
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cooperative reduction by Ln2+ and Cp*− ions: synthesis and properties of Sm, Eu, and Yb complexes with 3,6-di-tert-butyl-o-benzoquinone†
Nikolay A.
Pushkarevsky
*ab,
Mikhail A.
Ogienko
a,
Anton I.
Smolentsev
a,
Igor N.
Novozhilov
a,
Alexander
Witt
c,
Marat M.
Khusniyarov
c,
Vladimir K.
Cherkasov
de and
Sergey N.
Konchenko
ab
aNikolaev Institute of Inorganic Chemistry, Siberian Division of RAS, Akad. Lavrentieva str. 3, 630090 Novosibirsk, Russia. E-mail: nikolay@niic.nsc.ru
bDepartment of Natural Sciences, Novosibirsk State University, Pirogova Street 2, 630090 Novosibirsk, Russia
cDepartment of Chemistry and Pharmacy, Friedrich-Alexander-University of Erlangen-Nuremberg, Egerlandstr. 1, 91058 Erlangen, Germany
dG. A. Razuvaev Institute of Organometallic Chemistry of RAS, Tropinina St. 49, 603950 Nizhny Novgorod, Russia
eN. I. Lobachevsky Nizhny Novgorod State University, Gagarin Ave., 23, 603950, Nizhny Novgorod, Russia
First published on 1st December 2015
Abstract
The first examples of samarium, europium, and ytterbium complexes with 3,6-di-tert-butyl-o-benzoquinone (3,6-dbbq) in the form of catecholate have been obtained by reactions of the quinone with the corresponding lanthanocenes, 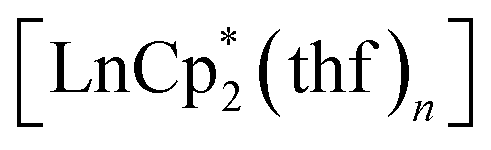 (n = 1 or 2) in solution. In the course of the reactions lanthanide ions lose one or two Cp* ligands, which take part in reduction of a quinone molecule into a catecholate anion (dbcat, 2−). As a result of the reactions, Sm and Yb clearly yield dimeric complexes [(LnCp*)2(dbcat)2], where each Ln ion loses one Cp* ligand. Eu forms a trimeric complex [(EuCp*)(Eu·thf)2(dbcat)3], in which one Eu ion is coordinated by one Cp* ligand, while two Eu ions have lost all Cp* ligands and are coordinated by THF molecules instead. Magnetic properties corroborate the assignment of oxidation states made on the basis of single-crystal X-ray diffraction: all the quinone ligands are present in the catecholate state; both Sm/Yb ions in the dimers are in the +3 oxidation state, whereas the Eu trimer contains two Eu(II) and one Eu(III) ions. Cyclovoltammetry studies show the presence of two reversible oxidation waves for all complexes, presumably concerned with the redox transitions of the dbcat ligands.
(n = 1 or 2) in solution. In the course of the reactions lanthanide ions lose one or two Cp* ligands, which take part in reduction of a quinone molecule into a catecholate anion (dbcat, 2−). As a result of the reactions, Sm and Yb clearly yield dimeric complexes [(LnCp*)2(dbcat)2], where each Ln ion loses one Cp* ligand. Eu forms a trimeric complex [(EuCp*)(Eu·thf)2(dbcat)3], in which one Eu ion is coordinated by one Cp* ligand, while two Eu ions have lost all Cp* ligands and are coordinated by THF molecules instead. Magnetic properties corroborate the assignment of oxidation states made on the basis of single-crystal X-ray diffraction: all the quinone ligands are present in the catecholate state; both Sm/Yb ions in the dimers are in the +3 oxidation state, whereas the Eu trimer contains two Eu(II) and one Eu(III) ions. Cyclovoltammetry studies show the presence of two reversible oxidation waves for all complexes, presumably concerned with the redox transitions of the dbcat ligands.
Introduction
Lanthanide-based single-molecule magnets (SMMs) currently attract considerable attention owing to the large spin values and high magnetic anisotropy of lanthanide ions.1–3 At the same time, lanthanide complexes with stable radical ligands, such as nitroxides, exhibit weak magnetic exchange coupling between metal and ligand spin carriers because of the deep lying f-orbitals.4–6 Recently, two groups of dinuclear lanthanide complexes were reported, nitrogen bridged [{(Me3Si)2(N)2(thf)Ln}2(μ-N2)]− (Ln = Gd, Tb, Dy, Ho, Er; thf = tetrahydrofuran),7,8 and bipyrimidyl-bridged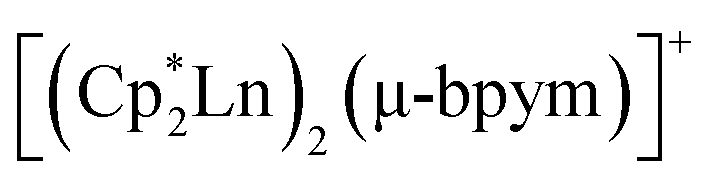 (Ln = Gd, Tb, Dy; Cp* = pentamethylcyclopentadyenyl; bpym = 2,2′-bipyrimidyl),9 of which those with Gd, Tb and Dy ions demonstrate remarkably high exchange coupling constants and blocking temperature values. In these compounds the radical ligands possess a strongly reducing behaviour. Calculations made for the related complexes showed that the magnetic coupling between the Ln centre and the paramagnetic ligand is likely to be more substantial in the case where both the lanthanide ion and the radical ligand have close redox potentials (i.e. Ln3+/2+ and L˙−/0).3 This conclusion is also supported by the case of substantial electronic and magnetic interactions between metal and ligand centres in heterospin complexes Cpx2YbL (Cpx = substituted cyclopentadienide), where L is 2,2′-bipyridine, 1,10-phenanthroline, 2,2′:6′,2′′-terpyridine or other pyridine-based ligands,10,11 in which the interaction depends on the correlation of electronegativities of the ytterbocene fragment and the ligand.
(Ln = Gd, Tb, Dy; Cp* = pentamethylcyclopentadyenyl; bpym = 2,2′-bipyrimidyl),9 of which those with Gd, Tb and Dy ions demonstrate remarkably high exchange coupling constants and blocking temperature values. In these compounds the radical ligands possess a strongly reducing behaviour. Calculations made for the related complexes showed that the magnetic coupling between the Ln centre and the paramagnetic ligand is likely to be more substantial in the case where both the lanthanide ion and the radical ligand have close redox potentials (i.e. Ln3+/2+ and L˙−/0).3 This conclusion is also supported by the case of substantial electronic and magnetic interactions between metal and ligand centres in heterospin complexes Cpx2YbL (Cpx = substituted cyclopentadienide), where L is 2,2′-bipyridine, 1,10-phenanthroline, 2,2′:6′,2′′-terpyridine or other pyridine-based ligands,10,11 in which the interaction depends on the correlation of electronegativities of the ytterbocene fragment and the ligand.
In the context of the synthesis and investigation of new lanthanide complexes with possibly high exchange constants between a lanthanide ion and a ligand, the lanthanide complexes with o-benzoquinone-based (bq) ligands are promising. Indeed, the o-quinone based ligands can exist in three charged states (quinone, semiquinone (sq, 1−), and catecholate (cat, 2−)), so the redox transitions can vary in a wide range.12,13 Besides, the ligands can be functionalized in a number of ways, including conversion to iminoquinones,14 thus providing variable donor and steric behaviours and a possibility to tune metal–ligand interactions. Despite this prominent ligand diversity, only a limited number of quinone-based Ln(III) complexes have been described and magnetically studied until now.4–6,15,16 A few complexes of Ln(II) with sq and cat ligands are known; to the best of our knowledge, the only examples are homoleptic [Ln(dbcat)] (Ln = Sm, Eu) and heteroleptic [EuLi4(dbsq)2(dbcat)2(LiI)2(thf)6] complexes obtained by Bochkarev and co-workers in the reaction of EuI2 or Ln(N(SiMe3)2 with 3,5-di-tert-butyl-o-benzoquinone (3,5-dbbq).17 Lately, a ligand containing 3,6-di-tert-butyl-o-benzoquinone (3,6-dbbq) coupled with tetrathiafulvalene was employed for syntheses of bridged dimeric Ln complexes (Ln = Dy, Er, Yb); in the case of dysprosium, each Dy3+ ion was seen to behave as an independent single-ion magnet.18,19 The possibility of tuning the redox properties of a ligand by changing its substituents or by using iminoquinone derivatives14 can be used to enhance the magnetic interactions between the Ln ion and the ligand in its radical-ion form.
Substituted cyclopentadienyl (Cpx) and indenyl (Ind) complexes of lanthanides(II) have been widely used for syntheses of complexes with radical ligands.20–29 In some cases, not only the Ln cation, but also Cpx and Ind ligands take part in the reduction process, and their role as a reducing agent is sufficiently induced by their bulkiness, in addition to the redox potential of the metal centre.
We were interested in finding new ways of producing the quinone complexes of lanthanides, for which easily accessible Cpx complexes can be good starting compounds, while using the 3,6-dbbq-based ligands could provide necessary solubility of the complexes and sterical protection owing to bulky butyl groups.
Results and discussion
Synthesis and crystal structures
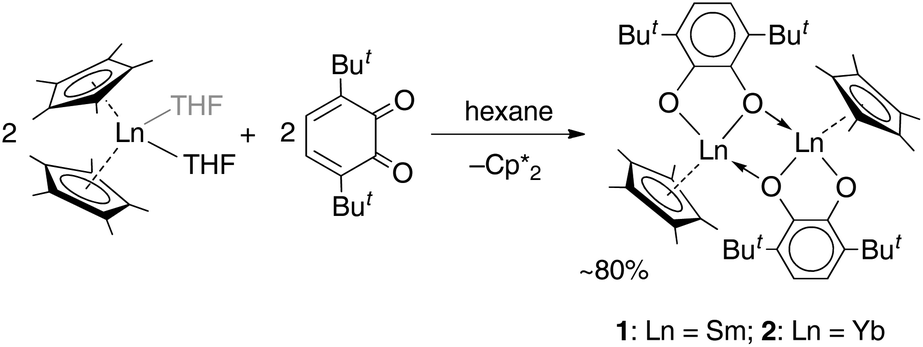
Cyclopentadienide complexes of lanthanides of either the LnCpx2 or LnCpx3 stoichiometry are well known starting materials, introduced largely by the work of Evans and coworkers.27,30,31 For o-benzoquinones, which possess a noticeable oxidative behaviour, the lanthanide complexes with Cp* ligands could serve as appropriate reducing counterparts owing to the reduced Ln(II) centre, as well as the Cp* ligand itself, which is readily oxidized to a neutral species, followed by dimerization.32The reactions of lanthanocenes(II) 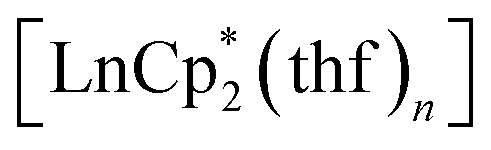 (Ln = Sm, n = 2; Ln = Yb, n = 1) with 3,6-dbbq were carried out in hexane solutions starting from deep-frozen mixtures. Upon warming to room temperature, the lanthanocene readily dissolves to give a green (Sm) or sky-blue (Yb) solution; upon successive dissolution of the quinone the reaction slowly proceeds to result in a yellow (Sm) or light-brown (Yb) clear solution and crystalline precipitate of the corresponding compound. The products [(SmCp*)2(dbcat)2] and [(YbCp*)2(dbcat)2] (1 and 2, correspondingly, Scheme 1) are well soluble in hexane and were crystallized from small volumes of this solvent; a by-product,
(Ln = Sm, n = 2; Ln = Yb, n = 1) with 3,6-dbbq were carried out in hexane solutions starting from deep-frozen mixtures. Upon warming to room temperature, the lanthanocene readily dissolves to give a green (Sm) or sky-blue (Yb) solution; upon successive dissolution of the quinone the reaction slowly proceeds to result in a yellow (Sm) or light-brown (Yb) clear solution and crystalline precipitate of the corresponding compound. The products [(SmCp*)2(dbcat)2] and [(YbCp*)2(dbcat)2] (1 and 2, correspondingly, Scheme 1) are well soluble in hexane and were crystallized from small volumes of this solvent; a by-product,  , is retained in the solution.
, is retained in the solution.
 | ||
| Scheme 1 | ||
Crystal structures were determined for both complexes 1 and 2, which possess close molecular structures and isostructural crystal lattices (Fig. 1). The binuclear complexes contain a crystallographic centre of inversion in the middle of the Ln–Ln vector with one quinone and one Cp* ligand per Ln atom. The quinone ligands are bound in the chelate-bridging mode: the two O atoms are coordinated to a Ln atom (2.3127(18) and 2.2269(17) Å for Sm, 2.2264(19) and 2.1285(18) Å for Yb), while one of them is additionally bound to a second Ln atom at a longer distance (by ca. 0.09 Å for Sm, 0.04 Å for Yb; see all distances in the captions to Fig. 1). It is noteworthy that the carbon atom C1 next to the μ-O atom is also located within a bonding distance to the Ln atom and the angles Ln–O1–C1 are slightly less than 90°. The mean-squared plane of the conjugated quinone ligand is nearly perpendicular to the plane formed by the Ln′, C1, and O1 atoms (84.4° for Sm, 82.1° for Yb), as well as to the flat Ln2O2 ring (84.8° for Sm, 89.5° for Yb). Out of more than a hundred structurally characterized complexes of transition metals (and several of lanthanides) with the 3,6-dbbq-based ligand, no analogous chelate-bridging mode has been documented to date (based on data from Cambridge Structural Database (CSD)33). The characteristic lengths of C–O and C–C bonds suggest that the quinone ligand acquires the catecholate (2−) state.17,34,35 The IR spectra of 1 and 2 are nearly identical, in accordance with the same coordination environment of the complexes (Fig. S1, ESI†). Consequently, both Ln ions in 1 and 2 are in the 3+ oxidation state, which is consistent with the rather light colour of the compounds. To obtain this composition, the 2e reduction of the neutral quinone requires 1e from the Ln ion and 1e from the Cp*− ligand, which corresponds to the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of the reagents:
:1 ratio of the reagents:
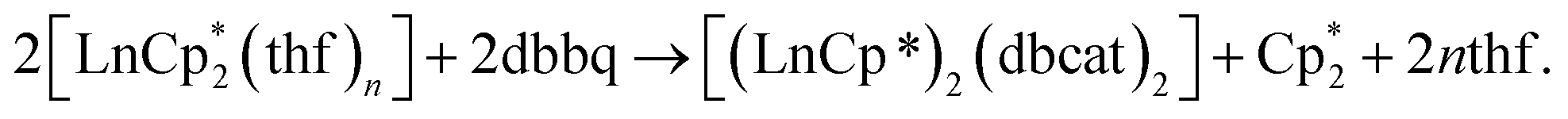
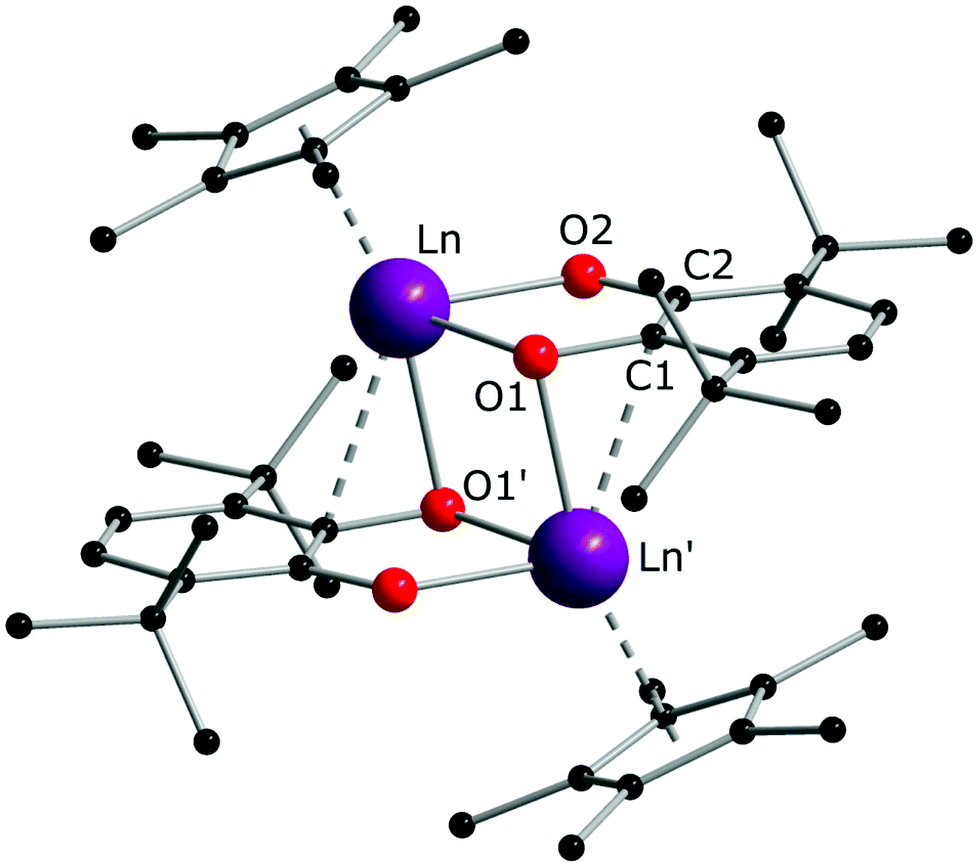 | ||
| Fig. 1 Molecular structure of complexes 1 and 2 in the crystal, exemplified by the structure of the Sm complex. Hydrogen atoms are omitted. Selected distances (Å) and angles (°) for Sm/Yb congeners: Ln–O1 2.3127(18)/2.2264(19); Ln–O2 2.2269(17)/2.1285(18); Ln′–O1 2.4052(16)/2.2686(16); C1–O1 1.375(3)/1.384(3); C2–O2 1.334(3)/1.334(3); C1–C2 1.429(3)/1.428(3); Ln′–C1 2.666(2)/2.612(2); O1–Ln–O2 69.77(6)/73.50(6); O1–Ln–O1′ 79.83(5)/78.58(6); Ln–O1–Ln′ 100.17(6)/101.42(6); Ln′–O1–C1 85.08(13)/87.83(13). | ||
The participation of the Cp* and related indenyl ligands in reduction processes of Ln complexes is well documented in cases of “sterically induced reduction” (SIR), that were described for the complexes with a sterically encumbered coordination sphere of a Ln ion.27,29 In our case, the addition of one quinone ligand to  is likely to proceed fast, concurrent with the oxidation of Ln2+ to Ln3+ and reduction of the dbbq to the semiquinone (1−) or catecholate (2−) state. Then, upon the formation of a dimer by coordination of the third O atom to the Ln3+ ion, the steric crowding becomes strong enough to facilitate the cleavage of the Cp* fragment accompanied (or followed) by further reduction of the quinone ligand to the catecholate state. This reaction sequence is especially noticeable during the synthesis of 1 or 2, where the characteristic colour of lanthanocene(II) does not appear in a cold solution, even before all of the quinone is dissolved; on the contrary, the blue colour of the reaction mixture is ascribed to the intermediate species [LnIII(Cp*)2(dbsq)], which then transforms into the brown product 2 upon stirring at room temperature, or, faster, upon slight warming. This sequence, i.e. a fast redox process connected with the oxidation of a metal centre followed by a slower process of sterically induced reduction, has been proven previously by Evans.27,36
is likely to proceed fast, concurrent with the oxidation of Ln2+ to Ln3+ and reduction of the dbbq to the semiquinone (1−) or catecholate (2−) state. Then, upon the formation of a dimer by coordination of the third O atom to the Ln3+ ion, the steric crowding becomes strong enough to facilitate the cleavage of the Cp* fragment accompanied (or followed) by further reduction of the quinone ligand to the catecholate state. This reaction sequence is especially noticeable during the synthesis of 1 or 2, where the characteristic colour of lanthanocene(II) does not appear in a cold solution, even before all of the quinone is dissolved; on the contrary, the blue colour of the reaction mixture is ascribed to the intermediate species [LnIII(Cp*)2(dbsq)], which then transforms into the brown product 2 upon stirring at room temperature, or, faster, upon slight warming. This sequence, i.e. a fast redox process connected with the oxidation of a metal centre followed by a slower process of sterically induced reduction, has been proven previously by Evans.27,36
The reaction of the europocene [Eu(Cp*)2(thf)] with 3,6-dbbq (Scheme 2) was carried out identically with Sm and Yb analogues, however, the colour of the resulting solution turned out to be much darker. The product [(EuCp*)(Eu·thf)2(dbcat)3] (3) is quite soluble in hexane, thus the yield of crystals was somewhat lower.
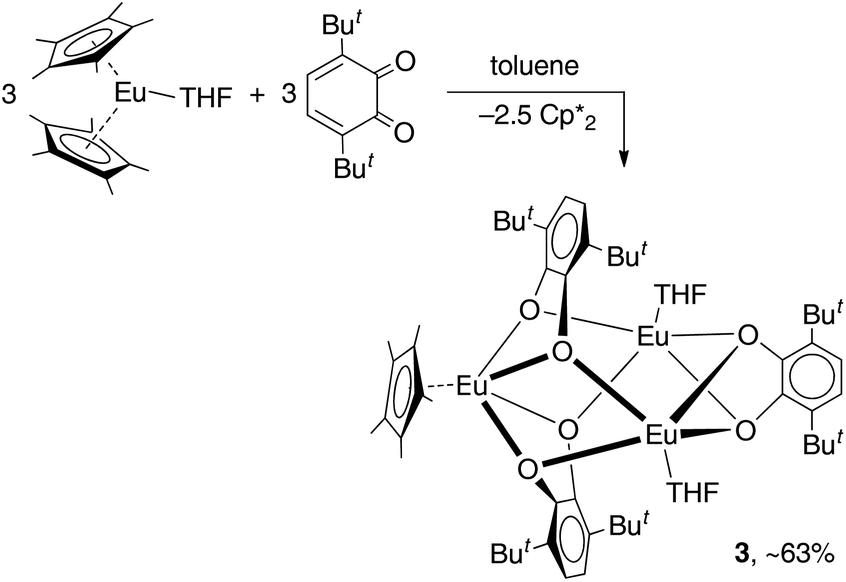 | ||
| Scheme 2 | ||
The crystal structure of 3 reveals a trinuclear complex (Fig. 2), where the three metal atoms are coordinated by three bridging quinone ligands. It can be conceived as a trigonal prism of six O atoms with all the square faces capped by Eu(L) vertices. Remarkably, the Eu centres have different ligand environments: all metal ions are bound to two bridging quinone ligands, while in the terminal position one Eu atom retains a Cp* ligand and the other two bear a thf molecule. There are two types of ligand bridging: the two dbbq ligands lying out of the Eu3 plane are of the μ3,η4 type, while the in-plane dbbq ligand acquires the μ,η4-bridging mode. The out-of-plane quinone ligands are located closer to the Eu1 atom: the Eu1–O bonds (2.280(2)–2.310(2) Å) are noticeably shorter than the bonds between the same O and the corresponding Eu2 or Eu3 atoms (2.487(2)–2.506(2) Å; see bond lengths in the Fig. 2 caption). Nevertheless, these ligands are not noticeably tilted towards Eu2 or Eu3; as opposed to the structures of 1 and 2, the angles between their mean-squared planes and the Eu3 plane are 79.2 and 77.4°, and all the distances Ln⋯C are much longer than corresponding Ln–O bonds. The in-plane quinone ligand is bound to the metal atoms nearly symmetrically with all the Eu–O bonds lying in the range 2.429(3)–2.457(3) Å and the mean-squared plane of the ligand being perpendicular (89.4°) to the Eu3 plane. All the C–O (1.356(4)–1.373(5) Å) and interjacent C–C bonds (1.419(5)–1.430(6) Å) in the quinone ligands point to their dianionic catecholate state. The IR spectrum of 3 indicates the same characteristic absorptions as those for 1 or 2 (Fig. S1, ESI†). Thus, Eu in 3 is present in two oxidation states, one cation of Eu3+ and two cations of Eu2+, which correspond to Eu–O bond distances and the darker colour, and are well reflected by magnetic properties of the complex (vide infra). The oxidation of Eu in the course of the reactions does not proceed to the full extent; instead, the Cp* ligands from two thirds of the starting europocene(II) are oxidized and lost. Again, the reagent stoichiometry is 1:1 as follows:
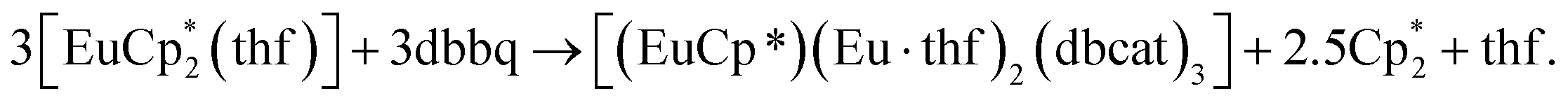
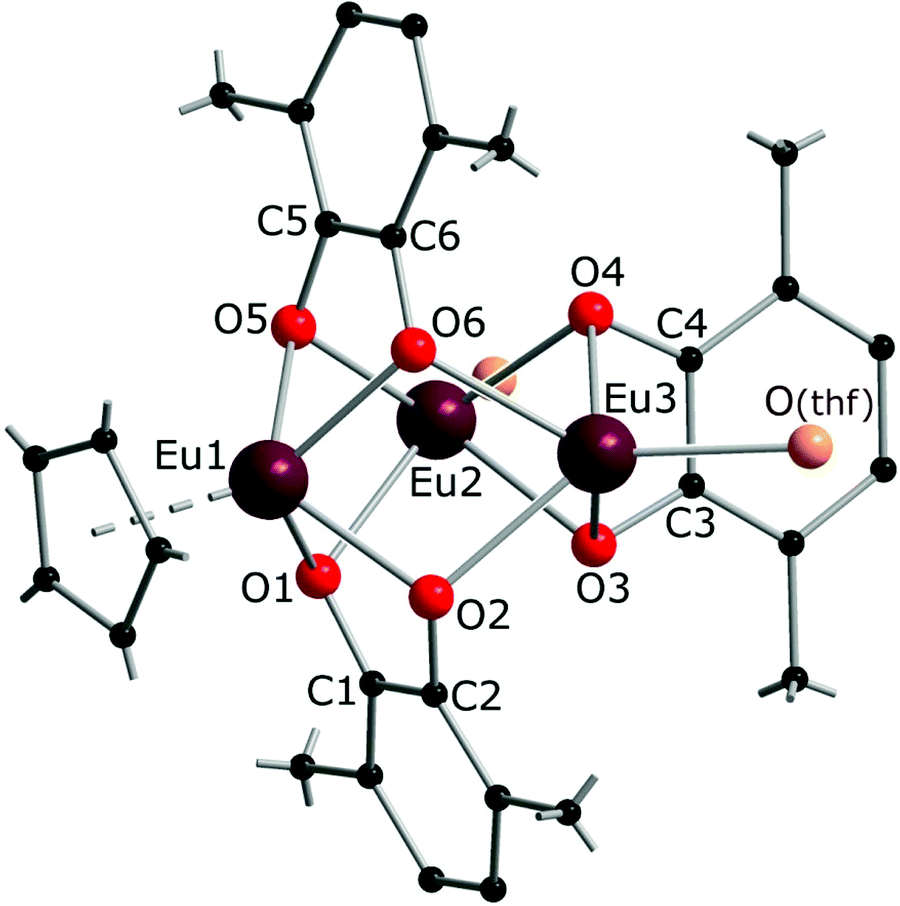 | ||
| Fig. 2 Molecular structure of complex 3 in the crystal. Hydrogen atoms are omitted, methyl groups and coordinated thf molecules are simplified. Selected distances (Å): Eu1–O1 2.280(2), Eu1–O2 2.289(2), Eu1–O5 2.300(2), Eu1–O6 2.311(2), Eu2–O1 2.506(2), Eu2–O5 2.504(2), Eu2–O3 2.429(3), Eu2–O4 2.439(2), Eu3–O2 2.494(2), Eu3–O6 2.487(2), Eu3–O3 2.457(2), Eu3–O4 2.457(3), C1–O1 1.373(5), C2–O2 1.371(4), C3–O3 1.356(4), C4–O4 1.359(5), C5–O5 1.368(4), C6–O6 1.362(5), C1–C2 1.420(5), C3–C4 1.430(6), C5–C6 1.419(5). | ||
The presence of unoxidized Eu2+ in the complex can be explained by its significantly lower reduction potential as compared to Sm and Yb low-valent metallocenes.27 It is noteworthy that in case the latter reaction is carried out in hexane, as in the case of compounds 1 and 2, small amounts of a pale-grey microcrystalline precipitate remain insoluble after recrystallization of the product 3. Changing the solvent from hexane to toluene results in a much cleaner reaction and the pale-grey by-product is formed in vanishingly small amounts. The IR spectrum of this by-product shows a very similar pattern to that of magnesium catecholate 4 (see below) and, according to elemental analysis, its composition is close to the formula [Eu(dbcat)(thf)]n (attempts to grow crystals suitable for X-ray structural analysis were unsuccessful; see the ESI† for further details). Presumably, the latter complex can be formed in the reaction of europocene and dbbq if only the Cp*− ligands take part in the reduction and Eu remains in the 2+ state.
Considering different reaction behaviours of lanthanocenes, it is not fully clear, which factors govern the partial or total loss of Cp* ligands from the Ln atoms, as well as Ln2+ → Ln3+ redox processes. Presumably, both the redox potentials of corresponding lanthanocenes and the steric bulkiness of the Cp* ligand in the coordination sphere of a given Ln ion have an influence. To make the reduction properties of the Cp*− anions more evident, one must exclude the possibility of a metal cation reducing the quinone. This can be achieved by involving in the reaction an analogous complex with a redox-innocent metal cation (2+), for which the alkaline-earth elements are a good choice. In the reaction of magnesocene 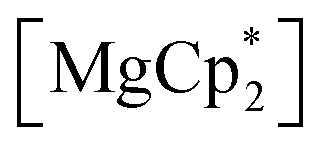 with dbbq (1:1) in thf carried out analogously to those of lanthanocenes, the solution appeared blue-green after mixing of the reagents and retained the same colour for several hours indicating the initial formation of semiquinone complexes. Considering that only Cp*− can act as a reductant, these complexes are supposedly [MgCp*(dbsq)(thf)n] species. After prolonged stirring the solution turned yellow and finally the already known complex [(Mg(thf)2)2(dbcat)2] (4) was isolated. This compound was initially obtained by Piskunov and co-workers in the reaction of amalgamated Mg with a quinone in THF.37 Substantial delay in the second step of the reduction process observed during the preparation of 4 is similar to that of the reactions with the lanthanocenes, and may be caused by the slow electron transfer between the Cp*− anion and the dbsq ligand in the same complex. However, to resolve whether this reductive action of Cp*− depends on the steric crowding in the Mg coordination sphere would require additional studies.
with dbbq (1:1) in thf carried out analogously to those of lanthanocenes, the solution appeared blue-green after mixing of the reagents and retained the same colour for several hours indicating the initial formation of semiquinone complexes. Considering that only Cp*− can act as a reductant, these complexes are supposedly [MgCp*(dbsq)(thf)n] species. After prolonged stirring the solution turned yellow and finally the already known complex [(Mg(thf)2)2(dbcat)2] (4) was isolated. This compound was initially obtained by Piskunov and co-workers in the reaction of amalgamated Mg with a quinone in THF.37 Substantial delay in the second step of the reduction process observed during the preparation of 4 is similar to that of the reactions with the lanthanocenes, and may be caused by the slow electron transfer between the Cp*− anion and the dbsq ligand in the same complex. However, to resolve whether this reductive action of Cp*− depends on the steric crowding in the Mg coordination sphere would require additional studies.
A dinuclear molecule of complex 4 (Fig. 3) resembles those of complexes 1 and 2 with regard to the coordination mode of the quinone ligand. The molecule is located in a common position of the crystal structure, so the bond lengths in both halves are not equal, but differ by no more than 0.01 Å (see the distances in the caption to Fig. 3). Unlike compounds 1 and 2, the Mg atoms are not bearing Cp* ligands, so the quinone ligands are present in the catecholate state, in agreement with the C–O and C1–C2 bond lengths,17,34,35 the IR spectral data,37 as well as its colourless appearance. The Mg2O2 ring is not flat (the angle between two Mg2O planes is 133.65(8)°), and both dbcat ligands are turned to the same side of the Mg2O2 butterfly, as opposed to the dimeric molecules of 1 and 2. Interestingly, the crystal structure of the similar dimeric pyridine complex [Mg2(3,6-dbbq)2(py)4]37 differs in the manner of coordination of quinone ligands: they acquire the same chelate-bridging mode as in 4, but both dbcat ligands chelate the same Mg ion and bind the second Mg ion with bridging O atoms to give 6- and 4-fold coordinated Mg centres in the same molecule. Apparently, both types of arrangements of catecholate ligands in dimeric complexes are close in energy and depend on the steric demand or rigidity of the second ligand (THF or pyridine). Hence, both Cp*− ligands of the initial Mg complex participate in the reduction of dbbq to dbcat ligands, and are replaced by THF molecules in the course of the reaction. The differences in the reaction behaviour between Mg and Ln complexes correlate with the ionic radii of the metal atoms involved: supposedly, the noticeably smaller Mg2+ (0.66 Å for a 5-fold coordination) constrains the Mg2O2 unit to acquire a bent geometry and does not provide enough space near the metal for a bulky Cp* ligand, unlike the much larger Sm3+ and Yb3+ ions (0.96 and 0.87 Å for a 6-fold coordination, correspondingly38). To corroborate this, experiments with larger alkaline-earth metals (Ca, Sr, Ba) will be useful, which could be considered for further work.
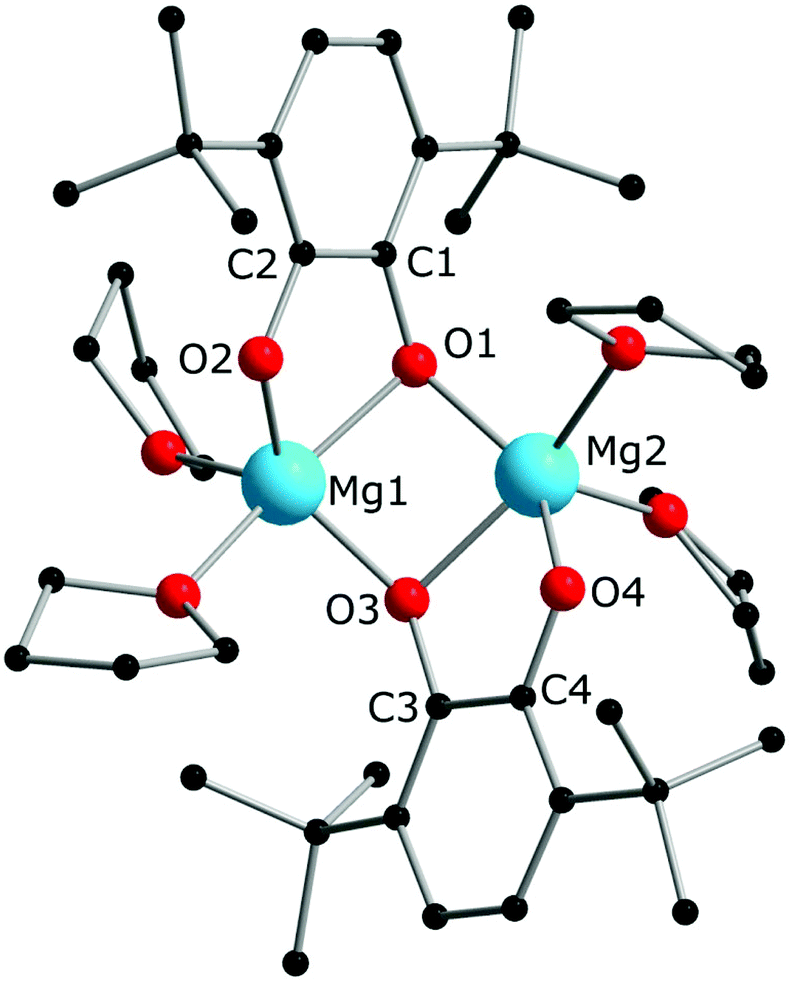 | ||
| Fig. 3 Molecular structure of complex 4 in the crystal. Hydrogen atoms are omitted. Selected distances (Å) and angles (°): Mg1–O1 2.0840(14); Mg1–O2 1.9374(15); Mg1–O3 1.9641(15); Mg2–O1 1.9745(15); Mg2–O3 2.0866(14); Mg2–O4 1.9360(14); C1–O1 1.369(2); C2–O2 1.341(2); C3–O3 1.369(2); C4–O4 1.338(2); C1–C2 1.429(3); C3–C4 1.430(3); O1–Mg1–O2 80.68(6); O3–Mg2–O4 80.63(6); O1–Mg1–O3 81.71(6); O1–Mg2–O3 81.40(6); Mg1–O1–Mg2 89.37(6); Mg1–O3–Mg2 89.58(6). | ||
Magnetic properties
Magnetic properties of paramagnetic species 1–3 were investigated in the solid state. The effective magnetic moment (μeff) of the Sm complex 1 is 2.17μB at room temperature (RT). Upon lowering the temperatures μeff decreases gradually reaching a value of 0.48μB at 2.6 K (Fig. 4). The typical values for the RT magnetic moment of mononuclear Sm(III) complexes are in the range μeff(SmIII) = 1.29–1.89μB.24 Thus, for a system with two non-coupled or weakly coupled Sm(III) centers, one expects to obtain μeff = 21/2μeff(SmIII) = 1.82–2.67μB. The experimentally observed value for 1 falls in the expected range, thus supporting the assignment of oxidation states based on crystallographic data (vide supra). Low-temperature variable-field measurements do not reveal saturation of magnetization at T = 2 K and magnetic fields up to 5 T (ESI†).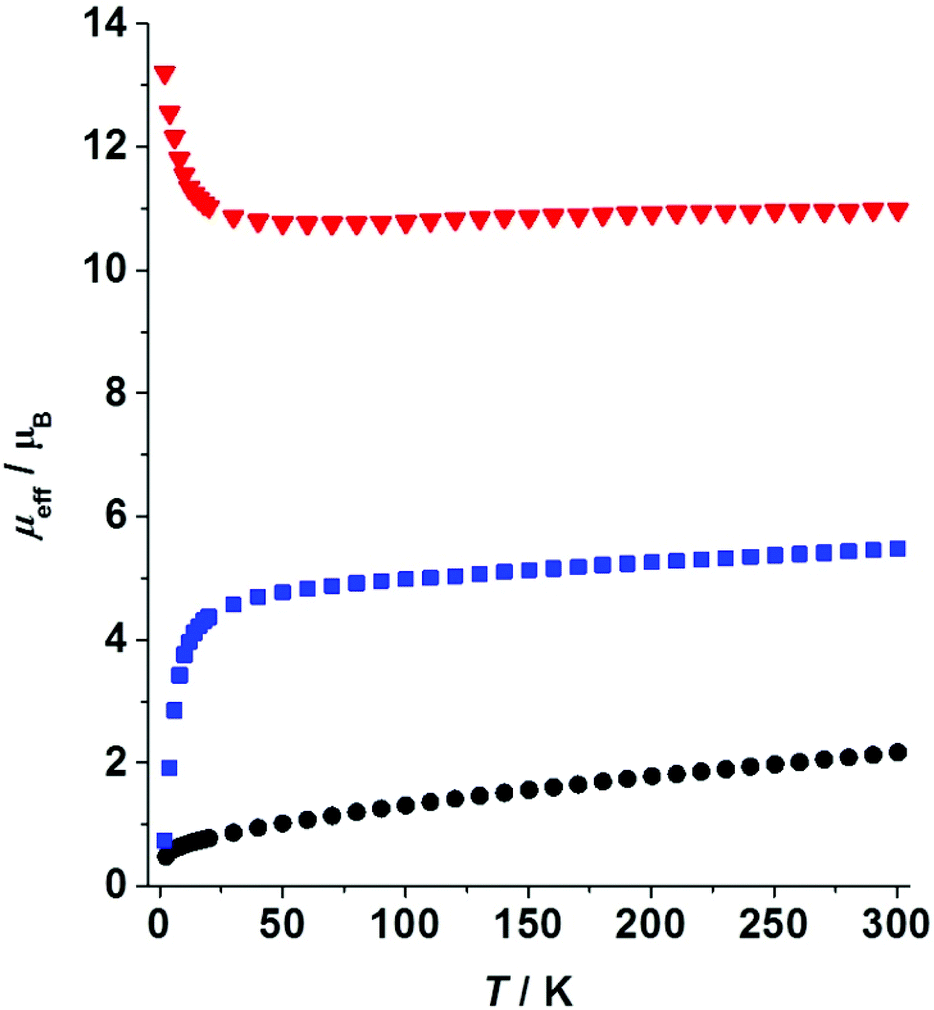 | ||
| Fig. 4 Variable-temperature effective magnetic moments measured at external magnetic field 0.1 T: 1 – black circles, 2 – blue squares, 3 – red triangles. | ||
The magnetic moment of Yb complex 2 measured at RT is 5.48μB. Upon lowering the temperature, the moment decreases gradually to a value of 4.69μB at 40 K followed by an abrupt decrease to 0.74μB recorded at 2 K (Fig. 4). The latter points to antiferromagnetic interactions present in the solid 2. A mononuclear Yb(III) complex is expected to have a RT moment in the range 4.3–4.9μB.39–41 Thus, for a dinuclear complex we expect to obtain μeff = 21/2μeff(YbIII) = 6.1–6.9μB. The experimental value is slightly lower than the predicted one. This is likely due to some diamagnetic or weakly paramagnetic impurities present in the solid 2. Alternatively, a quantum admixture of Yb(II) and Yb(III) states cannot be fully excluded.10,11,20 No saturation of magnetization was observed within 0–5 T at 2 K (ESI†).
Magnetic susceptibility measurements performed on a trinuclear Eu complex 3 reveal a RT magnetic moment of 10.98μB. This moment is nearly constant in the temperature range 30–300 K (Fig. 4). Upon cooling below 30 K the magnetic moment increases gradually reaching a value of 13.21μB at 2 K. This behaviour points to the presence of weak ferromagnetic interactions in the solid 3. However, the ferromagnetic behaviour can be suppressed at high magnetic fields (H = 5 T, see the ESI†). Taking into account the common RT magnetic moments for mononuclear complexes of Eu(II) (7.6–8.0μB) and Eu(III) (3.7–4.2μB),42 the moment for 3 is calculated as (2μeff2(EuII) + μeff2(EuIII))1/2 = 11.4–12.1μB. The experimentally observed value (10.98μB) is slightly lower than that predicted. This is likely due to the presence of a small amount of diamagnetic or weakly paramagnetic impurities in the highly oxygen-sensitive sample 3. Thus, the magnetic data confirm the presence of one Eu(III) and two Eu(II) ions in 3. Note that a much higher RT magnetic moment (13.3–14.0μB) would be expected for an uncoupled system with three Eu(II) ions and a ligand-radical. At very low temperatures magnetization saturates showing a plateau at 11–12NaμB in the variable-field series (ESI†). Due to the detected ferromagnetic interactions, 3 might exhibit SMM properties, which was investigated by ac susceptibility measurements. Unfortunately, no ac signal was observed, which precludes slow magnetic relaxation and thus SMM behaviour.
Cyclic voltammetry
It is well known that partial oxidation of catecholate ligands in the coordination sphere would potentially give complexes with radical anionic ligands, which should alter the magnetic interactions and can provide additional functionality to the complex.4,5 Moreover, two Eu2+ centres in complex 3 should be liable to oxidation; the approaches to oxidize the Ln centres in the paramagnetic complexes are well described for ytterbocene derivatives.10,11,21 It should be noted that the electrochemistry of 3,5-dbbq and its complexes with transition metals has been well studied,43–47 while for 3,6-dbbq only several complexes of main group elements are characterized.48 To study the redox behaviour, CV measurements of all complexes were performed. The lanthanide complexes 1–3 behave similarly at scan rates of 0.1–2.0 V s−1 and reveal two reversible oxidation waves in the positive region (Fig. 5 and Table 1). For highly related 3,5-dbbq complexes, including the quinone itself, most of the oxidation processes associated with cat to sq transitions proceed in the range from ca. −1.7 to −1.0 V, while those corresponding to sq to cat transitions proceed from ca. −1.0 to −0.2 V vs. Fc+/Fc.43,49–51 These values of potentials and corresponding peak currents were reported to be highly dependent on concentration of water and (for neat quinone) of metal ions, which form either protonated forms or metal complexes and thus shift the potentials to up to +0.3 V. The two reversible oxidation waves observed in our case possess somewhat higher half wave potentials (E1/2, ca. +0.5 and +0.9 V vs. Fc+/Fc); hence, they may not correspond to the purely cat–sq–quinone transitions in the coordination sphere. On the other hand, similar oxidation potentials are ascribed to such transitions in the complexes [(3,6-dbcat)SbAr3]48 (ca. +0.8 and +1.3 V vs. Ag/AgCl/KCl, which corresponds to +0.4 and +0.9 V vs. Fc+/Fc52), and to the 2e oxidation of the catechol 3,5-dbcatH243 (+1.19 V vs. a saturated calomel electrode, which corresponds to +0.81 V vs. Fc+/Fc52). Similarly, the Mg catecholate 4 shows two reversible waves at quite high potentials as well (E1/2 −0.31 and +0.66 V vs. Fc+/Fc), so these high values can be explained by the extensive coordination of catecholate ligands (the chelate-bridging coordination mode), as in all the complexes under study, or by more complex electrochemical reactions, accompanying the oxidation of catecholates. Peak heights (Ip) are proportional to the square root of the scan rate (according to the Randles–Sevcik equation), which points to the diffusion control of the processes. The values of ΔE are nearly independent of the scan rate but exceed 100 mV, which can stand for non-linear diffusion.
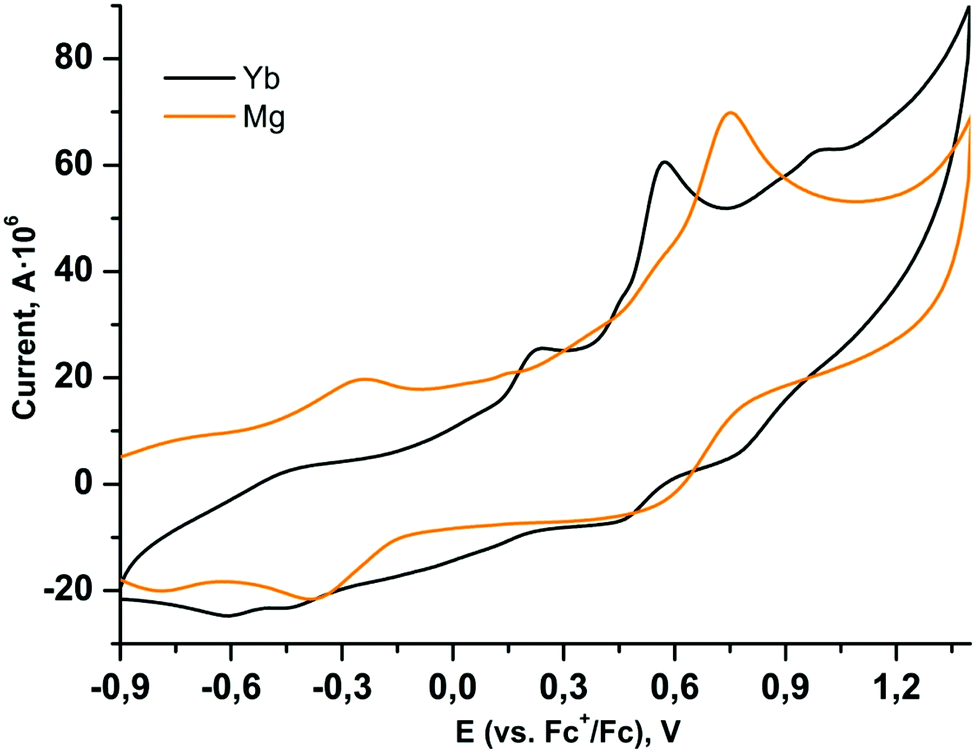 | ||
| Fig. 5 Cyclic voltammograms of complexes 2 (black curve) and 4 (orange curve), scan rate 0.5 V s−1. | ||
| Complex | E a, V | E 1/2, V | |
|---|---|---|---|
| Cp*− − e− = Cp* | dbcat2− − e− = dbsq−˙ | dbsq−˙ − e− = dbbq | |
| 1 | +0.25 | +0.52 | +0.89 |
| 2 | +0.23 | +0.50 | +0.88 |
| 3 | — | +0.51 | +0.88 |
| 4 | — | −0.31 | +0.65 |
Complexes 1 and 2 possess additional irreversible oxidation peaks (Eaca. +0.23 V) which probably correspond to the oxidation of the Cp*− ligand. Cp*− is expected to be oxidized at lower potentials than Cat2− and leaves as 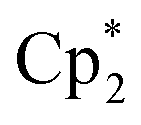 , in accordance with the chemical behaviour of these groups during the synthesis of 1–4. There are two irreversible peaks of smaller intensity in the negative region for 1 and 2 (−0.45 and −0.60 V vs. Fc+/Fc), which can correspond to the reduction of the species obtained upon oxidation of Cp* ligands.53 These irreversible oxidation and reduction peaks are absent in the case of 4, and, rather unexpectedly, in the case of 3; the latter fact cannot be ascribed to the insufficient concentration of Cp* ligands, since the concentration of complexes was maintained at the same level. Potentially, the reaction with the remaining water could lead to the partial removal of Cp* ligands owing to hydrolysis, but the preservation of the initial colour of the solutions during the CV experiments, and careful preparation of the solvent and solutions (similarly to the other complexes) does not support this supposition. More uncommonly, the Eu curves do not contain any waves attributable to the Eu2+/Eu3+ transition; presumably, this wave is outside of the measurement window (−2.6 to +1.4 V vs. Fc+/Fc).
, in accordance with the chemical behaviour of these groups during the synthesis of 1–4. There are two irreversible peaks of smaller intensity in the negative region for 1 and 2 (−0.45 and −0.60 V vs. Fc+/Fc), which can correspond to the reduction of the species obtained upon oxidation of Cp* ligands.53 These irreversible oxidation and reduction peaks are absent in the case of 4, and, rather unexpectedly, in the case of 3; the latter fact cannot be ascribed to the insufficient concentration of Cp* ligands, since the concentration of complexes was maintained at the same level. Potentially, the reaction with the remaining water could lead to the partial removal of Cp* ligands owing to hydrolysis, but the preservation of the initial colour of the solutions during the CV experiments, and careful preparation of the solvent and solutions (similarly to the other complexes) does not support this supposition. More uncommonly, the Eu curves do not contain any waves attributable to the Eu2+/Eu3+ transition; presumably, this wave is outside of the measurement window (−2.6 to +1.4 V vs. Fc+/Fc).
It is evident that the full explanation of the redox processes in the Ln–catecholate systems requires more systematical study.43,51 Chemical oxidation (e.g. with AgI or FcPF6) may be useful to obtain similar complexes with semiquinolate radicals; in the case of the compounds with Cp* ligands it may lead to the oxidation and substitution of the latter, before the oxidation of the dbcat ligands, which can serve as a method for introducing other ligands in the Ln coordination sphere.
Experimental
General remarks
All operations were carried out in evacuated vessels or ampules, the compounds were handled in an argon glove-box. The starting reagents, dbbq,54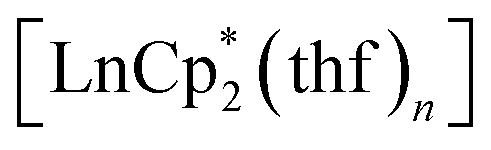 ,30,55 and
,30,55 and 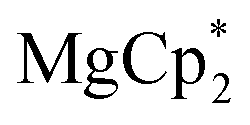 56,57 were prepared according to known methods. Solvents were distilled under inert atmospheres over common drying agents, stored with the addition of a Na–K alloy prior to use, and transferred under vacuum. The IR spectra were recorded in KBr pellets by using a FT-801 Fourier spectrometer (Simex). Elemental analysis for C, H was carried out by using an Euro EA 3000 analyzer (Eurovector). NMR spectra were recorded on an Avance 500 spectrometer (Bruker) and referenced to the solvent signals.
56,57 were prepared according to known methods. Solvents were distilled under inert atmospheres over common drying agents, stored with the addition of a Na–K alloy prior to use, and transferred under vacuum. The IR spectra were recorded in KBr pellets by using a FT-801 Fourier spectrometer (Simex). Elemental analysis for C, H was carried out by using an Euro EA 3000 analyzer (Eurovector). NMR spectra were recorded on an Avance 500 spectrometer (Bruker) and referenced to the solvent signals.
Syntheses of the compounds [(SmCp*)2(dbcat)2] (1) and [(YbCp*)2(dbcat)2] (2)
Synthesis was carried out in a 2-section ampoule, bent at a 90° angle. In one section the solid reagents,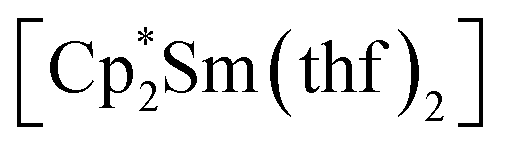 (0.340 g, 0.602 mmol) or
(0.340 g, 0.602 mmol) or 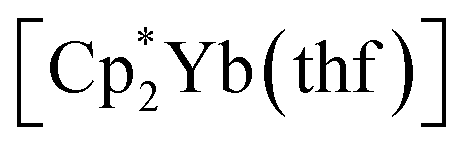 (0.310 g, 0.601 mmol) and 3,6-di-t-butyl-o-benzoquinone (0.132 g, 0.600 mmol) were placed. Under cooling with liquid N2 20 ml of hexane were condensed into the same section, and the ampoule was flame-sealed. The mixture was warmed with mixing to room temperature, then stirred at 60 °C for 10 h. After settling of the precipitate, the solution was decanted into the second section. Then the second section was positioned vertically and heated to ca. 10 °C above room temperature. Driven by the temperature gradient, slow extraction of the hexane soluble mixture components occurred, and the crystals (yellow for Sm, blue-green for Yb, suitable for XRD) slowly grew. Yields: 1, 0.233 g (77%); 2, 0.260 (82%).
(0.310 g, 0.601 mmol) and 3,6-di-t-butyl-o-benzoquinone (0.132 g, 0.600 mmol) were placed. Under cooling with liquid N2 20 ml of hexane were condensed into the same section, and the ampoule was flame-sealed. The mixture was warmed with mixing to room temperature, then stirred at 60 °C for 10 h. After settling of the precipitate, the solution was decanted into the second section. Then the second section was positioned vertically and heated to ca. 10 °C above room temperature. Driven by the temperature gradient, slow extraction of the hexane soluble mixture components occurred, and the crystals (yellow for Sm, blue-green for Yb, suitable for XRD) slowly grew. Yields: 1, 0.233 g (77%); 2, 0.260 (82%).
Synthesis of the complex [(EuCp*)(Eu·thf)2(dbcat)3] (3)
Solid reagents,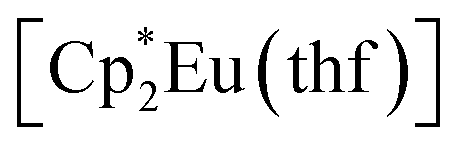 (0.130 g, 0.262 mmol), 3,6-di-t-butyl-o-benzoquinone (0.057 g, 0.26 mmol) were placed in a Schlenk tube with a Teflon stopper, and 10 ml of toluene were condensed under cooling with liquid N2. The mixture turned intense-blue upon warming, then vinous-red after stirring at room temperature for 1 h. The mixture was stirred overnight and then evaporated to dryness. The solids were washed with pentane to remove
(0.130 g, 0.262 mmol), 3,6-di-t-butyl-o-benzoquinone (0.057 g, 0.26 mmol) were placed in a Schlenk tube with a Teflon stopper, and 10 ml of toluene were condensed under cooling with liquid N2. The mixture turned intense-blue upon warming, then vinous-red after stirring at room temperature for 1 h. The mixture was stirred overnight and then evaporated to dryness. The solids were washed with pentane to remove 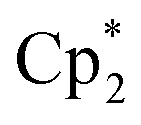 and extracted with pentane in a bent ampoule analogously to the procedure described above. Red crystals were separated by decantation and were suitable for XRD. Yield 0.075 g (63%).
and extracted with pentane in a bent ampoule analogously to the procedure described above. Red crystals were separated by decantation and were suitable for XRD. Yield 0.075 g (63%).
Reaction of 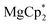 with dbbq
with dbbq
The solid reagents, 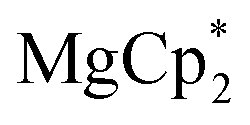
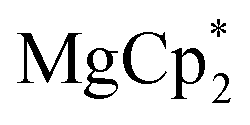 (0.104 g, 0.354 mmol) and 3,6-di-t-butyl-o-benzoquinone (0.075 g, 0.34 mmol), were placed in a Schlenk tube with a Teflon stopcock; 10 mL of THF were vacuum-transferred while cooling the reaction tube with liquid N2. The mixture was allowed to warm; successive mixing at room temperature resulted in colour change from emerald-green to yellow in ca. 4 h. The volume of the solution was reduced to 5 mL under vacuum, 15 mL of hexane was vacuum-transferred onto it, and the mixture was allowed to stand at −18 °C for a few days, to yield 4 as colourless crystals, suitable for XRD. The identity of the compound with the previously described complex37 was confirmed by NMR (C6D6) and elemental analysis. 1H NMR: 6.77 (dbcat CH), 3.79 (br.s., thf Hα), 1.67 (thf Hβ), 1.48 ppm (But). 13C NMR: 154.0 (dbcat C–O), 131.9 (dbcat C–But), 115.1 (dbcat CH), 70.9 (thf Cα), 34.9 (dbcat CMe3), 32.3 (dbcat CH3), 25.4 ppm (thf Cβ). The IR spectrum is somewhat different from the reported one measured in Nujol mull, νmax/cm−1: 3086m, 2952s, 2904s, 2870m sh, 1540m, 1483s, 1460w, 1442w, 1399s, 1383sh, 1359w, 1313w, 1295w, 1281m, 1239m sh, 1225s, 1205sh, 1177w, 1151s, 1026s, 976s, 940s, 878m, 812m, 789m, 684s, 655m, 565m. Yield 0.077 g (56%).
(0.104 g, 0.354 mmol) and 3,6-di-t-butyl-o-benzoquinone (0.075 g, 0.34 mmol), were placed in a Schlenk tube with a Teflon stopcock; 10 mL of THF were vacuum-transferred while cooling the reaction tube with liquid N2. The mixture was allowed to warm; successive mixing at room temperature resulted in colour change from emerald-green to yellow in ca. 4 h. The volume of the solution was reduced to 5 mL under vacuum, 15 mL of hexane was vacuum-transferred onto it, and the mixture was allowed to stand at −18 °C for a few days, to yield 4 as colourless crystals, suitable for XRD. The identity of the compound with the previously described complex37 was confirmed by NMR (C6D6) and elemental analysis. 1H NMR: 6.77 (dbcat CH), 3.79 (br.s., thf Hα), 1.67 (thf Hβ), 1.48 ppm (But). 13C NMR: 154.0 (dbcat C–O), 131.9 (dbcat C–But), 115.1 (dbcat CH), 70.9 (thf Cα), 34.9 (dbcat CMe3), 32.3 (dbcat CH3), 25.4 ppm (thf Cβ). The IR spectrum is somewhat different from the reported one measured in Nujol mull, νmax/cm−1: 3086m, 2952s, 2904s, 2870m sh, 1540m, 1483s, 1460w, 1442w, 1399s, 1383sh, 1359w, 1313w, 1295w, 1281m, 1239m sh, 1225s, 1205sh, 1177w, 1151s, 1026s, 976s, 940s, 878m, 812m, 789m, 684s, 655m, 565m. Yield 0.077 g (56%).
X-ray crystallography
Single-crystal X-ray diffraction data of 1–4 were collected on a Bruker-Nonius X8 Apex CCD diffractometer at 150(2) K using graphite monochromatized Mo Kα radiation (λ = 0.71073 Å). The standard technique was used (combined φ and ω scans of narrow frames). Data reduction and multi-scan absorption were carried out using SADABS.58–60 The structures were solved by direct methods and refined by full-matrix least-squares on F2 using the SHELXTL software package.58–60 All non-hydrogen atoms were refined anisotropically. Hydrogen atoms of organic ligands were located geometrically and refined as riding on their parent atoms. Crystallographic data and selected refinement details are given in Table 2. Crystallographic data have been deposited at the Cambridge Crystallographic Data Centre under the reference numbers CCDC 1409389–1409392 for 1–4, respectively.| 1 | 2 | 3 | 4 | |
|---|---|---|---|---|
| Empirical formula | C48H70O4Sm2 | C48H70O4Yb2 | C60H91Eu3O8 | C48H80Mg2O9 |
| Formula weight | 1011.74 | 1057.12 | 1396.21 | 849.74 |
| Temperature (K) | 150(2) | 150(2) | 150(2) | 150(2) |
| Crystal size (mm3) | 0.22 × 0.14 × 0.05 | 0.26 × 0.24 × 0.14 | 0.22 × 0.15 × 0.08 | 0.28 × 0.22 × 0.18 |
| Crystal system | Triclinic | Triclinic | Monoclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P21/n | P21/c |
| Z | 1 | 1 | 4 | 4 |
| a (Å) | 10.3712(2) | 10.3705(4) | 12.5855(3) | 21.0570(8) |
| b (Å) | 10.6440(2) | 10.5729(4) | 20.3033(6) | 12.0413(5) |
| c (Å) | 12.1789(3) | 12.1066(5) | 24.2945(7) | 19.0031(6) |
| α (°) | 70.4500(10) | 69.6750(10) | ||
| β (°) | 66.9300(10) | 65.6400(10) | 104.6510(10) | 95.7500(10) |
| γ (°) | 69.3720(10) | 70.2300(10) | ||
| V (Å3) | 1127.05(4) | 1104.04(8) | 6006.0(3) | 4794.1(3) |
| D calcd (g cm−3) | 1.491 | 1.590 | 1.544 | 1.177 |
| μ (Mo Kα) (mm−1) | 2.620 | 4.250 | 3.144 | 0.102 |
| θ range (°) | 2.10–27.53 | 1.90–27.59 | 1.73–27.69 | 1.94–27.51 |
| h, k, l indices range | −13 ≤ h ≤ 12; | −13 ≤ h ≤ 13; | −7 ≤ h ≤ 16; | −27 ≤ h ≤ 26; |
| −13 ≤ k ≤ 13; | −8 ≤ k ≤ 13; | −26 ≤ k ≤ 25; | −15 ≤ k ≤ 15; | |
| −15 ≤ l ≤ 15 | −11 ≤ l ≤ 15 | −31 ≤ l ≤ 29 | −12 ≤ l ≤ 24 | |
| F(000) | 514 | 530 | 2816 | 1856 |
| Reflections collected | 10517 |
8694 | 37990 |
36354 |
| Unique reflections | 5163 (Rint = 0.0291) | 5075 (Rint = 0.0140) | 13733 (Rint = 0.0251) |
10979 (Rint = 0.0522) |
| Observed reflections [I > 2σ(I)] | 4788 | 4771 | 10958 |
6770 |
| Parameters refined | 255 | 255 | 699 | 544 |
| R[F2 > 2σ(F2)] | R 1 = 0.0215 | R 1 = 0.0182 | R 1 = 0.0301 | R 1 = 0.0541 |
| wR2 = 0.0518 | wR2 = 0.0457 | wR2 = 0.0552 | wR2 = 0.1303 | |
| R(F2) (all data) | R 1 = 0.0247 | R 1 = 0.0205 | R 1 = 0.0482 | R 1 = 0.1039 |
| wR2 = 0.0530 | wR2 = 0.0466 | wR2 = 0.0595 | wR2 = 0.1438 | |
| GOOF on F2 | 1.051 | 1.048 | 1.025 | 1.046 |
| Δρmax, Δρmin (e Å−3) | 1.173, −0.764 | 1.305, −0.800 | 1.770, −0.858 | 0.564, −0.421 |
Magnetic measurements
Magnetic susceptibility data on solid samples were collected using a Quantum Design MPMS-XL SQUID magnetometer. The data were obtained for microcrystalline samples restrained within a polycarbonate gel capsule and corrected for underlying diamagnetism.Cyclic voltammetry
Cyclic voltammetry (CV) was performed on a Metrohm 797 VA Computrace instrument with a glassy carbon electrode as the working electrode and a saturated silver chloride reference electrode. The potentials were related to the standard platinum electrode. 0.15 M solution of tetrabutylammonium perchlorate (Bu4NClO4) in acetonitrile was used as an electrolyte. The cell was degassed by argon purging prior to CV measurements. Compounds were investigated within the potential window from −2 to 2 V at 25 °C; the value for the Fc+/Fc couple measured under the same conditions was +0.598 V. The formal half wave potentials (E1/2) were calculated as the midpoint between the anodic and cathodic peak potentials. Acetonitrile for electrochemical characterization was distilled under inert atmospheres over CaH2, then stored over CaH2 prior to use. The solutions (10−3 M) were prepared by vacuum transferring of freshly prepared CH3CN to the weighed compound, filled with purified argon after dissolution, and transferred into the cell by a Teflon pipe. Characteristic colours of the solutions remained unchanged during CV experiments indicating the absence of possible oxidation.Conclusions
We have found that lanthanocenes(II)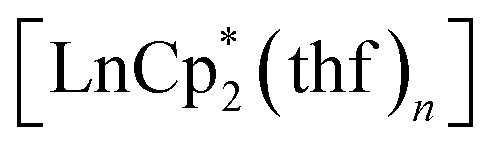 can serve as convenient precursors to the corresponding Ln(III) catecholate complexes. In their reactions with the o-quinone 3,6-dbbq, the initial reduction to the semiquinone complexes occurs fast, along with the oxidation of Ln(II) to Ln(III) for Ln = Sm and Yb, while the elimination of the Cp* ligand and formation of the catecholate complexes require longer times and can be dependent on steric crowding at the lanthanide centre. The reduction potential of the Ln ion plays an important role. Thus, in the reaction of europocene(II) the Cp* ligands are oxidized and lost before complete oxidation of the Eu centre, leading to the formation of a trinuclear mixed-valent complex containing one Eu(III) and two Eu(II) ions. The involvement of
can serve as convenient precursors to the corresponding Ln(III) catecholate complexes. In their reactions with the o-quinone 3,6-dbbq, the initial reduction to the semiquinone complexes occurs fast, along with the oxidation of Ln(II) to Ln(III) for Ln = Sm and Yb, while the elimination of the Cp* ligand and formation of the catecholate complexes require longer times and can be dependent on steric crowding at the lanthanide centre. The reduction potential of the Ln ion plays an important role. Thus, in the reaction of europocene(II) the Cp* ligands are oxidized and lost before complete oxidation of the Eu centre, leading to the formation of a trinuclear mixed-valent complex containing one Eu(III) and two Eu(II) ions. The involvement of 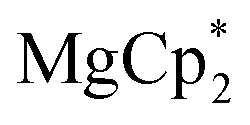 as a metallocene with a redox-silent metal into the reaction with 3,6-dbbq also shows successive two-step reduction processes, with the intermediate formation of semiquinolate species. It was shown earlier that substituted ytterbocenes display various reactivities with respect to the redox-active diazabutadiene ligands.29 Our results suggest that the use of lanthanocenes with a lower reduction potential (as for Eu vs. Yb) can extend this rich chemistry onto redox-active ligands with higher oxidation potentials, such as o-quinones.
as a metallocene with a redox-silent metal into the reaction with 3,6-dbbq also shows successive two-step reduction processes, with the intermediate formation of semiquinolate species. It was shown earlier that substituted ytterbocenes display various reactivities with respect to the redox-active diazabutadiene ligands.29 Our results suggest that the use of lanthanocenes with a lower reduction potential (as for Eu vs. Yb) can extend this rich chemistry onto redox-active ligands with higher oxidation potentials, such as o-quinones.
Acknowledgements
This work was supported by the Russian Foundation of Basic Research (grant no. 14-03-31268). MMK is grateful to the Fonds der Chemischen Industrie (Liebig Fellowship) and Deutsche Forschungsgemeinschaft (grant KH-279-2) for financial support. Prof. Karsten Meyer (Erlangen) is acknowledged for providing access to the magnetometer. The research is partly supported by a grant on the agreement between the Ministry of Education and Science of the Russian Federation and Lobachevsky State University of Nizhni Novgorod no. 02.B.49.21.0003 of August 27, 2013.Notes and references
- D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS PubMed.
- J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078–2085 RSC.
- W. W. Lukens, N. Magnani and C. H. Booth, Inorg. Chem., 2012, 51, 10105–10110 CrossRef CAS PubMed.
- A. Caneschi, A. Dei, D. Gatteschi, L. Sorace and K. Vostrikova, Angew. Chem., Int. Ed., 2000, 39, 246–248 CrossRef CAS.
- A. Dei, D. Gatteschi, J. Pécaut, S. Poussereau, L. Sorace and K. Vostrikova, C. R. Acad. Sci., Ser. IIc: Chim., 2001, 4, 135–141 CrossRef CAS.
- A. Caneschi, A. Dei, D. Gatteschi, S. Poussereau and L. Sorace, Dalton Trans., 2004, 1048–1055 RSC.
- J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, Nat. Chem., 2011, 3, 538–542 CrossRef CAS PubMed.
- J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, J. Am. Chem. Soc., 2011, 133, 14236–14239 CrossRef CAS PubMed.
- S. Demir, J. M. Zadrozny, M. Nippe and J. R. Long, J. Am. Chem. Soc., 2012, 134, 18546–18549 CrossRef CAS PubMed.
- M. Schultz, J. M. Boncella, D. J. Berg, T. D. Tilley and R. A. Andersen, Organometallics, 2002, 21, 460–472 CrossRef CAS.
- M. D. Walter, D. J. Berg and R. A. Andersen, Organometallics, 2006, 25, 3228–3237 CrossRef CAS.
- C. G. Pierpont, Coord. Chem. Rev., 2001, 216–217, 99–125 CrossRef CAS.
- C. G. Pierpont, Coord. Chem. Rev., 2001, 219–221, 415–433 CrossRef CAS.
- A. I. Poddel'sky, V. K. Cherkasov and G. A. Abakumov, Coord. Chem. Rev., 2009, 253, 291–324 CrossRef.
- G. A. Razuvaev, K. G. Shal'nova, L. G. Abakumova and G. A. Abakumov, Russ. Chem. Bull., 1977, 26, 1512–1515 CrossRef.
- A. V. Lobanov, G. A. Abakumov and G. A. Razuvaev, Dokl. Akad. Nauk, 1977, 235, 724–727 Search PubMed.
- D. M. Kuzyaev, D. L. Vorozhtsov, N. O. Druzhkov, M. A. Lopatin, E. V. Baranov, A. V. Cherkasov, G. K. Fukin, G. A. Abakumov and M. N. Bochkarev, J. Organomet. Chem., 2012, 698, 35–41 CrossRef CAS.
- F. Pointillart, S. Klementieva, V. Kuropatov, Y. Le Gal, S. Golhen, O. Cador, V. Cherkasov and L. Ouahab, Chem. Commun., 2012, 48, 714–716 RSC.
- F. Pointillart, V. Kuropatov, A. Mitin, O. Maury, Y. Le Gal, S. Golhen, O. Cador, V. Cherkasov and L. Ouahab, Eur. J. Inorg. Chem., 2012, 2012, 4708–4718 CrossRef CAS.
- C. H. Booth, D. Kazhdan, E. L. Werkema, M. D. Walter, W. W. Lukens, E. D. Bauer, Y.-J. Hu, L. Maron, O. Eisenstein, M. Head-Gordon and R. A. Andersen, J. Am. Chem. Soc., 2010, 132, 17537–17549 CrossRef CAS PubMed.
- G. Nocton, C. H. Booth, L. Maron and R. A. Andersen, Organometallics, 2013, 32, 5305–5312 CrossRef CAS.
- C. H. Booth, M. D. Walter, M. Daniel, W. W. Lukens and R. A. Andersen, Phys. Rev. Lett., 2005, 95, 267202 CrossRef CAS PubMed.
- T. Mehdoui, J.-C. Berthet, P. Thuéry, L. Salmon, E. Rivière and M. Ephritikhine, Chem. – Eur. J., 2005, 11, 6994–7006 CrossRef CAS PubMed.
- W. J. Evans and D. K. Drummond, J. Am. Chem. Soc., 1989, 111, 3329–3335 CrossRef CAS.
- W. J. Evans, D. K. Drummond, L. R. Chamberlain, R. J. Doedens, S. G. Bott, H. Zhang and J. L. Atwood, J. Am. Chem. Soc., 1988, 110, 4983–4994 CrossRef CAS.
- W. J. Evans, D. K. Drummond, S. G. Bott and J. L. Atwood, Organometallics, 1986, 5, 2389–2391 CrossRef CAS.
- W. J. Evans, J. Organomet. Chem., 2002, 647, 2–11 CrossRef CAS.
- N. A. G. Bandeira, C. Daniel, A. Trifonov and M. J. Calhorda, Organometallics, 2012, 31, 4693–4700 CrossRef CAS.
- A. A. Trifonov, Eur. J. Inorg. Chem., 2007, 2007, 3151–3167 CrossRef.
- W. J. Evans, J. W. Grate, H. W. Choi, I. Bloom, W. E. Hunter and J. L. Atwood, J. Am. Chem. Soc., 1985, 107, 941–946 CrossRef CAS.
- W. J. Evans, L. A. Hughes and T. P. Hanusa, Organometallics, 1986, 5, 1285–1291 CrossRef CAS.
- F. T. Edelmann, Lanthanocenes, in Metallocenes: Synthesis Reactivity Applications, ed. A. Togni and R. L. Halterman, Wiley-VCH Verlag GmbH, Weinheim, 1998 Search PubMed.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef ; CSD version 5.36, search date 01. Nov. 2015.
- O. Carugo, C. B. Castellani, K. Djinovic and M. Rizzi, J. Chem. Soc., Dalton Trans., 1992, 837–841 RSC.
- S. Bhattacharya, P. Gupta, F. Basuli and C. G. Pierpont, Inorg. Chem., 2002, 41, 5810–5816 CrossRef CAS PubMed.
- W. J. Evans, G. W. Nyce, M. A. Johnston and J. W. Ziller, J. Am. Chem. Soc., 2000, 122, 12019–12020 CrossRef CAS.
- A. V. Piskunov, A. V. Lado, G. A. Abakumov, V. K. Cherkasov, O. V. Kuznetsova, G. K. Fukin and E. V. Baranov, Russ. Chem. Bull., 2007, 56, 97–103 CrossRef CAS.
- R. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Cryst., 1976, 32, 751–767 CrossRef.
- J.-C. G. Bünzli, Acc. Chem. Res., 2006, 39, 53–61 CrossRef PubMed.
- M. A. AlDamen, S. Cardona-Serra, J. M. Clemente-Juan, E. Coronado, A. Gaita-Ariño, C. Martí-Gastaldo, F. Luis and O. Montero, Inorg. Chem., 2009, 48, 3467–3479 CrossRef CAS PubMed.
- J. Ruiz, G. Lorusso, M. Evangelisti, E. K. Brechin, S. J. A. Pope and E. Colacio, Inorg. Chem., 2014, 53, 3586–3594 CrossRef CAS PubMed.
- W. J. Evans, J. L. Shreeve and J. W. Ziller, Organometallics, 1994, 13, 731–733 CrossRef CAS.
- M. D. Stallings, M. M. Morrison and D. T. Sawyer, Inorg. Chem., 1981, 20, 2655–2660 CrossRef CAS.
- S. E. Jones, D. H. Chin and D. T. Sawyer, Inorg. Chem., 1981, 20, 4257–4262 CrossRef CAS.
- S. E. Jones, L. E. Leon and D. T. Sawyer, Inorg. Chem., 1982, 21, 3692–3698 CrossRef CAS.
- M. Haga, E. S. Dodsworth and A. B. P. Lever, Inorg. Chem., 1986, 25, 447–453 CrossRef CAS.
- J. P. Wilshire, L. Leon, P. Bosserman and D. T. Sawyer, J. Am. Chem. Soc., 1979, 101, 3379–3381 CrossRef CAS.
- A. I. Poddel'sky and I. V. Smolyaninov, Russ. J. Gen. Chem., 2010, 80, 538–540 CrossRef.
- M. W. Lehmann and D. H. Evans, J. Phys. Chem. B, 2001, 105, 8877–8884 CrossRef CAS.
- M. W. Lehmann and D. H. Evans, J. Electroanal. Chem., 2001, 500, 12–20 CrossRef CAS.
- A. René and D. H. Evans, J. Phys. Chem. C, 2012, 116, 14454–14460 Search PubMed.
- V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97–102 CrossRef CAS.
- F. Guyon, M. Fourmigué, P. Audebert and J. Amaudrut, Inorg. Chim. Acta, 1995, 239, 117–124 CrossRef CAS.
- V. A. Garnov, V. I. Nevodchikov, L. G. Abakumova, G. A. Abakumov and V. K. Cherkasov, Bull. Acad. Sci. USSR Div. Chem. Sci., 1987, 36, 1728 CrossRef.
- T. D. Tilley, R. A. Andersen, B. Spencer, H. Ruben, A. Zalkin and D. H. Templeton, Lanthanides and Actinides, in Synthetic Methods of Organometallic and Inorganic Chemistry, ed. F. T. Edelmann and W. A. Herrmann, Georg Thieme Verlag, Stuttgart, vol. 6, 1999 Search PubMed.
- J. Vollet, E. Baum and H. Schnöckel, Organometallics, 2003, 22, 2525–2527 CrossRef CAS.
- A. W. Duff, P. B. Hitchcock, M. F. Lappert, R. G. Taylor and J. A. Segal, J. Organomet. Chem., 1985, 293, 271–283 CrossRef CAS.
- Bruker, Bruker AXS Inc. (2000–2012), APEX2 (Version 2012.2-0), SAINT+ (Version 8.18c), SADABS (Version 2008/1), and SHELXTL (Version 6.14), Bruker Advanced X-ray Solutions, Madison, Wisconsin, USA, 2012 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional magnetic data, IR spectra, and X-ray crystallographic data. CCDC 1409389–1409392. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt03573b |
| This journal is © The Royal Society of Chemistry 2016 |