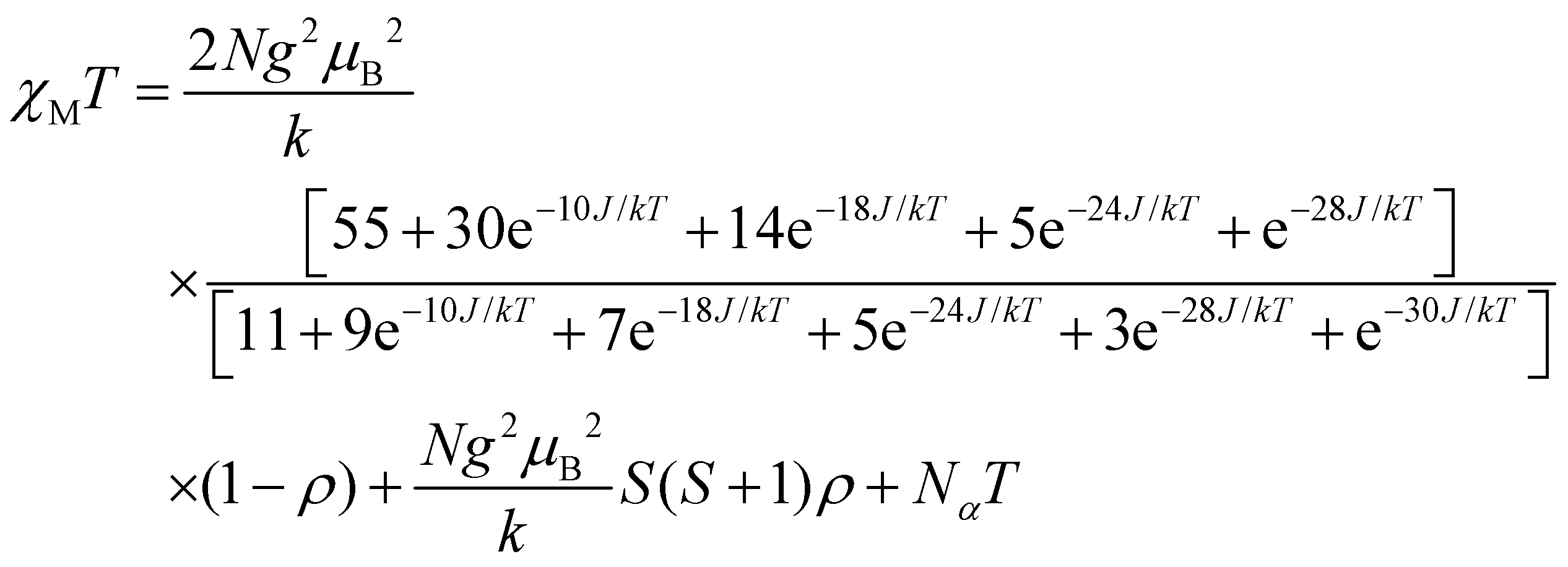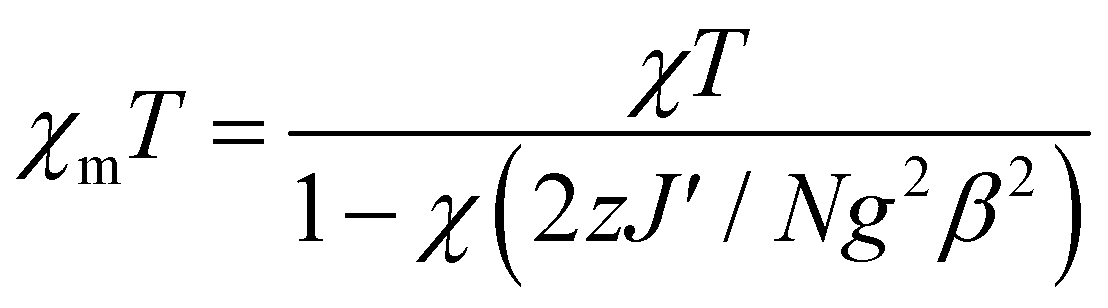DOI:
10.1039/C5DT03659C
(Paper)
Dalton Trans., 2016,
45, 742-752
Mn(II) complexes of different nuclearity: synthesis, characterization and catecholase-like activity†
Received
18th September 2015
, Accepted 12th November 2015
First published on 19th November 2015
Abstract
Two “end-off” compartmental ligands, 2-formyl-4-chloro-6-N-ethylmorpholine-iminomethyl-phenol (HL1) and 2-formyl-4-methyl-6-N-ethylpyrrolidine-iminomethyl-phenol (HL2) have been designed and three complexes of Mn(II), one mono-, one di- and a polynuclear, namely Mn(L1)(SCN)2(H2O)] (1), [Mn2(L1)(OAc)2](BPh4)] (2), and [Mn2(L2)(OAc)2(dca)]n (3) have been synthesized and structurally characterized. Variable temperature magnetic studies of 2 and 3 have been performed and data analyses reveal that Mn centers are antiferromagnetic coupled with J = −9.15 cm−1 and J = −46.89, respectively. Catecholase activity of all the complexes has been investigated using 3,5-di-tert-butyl catechol (3,5-DTBC). All are highly active and the activity order on the basis of the kcat value is 2 > 1 > 3. In order to unveil whether the metal centered redox participation or the radical pathway is responsible for the catecholase-like activity of the complexes, detailed EPR and cyclic voltammetric (CV) studies have been performed. In addition to the six-line EPR spectrum characteristic to Mn(II), an additional peak at g ∼ 2 is observed when the EPR study is done with the mixture of 3,5-DTBC and the catalyst, suggesting the formation of an organic radical, most likely ligand centered. The CV experiment with the mixture of 3,5-DTBC and the catalyst reveals ligand centered reduction rather than reduction of Mn(II) to Mn(I). It is thus inferred that complexes 1–3 show catecholase-like activity due to radical generation.
Introduction
The oxidation of organic substrates by metalloenzymes, that activate molecular oxygen under mild conditions, has received a vast deal of concentration in the arena of industrial and synthetic processes.1–4 As a result, many groups have tried to build up the finest model of transition metal complexes able to mimic the oxidase activity of metalloenzymes. One of the foremost enzymes that plays a key role in these oxidation reactions is catechol oxidase (CO), a less well known member of the type-III copper proteins that originate in plant tissues and crustaceans. This process, known as catecholase activity, allows catalysis of the oxidation of a wide range of o-diphenols (catechols) to the corresponding o-quinones (Scheme 1).5–8 Recently, our group has got admirable results while mimicking catecholase activity with phenol based compartmental ligand complexes of Ni(II),9 Cu(II)10,11 and Zn(II).12 In this manuscript we widen our work by using manganese, one of the most important trace transition elements in biological systems, which is present in numerous metalloenzymes of varied nuclearity. From a literature survey, it is possible to recognize that some complexes of Mn(III),13 Mn(IV),14 and a very few ones of Mn(II)15 are found to mimic catecholase-like activity, and these derivatives are generally made up from salen-type Schiff bases, tripodal and pyridine-containing ligands, etc., while phenol based compartmental ligands have been very rarely used. Our earlier studies with Mn(III) disclose metal-centered redox participation as the most probable pathway for the catecholase activity of the model compounds.13b In the case of Mn(II) complexes,15 although we found excellent catalytic activities, we failed to evaluate the plausible catalytic mechanism. That failure inspires us to synthesize new Mn(II) complexes to explore that untouched arena of catecholase activity study and thereby to unveil the most probable mechanistic pathway. In order to reach our goal, the ligands of our choice in the present study are two phenol based compartmental ligands, namely 2-formyl-4-chloro-6-N-ethylmorpholine-iminomethyl-phenol (HL1) and 2-formyl-4-methyl-6-N-ethylpyrrolidine-iminomethyl-phenol (HL2) (Fig 1). We have prepared three Mn(II) complexes having different nuclearities, i.e. [Mn(L1)(SCN)2(H2O)] (1), [Mn2(L1)(OAc)2](BPh4)] (2), and [Mn2(L2)(OAc)2(dca)]n (3), structurally characterized them and have studied their catecholase-like activity in DMSO solvent with 3,5-di-tert-butylcatechol (3,5-DTBC) as the substrate. EPR and cyclic voltammetric studies help us to understand the origin of the catecholase-like activity of our Mn(II) complexes. Variable temperature magnetic study on complexes 2 and 3 has also been performed since the compounds of phenol based compartmental ligands are well known for their stimulating magnetic properties.16–19 This examination of magnetic properties of such compounds helps not only in further understanding the relations between magnetic coupling centers in metalloproteins and enzymes, but also to extend the knowledge of molecular magnetism.
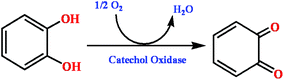 |
| | Scheme 1 Reaction pathway of the oxidation catalyzed by CO. | |
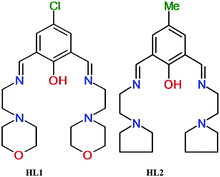 |
| | Fig. 1 Structure of the two synthesized compartmental ligands. | |
Results and discussion
Synthesis, rationalization, and characterization of the metal-complexes
Three manganese(II) complexes (1–3) were prepared by adopting the template synthesis technique by treating a methanolic solution of corresponding manganese salts with the Schiff base formed in situ between 2,6-diformyl-4-R-phenol (where R = chloro, methyl) and the diamines, and structurally characterized using single crystal X-ray diffraction (vide infra). FT-IR spectral study reveals that all complexes exhibit bands due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretch in the range of 1630–1648 cm−1 and skeletal vibration in the range of 1545–1555 cm−1. The presence of a coordinated SCN− anion in complex 1 has been assigned by a broad band in the region of 2065 cm−1. In complex 2, the OAc− anions show bands at 1483 cm−1 and in complex 3 OAc− anions and dicyanamide show bands at 1488 and 2165 cm−1, respectively. Electronic spectra of all complexes studied in DMSO medium display very similar absorption bands in the range of 375–425 nm. The observed strong higher energy single band (between 375 and 425 nm) is due to the combination of both phenoxido–Mn(II) and hydroxido–Mn(II) LMCT bands. Room temperature (300 K) magnetic moment calculations (5.96μB for complex 1, 8.37μB and 8.19μB for complex 2 and 3, respectively) reveal that Mn(II) in all complexes is in a high spin state.
N stretch in the range of 1630–1648 cm−1 and skeletal vibration in the range of 1545–1555 cm−1. The presence of a coordinated SCN− anion in complex 1 has been assigned by a broad band in the region of 2065 cm−1. In complex 2, the OAc− anions show bands at 1483 cm−1 and in complex 3 OAc− anions and dicyanamide show bands at 1488 and 2165 cm−1, respectively. Electronic spectra of all complexes studied in DMSO medium display very similar absorption bands in the range of 375–425 nm. The observed strong higher energy single band (between 375 and 425 nm) is due to the combination of both phenoxido–Mn(II) and hydroxido–Mn(II) LMCT bands. Room temperature (300 K) magnetic moment calculations (5.96μB for complex 1, 8.37μB and 8.19μB for complex 2 and 3, respectively) reveal that Mn(II) in all complexes is in a high spin state.
Crystal structure descriptions
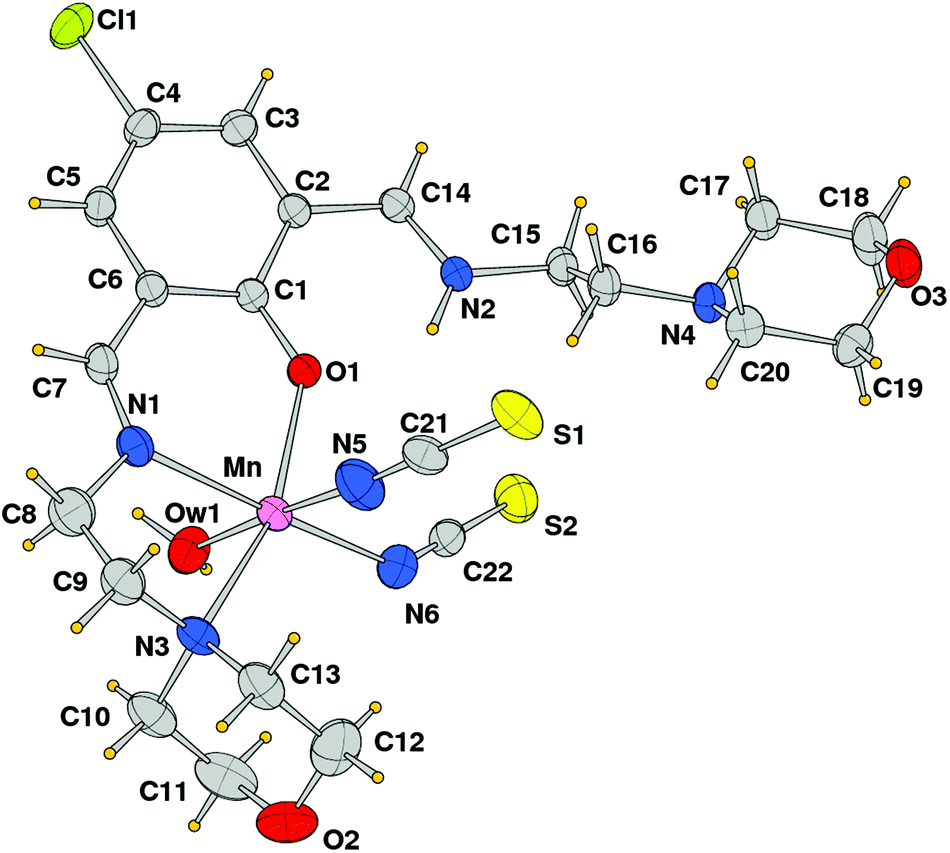
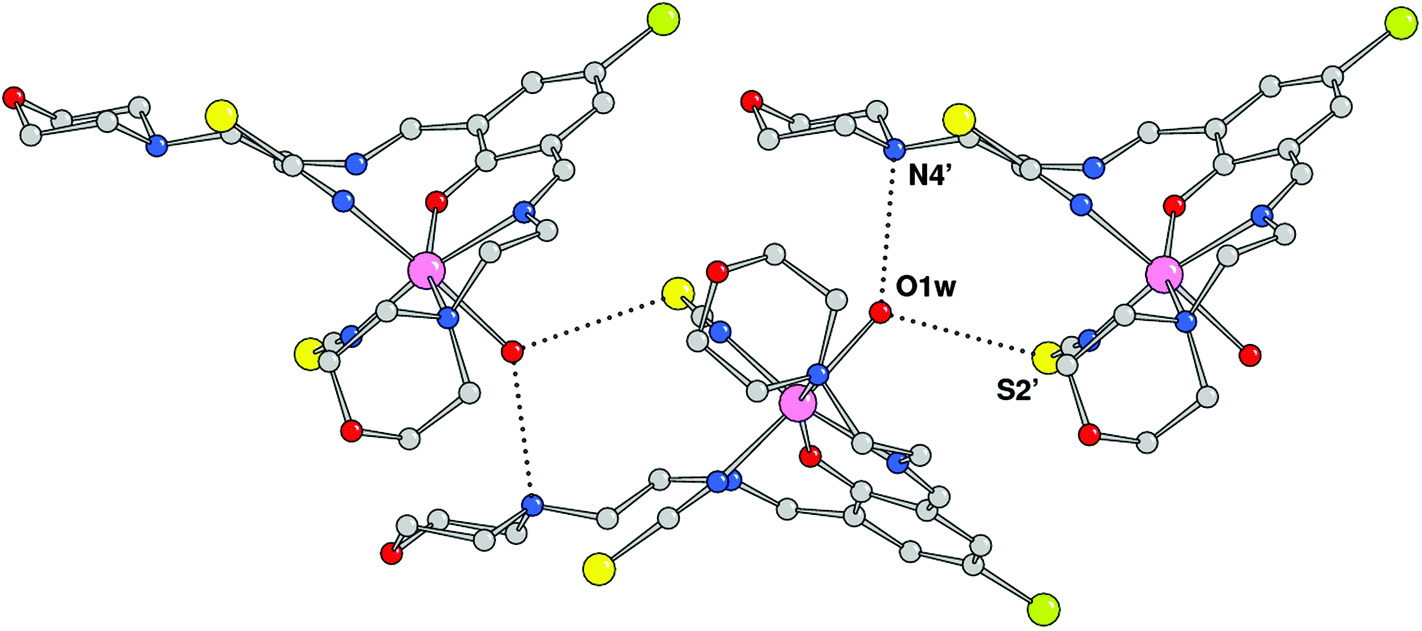
The crystal structure analysis showed the formation of a mononuclear neutral species, [Mn(HL1)(SCN)2(H2O)], due to the protonation of one of the imine nitrogen atoms. An ORTEP drawing is depicted in Fig. 2, and a selection of coordination bond distances and angles is reported in Table 1. The metal is chelated by ligand HL1 through the phenoxo oxygen atom, the imine–nitrogen and the amine donor of the morpholine ring with Mn–O and Mn–N bond distances of 2.1222(14), 2.2075(18), and 2.387(13) Å, respectively. The latter value refers to the coordinated morpholine moiety at a higher occupancy (see Experimental section). The distorted octahedral coordination sphere is completed by an aqua ligand and two isothiocyanate anions (Mn–O1w = 2.3692(18), Mn–NCS = 2.163(2) and 2.154(2) Å) (Fig. 3). The distortions in the coordination bond angles are attributable to the strength of the HL1 chelation and the value of O1–Mn–N3 (of 156.5(2)°) deviates more from ideal geometry (Table 1). The protonated imino nitrogen N1 forms an intramolecular H-bond with the phenolato oxygen O1 (N⋯O distance 2.598(2) Å), and the water molecule is engaged in weak H-bonding with N4 and S2 of a symmetry related complex at x, 1/2 − y, −1/2 + z (O1w⋯N4 = 3.130(3) and O1w⋯S2 = 3.330(2) Å) to form a polymeric species developed parallel to axis a. The structural data of complex 1 are comparable to those detected in the corresponding species having a pyridine replacing the morpholine ring, reported by us a few years ago.15
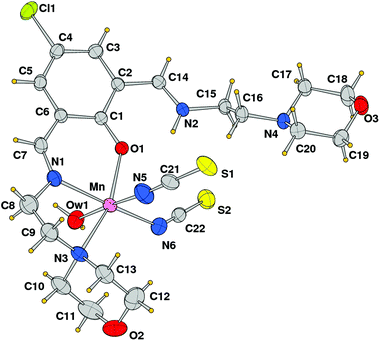 |
| | Fig. 2 ORTEP drawing (ellipsoid probability at 30%) with atom labels of complex 1 (of the disordered coordinated morpholine, only that at the higher occupancy is shown). | |
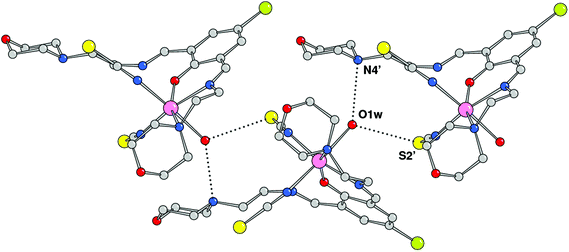 |
| | Fig. 3 Complexes 1 connected through H-bonds by the coordinated water molecules (primed atoms at x, 1/2 − y, −1/2 + z). | |
Table 1 Coordination bond lengths (Å) and angles (°) for complex 1
|
N3 and N3b represent the donor of the disordered coordinated morpholine. Only bond angles involving the higher occupancy are reported.
|
| Mn–O(1) |
2.1222(14) |
Mn–N(3)a |
2.387(13) |
| Mn–O(1w) |
2.3692(18) |
Mn–N(3b)a |
2.413(13) |
| Mn–N(1) |
2.2075(18) |
Mn–N(5) |
2.163(2) |
|
|
|
Mn–N(6) |
2.154(2) |
| |
| O(1)–Mn–N(6) |
89.73(7) |
N(5)–Mn–O(1w) |
176.52(8) |
| O(1)–Mn–N(5) |
89.35(8) |
N(1)–Mn–O(1w) |
82.69(7) |
| N(6)–Mn–N(5) |
93.29(9) |
O(1)–Mn–N(3) |
156.5(2) |
| O(1)–Mn–N(1) |
82.07(6) |
N(6)–Mn–N(3) |
111.3(2) |
| N(6)–Mn–N(1) |
163.57(8) |
N(5)–Mn–N(3) |
85.2(2) |
| N(5)–Mn–N(1) |
100.79(9) |
N(1)–Mn–N(3) |
76.6(2) |
| O(1)–Mn–O(1w) |
91.18(7) |
O(1w)–Mn–N(3) |
95.7(2) |
| N(6)–Mn–O(1w) |
83.28(8) |
|
|
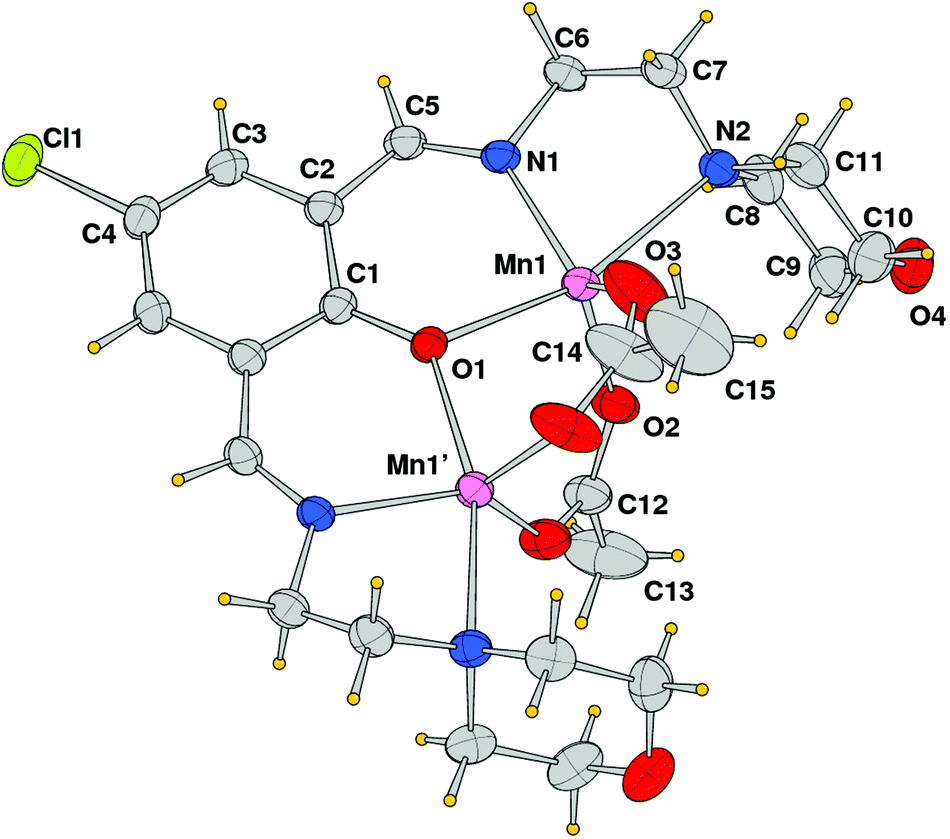
The structural analysis revealed that compound 2 is a dinuclear cationic species and the unit cell contains two independent complexes located on a crystallographic mirror plane, normal to the phenolato ring plane passing in between the manganese atoms, beside a BPh4 anion. An ORTEP view of one of the two μ-phenoxo-di-μ-acetato-dimanganese(II) complexes with the atom labeling scheme of independent atoms is shown in Fig. 4. In both species the metal ions exhibit a distorted trigonal bipyramidal coordination sphere, comprising in the equatorial plane the imine nitrogen donor N1 and acetate oxygen atoms O2 and O3, while the phenoxido-bridged oxygen O1, and morpholine amine nitrogen N2 occupy the axial positions (N4, O5, O6 and O4, N5 are the corresponding atoms in the second complex). The Mn–O and Mn–N bond distances reported in Table 2 indicate comparable values for the two independent complexes with the exception of the Mn–N(amino) bond that differs by ca. 0.1 Å (2.442(3) vs. 2.335(3) Å). It is worth noting the shorter Mn–O(acetate) bond lengths (mean value 2.176 Å with respect to the Mn–O(oxo) ones that average at 2.039 Å). Correspondingly a difference is observed in the intermetallic distance being 3.3455(9) and 3.2194(8) Å in the two complexes.
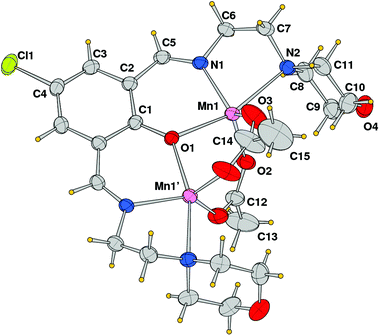 |
| | Fig. 4 ORTEP drawing (ellipsoid probability at 30%) with atom labels of one of the two independent cationic complexes of compound 2. The complex is located on a crystallographic symmetry plane. | |
Table 2 Coordination bond lengths (Å) and angles (°) for complex 2
| Primed atoms at x, 1/2 − y, z. |
| Mn(1)–O(1) |
2.197(2) |
Mn(2)–O(5) |
2.156(2) |
| Mn(1)–O(2) |
2.030(3) |
Mn(2)–O(6) |
2.058(3) |
| Mn(1)–O(3) |
2.020(3) |
Mn(2)–O(7) |
2.048(3) |
| Mn(1)–N(1) |
2.117(3) |
Mn(2)–N(4) |
2.092(3) |
| Mn(1)–N(2) |
2.442(3) |
Mn(2)–N(5) |
2.335(3) |
| Mn(1)–Mn(1)′ |
3.3455(9) |
Mn(2)–Mn(2)′ |
3.2194(8) |
| O(3)–Mn(1)–O(2) |
115.71(15) |
O(7)–Mn(2)–O(6) |
130.85(18) |
| O(3)–Mn(1)–N(1) |
123.69(15) |
O(7)–Mn(2)–N(4) |
115.63(13) |
| O(2)–Mn(1)–N(1) |
120.46(11) |
O(6)–Mn(2)–N(4) |
113.52(18) |
| O(3)–Mn(1)–O(1) |
95.80(13) |
O(7)–Mn(2)–O(5) |
92.16(12) |
| O(2)–Mn(1)–O(1) |
93.10(11) |
O(6)–Mn(2)–O(5) |
92.44(14) |
| N(1)–Mn(1)–O(1) |
85.12(9) |
N(4)–Mn(2)–O(5) |
84.96(11) |
| O(3)–Mn(1)–N(2) |
89.64(11) |
O(7)–Mn(2)–N(5) |
91.86(11) |
| O(2)–Mn(1)–N(2) |
99.89(10) |
O(6)–Mn(2)–N(5) |
95.69(13) |
| N(1)–Mn(1)–N(2) |
77.70(10) |
N(4)–Mn(2)–N(5) |
80.44(11) |
| O(1)–Mn(1)–N(2) |
162.09(9) |
O(5)–Mn(2)–N(5) |
165.19(10) |
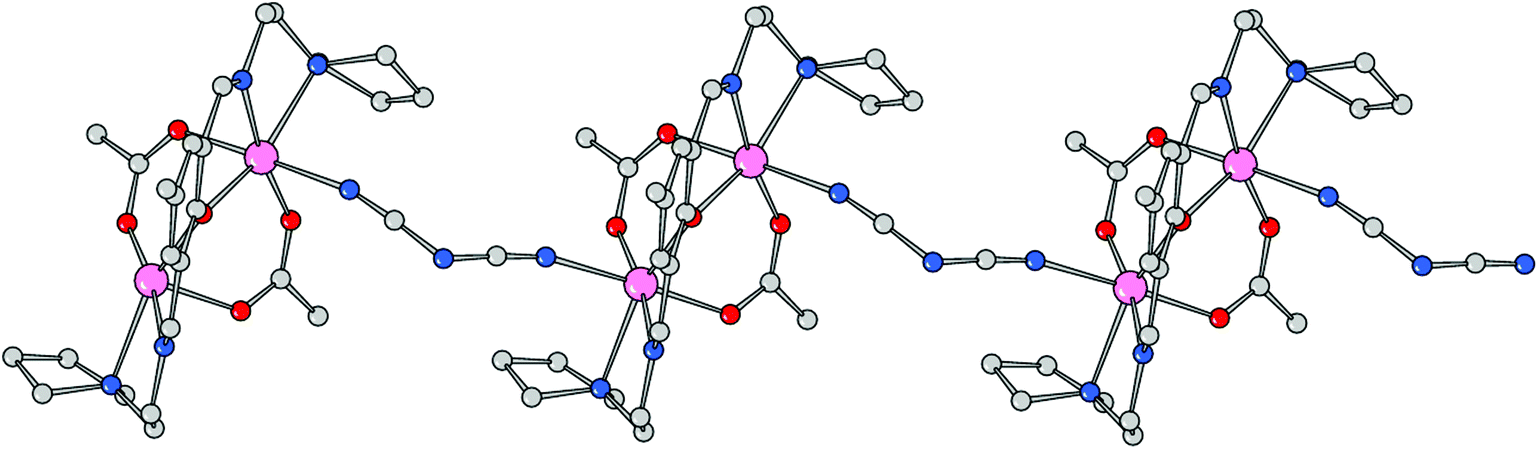
Using the coordinating dicyanoamide as the counteranion, the metals assume a distorted octahedral geometry in the dinuclear complex 3. Each metal is chelated by Schiff base ligand HL2 through the phenoxo oxygen atom, the imine–nitrogen and the amine donor of the pyrrolidine ring. In addition, the coordination sphere is filled by two oxygens from different acetate anions and a nitrogen donor from a bridging dca ligand. With respect to complex 2, the Mn–O and Mn–N bond distances are slightly longer here (Table 3). In particular, the Mn–N(imine) bond distances are 2.189(9) and 2.192(8) Å which must be compared to a mean value of 2.104 Å in 2. Here the acetate anions bridge the metals in an asymmetric fashion, with a long bond length with Mn1 (2.184(8) Å) and a shorter one with Mn2 (2.091(7) Å), and vice versa for the other anion (2.066(8) and 2.157(9) Å, respectively). The Mn–O(oxo) and Mn–N(amine) bond lengths are comparable to those measured in 2. As mentioned above, the dca anions bridge metals of different complexes giving rise to a coordination polymer running parallel to axis a. The dca CN groups coordinate the metals with different CN–Mn angles, of 148.4(12) and 166.3(11)°, where the latter shows an almost linear coordination. The intermetallic Mn1–Mn2 in this neutral complex, 3.389(3) Å, is slightly longer than the value measured in 2 (Fig. 5 and 6).
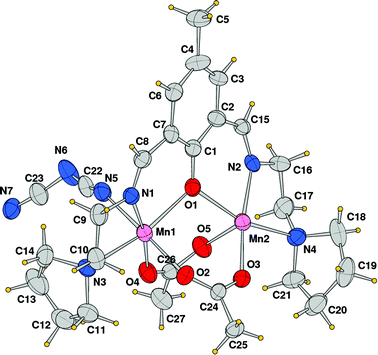 |
| | Fig. 5 ORTEP drawing (ellipsoid probability at 30%) with atom labels of the crystallographic independent unit of compound 3. | |
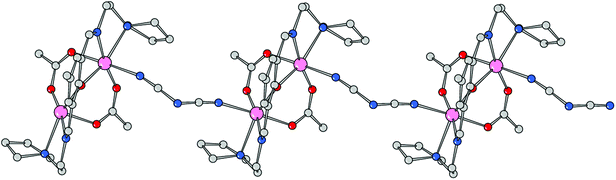 |
| | Fig. 6 Polymeric structure of compound 3 developed along axis a. | |
Table 3 Coordination bond lengths (Å) and angles (°) for complex 3
| Atom N(7)′ at x − 1, y, z. |
| Mn(1)–O(1) |
2.151(8) |
Mn(2)–O(1) |
2.162(7) |
| Mn(1)–O(2) |
2.184(8) |
Mn(2)–O(3) |
2.091(7) |
| Mn(1)–O(4) |
2.066(8) |
Mn(2)–O(5) |
2.157(9) |
| Mn(1)–N(1) |
2.189(9) |
Mn(2)–N(2) |
2.192(8) |
| Mn(1)–N(3) |
2.395(9) |
Mn(2)–N(4) |
2.421(9) |
| Mn(1)–N(5) |
2.256(11) |
Mn(2)–N(7)′ |
2.231(11) |
| Mn(1)–Mn(2) |
3.389(3) |
— |
— |
| O(4)–Mn(1)–O(1) |
106.6(3) |
O(3)–Mn(2)–O(1) |
103.3(3) |
| O(4)–Mn(1)–O(2) |
88.7(3) |
O(3)–Mn(2)–O(5) |
90.8(3) |
| O(1)–Mn(1)–O(2) |
93.1(3) |
O(5)–Mn(2)–O(1) |
92.4(3) |
| O(4)–Mn(1)–N(1) |
170.6(3) |
O(3)–Mn(2)–N(2) |
172.5(3) |
| O(1)–Mn(1)–N(1) |
82.6(3) |
O(5)–Mn(2)–N(2) |
93.9(3) |
| O(2)–Mn(1)–N(1) |
92.8(3) |
O(1)–Mn(2)–N(2) |
82.4(3) |
| O(4)–Mn(1)–N(5) |
87.8(4) |
O(3)–Mn(2)–N(7)′ |
88.7(4) |
| O(2)–Mn(1)–N(5) |
176.5(4) |
O(5)–Mn(2)–N(7)′ |
177.0(4) |
| O(1)–Mn(1)–N(5) |
88.1(3) |
O(1)–Mn(2)–N(7)′ |
90.6(3) |
| N(1)–Mn(1)–N(5) |
90.6(4) |
N(2)–Mn(2)–N(7)′ |
86.3(4) |
| O(4)–Mn(1)–N(3) |
94.8(4) |
O(3)–Mn(2)–N(4) |
99.4(3) |
| O(1)–Mn(1)–N(3) |
158.6(3) |
O(1)–Mn(2)–N(4) |
157.0(3) |
| O(2)–Mn(1)–N(3) |
87.7(3) |
O(5)–Mn(2)–N(4) |
83.9(4) |
| N(1)–Mn(1)–N(3) |
76.0(4) |
N(2)–Mn(2)–N(4) |
75.3(3) |
| N(5)–Mn(1)–N(3) |
92.4(4) |
N(7)′–Mn(2)–N(4) |
93.3(4) |
Catecholase-like activity
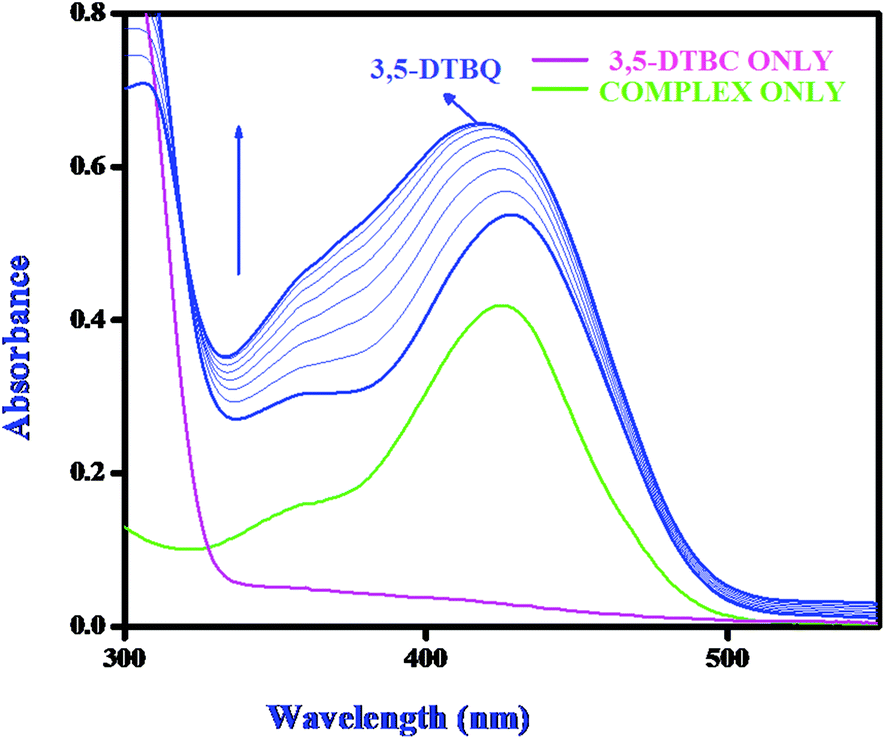
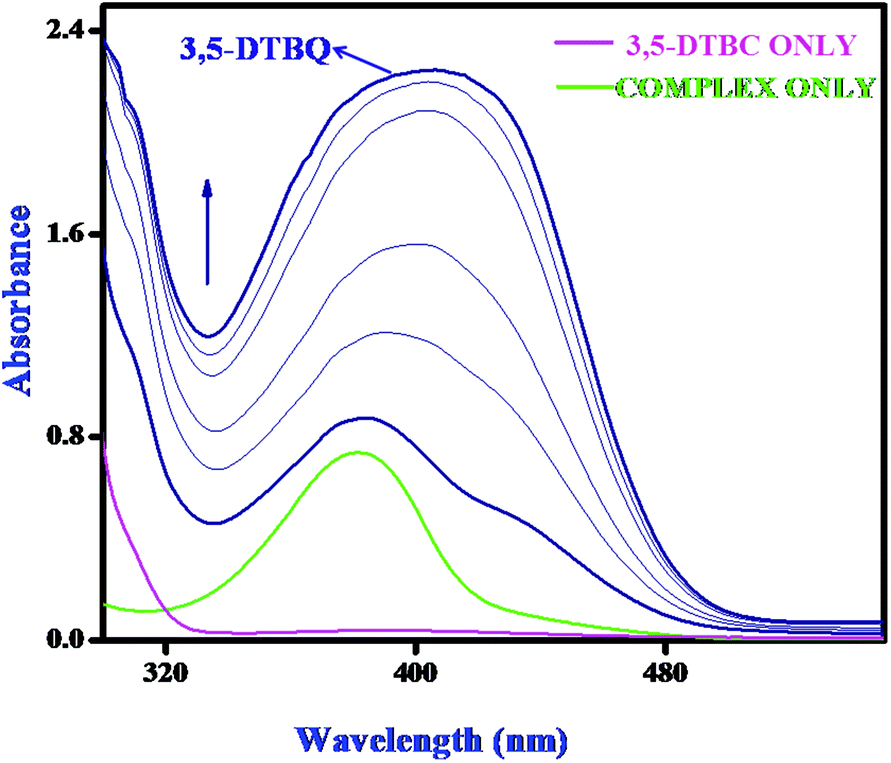
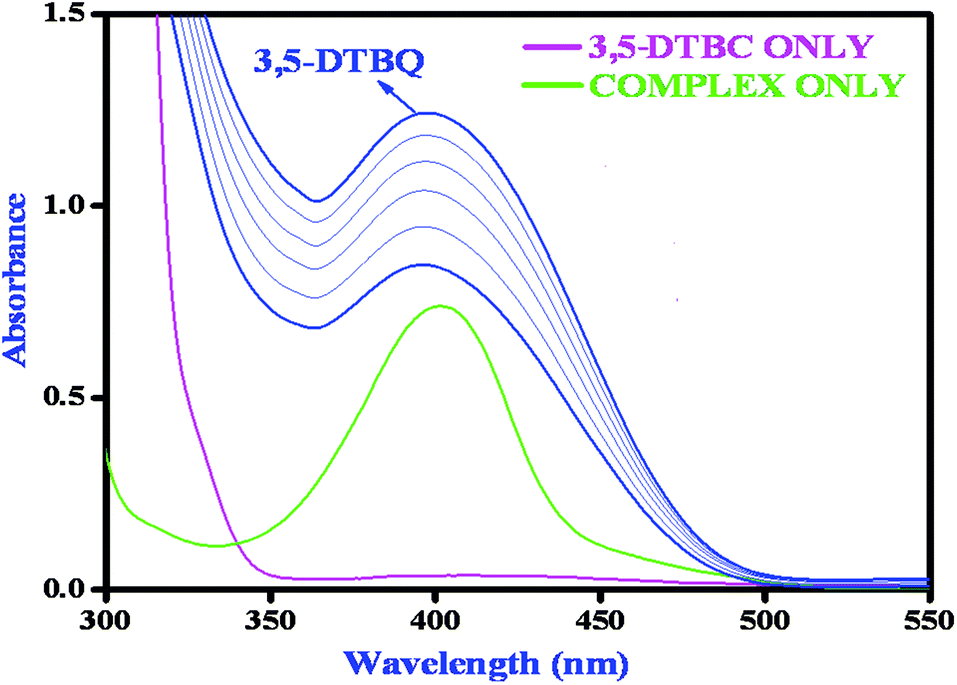
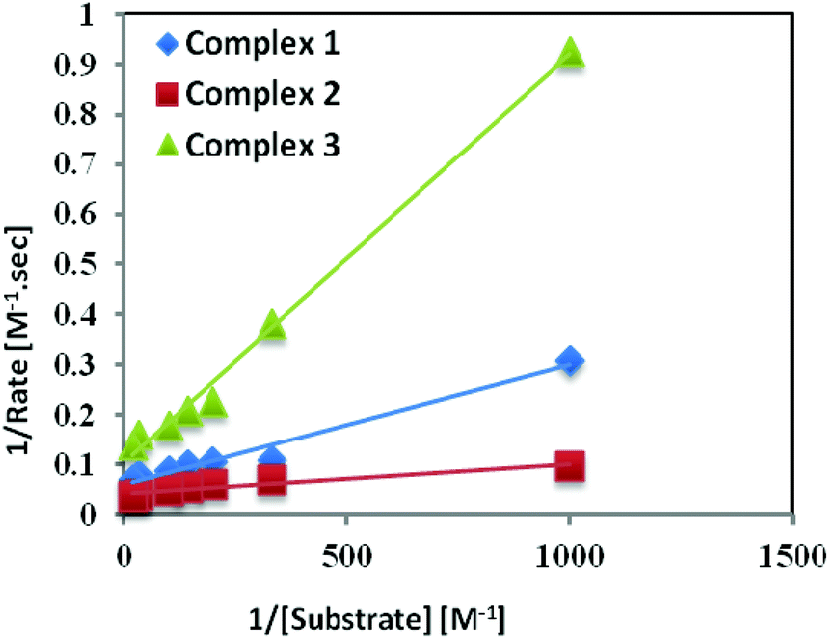
Catechol oxidase, a type-3 copper protein can bind oxygen reversibly at room temperature and so it can be used to oxidize catechols to respective quinones. As a model of the enzyme, here we have taken the three complexes of Mn(II). In order to mimic the functional property of the model compounds, the most widely accepted substrate is 3,5-di-tert-butylcatechol (3,5-DTBC) since its reduction potential for the quinone–catechol couple is low which makes it easy to be oxidized to the corresponding quinone, 3,5-di-tert-butyl-o-benzoquinone (3,5-DTBQ) and at the same time, due to the presence of bulky substituents, further oxidation and polymerization are stopped. Interestingly, all of the complexes exhibit extensive catalytic competence towards the oxidation of 3,5-DTBC to 3,5-DTBQ in the solvent DMSO. Moreover, complex 2, a binuclear Mn(II) Schiff base complex which shows a good catalytic efficiency towards the oxidation of catechols, is the first reported dinuclear Mn(II) catalyst derived from a compartmental Schiff base ligand. Before going into the detailed kinetic study we have checked the ability of the complexes to mimic the active site of catechol oxidase by treating 1 × 10−4 mol dm−3 solutions of the complexes with 1 × 10−2 mol dm−3 (100 equivalents) of 3,5-DTBC under aerobic conditions. The time-dependent UV–vis spectral scan was executed in pure DMSO. Fig. 7–9 show the spectral change for the complexes upon the addition of 100-fold 3,5-DTBC (1 × 10−2 M) observed at intervals of 5 minutes up to 30 minutes. The kinetics of the oxidation of 3,5-DTBC was determined by monitoring the increase of the concentration of the product 3,5-DTBQ and the experimental conditions were the same as reported earlier.5–8 All the complexes showed saturation kinetics and treatment based on the Michaelis–Menten model seemed to be appropriate. The binding constant (KM), maximum velocity (Vmax), and rate constant for the dissociation of substrates (i.e., turnover number, kcat) were calculated for the complex using the Lineweaver–Burk graph of 1/V vs. 1/[S], using the equation 1/V = {KM/Vmax}{1/[S]} + 1/Vmax} (Fig. 10), and the kinetic parameters are presented in Table 4 and in Table S1.† From the data presented in Table 4 it may be inferred that the catalytic efficiency of our present Mn(II) complexes is comparable with the highly active Mn based catalysts reported to date.
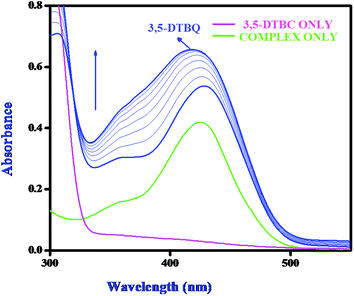 |
| | Fig. 7 Changes observed in the UV–vis spectra of complex 1 in DMSO up to 30 minutes (conc. 1 × 10−4 M) upon the addition of 100-fold 3,5-DTBC (1 × 10−2 M). | |
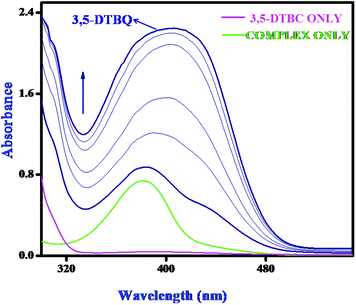 |
| | Fig. 8 Changes observed in the UV–vis spectra of complex 2 in DMSO up to 30 minutes (conc. 1 × 10−4 M) upon the addition of 100-fold 3,5-DTBC (1 × 10−2 M). | |
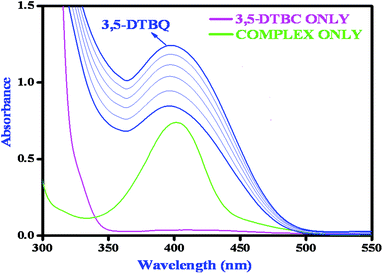 |
| | Fig. 9 Changes observed in the UV–vis spectra of complex 3 in DMSO up to 30 minutes (conc. 1 × 10−4 M) upon the addition of 100-fold 3,5-DTBC (1 × 10−2 M). | |
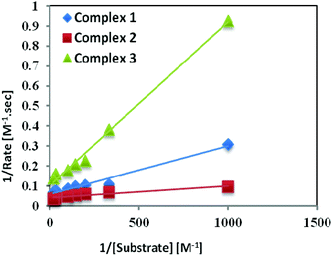 |
| | Fig. 10 Lineweaver–Burk plots for complexes 1, 2, and 3 in DMSO medium for the oxidation of catechol. | |
Table 4 First order rate constants for the oxidation of catechols by our complexes and the similar previously reported Mn(II/III/IV), Ni(II), Cu(II) and Zn(II) complexes using 3,5-DTBC as the substrate
| Complex |
Solvent |
Metal present |
Oxidation state |
k
cat (in h−1) |
Ref. |
|
L1 = 2-[(2-piperazin-1-ylethylimino)-methyl]phenol
L22 = 2,6-bis(R-iminomethyl)-4-methyl-phenolato, R = N-ethylpiperazine; L14 = 2-formyl-4-methyl-6R-iminomethyl-phenolato, R = N-ethylmorpholine; L11 = 2-formyl-4-methyl-6R-iminomethyl-phenolato. R = N-propylmorpholine; L23 = 2,6-bis(R-iminomethyl)-4-methyl-phenolato, R = N-ethylpyrrolidine; L21 = 2,6-bis(R-iminomethyl)-4-methyl-phenolato; R = N-propylmorpholine.
L11 = 2,6-bis(R-iminomethyl)-4-chloro-phenolato, R = N-ethylpiperidine; L12 = 2,6-bis(R-iminomethyl)-4-t-butyl-phenolato, R = N-ethylpiperidine; L21 = 2-formyl-4-methyl-6R-iminomethyl-phenolato, R = N-ethylpiperidine; L13 = 2,6-bis(R-iminomethyl)-4-chloro-phenolato, R = N-ethylmorpholine; L14 = 2,6-bis(R-iminomethyl)-4-t-butyl-phenolato, R = N-ethylmorpholine; L15 = 2,6-bis(R-iminomethyl)-4-chloro-phenolato, R = N-propylmorpholine; L16 = 2,6-bis(R-iminomethyl)-4-t-butyl-phenolato, R = N-propylmorpholine; L17 = 2,6-bis(R-iminomethyl)-4-chloro-phenolato, R = N-ethylpyrrolidine; L19 = 2,6-bis(R-iminomethyl)-4-t-butyl-phenolato, R = N-ethylpyrrolidine; L110 = 2,6-bis(R-iminomethyl)-4-chloro-phenolato, R = N-ethylpiperazine; L112 = 2,6-bis(R-iminomethyl)-4-t-butyl-phenolato, R = N-ethylpiperazine.
L1 = 2,6-bis(R-iminomethyl)-4-methyl-phenolato, R = N-ethylpiperazine; L2 = 2,6-bis(R-iminomethyl)-4-methyl-phenolato, R = N-ethylpyridine; L3 = 2,6-bis(R-iminomethyl)-4-methyl-phenolato, R = N-ethylpyrrolidine; L4 = 2,6-bis(R-iminomethyl)-4-methyl-phenolato, R = benzylamine.
L1 = 2,7-bis(2-hydroxyphenyl)-2,6-diazaocta-2,6-diene; L2 = 1,7-bis(2-hydroxyphenyl)-2,6-diazahepta-1,6-diene).
L2 = N,N′-1-methylethylenebis(3-formyl-5-methylsalicylaldimine); L3 = N,N′-1,1-dimethylethylenebis(3-formyl-5-methylsalicylaldimine); L4 = N,N′-cyclohexenebis(3-formyl-5-methylsalicylaldimine).
L1 = 1,3-bis(4,6-di-tert-butyl-2-iminophenol)benzene; L2 = 2-anilino-4,6-di-tert-butylphenol; L3 = 2-(3,5-di t-butyl-anilino)-4,6-di-tert-butylphenol; L4 = 2-(3,5-di-trifluromethane-anilino)-4,6-di-tert-butylphenol; L5 = 2-(3,5-di methyl-anilino)-4,6-di-tert-butylphenol; L6 = 2-(3,5-di chloro-anilino)-4,6-di-tert-butylphenol; L7 = 2-(3,5-di methoxy-anilino)-4,6-di-tert-butylphenol.
HL = 2,6-bis{2-(N-ethyl)pyridineiminomethyl}-4-methylphenolato.
|
| [Mn(L1)(SCN)2(H2O)] |
DMSO |
Mn |
(II) |
607.08 |
This work |
| [Mn2(L1)(OAc)2](BPh4)] |
DMSO |
Mn |
(II) |
853.38 |
This work |
| [Mn2(L2)(OAc)2(dca)]n |
DMSO |
Mn |
(II) |
255.19 |
This work |
| [NiL1(H2O)3]I2·H2Oa |
MeOH |
Ni |
(II) |
9.27 |
9
|
| [NiL1(H2O)3]Br2·H2Oa |
MeOH |
Ni |
(II) |
8.48 |
9
|
| [Cu2(H2L22)(OH)(H2O)(NO3)](NO3)3·2H2Ob |
MeOH |
Cu |
(II) |
32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 400 |
10
|
| [Cu(HL14)(H2O)(NO3)]2(NO3)2·2H2Ob |
MeOH |
Cu |
(II) |
14400 |
10
|
| [Cu(L11)(H2O)(NO3)]2b |
MeOH |
Cu |
(II) |
10800 |
10
|
| [Cu2(L23)(OH)(H2O)2](NO3)2b |
MeOH |
Cu |
(II) |
14400 |
10
|
| [Cu2(L21)(N3)3]b |
MeOH |
Cu |
(II) |
28800 |
10
|
| [Cu2(L11)(OH)(H2O)(NO3)2]c |
DMSO |
Cu |
(II) |
73.70 |
11
|
| [Cu2(L12)(OH)(H2O)(NO3)2]c |
DMSO |
Cu |
(II) |
50.50 |
11
|
| [Cu(L21)(H2O)(NO3)]2c |
DMSO |
Cu |
(II) |
84.60 |
11
|
| [Cu2(L13)(OH)(H2O)(NO3)2]c |
DMSO |
Cu |
(II) |
127.20 |
11
|
| [Cu2(L14)(OH)(H2O)(NO3)2]c |
DMSO |
Cu |
(II) |
49.60 |
11
|
| [Cu2(L15)(OH)(H2O)(NO3)2]c |
DMSO |
Cu |
(II) |
246.80 |
11
|
| [Cu2(L16)(OH)(H2O)(NO3)2]c |
DMSO |
Cu |
(II) |
50.70 |
11
|
| [Cu2(L17)(OH)(H2O)2(NO3)2]c |
DMSO |
Cu |
(II) |
26.90 |
11
|
| [Cu2(L19)(OH)(H2O)(NO3)2]c |
DMSO |
Cu |
(II) |
51.80 |
11
|
| [Cu2(L110)(OH)(H2O)(NO3)4]c |
DMSO |
Cu |
(II) |
68.30 |
11
|
| [Cu2(L112)(OH)(H2O)(NO3)2]c |
DMSO |
Cu |
(II) |
50.10 |
11
|
| [Zn2(H2L1)(OH)(H2O)(NO3)](NO3)3]d |
MeOH |
Zn |
(II) |
1060.00 |
12
|
| [Zn2L2Cl3]d |
MeOH |
Zn |
(II) |
882.0 |
12
|
| [Zn2L3Cl3]d |
MeOH |
Zn |
(II) |
297.00 |
12
|
| [Zn2(L4)2(CH3COO)2]d |
MeOH |
Zn |
(II) |
352.00 |
12
|
| [MnL1(OOCH)(OH2)]e |
MeCN |
Mn |
(III) |
936.64 |
13a
|
| [MnL2(OH2)2][Mn2(L22(NO2)3]e |
MeCN |
Mn |
(III) |
365.34 |
13a
|
| [Mn2L2(NO2)2]e |
MeCN |
Mn |
(III) |
1432.74 |
13a
|
| [MnL2Cl·4H2O]f |
MeOH |
Mn |
(III) |
247.00 |
13b
|
| [MnL3Cl·4H2O]f |
MeOH |
Mn |
(III) |
360.00 |
13b
|
| [MnL4Cl·4H2O]f |
MeOH |
Mn |
(III) |
720.00 |
13b
|
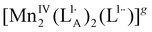
|
CH2Cl2 |
Mn |
(IV) |
12.60 |
14a
|
| [MnIV(L2A)(L2˙)2]g |
CH2Cl2 |
Mn |
(IV) |
21.30 |
14a
|
| [MnIV(L3A)(L3˙)2]g |
CH2Cl2 |
Mn |
(IV) |
55.20 |
14a
|
| [MnIV(L4A)(L4˙)2]g |
CH2Cl2 |
Mn |
(IV) |
14.80 |
14a
|
| [MnIV(L5A)(L5˙)2]g |
CH2Cl2 |
Mn |
(IV) |
34.14 |
14a
|
| [MnIV(L6A)(L6˙)2]g |
CH2Cl2 |
Mn |
(IV) |
16.32 |
14a
|
| [MnIV(L7A)(L7˙)2]g |
CH2Cl2 |
Mn |
(IV) |
15.60 |
14a
|
| [Mn(HL)(H2O)3](NO3)2·(H2O)]h |
MeOH |
Mn |
(II) |
2160.00 |
15
|
| [Mn(HL)(SCN)2(H2O)]·0.5H2O]h |
MeOH |
Mn |
(II) |
1440.00 |
15
|
| [Mn(HL)(N(CN)2)(H2O)2](NO3)·H2O]h |
MeOH |
Mn |
(II) |
720.00 |
15
|
Mechanistic interpretation of catecholase activity exhibited by the complexes
Earlier our group tried to explore the origin of the catecholase like activity exhibited by Mn(II) and Mn(III) with Schiff Base ligands.13b,15 In the case of Mn(III) we are able to prove that the origin of the activity was related to the metal center redox participation, where Mn(III) underwent reduction to Mn(II) with concomitant oxidation of catechol to quinone. However, in the case of the Mn(II) complex we failed to evaluate the plausible mechanism for the catecholase activity. The oxidation from catechol to quinone may have occurred via three pathways: (i) metal centered reduction, (ii) ligand centered imine radical formation, or (iii) phenoxy radical formation in the case of phenolic ligands. So in this present work we tried to evaluate the correct pathway with the help of EPR and electrochemical studies. As the three complexes are of the same kind, we selected the dinuclear one (2) as a model for EPR spectroscopic analysis. For this, we have prepared a 1:100 mixture of complex 2 and 3,5-DTBC (10−1 M) in DMSO solvent and recorded the EPR spectra within 2 min of mixing. A characteristic six-line EPR spectrum (Fig. 11) corroborates the Mn(II) species when the complex 2 is dissolved in DMSO. An additional sharp peak (g = 1.99) (Fig. 12) appears after mixing the complex with 3,5-DTBC which indicates the formation of the ligand-centered imine radical and that radical formation is most probably responsible for the catalytic oxidation of 3,5-DTBC to 3,5-DTBQ in the presence of aerial oxygen. We have also performed the EPR experiment at 77 K at power saturation conditions to find out if there is any signal at the half-field but our repeated attempts failed to distinguish any characteristic signal. However, our present EPR study implies that when the Mn(II) complex is mixed with 3,5-DTBC, the ligand part, most probably the imine bond of the coordinated ligand, undergoes reduction with concomitant oxidation of the catechol to quinone via the formation of a semibenzoquinone radical, a similar observation to what we found in our earlier study with our Zn complex.12
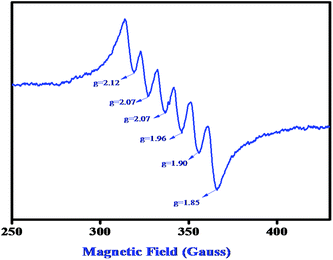 |
| | Fig. 11 EPR spectrum of complex 2 in DMSO medium at 77 K. | |
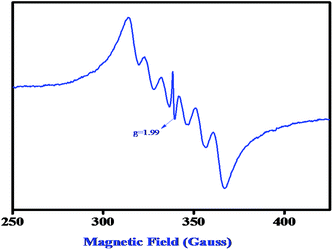 |
| | Fig. 12 EPR spectrum of a 1:100 mixture of complex 2 and 3,5-DTBC (10−1 M) in DMSO at 77 K. | |
Interestingly, when this catalytic oxidation was performed in an inert atmosphere, no 3,5-DTBQ formation was noticed. However, upon exposure of the reaction mixture to dioxygen atmosphere, 3,5-DTBQ formation was immediately observed. This observation indeed indicates that dioxygen is one of the active participants in the catalytic cycle; it converts the semibenzoquinone radical, formed in the first step of the catalysis, to the quinone with subsequent regeneration of the catalyst.
Electrochemical study
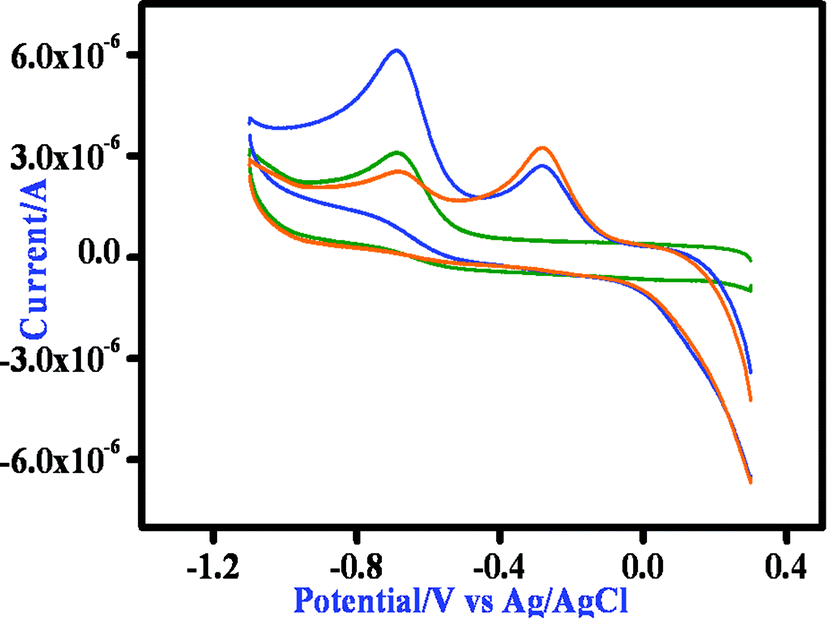
All the Mn(II) complexes show an irreversible reduction at −0.72 vs. Ag/AgCl, which we tentatively assign to imine reduction of the ligand moiety. On the oxidative side the mononuclear Mn(II) complex exhibits one irreversible oxidation at 0.89 V, which is probably due to the oxidation of the phenolate to a phenoxyl radical. The binuclear complexes show two distinctive oxidation peaks, both processes being irreversible in nature. For complex 2, the anodic peak potentials are at 0.96 and 1.12 V (Fig. S13†), whereas for 3, the corresponding values are 0.51 and 1.07 V (Fig. S15†). We tentatively assigned the first anodic peak to Mn(II)/Mn(III) oxidation and the second peak to the oxidation of the phenolate moiety of the ligand. Then we performed the cyclic voltammetric study of the complexes in the presence of 3,5-DTBC. All three complexes exhibit a nearly similar response and thus the behavior of complex 1 in the presence of 3,5-DTBC is stated here. On addition of 3,5-DTBC to a solution of complex 1 the current height of the imine reduction peak at −0.68 V decreases, while a new peak appears at −0.29 V which may be due to the reduction of a coordinated semiquinone to catechol (Fig. 13). On the oxidative side, a very strong peak at ∼0.8 V corresponding to the formation of semiquinone is observed without any signal responsible for the oxidation of Mn(II) to Mn(III) (Fig. S15 and S16†). Thus the cyclic voltammetry data confirms that the reduction of the complex during 3,5-DTBC oxidation can take place only at the imine part of the ligand and the metal center is in no way involved in this process.
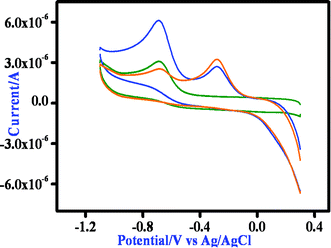 |
| | Fig. 13 Time dependent CV spectrum of complex 1 after the addition of DTBC at the GC electrode in DMSO medium at a 100 mV s−1 scan rate as representative. | |
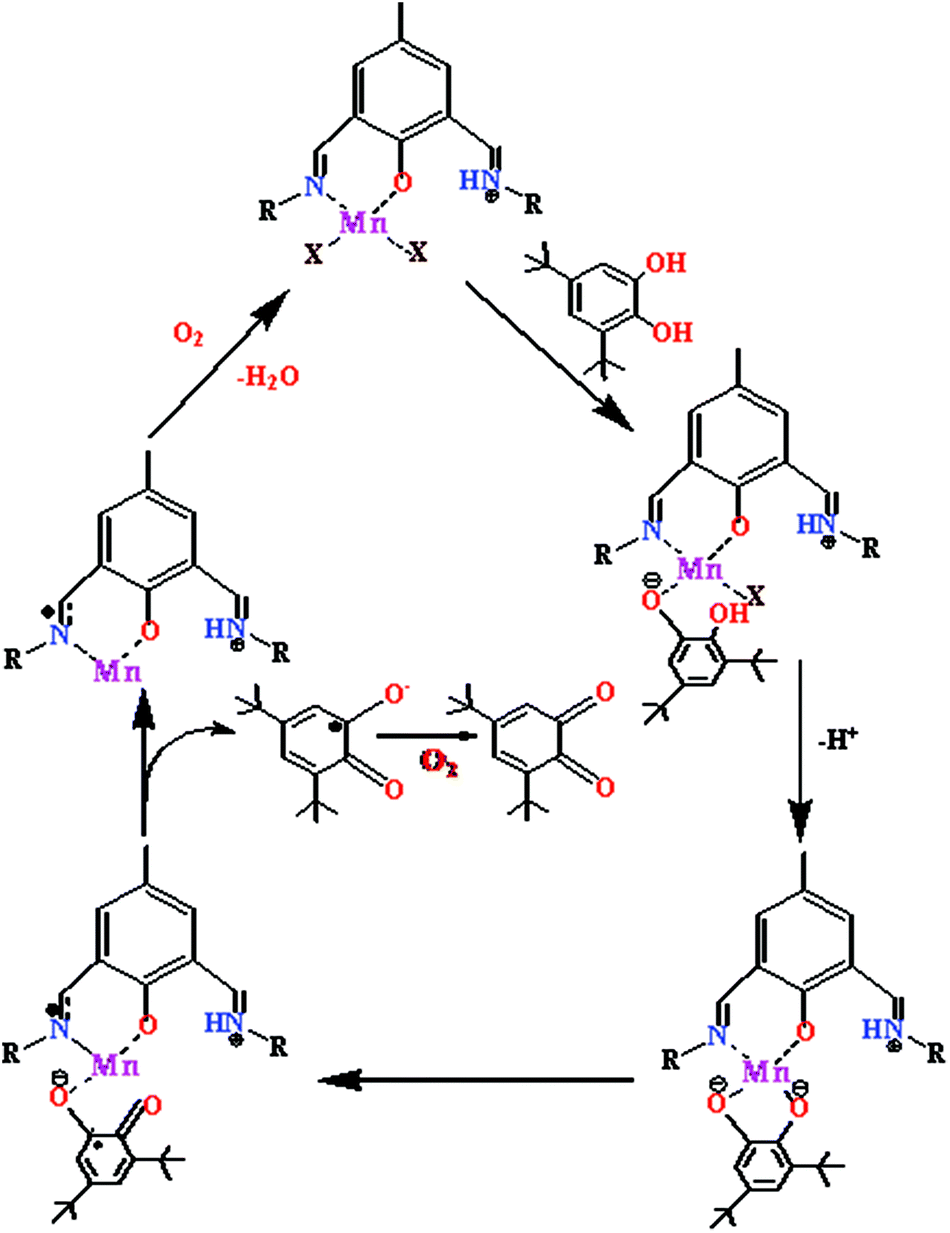
On the basis of the results obtained from EPR and cyclic voltammetric studies, a plausible mechanism has been proposed in Scheme 2. With the dinuclear complexes, two catechol molecules are oxidized per cycle, whereas only one is oxidized by the monomeric Mn(II) complex. Here the mononuclear complex 1 is presented for simplicity.
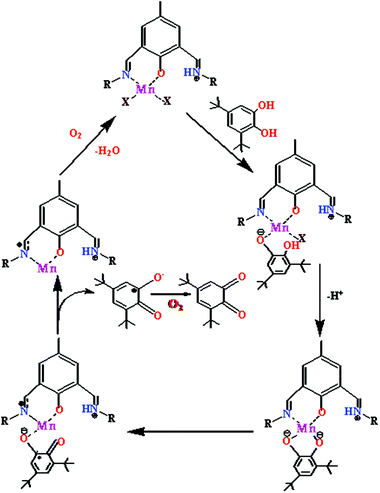 |
| | Scheme 2 Proposed mechanism for the catalytic cycle of the oxidation of 3,5-DTBC by the mononuclear Mn(II) complex. | |
Magnetic properties of complexes 2 and 3
The magnetic properties of complexes 2 and 3, in the form of χmT (χm is the susceptibility per dinuclear unit) vs. T plots, are shown in Fig. 14 in a temperature range of 5–300 K for 2 and 2–300 K for 3. The observed χmT values at room temperature, 8.76 emu K mol−1 (μeff = 8.37μB) and 8.39 emu K mol−1 (μeff = 8.19μB) for 2 and 3, respectively, are comparable with the spin-only value of 8.75 emu K mol−1 (μeff = 8.37μB) expected for two independent high-spin Mn(II) ions [(SMn, SMn) = (5/2, 5/2) assuming g = 2.0]. As the temperature is lowered, the χmT values decrease in a monotonous manner and become 0.39 emu K mol−1 for 2 at 5 K and 0.31 emu K mol−1 for 3 at 2 K. This result indicates the presence of an antiferromagnetic spin–exchange interaction between Mn(II) ions (Fig. 15).
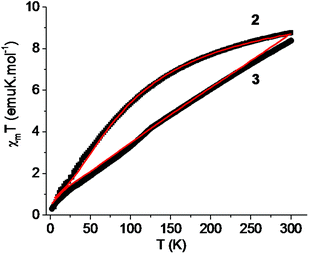 |
| | Fig. 14 Temperature variation of the magnetic susceptibilities of 2 and 3 as χmT versus T plots (the solid line represents the best fit of the experimental data based on the Heisenberg model). | |
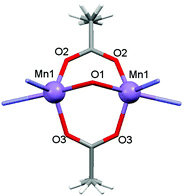 |
| | Fig. 15 Local coordination environments of Mn(II) atoms and the magnetic exchange coupling pathway for complex 2. | |
For dinuclear Mn(II) complexes (S1 = S2 = 5/2), the theoretical expression of the magnetic susceptibility based on the Heisenberg Hamiltonian (H = −2JS1S2) is:
| | 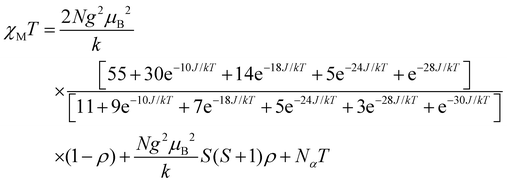 | (1) |
In this expression, ρ presents the fraction of paramagnetic impurity in the sample, Nα is the temperature independent paramagnetism and the other symbols have their usual meanings. To determine the exchange parameters,χmT was fitted using eqn (1) for the range of 5–300 K, giving the best agreement with the experimental data for J = −9.15 cm−1, g = 2.01, ρ = 0.09 and Nα = 0.00089 (R2 = 0.99802) for complex 2.
Complex 3 is a one-dimensional chain and its magnetic interactions should be expressed using alternating exchange interactions, namely, Mn–J–Mn–J′–Mn–J–Mn–J′–Mn, in which J represents the intradimer exchange interaction via the triple oxygen bridges, whereas J′ is the interdimer exchange interaction via the dicyanamide bridge in the chain (Fig. 16). To take into account the interaction between the dimer entities, the susceptibility can be corrected by the molecular field approximation20 (eqn (2)), where zJ′ denotes the intramolecular exchange parameter.
| | 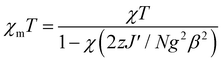 | (2) |
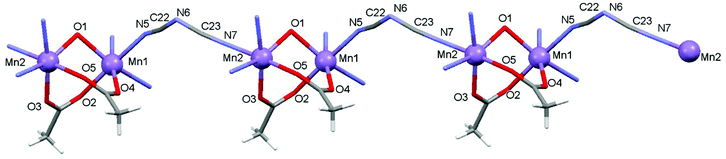 |
| | Fig. 16 Local coordination environments of Mn(II) atoms and the magnetic exchange coupling pathway for complex 3. | |
In this expression, z is the number of nearest-neighbouring dimers (in this case z = 2), J′ accounts for the presence of magnetic interactions between the neighboring dimers and the other symbols have their usual meanings. To determine the exchange parameters, χmT was fitted using eqn (2) for the range of 2–300 K, giving the best agreement with the experimental data for J = −46.89 cm−1, J′ = −2.38, g = 2.20, ρ = 0.025 and Nα = 0.00046 (R2 = 0.99852) for complex 3.
The overall magnetic behavior of complexes 2 and 3 is dominated by the antiferromagnetic interactions between the spin carriers and resembles that of related compounds.16–19
Experimental section
Physical methods and materials
3,5-Di-tert-butyl catechol was purchased from Sigma-Aldrich. Reaction solutions for 3,5-DTBC were prepared according to standard sterile techniques.
Elemental analyses (carbon, hydrogen, and nitrogen) were performed using a PerkinElmer 240C analyzer. Infrared spectra (4000–400 cm−1) were recorded at 28 °C on a Shimadzu FTIR-8400S and PerkinElmer Spectrum Express Version 1.03 using KBr pellets as mediums. UV–visible spectra and kinetic traces were monitored with a Shimadzu UV-2450PC spectrophotometer equipped with multiple cell-holders and a thermostat. Magnetic susceptibility measurements over the temperature range of 2–300 K were performed at a magnetic field of 0.0750 T using a Quantum Design SQUID MPMSXL-5 magnetometer. Correction for the sample holder as well as the diamagnetic correction, which was estimated from the Pascal constants, were considered.20,21 EPR experiments were performed at liquid nitrogen temperature (77 K) in methanol, using a JEOL JES-FA200 spectrometer at X band (9.13 GHz).
Cyclic voltammetric and DPV measurements were performed using a CH1106A potentiostat with glassy carbon (GC) as the working electrode, Pt-wire as the counter electrode and Ag, AgCl/sat KCl as the reference electrode. All solutions were purged with dinitrogen prior to measurements.
High-purity N-(2-aminoethyl)-pyrrolidine and N-(2-aminoethyl)morpholine, were purchased from commercial sources (Fluka, Lancaster Chemical Co. Inc., Aldrich) and used as received. The 2,6-diformyl-4-R-phenols (R = methyl, chloro) were prepared according to the literature method.22 MeOH was dried over CaH2 for 2 days and then distilled under reduced pressure prior to use. Water used in all physical measurements and experiments was Milli-Q grade. All other chemicals used were of AR grade.
Synthesis of the complexes
The following general template synthetic route was adopted for preparing all complexes. A methanolic solution of the manganese salt was added to the ligand solution formed in situ via the condensation of 2,6-diformyl-4-R-phenol (where R = methyl, chloro) with the corresponding amines, maintaining the same molar ratio. The preparation, composition, and other physicochemical characteristics of all complexes using the template technique are given below.
Syntheses
[Mn(L1)(SCN)2(H2O)] (1).
To a methanolic solution (5 mL) of N-(2-aminoethyl)morpholine (0.260 g, 2 mmol), a methanolic solution (10 mL) of 4-chloro-2,6-diformylphenol (0.180 g, 1 mmol) was added dropwise and the mixture was refluxed for 30 minutes. Then a methanolic solution (15 mL) of manganese chloride (0.604 g, 2.5 mmol) was added to it, and the resulting mixture was allowed to reflux for an additional 2 h. The deep brown solution was filtered and cooled to room temperature. Then an aqueous solution of sodium thiocyanate (0.32 g, 4 mmol) was added to the reaction mixture and stirred for about 30 minutes. The resulting solution was filtered and the filtrate was kept in a CaCl2 dessicator. Deep brown crystals suitable for X-ray data collection were obtained from the filtrate on keeping the solution overnight (yield 75%). Anal. Calcd for C22H31Mn1N6O4S2Cl: C (44.14%); H (5.5%); N (14.04%); Found: C (44.94%); H (5.89%); N (14.1%); FT-IR data (KBr pellet): I.R: ν(CN) 1641 cm−1; ν(skeletal vibration) 1533 cm−1; ν(SCN−) 2065 cm−1; UV/vis (MeOH): λmax(ε) = 425 nm (4200 L mol−1 cm−1).
[Mn2(L1)(OAc)2](BPh4)] (2).
To a methanolic solution (5 mL) of N-(2-aminoethyl)morpholine (0.260 g, 2 mmol), a methanolic solution (10 mL) of 4-chloro-2,6-diformylphenol (0.180 g, 1 mmol) was added in a dropwise manner and the mixture was refluxed for 30 min. Then a methanolic solution (15 mL) of manganese acetate (0.604 g, 2.5 mmol) was added to it and the resulting mixture was allowed to reflux for 2 h. The deep brown solution was filtered and cooled to room temperature. Then an aqueous solution of tetra phenyl borate (0.88 g, 4 mmol) was added to the reaction mixture and stirred for about 10 min. The resulting solution was filtered and the filtrate was kept in a CaCl2 dessicator. Deep brown crystals suitable for X-ray data collection were obtained from the filtrate after a few days (yield 70%).
Anal. Calcd for C48H34Mn2N4O7BCl: C (60.30%); H (3.55%); N (5.75%); Found: C (60.50%); H (3.95%); N (5.29%); FT-IR data (KBr pellet): I.R: ν(CN) 1643 cm−1; ν(skeletal vibration) 1537 cm−1; ν(OAc−) 1483 cm−1; UV/vis (MeOH): λmax(ε) = 381 nm (7390 L mol−1 cm−1).
[Mn2(L2)(OAc)2(dca)]n (3).
To a methanolic solution (5 mL) of N-(2-aminoethyl)pyrrolidine (0.228 g, 2 mmol), a methanolic solution (10 mL) of 4-methyl-2,6-diformylphenol (0.164 g, 1 mmol) was added in a dropwise manner and the mixture was refluxed for 30 min. Then a methanolic solution (15 mL) of manganese acetate (0.604 g, 2.5 mmol) was added to it and the resulting mixture was allowed to reflux for 2 h. The deep brown solution was filtered and cooled to room temperature. Then, to the reaction mixture, an aqueous solution of sodium dicyanamide (0.284 g, 4 mmol) was added and stirring was continued for about 30 min. The resulting solution was filtered and the filtrate was kept in a CaCl2 dessicator. Deep brown crystals suitable for X-ray data collection were obtained from the filtrate after keeping the solution overnight (yield 68%).
Anal. Calcd for C27H37Mn2N7O5: C (49.88%); H (5.69%); N (15.08%); Found: C (49.50%); H (5.95%); N (15.37%); FT-IR data (KBr pellet): I.R: ν(CN) 1641 cm−1; ν(skeletal vibration) 1533 cm−1; ν(OAc−) 1488 cm−1ν(N(CN)2−) 2165 cm−1; UV/vis (MeOH): λmax(ε) = 407 nm (7530 L mol−1 cm−1).
X-ray data collection and structure determination
Data collection of the structures reported was carried out at room temperature on a Bruker Smart CCD diffractometer (1–2) and on a Nonius DIP-1030H system (3), all equipped with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). Cell refinement, indexing and scaling of the data set were carried out using Bruker Smart Apex and Bruker Saint packages,23 Mosflm and Scala.24 All the structures were solved using direct methods and subsequent Fourier analyses25 and refined using the full-matrix least-squares method based on F2 with all observed reflections.25 In compound 1 the coordinated morpholine moiety was found disordered over two positions (occupancies refined at 0.537(6)/0.463(6) with a restrained geometry). All the calculations were performed using the WinGX System, Ver 1.80.05.26 Pertinent crystallographic data and refinement details are summarized in Table 5.
Table 5 Crystallographic data and details of refinements for complexes 1–3b
| |
1
|
2
|
3
|
|
R
1 = ∑|Fo| − |Fc||/∑|Fo|, wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2.
CCDC 1425706–1425708 contain the supplementary crystallographic data for this paper.
|
| Empirical formula |
C22H31ClMnN6O4S2 |
C48H54BClMn2N4O7 |
C27H37Mn2N7O5 |
| MW |
598.04 |
955.09 |
649.52 |
| Crystal system |
Monoclinic |
Monoclinic |
Triclinic |
| Space group |
P21/c |
P21/m |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
|
a/Å |
9.1058(4) |
11.1317(17) |
10.058(3) |
|
b/Å |
21.8162(9) |
24.757(4) |
12.348(3) |
|
c/Å |
14.3490(6) |
17.083(3) |
13.366(4) |
|
α/° |
— |
— |
78.53(2) |
|
β/° |
97.9080(10) |
93.100(2) |
84.51(3) |
|
γ/° |
— |
— |
70.55(2) |
|
V/Å3 |
2823.4(2) |
4700.9(12) |
1533.3(7) |
|
Z
|
4 |
4 |
2 |
|
D
calc/g cm−3 |
1.407 |
1.349 |
1.407 |
|
μ/mm−1 |
0.748 |
0.648 |
0.870 |
|
F(000) |
1244 |
1992 |
676 |
| Theta range/° |
1.71–25.57 |
1.19–26.39 |
1.56–23.26 |
| Total data |
32565 |
32933 |
11533 |
| Unique data |
5273 |
9482 |
4339 |
|
R
int
|
0.0325 |
0.0398 |
0.0779 |
| Reflections I > 2σ(I) |
4223 |
6350 |
1478 |
| Parameters |
395 |
592 |
370 |
| Goodness-of-fit |
1.022 |
1.038 |
0.825 |
|
R
1a |
0.0331 |
0.0557 |
0.0731 |
| wR2 (I > 2σ(I))a |
0.0849 |
0.1675 |
0.1723 |
| Residuals/e Å−3 |
0.285, −0.247 |
0.981, −0.399 |
0.364, −0.362 |
Conclusion
Three manganese(II) complexes having different nuclearities, one mono-, one di- and one dinuclear based polynuclear complex, have been synthesized using two “end-off” compartmental Schiff-base ligands, HL1 and HL2. Protonation of one of the imine nitrogen atoms of HL1 in the presence of SCN as a co-ligand cancels out the possibility of metalation in the second compartment of HL1, leading to mononuclearity. In the presence of acetate as a co-ligand and BPh4 as a counter anion, HL1 generates the dinuclear complex 2. HL2 in the presence of acetate and dca as co-ligands forms an acetate bridged dinuclear based polynuclear complex where dca bridges two dinuclear units. All three complexes have been structurally characterized and the oxidation state of each of the manganese atoms is +2 as is evidenced from magnetic susceptibility and EPR study. Variable temperature magnetic study implies the presence of an antiferromagnetic spin–exchange interaction between Mn(II) ions with J = −9.15 cm−1 and J = −46.89 cm−1 for complexes 2 and 3, respectively. All three complexes exhibit catecholase-like activity towards 3,5-DTBC as the model substrate with varying degrees, 2 > 1 > 3. In order to unveil the origin of the catecholase activity whether related to metal centered redox participation or radical generation, we have performed EPR and cyclic voltammetric studies. EPR investigation of the solution of 3,5-DTBC and the complexes suggests radical formation. Cyclic voltammetric study suggests ligand centered reduction rather than the reduction of Mn(II) to Mn(I). It is therefore concluded that the catecholase-like activity of the present Mn(II) complexes follows a radical pathway as is observed in analogous Zn(II) and Ni(II) complexes.
Acknowledgements
The authors wish to thank CSIR, New Delhi [01(2464)/11/EMR-II dt16-05-11 to D. D.] for financial support and the University of Calcutta for providing the facility of the single-crystal X-ray diffractometer from the DST FIST program and for collecting variable temperature magnetic study from SQUID-VSM in CRNN. Authors are also thankful to Prof. Shyamal Kumar Chattopadhyay of IIEST, Shibpur for providing the facilities for electrochemical studies. I. M is thankful to UGC [UGC/729/Jr Fellow(Sc)] for providing a fellowship.
References
- M. Fontecave and J.-L. Pierre, Coord. Chem. Rev., 1998, 170, 125 CrossRef CAS.
- L. Que Jr. and W. B. Tolman, Nature, 2008, 455, 333 CrossRef PubMed.
- J. Suh, Acc. Chem. Res., 1992, 25, 273 CrossRef CAS.
- P. Chaudhuri, M. Hess, U. Flörke and K. Wieghardt, Angew. Chem., Int. Ed., 1998, 37, 2217 CrossRef CAS.
- J. Reim and B. Krebs, J. Chem. Soc., Dalton Trans., 1997, 3793 RSC.
- S. Torelli, C. Belle, I. Gautier-Luneau, J. L. Pierre, E. Saint-Aman, J. M. Latour, L. Le Pape and D. Luneau, Inorg. Chem., 2000, 39, 3526 CrossRef CAS PubMed.
- A. Neves, L. M. Rossi, A. J. Bortoluzzi, B. Szpoganicz, C. Wiezbicki and E. Schwingel, Inorg. Chem., 2002, 41, 1788 CrossRef CAS PubMed.
- J. Mukherjee and R. Mukherjee, Inorg. Chim. Acta, 2002, 337, 429 CrossRef CAS.
- J. Adhikary, P. Chakraborty, S. Das, T. Chattopadhyay, A. Bauzá, S. K. Chattopadhyay, B. Ghosh, F. A. Mautner, A. Frontera and D. Das, Inorg. Chem., 2013, 52, 13442 CrossRef CAS PubMed.
- K. S. Banu, T. Chattopadhyay, A. Banerjee, S. Bhattacharya, E. Suresh, M. Nethaji, E. Zangrando and D. Das, Inorg. Chem., 2008, 47, 7083 CrossRef CAS PubMed.
- P. Chakraborty, J. Adhikary, B. Ghosh, R. Sanyal, S. K. Chattopadhyay, A. Bauzá, A. Frontera, E. Zangrando and D. Das, Inorg. Chem., 2014, 53, 8257 CrossRef CAS PubMed.
- A. Guha, T. Chattopadhyay, N. D. Paul, M. Mukherjee, S. Goswami, T. K. Mondal, E. Zangrando and D. Das, Inorg. Chem., 2012, 51, 8750 CrossRef CAS PubMed.
-
(a) P. Seth, M. G. B. Drew and A. Ghosh, J. Mol. Catal. A: Chem., 2012, 365, 154 CrossRef CAS;
(b) K. S. Banu, T. Chattopadhyay, A. Banerjee, M. Mukherjee, S. Bhattacharya, G. K. Patra, E. Zangrando and D. Das, Dalton Trans., 2009, 8755 RSC.
-
(a) S. Mukherjee, T. Weyhermüller, E. Bothe, K. Wieghardt and P. Chaudhuri, Dalton Trans., 2004, 3842 RSC;
(b) S. Mukherjee, E. Rentschler, T. Weyhermüller, K. Wieghardt and P. Chaudhuri, Chem. Commun., 2003, 1828 RSC.
- A. Guha, K. S. Banu, A. Banerjee, T. Ghosh, S. Bhattacharya, E. Zangrando and D. Das, J. Mol. Catal. A: Chem., 2011, 338, 51 CAS.
- C. S. Hong and Y. Do, Inorg. Chem., 1997, 36, 5684 CrossRef CAS PubMed.
- A. Escuer, R. Vicente, M. A. S. Goher and F. A. Mautner, Inorg. Chem., 1996, 35, 6386 CrossRef CAS PubMed.
- J. L. Manson, C. D. Incarvito, A. L. Rheingold and J. S. Miller, J. Chem. Soc., Dalton Trans., 1998, 3705 RSC.
- M. A. Kiskin, I. G. Fomina, G. G. Aleksandrov, A. A. Sidorov, V. M. Novotortsev, Y. V. Rakitin, Z. V. Dobrokhotova, V. N. Ikorskii, Y. G. Shvedenkov, I. L. Eremenko and I. I. Moiseev, Inorg. Chem. Commun., 2005, 8, 89 CrossRef CAS.
- C. J. O'Connor, J. Prog. Inorg. Chem., 1982, 29, 203 Search PubMed.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532 CrossRef CAS.
- R. R. Gagne, C. L. Spiro, T. J. Smith, C. A. Hamann, W. R. Thies and A. K. Shiemeke, J. Am. Chem. Soc., 1981, 103, 4073 CrossRef CAS.
-
SMART, SAINT. Software Reference Manual, Bruker AXS Inc., Madison, WI, 2000 Search PubMed.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235–242 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1425706–1425708. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt03659c |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a