
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sodalite-like rare-earth carbonates: a study of structural transformation and diluted magnetism†
Yanyan
Wang
a,
Tian
Han
a,
You-Song
Ding
a,
Zhiping
Zheng
b and
Yan-Zhen
Zheng
*a
aCentre for Applied Chemical Research, Frontier Institute of Science and Technology, and MOE Key Laboratory for Nonequilibrium Synthesis and Modulation of Condensed Matter, College of Science, Xi'an Jiaotong University, Xi'an 710054, China. E-mail: zheng.yanzhen@xjtu.edu.cn; Fax: (+)86-29-83395366
bDepartment of Chemistry and Biochemistry, University of Arizona, Tucson, Arizona 85721, USA
First published on 24th November 2015
Abstract
A series of novel rare earth carbonates, RE3(OH)6(CO3)Cl (RE = Dy, Er, Y), with sodalite-like (SOD-like) zeolite topologies have been successfully synthesized by introducing an appropriate amount of CO32− from NaCO3 or atmospheric carbon dioxide as a template. Single-crystal X-ray diffraction reveals that the structure consists of the RE3(OH)63+ cationic framework with a SOD-like topology built from vertex-sharing [RE4(μ3-OH)4] cubes. The CO32− anions seal the 6-ring opening and Cl− anions situated in the channels to achieve charge balance. After calcination at 370 °C, the compound RE3(OH)6(CO3)Cl in situ transforms into a new phase formulated as RE3O4Cl. Interestingly, the structure of RE3O4Cl represents a new SOD-like open framework, associated with the removal of the CO32− from RE3(OH)6(CO3)Cl. The samples are characterized by thermogravimetric analysis (TGA), elemental analysis, X-ray photoelectron spectroscopy and magnetic studies. Furthermore, single-molecule magnet behaviours can be observed for the diluted samples of Dy0.0068Y2.9932(OH)6(CO3)Cl and Dy0.0068Y2.9932O4Cl with a Dy/Y molar ratio of up to 1/440 as well as Er0.19Y2.81(OH)6(CO3)Cl with an Er/Y ratio of 1/15, showing dominant single-ion effects.
Introduction
Open framework materials with porous structures, especially with zeolitic topologies, have attracted considerable attention for their permanent porosity, excellent thermal and chemical stabilities, and are widely used in industrial applications.1 Among the 229 zeolitic topologies, the traditional zeolite is built exclusively from TO4 tetrahedra, with the T atoms being from the main group or transition metal group, such as Si4+, Al3+, Ge4+, Zn2+, etc.2 Rare earth (RE) elements are ideal candidates to incorporate into open framework materials for the exploration of new applications, since they contain unique properties in photoluminescence, magnetism and so on.3 Several microporous lanthanide silicates that are built from SiO4 tetrahedra and LnOn (n ≥ 6) polyhedra have been successfully prepared.4Moreover, RE ions have become attractive candidates for constructing new single-ion magnets (SIMs) as well as single-molecular magnets (SMMs), because most of them have a large unquenched orbital angular momentum, which may bring significant anisotropy to the system.5 Synthetic efforts along this line have led to the discovery of many materials exhibiting SIM behaviour, expanding to lanthanide clusters and even complexes with 3-dimensional (3D) metal–organic framework structures.6 However, few magnetic studies are focused on RE inorganic materials with 3D open framework structures.7
The synthesis of traditional open framework materials is typically carried out in a gel medium under solvothermal conditions by using alkali metal ions or organic amines as templates or structure-directing agents (SDAs).8 However, this strategy is not suitable for the synthesis of RE open frameworks, as the RE ions have variable and high coordination numbers as well as poor directionality, which is significantly different from transition metal ions.9
For the design of RE open frameworks, it is worthwhile to refer to the synthesis of transition or lanthanide-based clusters,10 in which the carbonate (CO32−) anion is an extremely versatile bridging ligand. Aside from balancing the positive charges of metal ions (3d or 4f), each oxygen atom of CO32− anion in the clusters may act as a monodentate, bifunctional, or trifunctional ligand to generate carbonato-bridged complexes.11 The CO32− anion affords various coordination modes, ranging from simple chelation to more intricate coordination modes (μ3, μ4, μ5 and μ6) to construct complicated metal-carbonate cores.12 Therefore, polycarbonate-lanthanoid cores with diverse structures are highly appropriate to construct special RE compounds with 3D open framework topologies.13 The CO32− anion may also act as a SDA in the design of RE compounds with unique structures, since the CO32− anion in the carbonates can be easily removed by calcinations.
Herein we present a series of novel rare earth carbonates RE3(OH)6(CO3)Cl (RE = Dy, 1; Er, 2; Y, 3) that exhibit the sodalite-like structure constructed from the building unit (BU) of [RE4(μ3-OH)4] cubes by introducing an appropriate amount of CO32− from Na2CO3 or atmospheric carbon dioxide. Remarkably, the calcination of 1 at 370 °C leads to the transformation of Dy3(OH)6(CO3)Cl to a new open framework compound, Dy3O4Cl (denoted as 1-c). The structures were solved using single-crystal X-ray diffraction. By diluting 1, 1-c and 2 with Y atoms, the interesting dynamic magnetism of three samples (denoted as 1d (Dy0.0068Y2.9932(OH)6(CO3)Cl), 1d-c (Dy0.0068Y2.9932O4Cl) and 2d (Er0.19Y2.81(OH)6(CO3)Cl)) have been studied.
Results and discussion
Syntheses
Compounds 1, 2 and 3 were prepared by the hydrothermal reaction of a mixture of RECl3·6H2O, 2.5 M NaOH, HCl (37 wt%) and H2O to get a pH value of 6. Finally, Na2CO3 was introduced to make a reaction gel of 1.0RECl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2HCl:2.5NaOH:0.33Na2CO3:332H2O. The gel was added into a 12 mL Teflon-lined stainless steel autoclave and reacted at 200 °C for 72 h. Many parameters including the pH value and reaction temperature were found to affect the formation of RE3(OH)6(CO3)Cl. The mixture of HCl and NaOH was used to reach pH = 6 and provide excessive chloride that is beneficial for a high yield. Notably, the addition of Na2CO3 is essential for the formation of the product. It has been previously shown that the structures of RE clusters and cages are able to be modified by incorporating CO32− anions as a template, which is critical for the further aggregation and growth of the product.14 In this system, the reaction without Na2CO3 leads to another RE compound with a 2D structure, RE(OH)2Cl, as well as a few target crystals. It suggests that the CO32− anion is necessary to generate the 3D structure in this reaction, and it may be introduced from atmospheric carbon dioxide. Contrarily, if Na2CO3 is added in a quantity greater than the stoichiometric ratio of 0.33, another known RE compound RE(OH)CO3 is obtained, and the yield of this by-product increases. By further increasing the ratio of Na2CO3 to 1, the pure phase of the by-product can be produced. Hence the presence of an appropriate amount of CO32− is vital for the syntheses of 1, 2 and 3.
:1.2HCl:2.5NaOH:0.33Na2CO3:332H2O. The gel was added into a 12 mL Teflon-lined stainless steel autoclave and reacted at 200 °C for 72 h. Many parameters including the pH value and reaction temperature were found to affect the formation of RE3(OH)6(CO3)Cl. The mixture of HCl and NaOH was used to reach pH = 6 and provide excessive chloride that is beneficial for a high yield. Notably, the addition of Na2CO3 is essential for the formation of the product. It has been previously shown that the structures of RE clusters and cages are able to be modified by incorporating CO32− anions as a template, which is critical for the further aggregation and growth of the product.14 In this system, the reaction without Na2CO3 leads to another RE compound with a 2D structure, RE(OH)2Cl, as well as a few target crystals. It suggests that the CO32− anion is necessary to generate the 3D structure in this reaction, and it may be introduced from atmospheric carbon dioxide. Contrarily, if Na2CO3 is added in a quantity greater than the stoichiometric ratio of 0.33, another known RE compound RE(OH)CO3 is obtained, and the yield of this by-product increases. By further increasing the ratio of Na2CO3 to 1, the pure phase of the by-product can be produced. Hence the presence of an appropriate amount of CO32− is vital for the syntheses of 1, 2 and 3.
Crystal structures
Single-crystal structure analyses reveal that 1, 2 and 3 crystallize in the cubic space group Im![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) , Imm and Imm, respectively (Tables S1–S3, ESI†). The asymmetric unit for each compound contains 0.5 crystallographically distinct RE sites and 0.16667 crystallographically distinct C sites (Fig. S1, ESI†). Every RE atom is coordinated with six μ3-O (O(2) and O(3)) atoms from OH− in the framework and four O (O(1)) atoms from two μ6-CO32−. Notably, the occupancy of each O(1) atom is half, due to the disorder of CO32−. Thus, the RE atoms are eight-coordinated. The bond distances of Dy–Oμ3 in 1 vary from 2.302(11) Å to 2.347(7) Å, those of Er–Oμ3 in 2 vary from 2.290(13) Å to 2.328(8) Å and those of Y–Oμ3 in 3 vary from 2.333(10) Å to 2.379(6) Å (Table S4, ESI†). The RE–O(1) bond is longer, with an average value of 2.538 Å for Dy–O(1), 2.522 Å for Er–O(1) and 2.565 Å for Y–O(1). The structure and purity of samples are verified using PXRD experiments (Fig. S2†).
, Imm and Imm, respectively (Tables S1–S3, ESI†). The asymmetric unit for each compound contains 0.5 crystallographically distinct RE sites and 0.16667 crystallographically distinct C sites (Fig. S1, ESI†). Every RE atom is coordinated with six μ3-O (O(2) and O(3)) atoms from OH− in the framework and four O (O(1)) atoms from two μ6-CO32−. Notably, the occupancy of each O(1) atom is half, due to the disorder of CO32−. Thus, the RE atoms are eight-coordinated. The bond distances of Dy–Oμ3 in 1 vary from 2.302(11) Å to 2.347(7) Å, those of Er–Oμ3 in 2 vary from 2.290(13) Å to 2.328(8) Å and those of Y–Oμ3 in 3 vary from 2.333(10) Å to 2.379(6) Å (Table S4, ESI†). The RE–O(1) bond is longer, with an average value of 2.538 Å for Dy–O(1), 2.522 Å for Er–O(1) and 2.565 Å for Y–O(1). The structure and purity of samples are verified using PXRD experiments (Fig. S2†).
1, 2 and 3 contain the same structure. Take 1 for example, it consists of a Dy3(OH)63+ cationic framework, with CO32− anions located in the centre of the 6-ring opening and Cl− anions in the channels to achieve charge balance (Fig. 1a). As shown in Fig. 1b, the Dy atoms are linked with each other via six μ3-OH−, and each Dy atom belongs to two [Dy4(OH)4] units. The [Dy4(OH)4] unit is the only BU of the framework. In the centre of the 6-ring opening, a μ6-CO32− ligand binds the six dysprosium ions via a μ6–η2:η2:η2 bridging mode, by coordinating every O atom of CO32− to two Dy atoms on the 6-ring. This coordination mode of CO32− is common in clusters.12d Because of the high symmetry of the structure, the O atoms here are disordered in two places, each one having an occupancy of one half. The six BUs on the 6-ring opening are linked with each other by vertex-sharing and they form a geometry similar to that of {Ni12} or {Mn12} clusters, which are also templated by CO32− anions.15
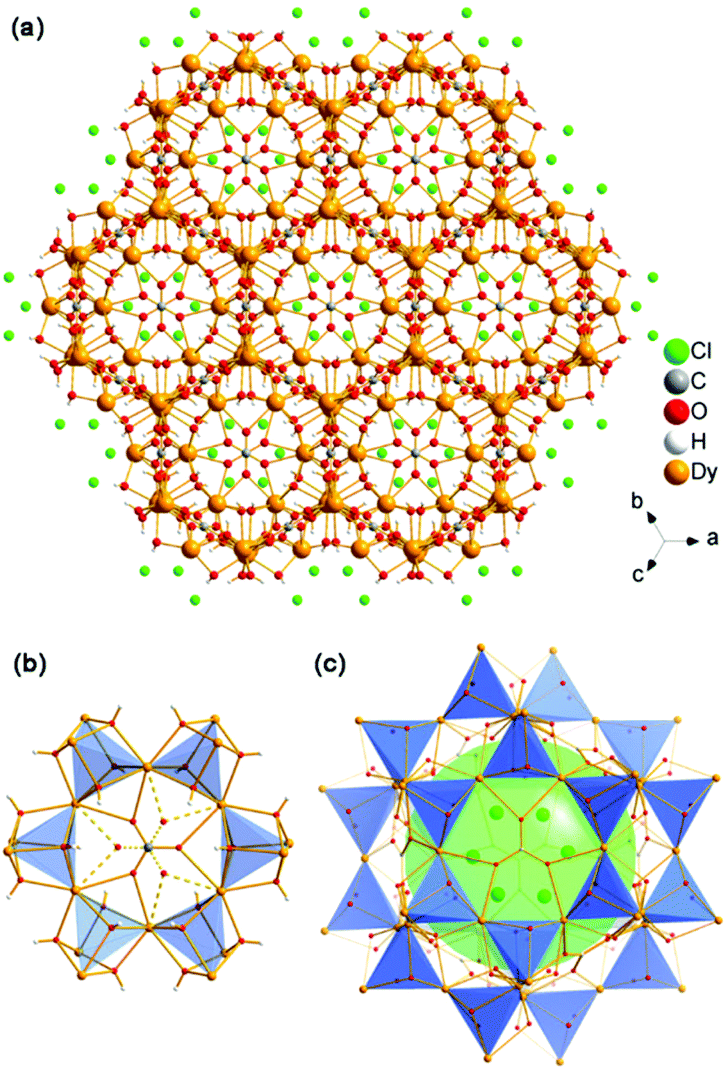 | ||
| Fig. 1 (a) 3D structure of 1 viewing along the [111] direction; (b) the 6-ring opening which templated by CO32− anions; (c) the SOD-like cage of the structure. | ||
Furthermore, an SOD cage can be observed through the vertex-sharing of 24 BUs (Fig. 1c). The cage shows six 4-ring openings and eight 6-ring openings, which are composed by four BUs and six BUs, respectively. Each 6-ring opening is sealed by a CO32− anion. Cl− anions in the cages act as the template here, which is similar to the role of alkali metals or organic amines in traditional zeolites. Interestingly, the structure of the super cage is the same as a 60-metal-atom cluster, [Er60(L-thre)34(μ6-CO3)8(μ3-OH)96(μ2-O)2(H2O)18]Br12(ClO4)18(H2O)40 (L-thre = L-threonine), that also possesses a sodalite cage structure featuring 24 vertex-sharing [Er4(μ3-OH)4] cubes.16 The cores of the 60-metal-atom clusters, as well as the {Ni12} or {Mn12} clusters, are encapsulated in a coordination sphere, making them independent. But in this framework, the cages combine with each other to give rise to a 3D structure. In general, the anions, such as CO32− and Cl−, may have a significant function in directing the structure of rare earth complexes. Besides, the triangular coordination geometry of CO32− may induce competing magnetic interactions between the metal centres, such as geometrical spin frustration, which is believed to be beneficial for enhancing magnetocaloric effects.17
The Dy3(OH)63+ open-framework structure of 1 is built up from DyO6 polyhedra. This framework features a characteristic body-centred building unit (BU), a [Dy4O4] cube, as shown in Fig. 2a. Each BU is composed of four Dy and four O atoms alternately connected. The Dy atoms are located on the vertices to form a [Dy4] tetrahedron. The [Dy4O4] cube turns out to be a [Dy4] tetrahedral node, like the SiO4 or AlO4 tetrahedron as the secondary building unit (SBU) in the zeolite (Fig. 2b). The [Dy4] tetrahedra are linked with each other by the vertices (Fig. 2c).
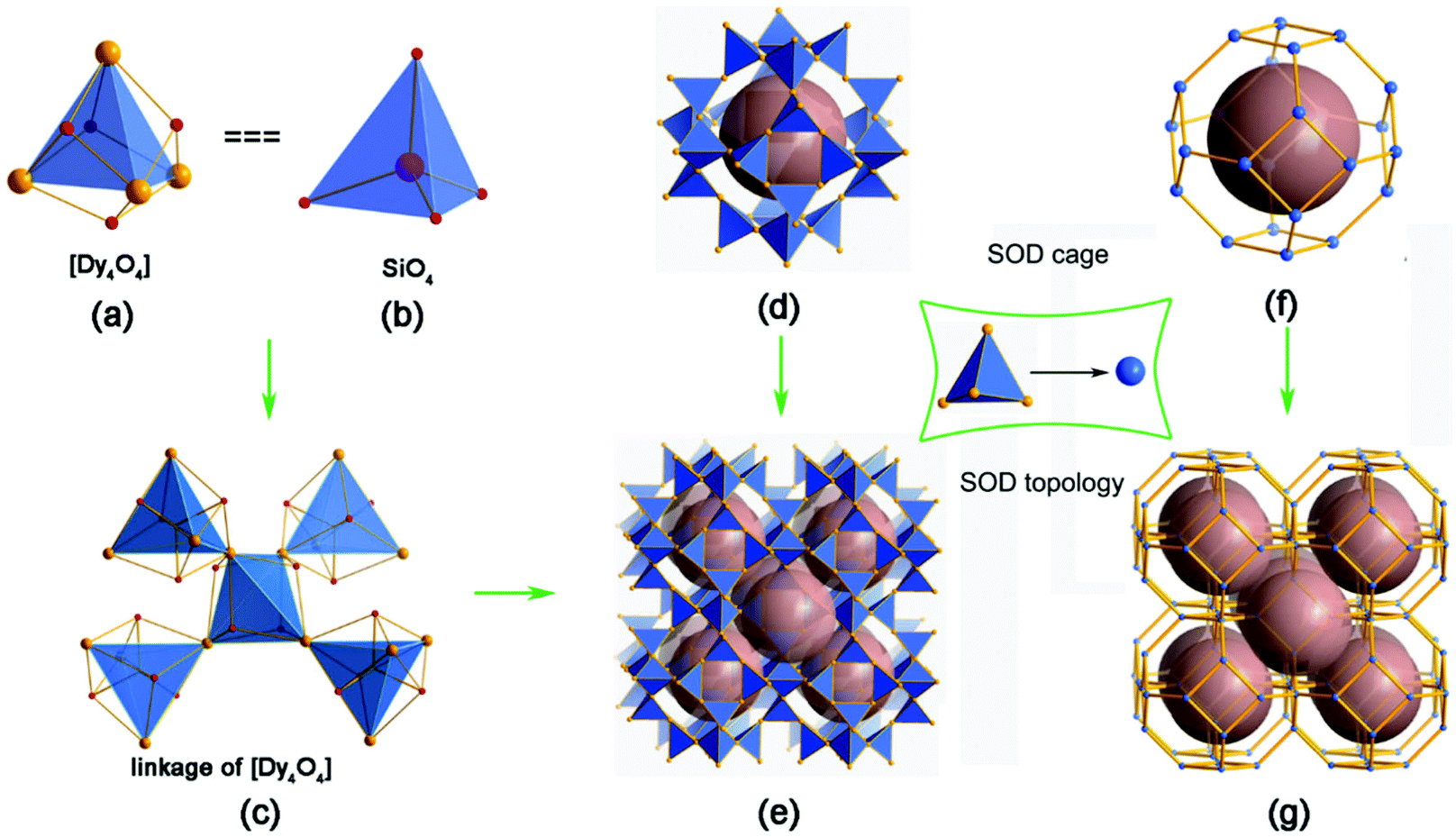 | ||
| Fig. 2 (a) The building unit (BU) for the 3D framework of 1, [Dy4O4]; (b) the BU for most zeolites, [SiO4]; (c) the linkage of [Dy4O4] BUs in the 3D framework of 1; (d) the SOD cage linked by 24 [Dy4] tetrahedral nodes; (e) the 3D framework of 1 connected by sharing 6-ring and 4-ring faces from the SOD cages; (f) the SOD cage derived from taking the [Dy4] tetrahedron as one tetrahedral node; (g) the SOD topology derived from the structure shown in (e). | ||
To simplify the structure, all of the Oμ3 atoms are omitted, as shown in Fig. 2d–g. Firstly, the [Dy4] tetrahedra are linked with each other to give rise to a super cage, which contains 24 [Dy4] tetrahedra (Fig. 2d). Through the face-sharing of the SOD-like cages, a 3D open framework of 1 (Fig. 2e) is constructed, which contains 3D intersection channels along the [111], [1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 1], [11] and [11] directions. Considering the [Dy4] tetrahedron as a tetrahedral node, the cage is built up through the vertex-sharing of the [Dy4] tetrahedra, which is very similar to the typical sodlite cage (Fig. 2f). The framework of 1 shows the topology of SOD, as shown in Fig. 2g. The free diameters are 4.1 × 4.3 Å2 (Dy⋯Dy distance) for the 6-ring channel, which is comparable to those of 8-ring channels in aluminosilicate zeolites, such as CHA, LEV, LTA, etc. The framework density (FD) of 1 is 12.3 Dy atoms per 1000 Å3. That is lower than SOD with FDSi of 16.7 T/1000 Å3 and comparable to some zeolites with 8-ring channels due to the long distance of the Dy–O bond.18 The SOD topology was firstly discovered for the mineral sodalite in 1930, and used to be studied in zeolites.19 Recently, the SOD topology has also been exhibited in several ZMOFs or ZIFs.20 This is one of the rare examples of a rare earth complex with a zeolite topology.21 It may act as a guidance to explore new RE compounds with zeolite topology by using a [RE4] tetrahedron as building unit.
1], [11] and [11] directions. Considering the [Dy4] tetrahedron as a tetrahedral node, the cage is built up through the vertex-sharing of the [Dy4] tetrahedra, which is very similar to the typical sodlite cage (Fig. 2f). The framework of 1 shows the topology of SOD, as shown in Fig. 2g. The free diameters are 4.1 × 4.3 Å2 (Dy⋯Dy distance) for the 6-ring channel, which is comparable to those of 8-ring channels in aluminosilicate zeolites, such as CHA, LEV, LTA, etc. The framework density (FD) of 1 is 12.3 Dy atoms per 1000 Å3. That is lower than SOD with FDSi of 16.7 T/1000 Å3 and comparable to some zeolites with 8-ring channels due to the long distance of the Dy–O bond.18 The SOD topology was firstly discovered for the mineral sodalite in 1930, and used to be studied in zeolites.19 Recently, the SOD topology has also been exhibited in several ZMOFs or ZIFs.20 This is one of the rare examples of a rare earth complex with a zeolite topology.21 It may act as a guidance to explore new RE compounds with zeolite topology by using a [RE4] tetrahedron as building unit.
The framework of 1 can be described as a three-period net with the highest symmetry of Imm containing only one type of T site as determined by the Systre software.22 The vertex symbol for the T site in the net is 36·4·54·64. This three-period net features a unique natural tiling with a transitivity of (1 2 3 2). There are two different tiles in this tiling with face symbols of [34] and [324·46·68]. The signature of this tiling is 6[34] + [324·46·68]. Considering the building unit of [Dy4(O)4], the framework of 1 also can be described as a three-period net of SOD topology with the highest symmetry of Imm. The vertex symbol for the T site in the net is 42·64. This three-period net is carried by a unique natural tiling with a transitivity of (1 1 2 1). There is one type of tile in this tiling with a face symbol of [46·68].
TG analysis has been performed to study the thermal stability of 1. It shows a total weight loss of 18.6% from 200 to 800 °C with two stages with the first one occurring between 300–450 °C (Fig. S3†). Under heat treatment at 370 °C for 4 h, the structure will transform into 1-c. Surprisingly, the structure of 1-c can be solved using single-crystal X-ray diffraction. This phenomenon is rarely observed in RE inorganic compounds. In the analysis of the single-crystal X-ray diffraction data, we noticed that the CO32− anions are removed. The space group has transformed into trigonal R for 1-c from cubic Im for 1. Besides, some micro variation occurs in the structure. Here, we consider the CO32− anions are removed by forming CO2 during the calcinations. From the TG curve, the first stage weight loss of 13.6 wt% occurs at around 370 °C, which corresponds with the removal of CO2 and H2O. The complex then has a molecular formula of Dy3O4Cl. The possible chemical equation is shown as follows:
| Dy3(OH)6(CO3)Cl → Dy3O4Cl + CO2 + 3H2O |
The PXRD pattern of 1-c corresponds well with the simulated one based on the single-crystal structure of 1-c, indicating the pure phases of the as-synthesized sample (Fig. S4†). The formula of 1-c is confirmed by ICP, elemental analyses and anion chromatography analysis (Dy, 82.7; C, 0.41; H, 0.92; Cl, 5.95; Anal. Calcd Dy, 83.0; C, 0; H, 0; Cl, 6.04 wt%). In addition, X-ray photoelectron spectroscopy (XPS) of 1 and 1-c are also measured to verify the existence of Cl. Fig. S5† shows the XPS survey spectra and narrow scan XPS spectra of 1 and 1-c. The signal at around 198 eV is consistent with the binding energy of Cl 2p, indicating the existence of Cl− in the structures.23 The content ratios of Dy/Cl for 1 and 1-c derived from the XPS analysis are close to 3, corresponding to the formula of Dy3(OH)6(CO3)Cl and Dy3O4Cl, respectively. Additionally, the calcination derivatives (2-c and 3-c) with similar structures can be obtained following the same treatment for 2, and 3.
Single-crystal structure analysis of 1-c reveals that the asymmetric unit contains three crystallographically distinct Dy sites and four crystallographically distinct O sites (Fig. S6, ESI†). Dy(1) and Dy(2) are six-coordinated with six μ4-O (O(1), O(2), O(3) and O(4)), Dy(3) is seven-coordinated with four μ4-O and three Cl atoms. The Dy–Oμ4 bond lengths have a reasonable range of 2.13(4)–2.41(6) Å.
The structure of 1-c may be described as a cationic framework of Dy3O4+ units with 3D intersection 6-ring channels along the [001], [11], [211] and [12] directions (Fig. 3a and S7†). Cl− anions are situated in the channels to balance the charge. Compared with 1, all of the CO32− anions have been removed. The 3D open framework of 1-c is constructed by face-sharing of the super cages, which is similar to 1, as shown in Fig. 3b. However, all of the μ3-OH in 1 have become μ4-O in 1-c. The μ4-O connect to one Dy(3) atom and three other Dy atoms (Dy(1) and/or Dy(2)) to form anion-centered ODy4 tetrahedra, which is common in inorganic compounds.24 By ignoring the Dy(3) atom, the framework of 1-c also contains vertex-sharing linkage of the [Dy4] cubes with the same framework of 1. In the SOD-like cage, the [Dy4] cubes are comprised of all of the Dy(1) and Dy(2) atoms with Dy(3) situated nearby the six 4-ring openings. Inside and outside the 4-ring opening, there is a central Dy(3) atom that links to the four [Dy4O4] cubes by coordinating with four O atoms from the four cubes (Fig. 3c). As shown in Fig. 3d, the high-resolution TEM images (HRTEM) of 1-c clearly show a hexagonal lattice with void distances of 5.9 Å, which corresponds to the distances of the crystallographic planes of (021), (![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 01) and (21) (5.883 Å). This further confirms the structure of 1-c.
01) and (21) (5.883 Å). This further confirms the structure of 1-c.
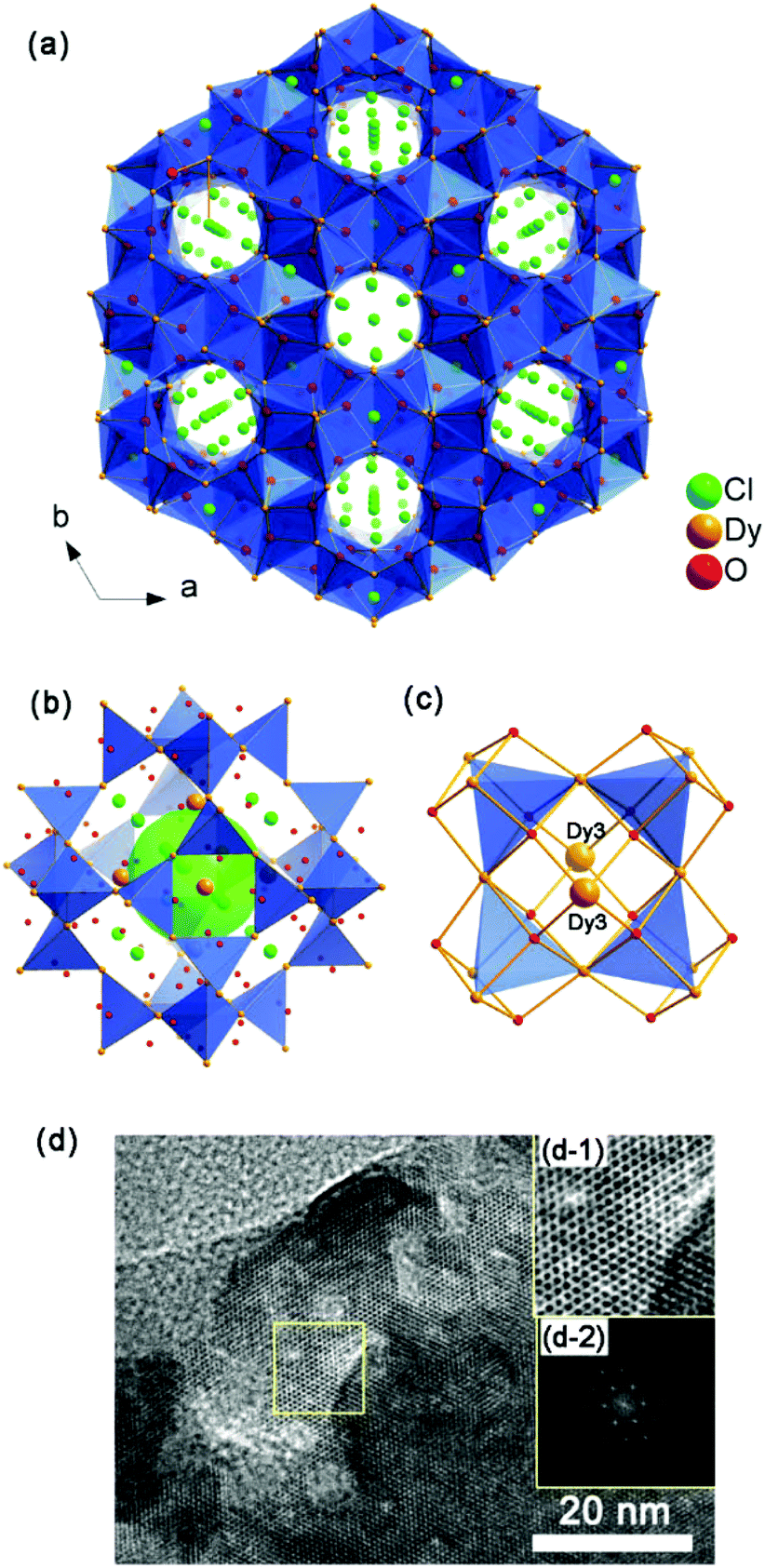 | ||
| Fig. 3 (a) Structure of 1-c viewed along the [001] direction; (b) super cage linked by 24 [Dy4] tetrahedra; (c) the coordination between Dy(3) atoms and the 4-ring opening of the super cage; (d) HRTEM micrograph of 1-c along the [010] zone axis. Inset: (d-1) enlarged image of the yellow box and corresponding FFT; (d-2) enlarged zones of the crystallographic planes of (021), (01) and (21) with inter-plane distances of 5.883 Å. | ||
Magnetic analyses
Dc magnetic susceptibility data of 1, 1-c and the corresponding diluted samples (1d and 1d-c, respectively) are measured in the temperature range of 2–300 K (Fig. 4). At 300 K, the χmT products for the samples are 13.90, 13.81, 13.29 and 13.39 cm3 K mol−1 for 1, 1d, 1-c and 1d-c, respectively, which are compatible with the calculated value of 14.17 cm3 K mol−1 for the ground state of one DyIII ion (4f9, S = 5/2, L = 5, J = 15/2 and g = 4/3) in the free ion approximation. Upon cooling, the χmT products decrease gradually at first and then more rapidly to the minimum at 2 K for all of the compounds.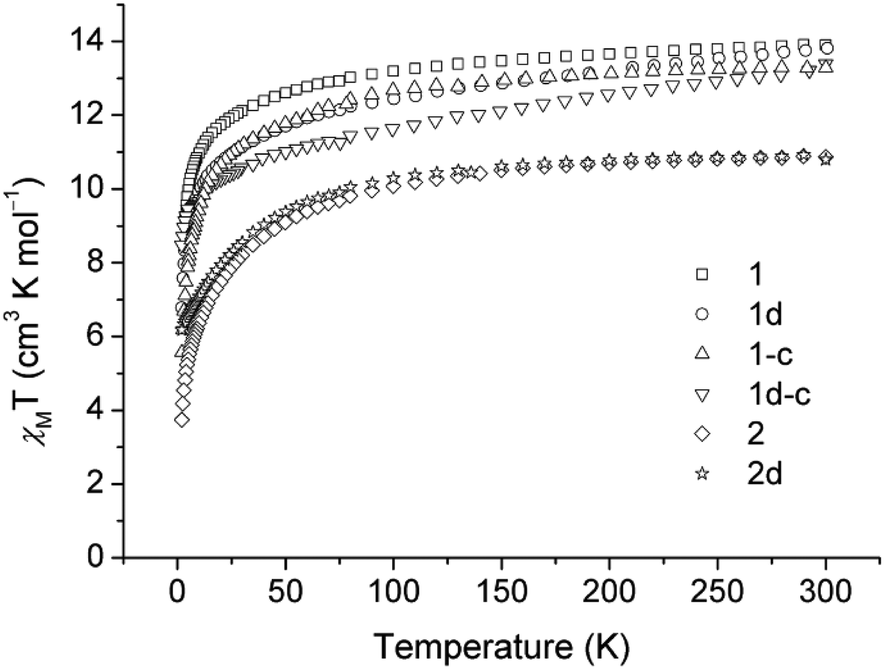 | ||
| Fig. 4 Experimental χmT products of 1, 1d, 1-c, 1d-c, 2 and 2d in an applied field of 2 kOe, 1 kOe, 1 kOe, 1 kOe, 0.5 KOe and 1 KOe for 1, 1d, 1-c, 1d-c, 2 and 2d, respectively. The χmT curve has been rescaled for one REIII ion. | ||
The χmT versus T plots for compounds 2 and 2d are recorded between 2 and 300 K under a dc field of 500 Oe and 1 kOe, respectively (Fig. 4). The χmT values at room temperature are 10.87 and 10.80 for 2 and 2d, respectively, which are close to the expected value of 11.48 cm3 K mol−1 for one ErIII ion (4f12, S = 3/2, L = 6, J = 15/2 and g = 6/5). In the high-temperature range, the χmT product decreases slowly upon cooling for both compounds; below 75 K, the χmT product drops quickly to a minimum value of 3.74 cm3 K mol−1 at 2 K for 2, and a minimum value of 6.17 cm3 K mol−1 for the diluted sample of 2d.
The magnetization versus field plots are determined at low temperatures in the range of 0–70 kOe, as shown in Fig. S8.† For 1 and 1-c, the magnetization at 2 K increases continuously upon an increase of the magnetic field, reaching the maxima of 5.57Nβ and 5.04Nβ for 1 and 1-c, respectively. These obtained values are far from the saturated magnetization for one DyIII ion, but are in agreement with the data reported previously as a result of the large magneto-anisotropy and/or the low lying excited states of the DyIII ion. For 2, similar behavior of the field-dependent magnetization is observed, with a highest value of 4.46Nβ at 2 K, which again does not exhibit saturation, possibly due to the presence of large magnetic anisotropy.
Alternating current (ac) susceptibility measurements were performed on the microcrystalline samples to investigate the dynamic behaviour of the magnetization at low temperatures, as displayed in Fig. S9.† For 1, 1-c and 2, curves based on the in-phase and out-of-phase signals of the ac susceptibility are all superimposed on a nearly single-master curve, either in the absence of an external dc field or at an applied dc field of 1 kOe. This phenomenon often exists in high-nuclearity lanthanide cages, and is probably due to the crowded distribution of metal ions in high-nuclearity clusters, as well as in titled complexes.25
Magnetic dilution may enhance the slow magnetic relaxation Ueff originating from SIM behaviour in mononuclear and polymeric compounds, such as 1D dysprosium chains, 2D layers, and even 3D LiYF4 compounds.26 It is an advantage here that we can study the magnetism of samples with very large dilution ratios of up to 0.227%, for which the magnetic signal is still precluded by the instrument sensitivity for 1 and 1-c, while the same cannot be detected for most diluted samples reported. Therefore, the doped samples 1d and 1d-c in Dy:Y molar ratios of 1:440 are prepared to examine the nature of the magnetism. As shown in Fig. 5, ac measurements for both diluted samples in zero dc field show obvious frequency dependence below 8 K, an indication of slow relaxation of the magnetization. Unfortunately, no maximum is observed in both the in-phase and out-of-phase signals of the ac susceptibility, possibly due to the presence of a fast relaxation of the magnetisation through a quantum tunnelling mechanism. Thus, ac measurements are taken under an applied small external dc field of 500 Oe to mitigate the quantum tunnelling (Fig. S10†). Significant frequency-dependent χ′ and χ′′ signals appear for both compounds and a maximum in χ′ is clearly observed moving to higher temperatures towards higher frequencies especially for 1d, indicating SIM behaviour.
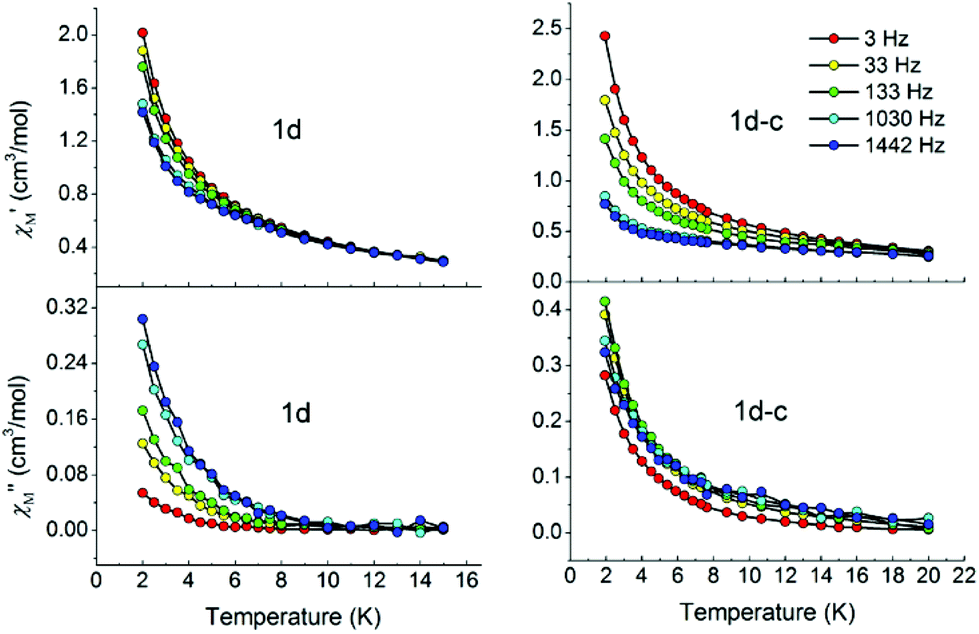 | ||
| Fig. 5 In-phase (top) and out-of-phase (bottom) dynamic susceptibilities of 1d and 1d-c without external dc magnetic fields. Solid lines are included to guide the eyes. | ||
For 2, a similar phenomenon has occurred (Fig. S11†). The doped sample 2d with a Dy:Y molar ratio of 1:15 shows slow relaxation of its magnetization under a dc field of 1 kOe, proving the position effect that the dilution of open framework materials has on the SIM behaviour once again. All the information points out that such a slow relaxation is likely dominated by single ion effects, and vanishes when more of the lanthanide ions are incorporated. The magnetic exchange interaction, although it is weak between lanthanides, and the dipolar interaction, as well as the orientation of the easy axes of magnetization work together in the molecular architecture, leading to a complicated situation which is not beneficial to the SIM behaviour.
Conclusions
In conclusion, we have successfully synthesized a series of novel rare earth carbonates RE3(OH)6(CO3)Cl (RE = Dy (1), Er (2), Y (3)) with SOD-like structures built from [RE4(μ3-OH)4] cubes by introducing CO32− for the first time. Interestingly, the CO32− can be the removed after calcination at 370 °C. In addition, the single-crystal of 1 can be in situ transformed into a new zeolitic compound Dy3O4Cl (1-c) with two Dy atoms located at both sides of the 4-ring on the SOD-like structure. Magnetic studies of both diluted samples of 1 and 1-c show that they exhibit SIM-type behaviours. As such, both diluted compounds can be safely labelled as M0U0S3 according to their magneto-structural correlations.27 These results provide guidance on the syntheses of RE-based open frameworks, and promisingly open a new door towards diluted magnets.Experimental section
Reagents and solvents were commercially available and used as received without further purification.Synthetic procedures
:1.2HCl:2.5NaOH:0.33Na2CO3:332H2O. About 1 h later, the gel was added into a 12 mL Teflon-lined stainless steel autoclave and heated at 200 °C for 72 h, followed by cooling to room temperature for 20 h. The white pure crystals were obtained by washing in distilled water several times and dried at room temperature overnight after filtration. Yield: 176 mg (77% based on Dy). Elemental analysis of the product gave (in wt%) Dy: 70.9 (Calcd Dy: 71.2), C: 1.84 (Calcd C: 1.75), H: 1.21 (Calcd H: 0.876), and anion chromatography analysis gave (in wt%) Cl: 5.11 (Calcd Cl: 5.18), which were consistent with the elemental content calculated from the molecular formula of Dy3(OH)6(CO3)Cl.
:1.2HCl:2.5NaOH:0.33Na2CO3:332H2O. The product was obtained with a yield of 53% based on Er with the molecular formula of Er3(OH)6(CO3)Cl.
:1.2HCl:2.5NaOH:0.33Na2CO3:332H2O. The product was obtained with a yield of 80% based on Y with the molecular formula of Y3(OH)6(CO3)Cl.
:449 molar ratio; 10 mmol in total) was dispersed into 50 mL water, and 1 mL hydrochloric acid (37 wt%, 12 mmol) was added. Then 10 mL NaOH (2.5 mol L−1) was added to the mixture with stirring for 1 h to get a pH value of 6. Finally, Na2CO3 was introduced to make a reaction gel of 0.002DyCl3:0.998YCl3:1.2HCl:2.5NaOH:0.33Na2CO3:332H2O. About 1 h later, the gel was added into a 100 mL Teflon-lined stainless steel autoclave and heated at 200 °C for 72 h, followed by cooling to room temperature for 20 h. Pure crystals of 1d were obtained after washing the product in distilled water and were dried at room temperature overnight. The Dy/Y ratio of the product based on ICP analysis (in wt%) Dy: 0.238, Y: 57.2, gave the molecular formula Dy0.0068Y2.9932(OH)6(CO3)Cl.
:0.94YCl3:1.2HCl:2.5NaOH:0.33Na2CO3:332H2O. The Er/Y ratio of the product based on ICP analysis (in wt%) Er: 6.63, Y: 52.1, gave the molecular formula of Er0.19Y2.81(OH)6(CO3)Cl.
Characterization methods
PXRD data were collected on a Rigaku SmartLab(3) diffractometer with Cu Kα radiation (λ = 1.5418 Å). Inductively coupled plasma (ICP) analysis was performed on a Perkin-Elmer Optima 3300DV spectrometer. Elemental analysis was conducted on a Vario MICRO elemental analyzer. Anion chromatography analysis was performed on a DX-500 Ion Chromatography (IC) System of Dionex. Thermogravimetric analysis (TGA) was carried out on a TA Q500 analyzer in air with a heating rate of 10 °C min−1 from RT to 800 °C. X-ray photoelectron spectroscopy (XPS) measurements were performed using a Thermo Escalab 250 spectrometer with monochromatized Al Kα excitation. The TEM and HRTEM images were taken on a JEOL JEM-3010 transmission electron microscope operating at an accelerating voltage of 300 kV. Magnetic measurements were performed on a Quantum Design MPMS XL-7 SQUID magnetometer on polycrystalline samples. Diamagnetic corrections were made with Pascal's constants for all the constituent atoms and sample holder. Alternating-current (ac) magnetic susceptibility data were collected on the same instrument employing a 3.5 Oe oscillating field at frequencies up to 1442 Hz.X-ray crystallographic analyses
Suitable single crystals of Dy3(OH)6(CO3)Cl (1), Er3(OH)6(CO3)Cl (2), Y3(OH)6(CO3)Cl (3) and Dy3O5Cl0.5H1.5 (1-c) with dimensions of 0.32 × 0.21 × 0.15, 0.07 × 0.07 × 0.05, 0.05 × 0.05 × 0.05 and 0.06 × 0.06 × 0.03 mm3, respectively, were selected for single-crystal X-ray diffraction analyses. The intensity data were collected on a Bruker Apex II DUO area-detector diffractometer with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) at a temperature of 23 ± 2 °C. Cell refinement and data reduction were accomplished with the SAINT processing program.28 The structures were solved by direct methods and refined by a full matrix least-squares technique with the SHELXTL crystallographic software package.29 The heaviest atoms of Dy, Er, Y, O, Cl and C could be unambiguously located. The H atoms in the structures of 1, 2 and 3 were added theoretically, and that of 1-c were not added. All non-hydrogen atoms were refined anisotropically. CCDC 1416221–1416224 contain the supplementary crystallographic data for this paper. Crystal data and refinement parameters for the structure determination are presented in Tables S1–3 and S5.† Selected bond distances are listed in the Table S4.†Acknowledgements
Financial support from “973 projects” (2012CB619401 and 2012CB619402), NSFC (21473129, 21201137 and IRT13034), the China Postdoctoral Science Foundation (Grant 2014M562393), the “National 1000-Plan” program, and the Fundamental Research Funds for the Central Universities. We thank the MOF team from Jilin University (Dr Ying Mu, Prof. Jiyang Li and Prof. Jihong Yu) for TEM and elemental analyses. We thank the team from Xi'an Jiaotong University (Dr Yali Lei and Prof. Zhenxing Shen) for anion chromatography analysis.Notes and references
- (a) A. Corma, Chem. Rev., 1997, 97, 2373–2420 CrossRef CAS PubMed; (b) J. Li, A. Corma and J. Yu, Chem. Soc. Rev., 2015, 44, 7112–7127 RSC; (c) Y. Li and J. Yu, Chem. Rev., 2014, 114, 7268–7316 CrossRef CAS PubMed.
- C. Baerlocher and L. B. McCusker, Database of Zeolite Structures: http://www.iza-structure.org/databases/.
- (a) G. Yang, Y. Wang, D. Zhou, J. Zhuang, X. Liu, X. Han and X. Bao, J. Chem. Phys., 2003, 119, 9765–9770 CrossRef CAS; (b) N. Rahimi and R. Karimzadeh, Appl. Catal., A, 2011, 398, 1–17 CrossRef CAS; (c) M. Iwasaki and H. Shinjoh, Chem. Commun., 2011, 47, 3966–3968 RSC; (d) S. Kim, E. Sasmaz and J. Lauterbach, Appl. Catal., B, 2015, 168–169, 212–219 CrossRef CAS.
- (a) D. Ananias, F. A. Almeida Paz, L. D. Carlos, C. F. G. C. Geraldes and J. Rocha, Angew. Chem., Int. Ed., 2006, 45, 7938–7942 CrossRef CAS PubMed; (b) X. Wang, J. Li, Y. Han, Y. Li, J. Yu and R. Xu, Chem. Mater., 2011, 23, 2842–2847 CrossRef CAS.
- (a) N. Ishikawa, M. Sugita, T. Ishikawa, S. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694–8695 CrossRef CAS PubMed; (b) L. Sorace, C. Benelli and D. Gatteschi, Chem. Soc. Rev., 2011, 40, 3092–3104 RSC; (c) Y.-L. Wang, Y. Ma, X. Yang, J. Tang, P. Cheng, Q.-L. Wang, L.-C. Li and D.-Z. Liao, Inorg. Chem., 2013, 52, 7380–7386 CrossRef CAS PubMed.
- (a) M. Mannini, F. Pineider, C. Danieli, F. Totti, L. Sorace, P. Sainctavit, M. A. Arrio, E. Otero, L. Joly, J. C. Cezar, A. Cornia and R. Sessoli, Nature, 2010, 468, 417–421 CrossRef CAS PubMed; (b) S. Cardona-Serra, J. M. Clemente-Juan, E. Coronado, A. Gaita-Ariño, A. Camón, M. Evangelisti, F. Luis, M. J. Martínez-Pérez and J. Sesé, J. Am. Chem. Soc., 2012, 134, 14982–14990 CrossRef CAS PubMed; (c) D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS PubMed; (d) H. Miyasaka, K. Nakata, K.-i. Sugiura, M. Yamashita and R. Clérac, Angew. Chem., Int. Ed., 2004, 43, 707–711 CrossRef CAS PubMed; (e) I.-R. Jeon and R. Clerac, Dalton Trans., 2012, 41, 9569–9586 RSC; (f) J. J. Baldoví, E. Coronado, A. Gaita-Ariño, C. Gamer, M. Giménez-Marqués and G. Mínguez Espallargas, Chem. – Eur. J., 2014, 20, 10695–10702 CrossRef PubMed.
- (a) R. Giraud, W. Wernsdorfer, A. M. Tkachuk, D. Mailly and B. Barbara, Phys. Rev. Lett., 2001, 87, 057203 CrossRef CAS PubMed; (b) M. Wu, F. Jiang, X. Kong, D. Yuan, L. Long, S. A. Al-Thabaiti and M. Hong, Chem. Sci., 2013, 4, 3104–3109 CAS; (c) M. Wu, F. Jiang, D. Yuan, J. Pang, J. Qian, S. A. Al-Thabaiti and M. Hong, Chem. Commun., 2014, 50, 1113–1115 RSC.
- J. Yu and R. Xu, Acc. Chem. Res., 2010, 43, 1195–1204 CrossRef CAS PubMed.
- M. Wu, F. Jiang, X. Kong, D. Yuan, L. Long, S. A. Al-Thabaiti and M. Hong, Chem. Sci., 2013, 4, 3104–3109 RSC.
- (a) X.-B. Han, Y.-G. Li, Z.-M. Zhang, H.-Q. Tan, Y. Lu and E.-B. Wang, J. Am. Chem. Soc., 2015, 137, 5486–5493 CrossRef CAS PubMed; (b) J. C. G. Bunzli, Coord. Chem. Rev., 2015, 293, 19–47 CrossRef; (c) V. Velasco, D. Aguila, L. A. Barrios, I. Borilovic, O. Roubeau, J. Ribas-Arino, M. Fumanal, S. J. Teat and G. Aromi, Chem. Sci., 2015, 6, 123–131 RSC; (d) Y. Zhang, L. Huang, H. Miao, H. X. Wan, H. Mei, Y. Liu and Y. Xu, Chem. – Eur. J., 2015, 21, 3234–3241 CrossRef CAS PubMed.
- A. S. R. Chesman, D. R. Turner, B. Moubaraki, K. S. Murray, G. B. Deacon and S. R. Batten, Chem. – Eur. J., 2009, 15, 5203–5207 CrossRef CAS PubMed.
- (a) A. S. R. Chesman, D. R. Turner, B. Moubaraki, K. S. Murray, G. B. Deacon and S. R. Batten, Chem. – Eur. J., 2009, 15, 5203–5207 CrossRef CAS PubMed; (b) B. Zhang, X. Zheng, H. Su, Y. Zhu, C. Du and M. Song, Dalton Trans., 2013, 42, 8571–8574 RSC; (c) K.-C. Xiong, F.-L. Jiang, Y.-L. Gai, D.-Q. Yuan, D. Han, J. Ma, S.-Q. Zhang and M.-C. Hong, Chem. – Eur. J., 2012, 18, 5536–5540 CrossRef CAS PubMed; (d) R. McLellan, J. Reze, S. M. Taylor, R. D. McIntosh, E. K. Brechin and S. J. Dalgarno, Chem. Commun., 2014, 50, 2202–2204 RSC.
- (a) S. K. Langley, B. Moubaraki and K. S. Murray, Inorg. Chem., 2012, 51, 3947–3949 CrossRef CAS PubMed; (b) R. Sen, D. K. Hazra, M. Mukherjee and S. Koner, Eur. J. Inorg. Chem., 2011, 2011, 2826–2831 CrossRef CAS.
- P. C. Andrews, T. Beck, C. M. Forsyth, B. H. Fraser, P. C. Junk, M. Massi and P. W. Roesky, Dalton Trans., 2007, 5651–5654 RSC.
- G. J. T. Cooper, G. N. Newton, P. Kögerler, D.-L. Long, L. Engelhardt, M. Luban and L. Cronin, Angew. Chem., Int. Ed., 2007, 46, 1340–1344 CrossRef CAS PubMed.
- X.-J. Kong, Y. Wu, L.-S. Long, L.-S. Zheng and Z. Zheng, J. Am. Chem. Soc., 2009, 131, 6918–6919 CrossRef CAS PubMed.
- (a) Y. Zheng, A. Ellern and P. Kogerler, Acta Crystallogr., 2011, 67, i56–i58 CrossRef CAS PubMed; (b) Z. Fu, Y. Zheng, Y. Xiao, S. Bedanta, A. Senyshyn, G. G. Simeoni, Y. Su, U. Rücker, P. Kögerler and T. Brückel, Phys. Rev. B: Condens. Matter, 2013, 87, 214406 CrossRef; (c) Y.-C. Chen, L. Qin, Z.-S. Meng, D.-F. Yang, C. Wu, Z. Fu, Y.-Z. Zheng, J.-L. Liu, R. Tarasenko, M. Orendac, J. Prokleska, V. Sechovsky and M.-L. Tong, J. Mater. Chem. A, 2014, 2, 9851–9858 RSC; (d) Y.-Z. Zheng, G.-J. Zhou, Z. Zheng and R. E. P. Winpenny, Chem. Soc. Rev., 2014, 43, 1462–1475 RSC.
- (a) X. Song, Y. Li, L. Gan, Z. Wang, J. Yu and R. Xu, Angew. Chem., Int. Ed., 2009, 48, 314–317 CrossRef CAS PubMed; (b) Y. Wang, Y. Li, Y. Yan, J. Xu, B. Guan, Q. Wang, J. Li and J. Yu, Chem. Commun., 2013, 49, 9006–9008 RSC.
- L. Pauling, Z. Kristallogr., 1930, 74, 213–225 CAS.
- (a) L. Sun, H. Xing, Z. Liang, J. Yu and R. Xu, Chem. Commun., 2013, 49, 11155–11157 RSC; (b) Y. Ban, Y. Li, Y. Peng, H. Jin, W. Jiao, X. Liu and W. Yang, Chem. – Eur. J., 2014, 20, 11402–11409 CrossRef CAS PubMed; (c) C.-T. He, L. Jiang, Z.-M. Ye, R. Krishna, Z.-S. Zhong, P.-Q. Liao, J. Xu, G. Ouyang, J.-P. Zhang and X.-M. Chen, J. Am. Chem. Soc., 2015, 137, 7217–7223 CrossRef CAS PubMed.
- L. F. Wang, H. Li, G. S. Zhu, H. Ren, F. X. Sun and S. L. Qiu, Chem. J. Chin. Univ., 2008, 29, 2127–2130 CAS.
- O. Delgado-Friedrichs and M. O'Keeffe, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2003, 59, 351 CrossRef.
- (a) The Handbook of X-ray Photoelectron Spectroscopy, Perkin Elmer Corporation, 1979 Search PubMed; (b) H. Xing, W. Yang, T. Su, Y. Li, J. Xu, T. Nakano, J. Yu and R. Xu, Angew. Chem., Int. Ed., 2010, 49, 2328–2331 CrossRef CAS PubMed.
- S. V. Krivovichev, O. Mentré, O. I. Siidra, M. Colmont and S. K. Filatov, Chem. Rev., 2013, 113, 6459–6535 CrossRef CAS PubMed.
- M. Wu, F. Jiang, X. Kong, D. Yuan, L. Long, S. A. Al-Thabaiti and M. Hong, Chem. Sci., 2013, 4, 3104–3109 RSC.
- (a) Q. Chen, Y.-S. Meng, Y.-Q. Zhang, S.-D. Jiang, H.-L. Sun and S. Gao, Chem. Commun., 2014, 50, 10434–10437 RSC; (b) D.-D. Yin, Q. Chen, Y.-S. Meng, H.-L. Sun, Y.-Q. Zhang and S. Gao, Chem. Sci., 2015, 6, 3095–3101 RSC; (c) B. Monteiro, J. T. Coutinho, C. C. L. Pereira, L. C. J. Pereira, J. Marçalo, M. Almeida, J. J. Baldoví, E. Coronado and A. Gaita-Ariño, Inorg. Chem., 2015, 54, 1949–1957 CrossRef CAS PubMed; (d) T. Han, J.-D. Leng, Y.-S. Ding, Y. Wang, Z. Zheng and Y.-Z. Zheng, Dalton Trans., 2015, 44, 13480–13484 RSC; (e) J. A. Quilliam, S. Meng and J. B. Kycia, Phys. Rev. B: Condens. Matter, 2012, 85, 184415 CrossRef.
- Y.-Z. Zheng, Z. Zheng and X.-M. Chen, Coord. Chem. Rev., 2014, 258–259, 1–15 CrossRef CAS.
- SAINT, Bruker AXS Inc., 5465 East Cheryl Parkway, Madison, WI, 53711-5373, USA, 2000 Search PubMed.
- SHELXTL, Bruker AXS Inc., 5465 East Cheryl Parkway, Madison, WI, 53711-5373, USA, 2000 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal data and structure refinement and selected bond lengths [Å] for 1–3 and 1-c; thermal ellipsoids for 1–3 and 1-c; 3-dimensional structure of 1-c; PXRD patterns for all samples; TGA curve for 1; XPS for 1 and 1-c; temperature dependence of M vs. H, in-phase (χ′) and out-of-phase (χ′′) ac susceptibility curves of 1, 2, 1d, 2d, 1-c and 1d-c; crystallographic data for 1–3 and 1-c (CIF). CCDC 1416221–1416224 for 1–3 and 1-c. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt03314d |
| This journal is © The Royal Society of Chemistry 2016 |