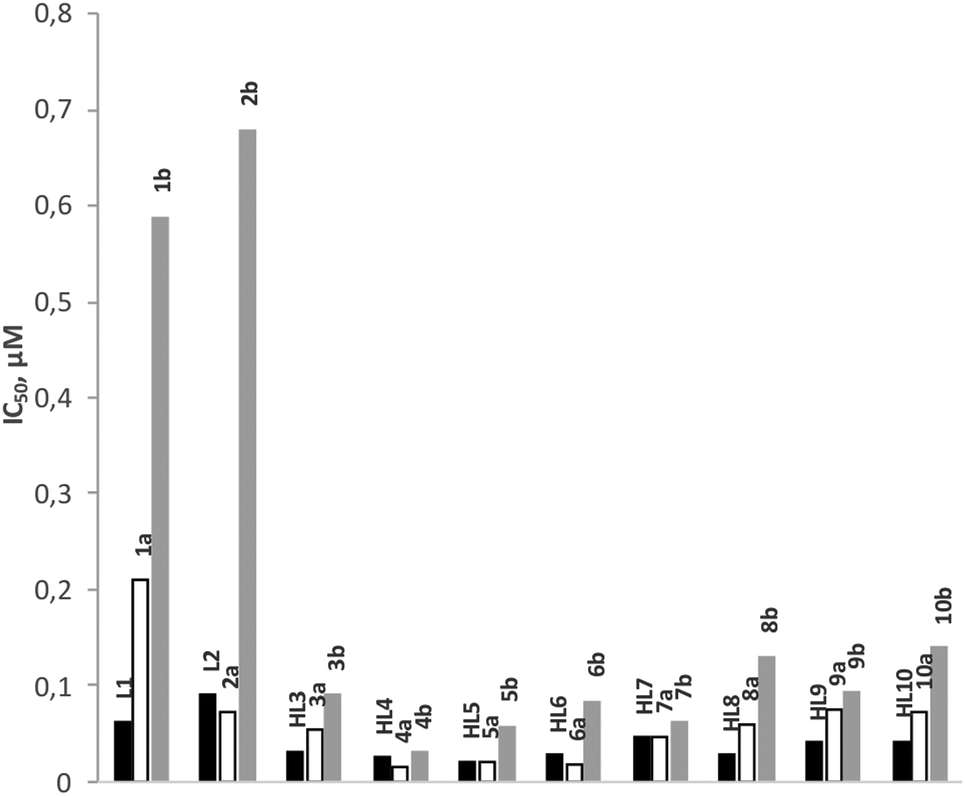 | ||
| Fig. 5 IC50 values of ligands (L1–HL10, black bars), rhodium (1a–10a, hollow bars) and iridium complexes (1b–10b, grey bars) tested in vitro for antiplasmodial activity against the chloroquine sensitive NF54 strain of P. falciparum. | ||
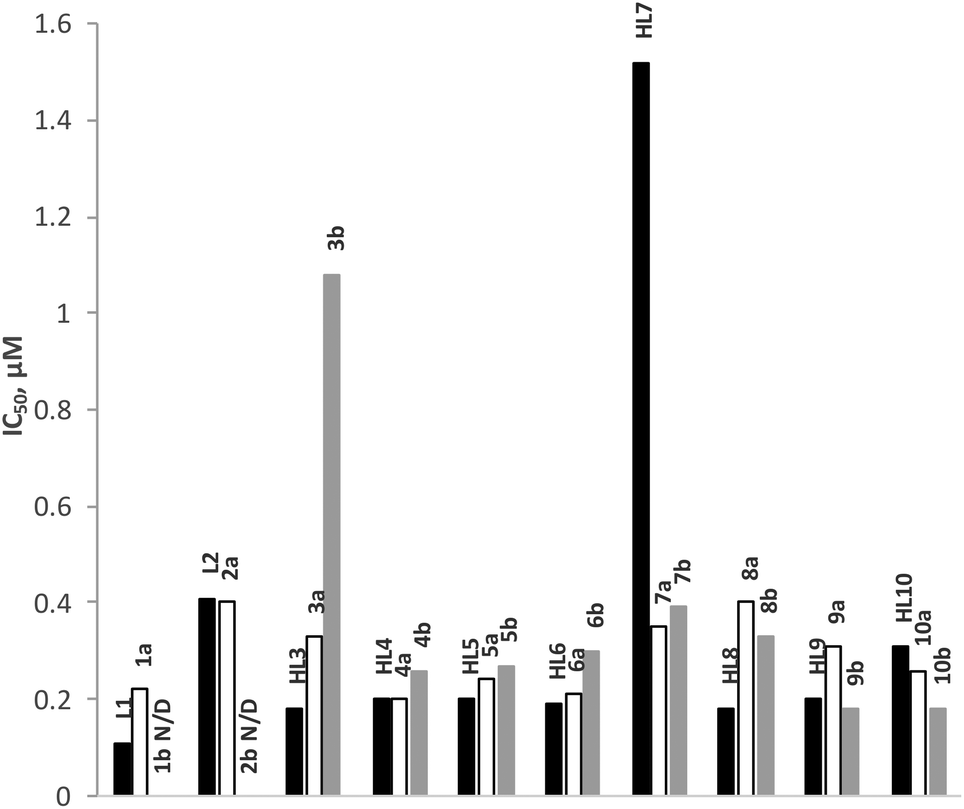 | ||
| Fig. 6 IC50 values of ligands (L1–HL10, black bars), rhodium (1a–10a, hollow bars) and iridium complexes (1b–10b, grey bars) tested in vitro for antiplasmodial activity against the chloroquine resistant Dd2 strains of P. falciparum. N/D = not determined. | ||
| Compound | NF54: IC50 (μM) | Dd2: IC50 (μM) | RI |
|---|---|---|---|
| ND (not determined), RI (resistance index) = IC50(CQR)/IC50(CQS). | |||
| L1 | 0.063 ± 0.002 | 0.11 ± 0.04 | 1.7 |
| L2 | 0.091 ± 0.016 | 0.41 ± 0.01 | 4.5 |
| HL3 | 0.032 ± 0.004 | 0.18 ± 0.02 | 5.5 |
| HL4 | 0.025 ± 0.007 | 0.20 ± 0.02 | 8 |
| HL5 | 0.021 ± 0.003 | 0.20 ± 0.04 | 9.5 |
| HL6 | 0.027 ± 0.009 | 0.19 ± 0.03 | 7.2 |
| HL7 | 0.046 ± 0.006 | 1.52 ± 0.27 | 33 |
| HL8 | 0.028 ± 0.008 | 0.18 ± 0.01 | 6.5 |
| HL9 | 0.040 ± 0.002 | 0.20 ± 0.03 | 5.2 |
| HL10 | 0.041 ± 0.002 | 0.31 ± 0.03 | 7.8 |
| 1a | 0.209 ± 0.038 | 0.22 ± 0.13 | 1.1 |
| 2a | 0.072 ± 0.021 | 0.40 ± 0.035 | 5.7 |
| 3a | 0.054 ± 0.002 | 0.33 ± 0.08 | 6.1 |
| 4a | 0.016 ± 0.002 | 0.20 ± 0.004 | 12 |
| 5a | 0.020 ± 0.006 | 0.24 ± 0.01 | 12 |
| 6a | 0.018 ± 0.006 | 0.21 ± 0.03 | 12 |
| 7a | 0.047 ± 0.011 | 0.35 ± 0.08 | 7.5 |
| 8a | 0.060 ± 0.014 | 0.40 ± 0.02 | 6.6 |
| 9a | 0.076 ± 0.012 | 0.31 ± 0.04 | 4.1 |
| 10a | 0.073 ± 0.009 | 0.26 ± 0.07 | 3.5 |
| 1b | 0.587 ± 0.143 | >1.4 ± ND | ND |
| 2b | 0.677 ± 0.029 | >1.4 ± ND | ND |
| 3b | 0.091 ± 0.006 | 1.08 ± 0.35 | 12 |
| 4b | 0.030 ± 0.007 | 0.26 ± 0.02 | 8.8 |
| 5b | 0.058 ± 0.017 | 0.27 ± 0.05 | 4.7 |
| 6b | 0.084 ± 0.013 | 0.30 ± 0.03 | 3.6 |
| 7b | 0.061 ± 0.016 | 0.39 ± 0.06 | 6.3 |
| 8b | 0.13 ± 0.03 | 0.33 ± 0.12 | 2.5 |
| 9b | 0.093 ± 0.002 | 0.18 ± 0.05 | 1.9 |
| 10b | 0.14 ± 0.04 | 0.18 ± 0.02 | 1.3 |
| CQ | 0.027 ± 0.011 | 0.22 ± 0.05 | 8.1 |
| Artesunate | <0.005 | 0.011 ± 0.001 | — |
In this study, the rhodium complexes 1a to 10a exhibited lower IC50 values against the CQS NF54 strain relative to their iridium congeners. The same holds for the activity against the CQR Dd2 strain, with the exception of complexes 8a, 9a and 10a, which showed slightly lower activity than their iridium analogues. The N^N-coordinating cationic complexes 1b and 2b were found to be inactive against the CQR Dd2 strain. In almost all cases there was no significant difference in activity between the free ligands and the rhodium complexes, while the activities of the iridium complexes were statistically different from both the free ligands and the rhodium complexes.
Activity against both the CQS-NF54 and CQR-Dd2 strains
It is interesting to note that electronic variation in the Schiff base (N^O) ligand systems also has a significant effect on the antiplasmodial activity of the ligands as well as the rhodium and iridium complexes. The ligands having electron-withdrawing groups (F, Cl, Br, I and NO2) in para position to the phenolic OH and their rhodium complexes showed good antiplasmodial activity. Indeed, some of these rhodium complexes (4a, 5a and 6a) are more active than chloroquine against the CQS NF54 strain. If the IC50 values for ligands HL3 to HL10 and complexes 3a to 10a and 3b to 10b are plotted as a function of Hammett's σp parameter (Fig. 7), a clear trend that the compounds bearing electron-withdrawing groups are more active against the CQS strain can be seen, but the same trend is not evident for the CQR strain. The reason for the observed correlation in the case of the CQS strain remains unclear.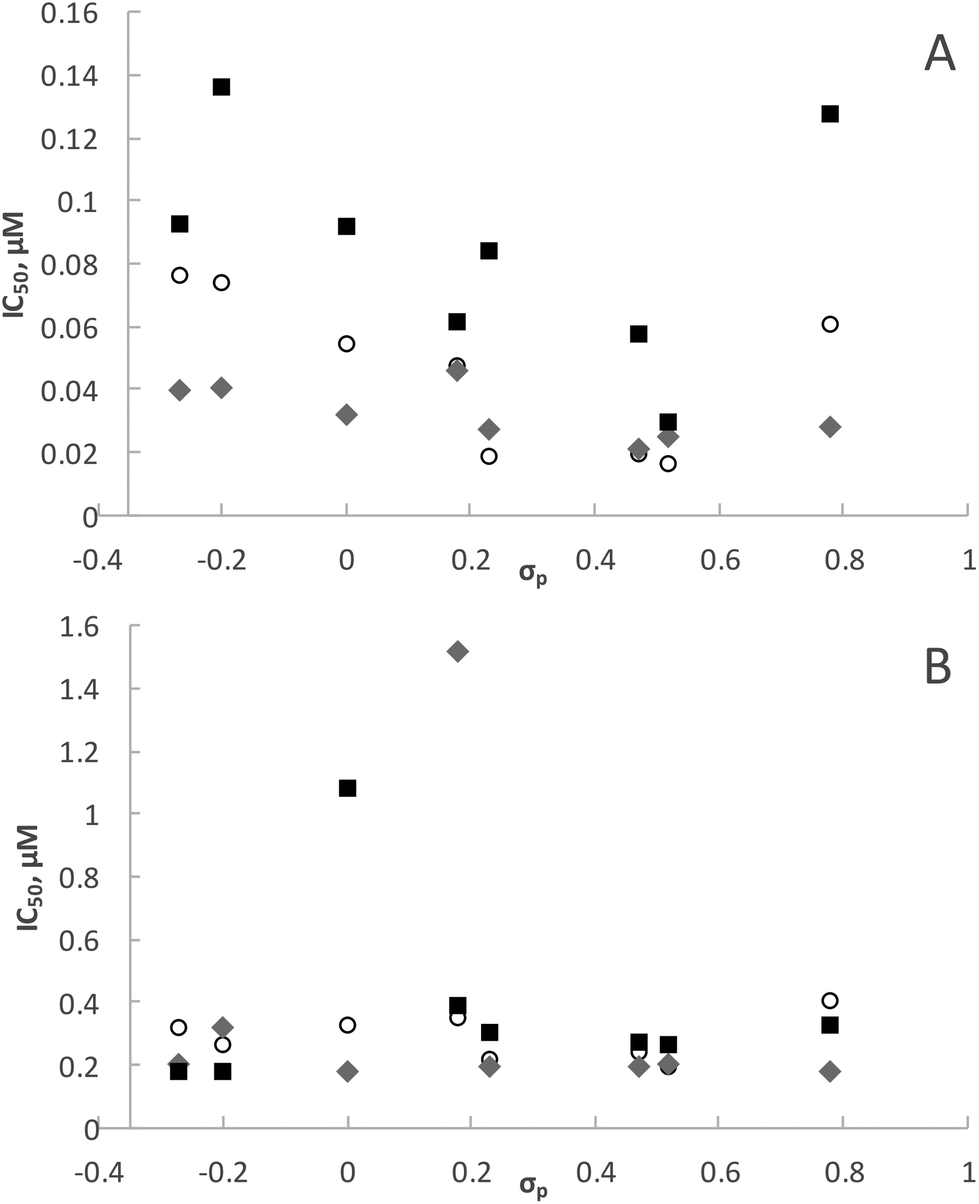 | ||
| Fig. 7 A plot of the IC50 values vs. Hammett's σp parameter for the substituent para to the phenolic group in HL3 to HL10. (A) CQS NF54 strain, (B) CQR Dd2 strain. Ligands HL3 to HL10: grey diamonds. Rhodium complexes 3a to 10a: hollow circles. Iridium complexes 3b to 10b: black squares. | ||
Experimental
All synthetic procedures were performed under dry nitrogen using standard Schlenk and vacuum-line techniques. Solvents used were dried by distillation over appropriate drying reagents and stored over molecular sieves under nitrogen. All chemicals were purchased from Sigma-Aldrich and used as received. N1-(7-chloroquinolin-4-yl)ethane-1,2-diamine was prepared according to a literature method.40 Ligands L1, L2 and HL3–10 were synthesized according to published procedures.22,23 NMR spectra were recorded on a Varian Inova 500 MHz spectrometer using the solvent resonance as internal standard for 1H NMR and 13C NMR shifts. Infrared spectra were recorded on a Nicolet Avatar 360 FT-IR spectrometer. Electrospray ionization (ESI) mass spectra were recorded using a Waters Micromass Q-Tof micro mass spectrometer or an LC-MS Agilent 6220-TOF spectrometer coupled with a 1200 series HPLC. Elemental analysis was performed by Mikroanalytische Laboratorium Kolbe, Mülheim an der Ruhr.Synthesis of complexes
:1 v/v) to afford compound 3a as an orange solid. Yield: 22 mg (∼61%). 1H NMR (500 MHz, CDCl3, δ ppm): 8.23 (s, 1H), 8.01 (s, 1H), 7.91 (s, 1H), 7.75 (s, 1H), 7.29 (d, 1H, J = 9.0 Hz), 7.15 (td, 1H, J = 1.89, J = 7.3 Hz), 6.86 (d, 1H, J = 8.3 Hz), 6.78 (d, 1H, J = 6.8 Hz), 6.51 (d, 1H, J = 4.1 Hz), 6.37 (t, 1H, J = 7.1 Hz), 4.40 (m, 1H), 4.29 (m, 1H), 4.20 (m, 1H), 3.81 (m, 1H), 1.58 (s, 15H, Cp*CH3). 13C NMR (125 MHz, CDCl3, δ ppm): 166.17, 166.12, 151.62, 148.61, 145.82, 136.28, 135.03, 134.45, 126.00, 125.36, 124.06, 123.59, 120.94, 116.83, 114.84, 98.35, 93.31 (d, Jc-Rh = 8.2 Hz, Cp*), 61.60, 42.88, 8.81. IR (KBr) vmax/cm−1: 3434 (br), 2920w, 1620sh (CN), 1615s (7-chloroquinoline), 1580s (7-chloroquinoline), 1537w (7-chloroquinoline), 1447m, 1330m, 1141w, 1023m, 810m 759w. MS (ES+, m/z): 562 ([M − Cl]+) 100%, 598 ([M + H]+) 68%.
N), 1623s (7-chloroquinoline), 1578s (7-chloroquinoline), 1541w (7-chloroquinoline), 1460s, 1376w, 1307w, 1261w, 1243w, 1210w, 1139s, 1081s, 804w. MS (ES+, m/z): 580 ([M − Cl]+) 100%, 616 ([M + H]+) 34%.
N), 1619s (7-chloroquinoline), 1578s (7-chloroquinoline), 1542w (7-chloroquinoline), 1509w, 1459s, 1375w, 1261w, 1024m, 800w. MS (ES+, m/z): 678 ([M + H]+) 100%, 642 ([M − Cl]+) 91%.
N), 1618s (7-chloroquinoline), 1588s (7-chloroquinoline), 1541w (7-chloroquinoline), 1509w, 1458m, 1375w, 1318w, 1023m, 806w. MS (ES+, m/z): 688 ([M − Cl]+) 100%, 724 ([M + H]+) 24%.
N), 1627s (7-chloroquinoline), 1598s (7-chloroquinoline), 1542w (7-chloroquinoline), 1473w, 1456m, 1138w, 1101w, 1025w, 1019w. MS (ES+, m/z): 607 ([M − Cl]+) 100%, 643 ([M + H]+) 40%.
N), 1610s (7-chloroquinoline), 1578s (7-chloroquinoline), 1512w (7-chloroquinoline), 1459s, 1374w, 1290w, 1261s, 1213w, 1136w, 1093w, 1022m, 818w. MS (ES+, m/z): 592 ([M − Cl]+) 96%, 628 ([M + H]+) 100%.
N), 1618s (7-chloroquinoline), 1580s (7-chloroquinoline), 1528w (7-chloroquinoline), 1458w, 1362w, 1323w, 1075m, 835w. MS (ES+, m/z): 618 ([M − Cl]+) 100%, 654 ([M + H]+) 57%.
N), 1618s (7-chloroquinoline), 1578s (7-chloroquinoline), 1560w (7-chloroquinoline), 1541w, 1508w, 1449m, 1325w, 1143w, 1029w, 809w, 758w. MS (ES+, m/z): 652 ([M − Cl]+) 100%, 688 ([M + H]+) 15%.
N), 1614s (7-chloroquinoline), 1581s (7-chloroquinoline), 1538w (7-chloroquinoline), 1464s, 1384w, 1313w, 1140w, 1028m, 813w. MS (ES+, m/z): 670 ([M − Cl]+) 100%, 706 ([M + H]+) 7%.
N), 1615s (7-chloroquinoline), 1580s (7-chloroquinoline), 1558w (7-chloroquinoline), 1520w 1456s, 1383w, 1314m, 1173w, 1138w, 1079w, 1028w, 876w, 821w. MS (ES+, m/z): 686 ([M − Cl]+) 100%, 722 ([M + H]+) 18%.
N), 1615s (7-chloroquinoline), 1580s (7-chloroquinoline), 1557w (7-chloroquinoline), 1520w, 1456s, 1384w, 1315w, 1171w, 1137w, 1029m, 875w, 820w. MS (ES+, m/z): 732 ([M − Cl]+) 100%, 768 ([M + H]+) 12%.
N), 1612s (7-chloroquinoline), 1578s (7-chloroquinoline), 1541w (7-chloroquinoline), 1522w, 1458s, 1382m, 1315m, 1170w, 1030m, 875w, 820w. MS (ES+, m/z): 778 ([M − Cl]+) 100%, 814 ([M + H]+) 15%.
N), 1623s (7-chloroquinoline), 1603s, 1578s (7-chloroquinoline), 1547w (7-chloroquinoline), 1473m, 1458w, 1382w, 1363w, 1313s, 1130w, 1100m, 1029m, 947w, 904w, 875w, 831w, 804w. MS (ES+, m/z): 697 ([M − Cl]+) 100%, 733 ([M + H]+) 16%.
N), 1611s (7-chloroquinoline), 1580s (7-chloroquinoline), 1539m (7-chloroquinoline), 1469s, 1377w, 1309w, 1263w, 1219w, 1155w, 1140w, 1031m, 875w, 818w, 775w. MS (ES+, m/z): 682 ([M − Cl]+) 100%, 718 ([M + H]+) 25%.
N), 1618s (7-chloroquinoline), 1578s (7-chloroquinoline), 1533w (7-chloroquinoline), 1474m, 1458w, 1380w, 1363w, 1320w, 1256w, 1178w, 1141w, 1030m, 829w. MS (ES+, m/z): 708 ([M − Cl]+) 100%, 744 ([M + H]+) 12%.
X-ray structure determinations
Determination of the antiplasmodial activity
The test samples were tested in triplicate on one occasion against chloroquine-sensitive (CQS) NF54 and chloroquine-resistant (CQR) Dd2 strains of Plasmodium falciparum. Continuous in vitro cultures of asexual erythrocyte stages of P. falciparum were maintained using a modified method of Trager and Jensen.46 Quantitative assessment of antiplasmodial activity in vitro was determined via the parasite lactate dehydrogenase assay using a modified method described by Makler.47 The test samples were prepared to a 20 mg ml−1 stock solution in 100% DMSO and sonicated to enhance solubility. Samples were tested as a suspension if not completely dissolved. Stock solutions were stored at −20 °C. Further dilutions were prepared on the day of the experiment. Chloroquine (CQ) and artesunate were used as the reference standards in all experiments. A full dose–response was performed for all compounds to determine the concentration inhibiting 50% of parasite growth (IC50-value). Test samples were tested at a starting concentration of 10 μg ml−1, which was then serially diluted 2-fold in complete medium to give 10 concentrations, with the lowest concentration being 0.02 μg ml−1. The same dilution technique was used for all samples. Samples were also tested at a starting concentration of 1000 ng ml−1. Reference standards were tested at a starting concentration of 1000 ng ml−1. The highest concentration of solvent to which the parasites were exposed had no measurable effect on the parasite viability (data not shown). The IC50-values were obtained using a non-linear dose–response curve fitting analysis via Graph Pad Prism v.4.0 software.Conclusions
New pentamethylcyclopentadienyl-rhodium and -iridium complexes with chloroquine analogue ligands have been synthesized and fully characterized. Molecular structures of two complexes, [Rh(L1)Cp*Cl]Cl(1a), and [Rh(L5)Cp*Cl] (5a) have also been authenticated by X-ray crystallography. The iridium complex [Ir(L1)Cp*Cl]Cl (1b) underwent a ligand dehydrogenation reaction to yield the imine complex 11 during crystallization from acetone, and the structure of [Ir(L1ox)Cp*Cl]Cl 11 was elucidated by X-ray crystallography.All complexes have been evaluated for antimalarial activity against the CQS-NF54 and CQR-Dd2 strains of the P. falciparum malaria parasite. The rhodium complexes showed good antimalarial activity against both strains. The rhodium complexes [Rh(L4)Cp*Cl] (4a), [Rh(L5)Cp*Cl] (5a) and [Rh(L6)Cp*Cl] (6a) showed higher antimalarial activity than chloroquine against the CQS-NF54 strain. To the best of our knowledge, 4a is the rhodium complex with the highest anti-plasmodial activity reported thus far that is not a ferroquine derivative. However, the iridium complexes showed only moderate activity against the CQS strain. The iridium complexes 1b and 2b are inactive against the CQR-Dd2 strain. A correlation between the nature (Hammett's σp parameter) of the electron-withdrawing groups on the salicylaldimine ligands and the anti-plasmodial activity against the CQSNF54 strain could be detected, but no such trend could be seen with the activities against the CQR-Dd2 strain.
Acknowledgements
EE thanks FLÄK, the Research School in Pharmaceutical Sciences at Lund University, for a PhD studentship. KK thanks the European Commission (Erasmus Mundus Europe Asia, EMEA) for a postdoctoral fellowship. Professor Steve Colbran, University of New South Wales, Australia, is thanked for inspiring the start of this project and for a gift of [RhCp*Cl2]2 and [IrCp*Cl2]2.References
- World Malarial Report 2014, World Health Organization, 2014.
- L. H. Bannister, J. M. Hopkins, R. E. Fowler, S. Krishna and G. H. Mitchell, Parasitol. Today, 2000, 16, 427–433 CrossRef PubMed.
- M. Schlitzer, Arch. Pharm., 2008, 341, 149–163 CrossRef CAS PubMed.
- P. I. Trigg and A. V. Kondrachine, in Malaria: Parasite Biology, Pathogenesis and Protection, ASM Press, Washington DC, 1998 Search PubMed.
- C. Biot, W. Castro, C. Y. Botte and M. Navarro, Dalton Trans., 2012, 41, 6335–6349 RSC.
- L. M. Ursos and P. D. Roepe, Med. Res. Rev., 2002, 22, 465–491 CrossRef CAS PubMed.
- K. A. de Villiers and T. J. Egan, Molecules, 2009, 14, 2868–2887 CrossRef CAS PubMed.
- T. J. Egan and H. M. Marques, Coord. Chem. Rev., 1999, 190–192, 493–517 CrossRef CAS.
- A. Leed, K. DuBay, L. M. B. Ursos, D. Sears, A. de Dios and C. P. D. Roepe, Biochemistry, 2002, 31, 10245–10255 CrossRef.
- P. J. Rosenthal, Antimalarial chemotherapy : mechanisms of action, resistance, and new directions in drug discovery, Humana Press, New Jersey, 2001 Search PubMed.
- C. R. Chong and D. J. Sullivan Jr., Biochem. Pharmacol., 2003, 66, 2201–2212 CrossRef CAS PubMed.
- T. J. Egan, R. Hunter, C. H. Kaschula, H. M. Marques, A. Misplon and J. Walden, J. Med. Chem., 2000, 43, 283–291 CrossRef CAS PubMed.
- B. Rosenberg, L. Van Camp and T. Krigas, Nature, 1965, 205, 698–699 CrossRef PubMed.
- B. Rosenberg, L. Van Camp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385–386 CrossRef CAS PubMed.
- C. Biot and D. Dive, in Medicinal Organometallic Chemistry, ed. G. Jaouen and N. Metzler-Nolte, Springer, Berlin Heidelberg, 2010, ch. 7, vol. 32, pp. 155–193 Search PubMed.
- C. G. Hartinger and P. J. Dyson, Chem. Soc. Rev., 2009, 38, 391–401 RSC.
- C. Biot, G. Glorian, L. A. Maciejewski and J. S. Brocard, J. Med. Chem., 1997, 40, 3715–3718 CrossRef CAS PubMed.
- M. Henry, S. Briolant, A. Fontaine, J. Mosnier, E. Baret, R. Amalvict, T. Fusai, L. Fraisse, C. Rogier and B. Pradines, Antimicrob. Agents Chemother., 2008, 52, 2755–2759 CrossRef CAS PubMed.
- C. Biot, F. Nosten, L. Fraisse, D. Ter-Minassian, J. Khalife and D. Dive, Parasite, 2011, 18, 207–214 CrossRef CAS PubMed.
- R. A. Sánchez-Delgado, M. Navarro, H. Pérez and J. A. Urbina, J. Med. Chem., 1996, 39, 1095–1099 CrossRef PubMed.
- C. S. Rajapakse, A. Martinez, B. Naoulou, A. A. Jarzecki, L. Suarez, C. Deregnaucourt, V. Sinou, J. Schrevel, E. Musi, G. Ambrosini, G. K. Schwartz and R. A. Sanchez-Delgado, Inorg. Chem., 2009, 48, 1122–1131 CrossRef CAS PubMed.
- L. Glans, A. Ehnbom, C. de Kock, A. Martinez, J. Estrada, P. J. Smith, M. Haukka, R. A. Sanchez-Delgado and E. Nordlander, Dalton Trans., 2012, 41, 2764–2773 RSC.
- E. Ekengard, L. Glans, I. Cassells, T. Fogeron, P. Govender, T. Stringer, P. Chellan, G. C. Lisensky, W. H. Hersh, I. Doverbratt, S. Lidin, C. de Kock, P. J. Smith, G. S. Smith and E. Nordlander, Dalton Trans., 2015, 44, 19314–19329 RSC.
- M. Navarro, S. Pekerar and H. A. Pérez, Polyhedron, 2007, 26, 2420–2424 CrossRef CAS.
- D. A. van Schalkwyk and T. J. Egan, Drug Resist. Updates, 2006, 9, 211–226 CrossRef CAS PubMed.
- D. C. Warhurst, Malar. J., 2003, 2, 31 CrossRef PubMed.
- D. C. Warhurst, J. C. Craig, I. S. Adagu, D. J. Meyer and S. Y. Lee, Malar. J., 2003, 2, 26 CrossRef PubMed.
- M. Adams, C. de Kock, P. J. Smith, K. M. Land, N. Liu, M. Hopper, A. Hsiao, A. R. Burgoyne, T. Stringer, M. Meyer, L. Wiesner, K. Chibale and G. S. Smith, Dalton Trans., 2015, 44, 2456–2468 RSC.
- P. Chellan, K. M. Land, A. Shokar, A. Au, S. H. An, D. Taylor, P. J. Smith, T. Riedel, P. J. Dyson, K. Chibale and G. S. Smith, Dalton Trans., 2014, 43, 513–526 RSC.
- W. Nkoana, D. Nyoni, P. Chellan, T. Stringer, D. Taylor, P. J. Smith, A. T. Hutton and G. S. Smith, J. Organomet. Chem., 2014, 752, 67–75 CrossRef CAS.
- Y. Li, C. de Kock, P. J. Smith, K. Chibale and G. S. Smith, Organometallics, 2014, 33, 4345–4348 CrossRef CAS.
- Y. Li, C. de Kock, P. J. Smith, H. Guzgay, D. T. Hendricks, K. Naran, V. Mizrahi, D. F. Warner, K. Chibale and G. S. Smith, Organometallics, 2013, 32, 141–150 CrossRef CAS.
- D. Carmona, C. Vega, F. J. Lahoz, S. Elipe, L. A. Oro, M. P. Lamata, F. Viguri, R. García-Correas, C. Cativiela and M. P. López-Ram de Víu, Organometallics, 1999, 18, 3364–3371 CrossRef CAS.
- R. Payne, P. Govender, B. Therrien, C. M. Clavel, P. J. Dyson and G. S. Smith, J. Organomet. Chem., 2013, 729, 20–27 CrossRef CAS.
- H. Brunner, A. Köllnberger, T. Burgemeister and M. Zabel, Polyhedron, 2000, 19, 1519–1526 CrossRef CAS.
- D. Carmona, F. J. Lahoz, S. Elipe, L. A. Oro, M. P. Lamata, F. Viguri, C. Mir, C. Cativiela and M. P. L. R. de Viu, Organometallics, 1998, 17, 2986–2995 CrossRef CAS.
- F. Richard Keene, Coord. Chem. Rev., 1999, 187, 121–149 CrossRef.
- T. Jerphagnon, A. J. A. Gayet, F. Berthiol, V. Ritleng, N. Mršić, A. Meetsma, M. Pfeffer, A. J. Minnaard, B. L. Feringa and J. G. de Vries, Chem. – Eur. J., 2009, 15, 12780–12790 CrossRef CAS PubMed.
- L. Barloy, J.-T. Issenhuth, M. G. Weaver, N. Pannetier, C. Sirlin and M. Pfeffer, Organometallics, 2011, 30, 1168–1174 CrossRef CAS.
- K. Yearick, K. Ekoue-Kovi, D. P. Iwaniuk, J. K. Natarajan, J. Alumasa, A. de Dios, C. P. D. Roepe and C. Wolf, J. Med. Chem., 2008, 51, 1995–1998 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, SADABS, program for empirical absorption correction, University of Göttingen, Göttingen, Germany, 1996 Search PubMed.
- SMART & SAINT Software Reference Manuals, version 5.051 (Windows NT Version), Bruker Analytical X-ray Instruments Inc., Madison, WI, 1998 Search PubMed.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS.
- G. M. Sheldrick, SHELXTLplus (Windows NT Version) Structure Determination Package, Bruker Analytical X-ray Instruments Inc., 5.1 edn., 1998 Search PubMed.
- W. Trager and J. B. Jensen, Science, 1976, 193, 673–675 CAS.
- M. T. Makler and D. J. Hinrichs, Am. J. Trop. Med. Hyg., 1993, 48, 205–210 CAS.
Footnotes |
| † CCDC 1426906–1426908. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt03739e |
| ‡ These authors contributed equally to the paper. |
| This journal is © The Royal Society of Chemistry 2016 |