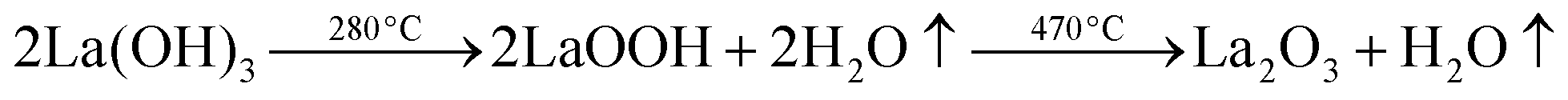DOI:
10.1039/C5DT03190G
(Paper)
Dalton Trans., 2016,
45, 121-133
Neutron diffraction and multinuclear solid state NMR investigation into the structures of oxide ion conducting La9.6Si6O26.4 and La8Sr2Si6O26, and their hydrated phases
Received
18th August 2015
, Accepted 6th November 2015
First published on 17th November 2015
Abstract
Apatite silicates are attracting significant interest as potential SOFC electrolyte materials. They are non-conventional oxide ion conductors in the sense that oxide ion interstitials, rather than vacancies, are the key defects. In this work we compare the structures of La9.6Si6O26.4 and La8Sr2Si6O26, both before and after hydration in order to gather information about the location of the interstitial oxide ion site. Neutron diffraction structural studies suggest that in the as-prepared La8Sr2Si6O26 and hydrated La8Sr2Si6O26, the interstitial oxide ion sites are close to the apatite channel centre. For La9.6Si6O26.4, a similar site close to the channel centre is observed, but on hydration of this particular sample, the interstitial site is shown to be significantly displaced away from the channel centre towards the SiO4 units. This can be explained by the need for additional displacement from the channel centre to accommodate the large amount of interstitial anions in this hydrated phase. The solid state 29Si MAS NMR spectra are shown to be very sensitive to the different speciation exhibited by the La8Sr2Si6O26 and La9.6Si6O26.4 systems, with the former being dominated by regular SiO4 framework species and the latter being dominated by interruptions to this network caused by cation vacancies and interstitials. The corresponding 17O MAS NMR study identifies a strong signal from the O atoms of the SiO4 groups, thus demonstrating that all of the O species in these systems are exchangeable O under heterogeneous gas phase conditions. In addition, interstitial O species attributed to pendant OH linkages on the Si positions are clearly identified and resolved, and these are removed on dehydration. This observation and assignment is corroborated by corresponding 1H MAS NMR measurements. Overall the neutron diffraction work indicates that the interstitial site location in these apatite silicates depends on the anion content with progressive displacement towards the SiO4 tetrahedra on increasing anion content, while the observation of exchangeable O on the SiO4 groups is consistent with prior modelling predictions as to the importance on the silicate units in the conduction process.
1. Introduction
The increasing concerns regarding greenhouse gas emissions and diminishing fuel reserves are driving considerable interest in the development of fuel cell technology. In terms of such technology, polymer based fuel cells are the dominant system for transport and portable applications, while Solid Oxide Fuel Cells (SOFCs) are being targeted for stationary power generation. In terms of the electrolyte adopted for SOFC systems, traditionally research has focused on perovskite and fluorite systems, with the most widely researched in terms of applications being the fluorite-type materials ZrO2 doped with Y2O3 (YSZ) or Sc2O3 (ScSZ), and CeO2 doped with Gd2O3 (CGO).1,2 The key defects in these fluorite systems are oxide ion vacancies which are introduced on partial substitution of Zr4+/Ce4+ by a trivalent rare earth, and conduction then proceeds via a vacancy hopping mechanism. More recently there has been growing interest in new structure-types displaying high oxide ion conduction. In this respect, apatite-type rare earth silicates and germanates have attracted considerable interest, following the identification of high oxide ion conductivity in the silicates by Nakayama et al., and subsequently in analogous germanates by Arikawa et al.3–5 Since the discovery of high oxide ion conductivity in these apatite systems, there has been considerable interest in understanding their conduction mechanisms as well as in doping strategies to optimize the conductivities.6–60 The ideal apatite stoichiometry can be written A10(MO4)6O2 (A = rare earth/alkaline earth; M = Ge, Si), and their structure can be viewed as a A4(MO4)6 framework (consisting of corner linked MO4 tetrahedra and AO6 trigonal metaprisms), with the remaining A6O2 units within the “channel” of this framework (Fig. 1).23,24 The variation in the size of the A6O2 “channels” can be correlated with variations in the AO6 metaprism twist angle.23 For high oxide ion conductivity, interstitial oxide ion defects are required, which can be achieved through increasing the oxygen content or the introduction of cation vacancies (the latter enhancing Frenkel defect (i.e. displacement of ions off their normal site into interstitial sites) formation). While for the apatite germanates, there is general consensus regarding the location of the interstitial site (neighbouring the GeO4 units),24,25,40,43,49,50 for the silicates, a range of sites have been reported,7,11,15,29–31,35,39 although there is growing support for a location closer to the channel centre.7,35,39,46 One of the difficulties with locating these interstitial sites is the generally low levels of oxygen in these sites, especially for the silicates, where prior studies have suggested a maximum limit close to 0.5 O per formula unit.35 In previous work on germanate apatites, we have shown that the interstitial oxygen content can be increased through water incorporation,33 and we have been extending this work to examine the ability of the silicate apatites to accommodate water. These initial studies suggested low levels of water incorporation in cation stoichiometric apatite silicates, e.g. La8+xA2−xSi6O26+x/2 (A = Ca, Sr, Ba), while higher levels were observed in systems which contain oxygen excess and cation vacancies, i.e. La9.33+xSi6O26+3x/2. This initial work suggested, in particular, that for x > 0.17 significant (>0.5 O per formula unit) water incorporation was observed. In this paper, we report a neutron diffraction and solid state NMR study examining the effect of such water incorporation in the two systems, La8Sr2Si6O26 and La9.6Si6O26.4. In particular, we have analyzed the effect of water incorporation on the structure, and the location of the interstitial oxide ion site.
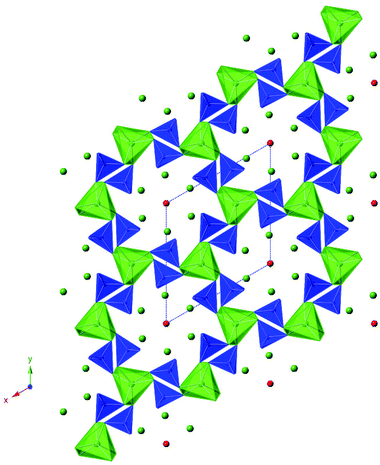 |
| | Fig. 1 Illustration of the apatite structure (A10(MO4)6O2, showing a A4(MO4)6 framework (consisting of corner linked MO4 tetrahedra (blue) and AO6 trigonal metaprisms (green)) with the remaining A6O2 unit within the “channels” of this framework (A green spheres, O red spheres). | |
2. Experimental
La9.6Si6O26.4 and La8Sr2Si6O26 were prepared as follows. High purity La2O3, SrCO3 and SiO2 were ground together in the correct stoichiometric ratio and heated for 12 hours at 1350 °C, with a second firing at 1350 °C–1400 °C for a further 12 hours. Between firings the sample was reground to ensure a homogeneous mixture. Phase purity was established through X-ray powder diffraction (Bruker D8 diffractometer with Cu Kα1 radiation = 1.5406 Å).
For the water incorporation, half of each sample was heated in water in a hydrothermal vessel (model 4749 Parr digestion vessel with 23 ml capacity) at 200 °C for 48 hours, as described previously.33 The water contents were assessed through thermogravimetric analysis (Netzsch STA 449 F1 Jupiter Thermal Analyser). The TGA experiments were carried out in N2 with a heating rate of 10 °C min−1 up to 700 °C.
In order to gain additional information on the thermal stability of the water in the apatite structure, the hydrated La9.6Si6O26.4 sample was investigated further through high temperature X-ray diffraction, utilising a Bruker D8 diffractometer. Measurements were made between 50 and 550 °C in air.
The structures of both as prepared and hydrated La9.6Si6O26.4 and La8Sr2Si6O26 samples were determined by Rietveld refinement using neutron diffraction data. Room temperature data for as prepared La9.6Si6O26.4 and La8Sr2Si6O26, and hydrated La9.6Si6O26.4 samples were collected on diffractometer HRPT at the SINQ, Paul Scherrer Institut while room temperature data for hydrated La8Sr2Si6O26 were collected on the HRPD diffractometer, ISIS, Rutherford Appleton Laboratory. All structural refinements employed the GSAS suite of Rietveld refinement software.61
All 29Si MAS and CPMAS NMR measurements were performed at an external B0 field of 9.4 T using a Bruker DSX-400 spectrometer operating at a 29Si Larmor frequency of 59.61 MHz. Each MAS and CPMAS NMR experiment was undertaken using a Bruker 4 mm dual channel (HX) MAS probe in which MAS frequencies (νr) of 12 kHz were achieved. The 29Si pulse time calibration was performed on a sample of solid kaolinite where a π/2 pulse width of 4 μs was measured, and the reported 29Si MAS NMR data were acquired using single pulse (Bloch decay) experiments with high power 1H decoupling (B1 = 50 kHz) during acquisition. A π/4 excitation pulse of 2 μs and a recycle delay of 30 s were common to all measurements and provided a quantitative description of the Si speciation, although checks with longer recycle delays of up to 120 s were also undertaken. For the 29Si CPMAS measurements an initial 1H π/2 time of 4 μs and a Hartmann-Hahn contact period of 5 ms were also calibrated on the kaolinite sample, with a recycle delay of 5 s being used. All 29Si chemical shifts are reported against the primary TMS solution reference (δ 0.0 ppm) via the secondary kaolinite solid reference (δ −92.0 ppm).
The corresponding 17O and 1H MAS NMR measurements were performed on 17O enriched samples at an external B0 field of 14.1 T using a Bruker Avance II-600 spectrometer operating at 17O and 1H Larmor frequencies of 81.30 and 600.13 MHz, respectively. Since, the natural abundance of 17O is very low, it is important to enrich the samples prior to measurement of the 17O NMR. To achieve 17O enrichment, the samples (1 g) were initially hydrated (0.5 cm3 90% 17O enriched water) under hydrothermal conditions (200 °C, 48 hours) as described earlier. These 17O MAS NMR experiments were undertaken using a Bruker 2.5 mm triple channel (HXY) MAS probe in which MAS frequencies (νr) of 31.25 kHz were achieved. The 17O pulse time calibration was performed on a water sample where a ‘non-selective’ (solution) π/2 pulse width of 3 μs was measured, and the reported 17O MAS NMR data were acquired using a rotor synchronized spin echo (θ − τ − 2θ − τ − acq.) experiment. The ‘selective’ (solids) pulses used θ and 2θ pulses of 1 and 2 μs duration (representing flip angles of π/2 and π), respectively, and a recycle delay of 10 s was employed. All 17O chemical shifts are reported against the primary solution reference of water (δ 0.0 ppm). The 1H pulse time calibration was performed on a water sample where a π/2 pulse width of 3 μs was measured, and the reported 1H MAS NMR data were acquired using a rotor synchronized spin echo (θ − τ − 2θ −τ − acq.) experiment. All reported 1H chemical shifts are referenced to the TMS primary reference (δ 0.0 ppm).
3. Results and discussion
3.1 As prepared and hydrated La8Sr2Si6O26
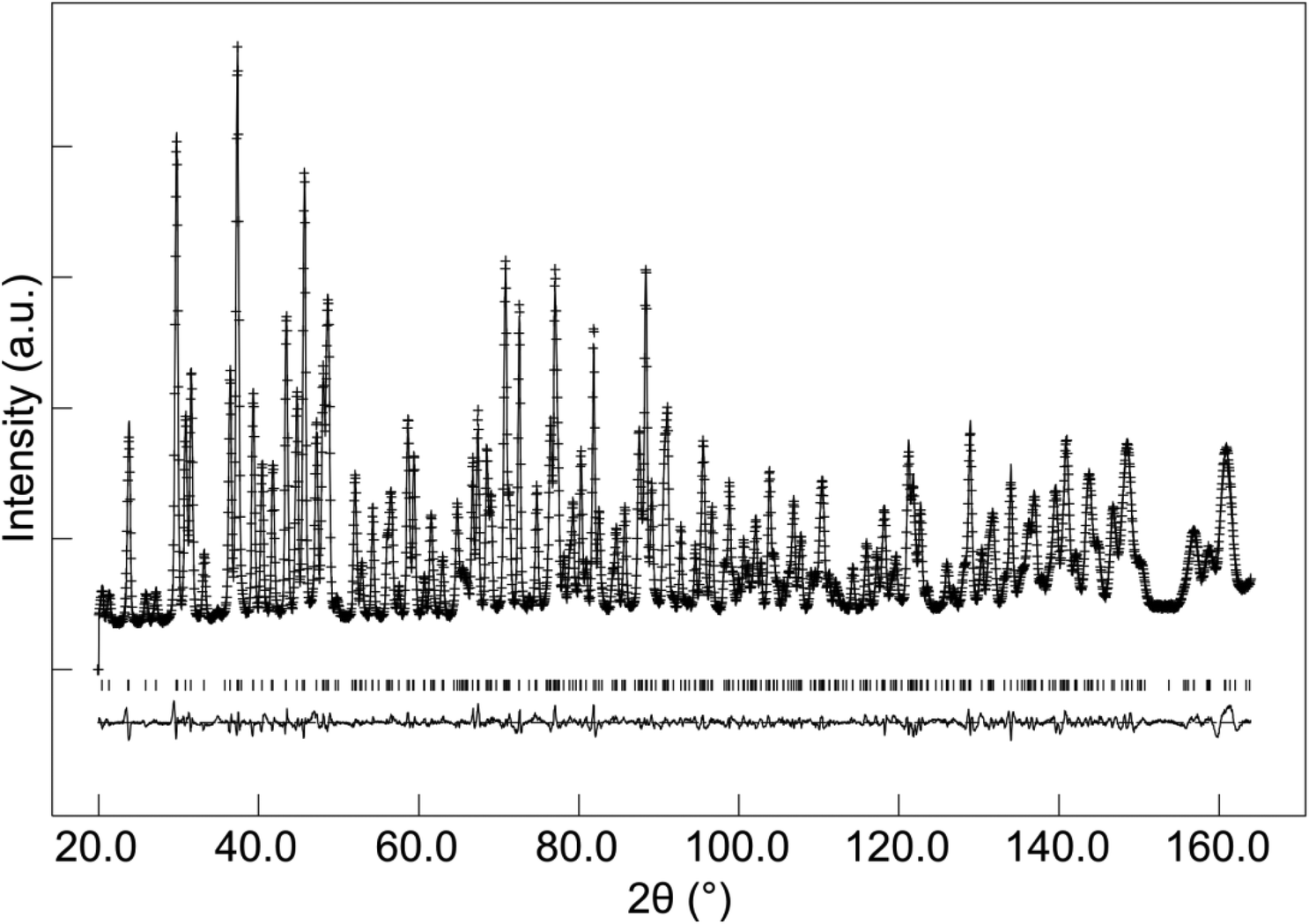
In the case of as-prepared La8Sr2Si6O26, the sample should nominally have no oxygen excess, and therefore negligible interstitial oxide ion content. In agreement with this, the neutron diffraction structural study found no evidence for any interstitial oxide ions, along with a low thermal displacement parameter for the channel oxide ion site. The final refined structural parameters (space group P63/m was used in line with prior single crystal structural studies of the related Nd8Sr2Si6O26 system60) and bond distances are given in Tables 1 and 2, while the observed calculated and difference profiles are given in Fig. 2.
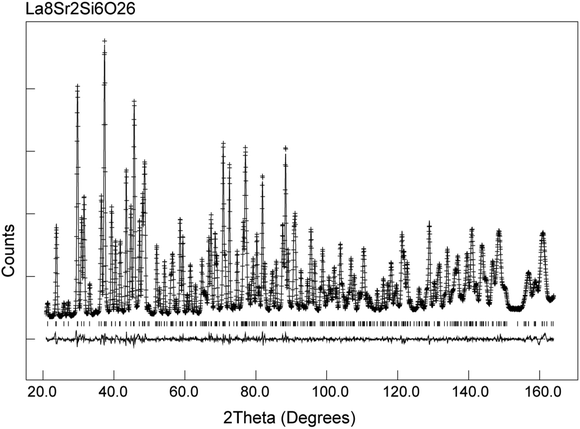 |
| | Fig. 2 Observed, calculated and difference neutron diffraction profiles for as-prepared La8Sr2Si6O26. | |
Table 1 Structural parameters of as prepared La8Sr2Si6O26
| Space group |
a/b (Å) |
c (Å) |
R
wp
|
R
p
|
χ
2
|
|
P63/m |
9.70680 (8) |
7.23791 (7) |
2.54 |
1.92 |
5.884 |
| Atom |
Site |
x
|
y
|
z
|
U
iso × 100 (Å) |
SOF |
| La(1) |
2b |
1/3 |
2/3 |
−0.0006(2) |
0.597(20) |
0.5 |
| Sr(1) |
2b |
1/3 |
2/3 |
−0.0006(2) |
0.597(20) |
0.5 |
| La(2) |
2b |
0.01292(11) |
0.24500(9) |
1/4 |
0.436(15) |
1 |
| Si |
6c |
0.40092(18) |
0.37049(17) |
1/4 |
0.160(30) |
1 |
| O(1) |
6c |
0.32170(13) |
0.48327(14) |
1/4 |
|
1 |
| O(2) |
6c |
0.59401(13) |
0.47124(15) |
1/4 |
|
1 |
| O(3) |
6c |
0.34263(11) |
0.25364(10) |
0.07024(11) |
|
1 |
| O(4) |
2a |
0 |
0 |
1/4 |
|
1 |
| 100× |
U
11
|
U
22
|
U
33
|
U
12
|
U
13
|
U
23
|
| O(1) |
1.40(6) |
1.04(6) |
0.867(67) |
1.049(56) |
0 |
0 |
| O(2) |
0.43(6) |
0.42(5) |
1.229(58) |
0.116(50) |
0 |
0 |
| O(3) |
2.02(5) |
0.77(4) |
0.552(34) |
0.739(38) |
−0.573(33) |
−0.287(30) |
| O(4) |
0.67(5) |
0.67(5) |
2.427(107) |
0.335(26) |
0 |
0 |
Table 2 Selected bond distances for as prepared La8Sr2Si6O26
| Bond |
Bond distance (Å) |
| La(1)/Sr(1)–O(1) |
2.5048(12), 2.5045(12), 2.5041(12) |
| La(1)/Sr(1)–O(2) |
2.5474(12), 2.5480(12), 2.5475(12) |
| La(1)/Sr(1)–O(3) |
2.8835(10), 2.8825(10), 2.8832(10) |
| La(2)–O(1) |
2.7204(15) |
| La(2)–O(2) |
2.4970(15) |
| La(2)–O(3) (×2) |
2.4731(8) |
| La(2)–O(3) (×2) |
2.6005(12) |
| La(2)–O(4) |
2.3180(7) |
| Si(1)–O(1) |
1.6222(18) |
| Si(1)–O(2) |
1.6236(18) |
| Si(1)–O(3) (×2) |
1.6302(11) |
On hydration, a small expansion in the cell volume was observed, and TGA studies indicated a water content of 0.18 molecules per formula unit. The presence of water means the occupancy of interstitial sites by the extra oxide ions from the water. The neutron diffraction structural studies were in agreement with this, indicating the presence of interstitial oxide ions at a position of (−0.0247, 0.1416, 0.6617). It was not possible to locate the proton site, most likely due to the low occupancy, thermal motion of these protons, and the presence of a range of different H sites with significant local displacement in these positions. The final refined structural parameters and bond distances are given in Tables 3 and 4, while the observed calculated and difference profiles are given in Fig. 3.
 |
| | Fig. 3 Observed, calculated and difference neutron diffraction profiles for hydrated La8Sr2Si6O26. | |
Table 3 Structural parameters of hydrated La8Sr2Si6O26
| Space group |
a/b (Å) |
c (Å) |
R
wp
|
R
p
|
χ
2
|
|
P63/m |
9.72828(11) |
7.25273(9) |
1.58 |
2.23 |
7.773 |
| Atom |
Site |
x
|
y
|
z
|
U
iso × 100 (Å) |
SOF |
| La(1) |
2b |
1/3 |
2/3 |
−0.0009(1) |
0.531(16) |
0.5 |
| Sr(1) |
2b |
1/3 |
2/3 |
−0.0009(1) |
0.531(16) |
0.5 |
| La(2) |
2b |
0.01320(9) |
0.24494(8) |
1/4 |
0.454(12) |
1 |
| Si |
6c |
0.40086(16) |
0.3709(2) |
1/4 |
0.301(22) |
1 |
| O(1) |
6c |
0.32136(12) |
0.4831(2) |
1/4 |
|
1 |
| O(2) |
6c |
0.59393(11) |
0.4711(3) |
1/4 |
|
1 |
| O(3) |
6c |
0.34321(9) |
0.2541(1) |
0.07047(8) |
|
1 |
| O(4) |
2a |
0 |
0 |
1/4 |
|
0.981(6) |
| O(i) |
6c |
−0.0246(33) |
0.1416(32) |
0.6617(35) |
|
0.031(2) |
| 100× |
U
11
|
U
22
|
U
33
|
U
12
|
U
13
|
U
23
|
| O(1) |
1.49(5) |
1.11(6) |
0.496(47) |
1.084(49) |
0 |
0 |
| O(2) |
0.24(5) |
0.37(4) |
1.260(57) |
−0.001(41) |
0 |
0 |
| O(3) |
2.39(5) |
0.68(4) |
0.404(26) |
0.774(34) |
−0.798(31) |
−0.234(27) |
| O(4) |
0.84(6) |
0.84(6) |
2.60(12) |
0.418(29) |
0 |
0 |
| O(i) |
2.5 |
2.5 |
2.5 |
0 |
0 |
0 |
Table 4 Selected bond distances for hydrated La8Sr2Si6O26
| Bond |
Bond distance (Å) |
| La(1)/Sr(1)–O(1) |
2.5111(13), 2.5108(13), 2.5105(13) |
| La(1)/Sr(1)–O(2) |
2.5519(13), 2.5525(13), 2.5520(13) |
| La(1)/Sr(1)–O(3) |
2.8876(11), 2.8866(11), 2.8873(11) |
| La(2)–O(1) |
2.7233(17) |
| La(2)–O(2) |
2.5078(16) |
| La(2)–O(3) (×2) |
2.4810(9) |
| La(2)–O(3) (×2) |
2.6081(13) |
| La(2)–O(4) |
2.3202(8) |
| La(2)–O(i) |
2.40(4), 1.843(34), 2.40(4) |
| Si(1)–O(1) |
1.6207(20) |
| Si(1)–O(2) |
1.6258(21) |
| Si(1)–O(3) (×2) |
1.6331(13) |
| Si(1)–O(i) (×2) |
2.24(4) |
Accompanying the water incorporation, there was a small decrease in the AO6 metaprism twist angle from 23.23 to 23.20°, leading to a small expansion of the channels to accommodate this water.
3.2 As prepared La9.6Si6O26.4
Among the various space groups reported to be exhibited by apatite systems containing cation vacancies/oxygen excess,16 space groups P63 and P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) were analysed initially, as these are the most widely reported space groups used to describe oxygen excess apatite-type silicates.17 The initial structural refinement using both symmetries showed similar R-factor values, and thus the higher symmetry space group P63 was chosen for the full structural refinement. Since there is no special position in terms of the z coordinate in this space group, it was required to constrain the z coordinate of the channel oxygen to a value of 1/4 to provide a fixed origin in z. In the initial structure refinement, there was no evidence of any cation vacancy ordering phenomena involving the La(1) and La(2) sites,18 and so to avoid errors involving the high degree of correlation between these sites, their fractional occupancy and atomic displacement parameters were constrained to be equal. Among the various interstitial oxide ion sites reported in apatite silicates, two sites at channel periphery (0.037, 0.277, 0.584)17 and near channel centre (0.018, 0.050, 0.573)12 were tested initially. The refinement for both sites showed an improved fit compared to the model without any interstitial oxide ion (Rwp = 2.18) but the interstitial oxide ion positioned near the channel centre resulted in a small improvement in fit (Rwp = 2.08) over the channel periphery site (Rwp = 2.13). Considering both models have the same number of variables, the full structural refinement was continued therefore with the interstitial oxide ion near the channel centre, although the fact that the difference in the two models is relatively small may indicate that there are a range of interstitial oxide ion positions, with a greater occupancy of sites closer to the channel centre than the periphery.
were analysed initially, as these are the most widely reported space groups used to describe oxygen excess apatite-type silicates.17 The initial structural refinement using both symmetries showed similar R-factor values, and thus the higher symmetry space group P63 was chosen for the full structural refinement. Since there is no special position in terms of the z coordinate in this space group, it was required to constrain the z coordinate of the channel oxygen to a value of 1/4 to provide a fixed origin in z. In the initial structure refinement, there was no evidence of any cation vacancy ordering phenomena involving the La(1) and La(2) sites,18 and so to avoid errors involving the high degree of correlation between these sites, their fractional occupancy and atomic displacement parameters were constrained to be equal. Among the various interstitial oxide ion sites reported in apatite silicates, two sites at channel periphery (0.037, 0.277, 0.584)17 and near channel centre (0.018, 0.050, 0.573)12 were tested initially. The refinement for both sites showed an improved fit compared to the model without any interstitial oxide ion (Rwp = 2.18) but the interstitial oxide ion positioned near the channel centre resulted in a small improvement in fit (Rwp = 2.08) over the channel periphery site (Rwp = 2.13). Considering both models have the same number of variables, the full structural refinement was continued therefore with the interstitial oxide ion near the channel centre, although the fact that the difference in the two models is relatively small may indicate that there are a range of interstitial oxide ion positions, with a greater occupancy of sites closer to the channel centre than the periphery.
The structural parameters and bond distances for the as prepared La9.6Si6O26.4 sample using neutron diffraction data are given Table 5 and 6, with the observed, calculated and difference profiles in Fig. 4. The data indicated a refined composition of La9.50Si6O26.26, close to that expected from the starting stoichiometry. The position of the interstititial oxide ions is similar to the position observed by Bechade et al. and others.38,44,45 Furthermore, as in the prior studies, the presence of oxide ion interstitials is accompanied by some vacancies in the ideal channel oxide ion site. In their work, Bechade et al. proposed a defect complex (O′′i– 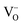 –O′′i (Kröger–Vink notation, O′′i = interstitial oxide ion with a double effective negative charge,
–O′′i (Kröger–Vink notation, O′′i = interstitial oxide ion with a double effective negative charge, 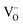 = oxide ion vacancy with a double effective positive charge)), and in the present study, a similar complex can be proposed. The observed length scale in this study for this complex is 2.27 Å which is smaller than the predicted value of 2.93 Å from the modelling work.10 However, it should be noted that the atomic displacement parameter for O(6) is very high perpendicular to the channel (100 × U22 = 34 Å2). This suggests significant local displacement from the refined position, which may hence allow an increase in the O′′i–
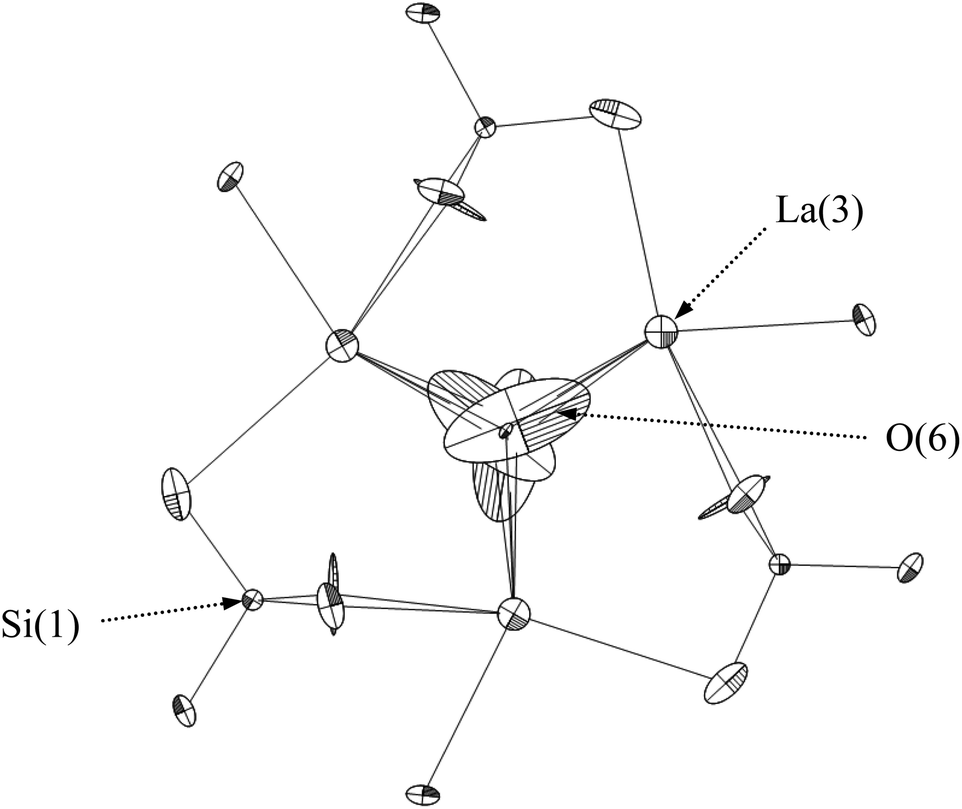
= oxide ion vacancy with a double effective positive charge)), and in the present study, a similar complex can be proposed. The observed length scale in this study for this complex is 2.27 Å which is smaller than the predicted value of 2.93 Å from the modelling work.10 However, it should be noted that the atomic displacement parameter for O(6) is very high perpendicular to the channel (100 × U22 = 34 Å2). This suggests significant local displacement from the refined position, which may hence allow an increase in the O′′i– 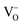 length. Overall it would suggest the presence of various interstitial oxide ion sites with differing displacements from the channel centre. In particular, the anisotropic thermal ellipsoids for the O(6) sites are directed towards channel lanthanum, La(3) (Fig. 5), which is not unexpected as O(6) is highly underbonded (bond valence sum calculations for this oxygen site give a value of only −1.08), and so some local displacement might be expected to aid the stability of this oxide ion site.
length. Overall it would suggest the presence of various interstitial oxide ion sites with differing displacements from the channel centre. In particular, the anisotropic thermal ellipsoids for the O(6) sites are directed towards channel lanthanum, La(3) (Fig. 5), which is not unexpected as O(6) is highly underbonded (bond valence sum calculations for this oxygen site give a value of only −1.08), and so some local displacement might be expected to aid the stability of this oxide ion site.
 |
| | Fig. 4 Observed, calculated and difference neutron diffraction profiles for as-prepared La9.6Si6O26.4. | |
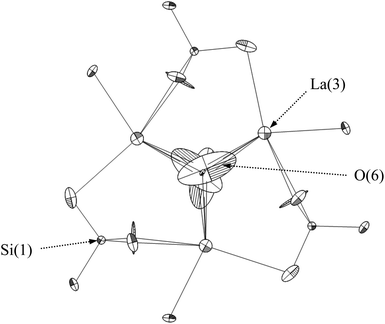 |
| | Fig. 5 The anisotropic thermal ellipsoids around the apatite channel position of as prepared La9.6Si6O26.4, viewed down the c-axis. | |
Table 5 Structural parameters of as prepared La9.6Si6O26.4
| Space group |
a/b (Å) |
c (Å) |
R
wp
|
R
p
|
χ
2
|
|
P63 |
9.72441(4) |
7.18726(5) |
2.00 |
1.58 |
3.299 |
| Atom |
Site |
x
|
y
|
z
|
U
iso × 100 (Å) |
SOF |
| La(1) |
2b |
1/3 |
2/3 |
−0.0161(5) |
1.28(2) |
0.874(2) |
| La(2) |
2b |
2/3 |
1/3 |
−0.0139(5) |
1.28(2) |
0.874(2) |
| La(3) |
6c |
0.2286(1) |
−0.0119(1) |
0.2427(5) |
1.09(1) |
1 |
| Si |
6c |
0.4028(2) |
0.3725(1) |
0.2488(5) |
0.69(2) |
1 |
| O(1) |
6c |
0.3235(2) |
0.4847(2) |
0.2429(9) |
|
1 |
| O(2) |
6c |
0.5948(1) |
0.4729(2) |
0.2438(10) |
|
1 |
| O(3) |
6c |
0.3531(4) |
0.2580(5) |
0.0592(8) |
|
1 |
| O(4) |
6c |
0.6643(6) |
0.7466(5) |
0.9204(7) |
|
1 |
| O(5) |
2a |
0 |
0 |
1/4 |
|
0.853(5) |
| O(i) |
6c |
0.0033(23) |
0.0193(41) |
0.4049(21) |
|
0.092(3) |
| 100× |
U
11
|
U
22
|
U
33
|
U
12
|
U
13
|
U
23
|
| O(1) |
2.98(7) |
2.51(7) |
2.43(9) |
2.38(7) |
−1.64(20) |
−0.52(21) |
| O(2) |
1.01(6) |
0.98(5) |
2.47(7) |
0.37(5) |
0.37(20) |
0.30(21) |
| O(3) |
2.22(11) |
1.84(14) |
1.75(14) |
1.29(11) |
−0.80(9) |
−0.51(14) |
| O(4) |
8.27(20) |
1.18(14) |
1.59(15) |
2.08(16) |
−2.73(12) |
−0.84(15) |
| O(5) |
0.28(5) |
0.27(5) |
13.4(3) |
0.14(3) |
0 |
0 |
| O(i) |
8.9(18) |
33.8(36) |
8.3(15) |
13.7(24) |
15.0(16) |
19.3(24) |
Table 6 Selected bond distances for as prepared La9.6Si6O26.4
| Bond |
Bond distance (Å) |
| Si–O(1) |
1.620(1) |
| Si–O(2) |
1.618(1) |
| Si–O(3) |
1.674(1) |
| Si–O(4) |
1.587(1) |
| La(1)–O(1) (×3) |
2.535(1) |
| La(1)–O(2) (×3) |
2.503(1) |
| La(1)–O(4) (×3) |
2.944(1) |
| La(2)–O(1) (×3) |
2.453(1) |
| La(2)–O(2) (×3) |
2.591(1) |
| La(2)–O(3) (×3) |
2.806(1) |
| La(3)–O(1) |
2.762(1) |
| La(3)–O(2) |
2.519(1) |
| La(3)–O(3) |
2.630(1), 2.476(1) |
| La(3)–O(4) |
2.464(1), 2.586(1) |
| La(3)–O(5) |
2.284(1) |
| O(i)–La(3) |
2.632(1), 2.677(1), 2.405(1) |
3.3 Hydrated La9.6Si6O26.4
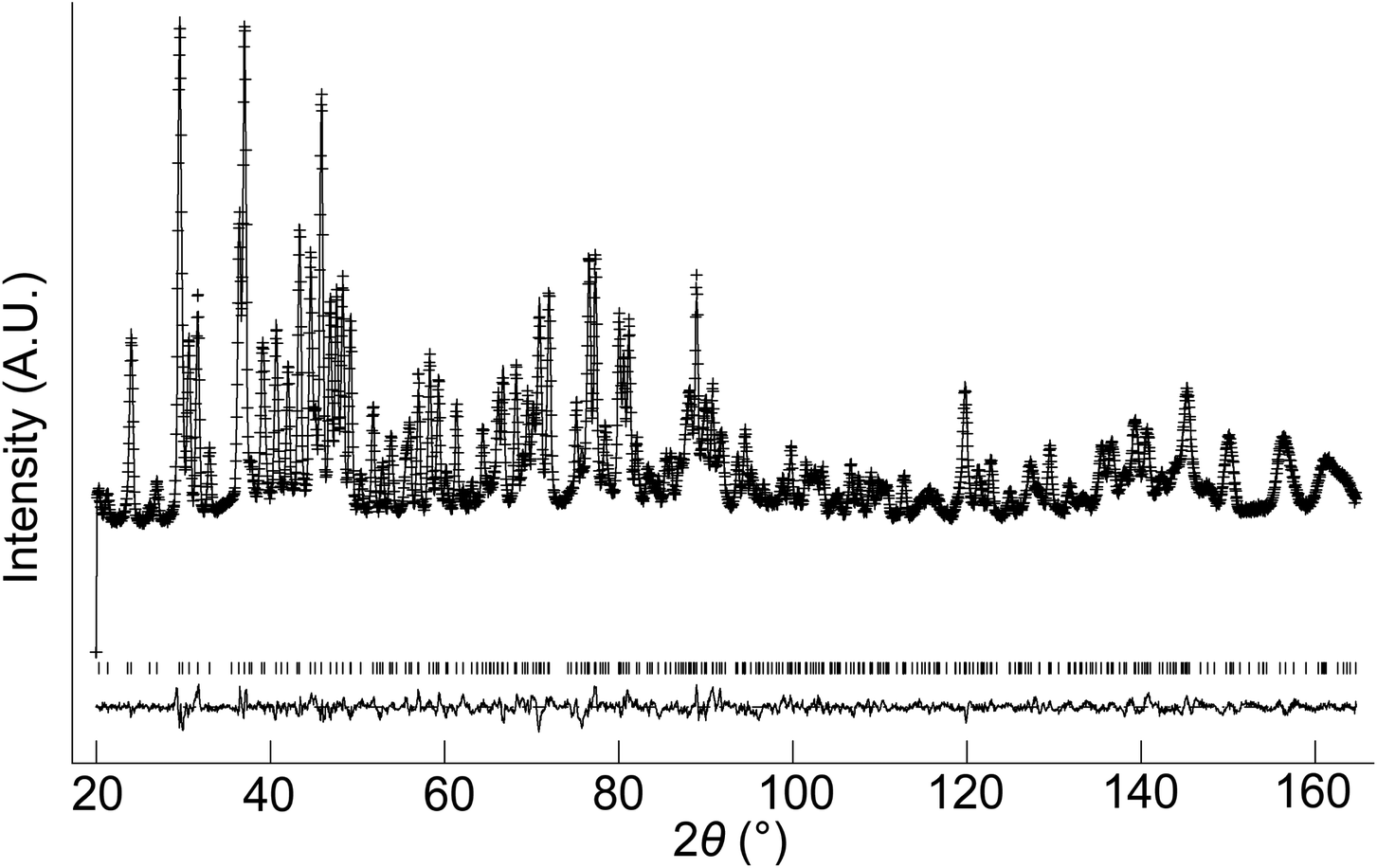
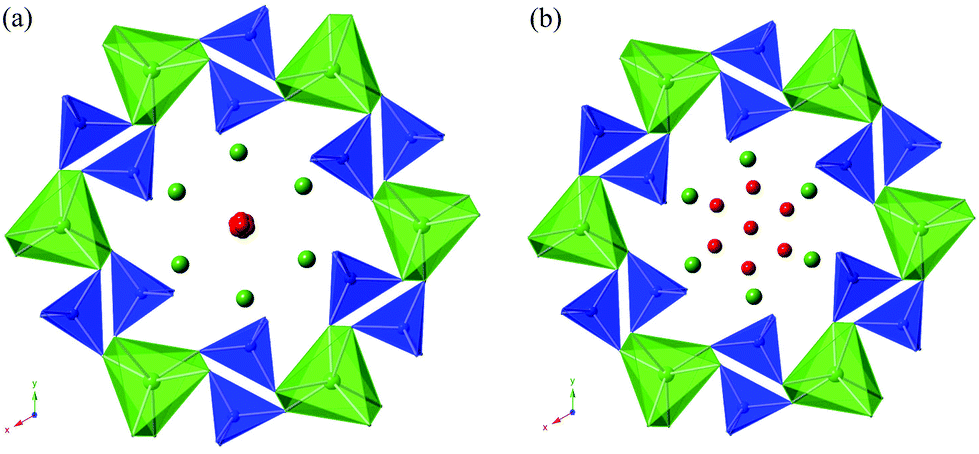
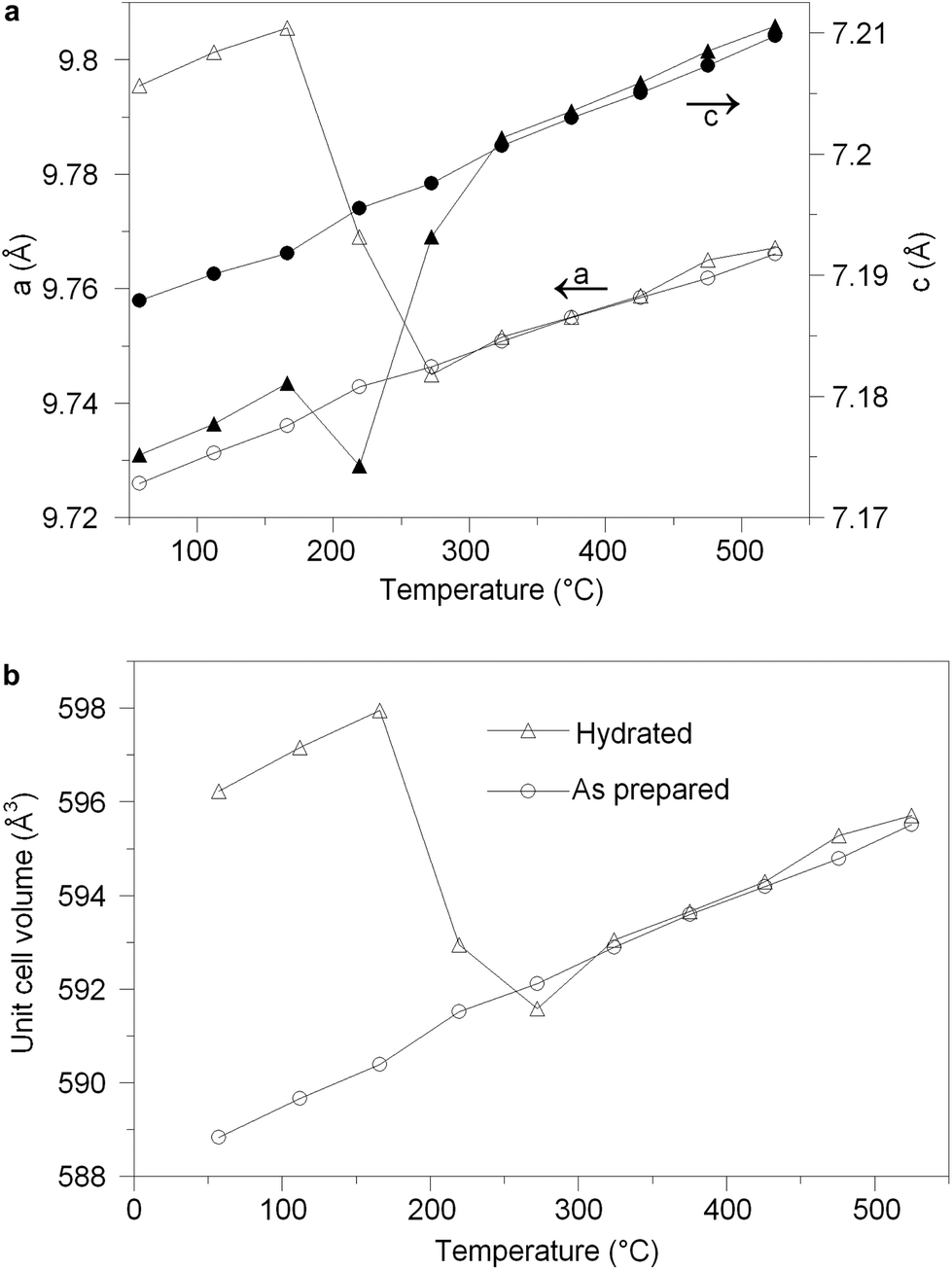
The structural parameters and bond distances for hydrated La9.6Si6O26.4 are given in Tables 7 and 8 with the observed, calculated and difference profiles in Fig. 6. It was not possible to locate the proton site, most likely due to the presence of a range of different H sites, thermal motion of the H, and significant local displacement in these positions. In this respect, further studies at low temperature (≈4 K) would be required, as has been performed to locate the proton sites in perovskite systems. The data indicated a refined composition of La9.53Si6O26.98 (excluding protons) showing a significant increase in the O content. If we assume that this extra O is charge balanced by protons, the composition of La9.53Si6O26.98H1.37 (La9.53Si6O26.295·0.685H2O) is obtained. Apart from the increased O content, the major difference compared to the as prepared sample is the significant deviation of the interstitial oxide ion site away from the channel centre (0.133, 0.143, 0.419) (Fig. 7). It can be proposed that the additional displacement from the channel centre is required to accommodate more anions in the structure, and the data showed that further structure distortion accompanied the water incorporation. In particular, the calculated average metaprism twist angle of LaO6 was decreased from 22.24° to 21.68°, in order to expand the channel size to allow the accommodation of the extra oxide ions, resulting in a significant increase (0.70%) in the cell size along a/b and decrease (0.17%) in c. These cell parameter changes can be elegantly demonstrated using high temperature X-ray diffraction data (see Fig. 8). Such data show that there is a steep decrease in the size of a/b and increase in c at around 210 °C, on heating the hydrated sample (due to water loss), while the as prepared sample showed a linear increase over the temperature range. This single stage dehydration process can also be easily noticed on the plot of cell volume change with temperature.
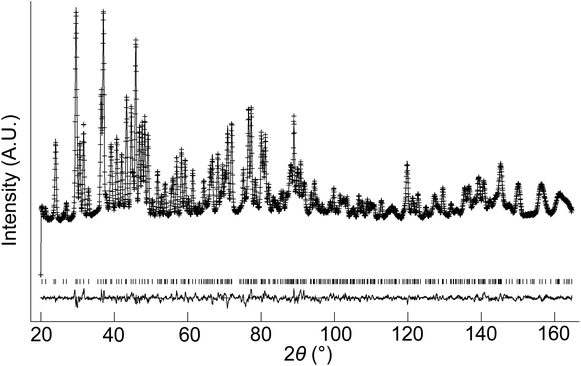 |
| | Fig. 6 Observed, calculated and difference neutron diffraction profiles for hydrated La9.6Si6O26.4. | |
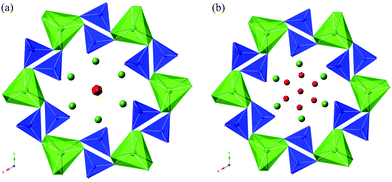 |
| | Fig. 7 Illustration of the structures of (a) as-prepared and (b) hydrated La9.6Si6O26.4 (blue tetrahedra = SiO4, green trigonal metaprisms = LaO6, green spheres = La, red spheres = O) showing the presence of the interstitial oxide ion site close to the centre of the oxide ion channel for the former, while there is a significant deviation of this interstitial site away from the channel centre for the latter. | |
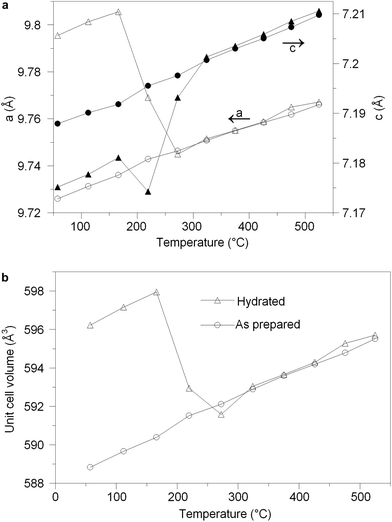 |
| | Fig. 8 (a) Cell parameter variation versus temperature for the as prepared (circle) and hydrated (triangle) La9.6Si6O26.4 on heating. (b) Cell volume variation versus temperature for the as prepared (circle) and hydrated (triangle) La9.6Si6O26.4 on heating. | |
Table 7 Structural parameters for hydrated La9.6Si6O26.4
| Space group |
a/b (Å) |
c (Å) |
R
wp
|
R
p
|
χ
2
|
|
P63 |
9.79242(10) |
7.17565(9) |
2.47 |
1.93 |
5.309 |
| Atom |
Site |
x
|
y
|
z
|
U
iso × 100 (Å) |
SOF |
| La(1) |
2b |
1/3 |
2/3 |
0.0058(9) |
1.93(4) |
0.882(3) |
| La(2) |
2b |
2/3 |
1/3 |
0.0028(9) |
1.93(4) |
0.882(3) |
| La(3) |
6c |
0.2360(1) |
−0.0077(2) |
0.2438(6) |
1.28(2) |
1 |
| Si |
6c |
0.4032(3) |
0.3757(3) |
0.2475(12) |
1.51(5) |
1 |
| O(1) |
6c |
0.3269(3) |
0.4877(3) |
0.24941(8) |
|
1 |
| O(2) |
6c |
0.5939(2) |
0.4724(3) |
0.2515(9) |
|
1 |
| O(3) |
6c |
0.3624(4) |
0.2649(6) |
0.0628(6) |
|
1 |
| O(4) |
6c |
0.6664(6) |
0.7494(6) |
0.9169(6) |
|
1 |
| O(5) |
2a |
0 |
0 |
1/4 |
|
0.890(11) |
| O(i) |
6c |
0.1334(20) |
0.1425(12) |
0.4192(17) |
|
0.200(7) |
| 100× |
U
11
|
U
22
|
U
33
|
U
12
|
U
13
|
U
23
|
| O(1) |
6.09(17) |
4.29(17) |
0.71(10) |
4.70(15) |
−1.62(25) |
−2.36(18) |
| O(2) |
2.06(12) |
1.74(11) |
2.25(12) |
0.04(10) |
−0.99(30) |
−0.97(31) |
| O(3) |
5.29(24) |
4.75(27) |
0.64(16) |
4.03(23) |
0.79(14) |
0.17(16) |
| O(4) |
8.46(29) |
1.82(20) |
2.18(18) |
2.34(23) |
−3.68(15) |
−1.09(19) |
| O(5) |
1.64(15) |
1.64(15) |
22.5(8) |
0.82(7) |
0 |
0 |
| O(i) |
8.5(13) |
0.20(58) |
3.09(71) |
3.07(66) |
0.52(70) |
−2.21(51) |
Table 8 Selected bond distances for hydrated La9.6Si6O26.4
| Bond |
Bond distance (Å) |
| Si–O(1) |
1.609(1) |
| Si–O(2) |
1.618(1) |
| Si–O(3) |
1.628(1) |
| Si–O(4) |
1.614(1) |
| La(1)–O(1) (×3) |
2.455(1) |
| La(1)–O(2) (×3) |
2.580(1) |
| La(1)–O(4) (×3) |
3.010(1) |
| La(2)–O(1) (×3) |
2.505(1) |
| La(2)–O(2) (×3) |
2.552(1) |
| La(2)–O(3) (×3) |
2.740(1) |
| La(3)–O(1) |
2.830(1) |
| La(3)–O(2) |
2.479(1) |
| La(3)–O(3) |
2.651(1), 2.520(1) |
| La(3)–O(4) |
2.483(1), 2.538(1) |
| La(3)–O(5) |
2.350(1) |
| O(i)–La(3) |
2.489(1), 2.302(1), 2.538(1) |
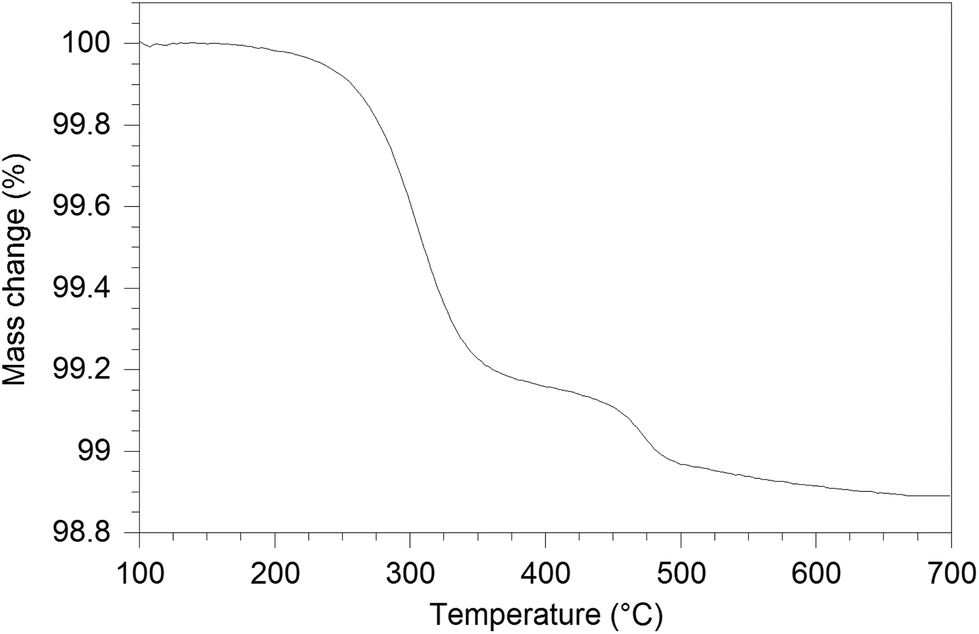
The water content of the hydrated sample was determined by TGA measurements (see Fig. 9). Unlike the single stage dehydration suggested by the high temperature X-ray diffraction data, there was evidence for a two stage water loss from the TGA data: firstly there was an abrupt loss in mass at around 280 °C with a second mass loss at around 470 °C. Since high temperature X-ray diffraction data did not show any change at higher temperature, the second mass loss was attributed to the decomposition of an impurity phase (most likely amorphous). As the structure refinement indicated a lower La content than the starting ratio, it was presumed that this may be amorphous La(OH)3. La(OH)3 is known to decompose in two steps, via an LaOOH intermediate as outlined below, and so it was proposed that the second mass loss observed in the TGA plots was due to the LaOOH dehydrating. This would suggest that part of the first mass loss was due to La(OH)3 dehydrating to LaOOH.
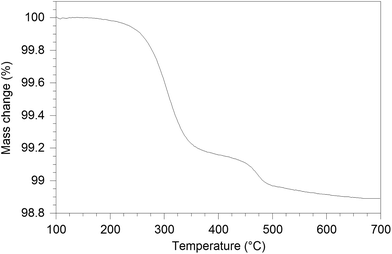 |
| | Fig. 9 TG profile of hydrated La9.6Si6O26.4. | |
In order to estimate the contribution from this proposed amorphous La(OH)3 impurity phase, firstly, the mass of LaOOH was estimated from the mass loss at 470 °C and subsequently, the mass of La(OH)3 was calculated (1.7(2) wt%). Eliminating the contribution of the above process, the calculated level of water incorporated was 0.75 H2O per formula unit, which is similar to the composition La9.53Si6O26.295·0.685H2O and the calculated interstitial content (≈1 O per formula unit) from the diffraction studies. The dehydration temperature difference between X-ray diffraction study and the TGA result is due to the fact that the TGA measurement was performed with a 10 °C min−1 ramp rate, and so the experiment is performed under non-equilibrium conditions.
3.4 MAS NMR studies
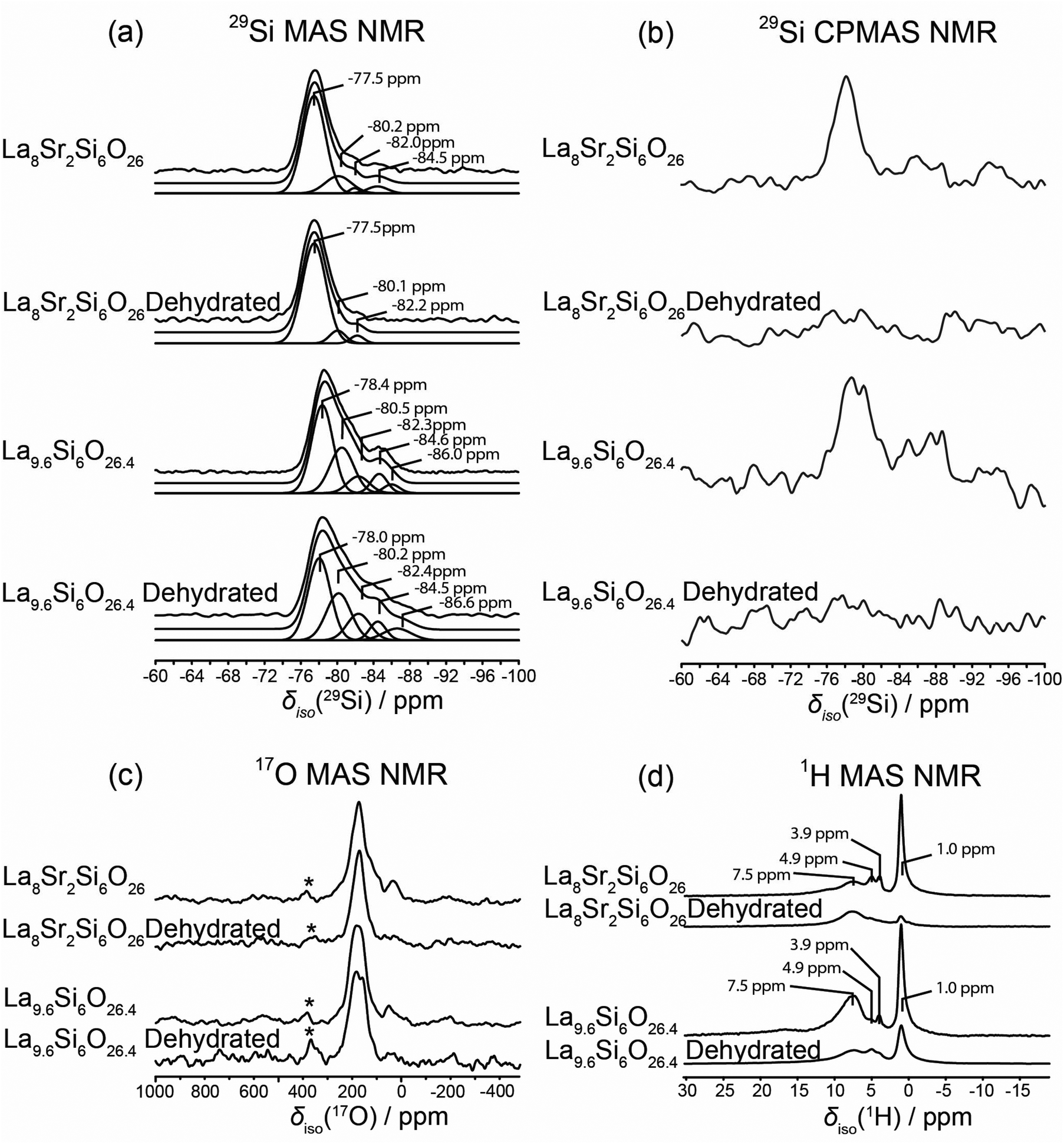
All 29Si MAS and CPMAS NMR data acquired from the La8Sr2Si6O26 and La9.6Si6O26.4 systems are presented in Fig. 10(a) and (b), respectively. The 29Si MAS NMR data demonstrate that the Si speciation comprising the stoichiometric La8Sr2Si6O26 system is dominated by monomeric framework SiO4 environments which are influenced by Sr substitution on the La position; this is evidenced by broadening of the dominant 29Si resonance at an isotropic chemical shift at δiso −77.5 ppm. The much less intense resonances at δiso ∼ −80.2 and −82.2 ppm are ascribed to low levels of cation vacancies and interstitial species that originate from substitutional arrangements and charge balancing. In comparison, the multiple resonances from the 29Si MAS NMR data describing the non-stoichiometric La9.6Si6O26.4 system reveal more complex Si speciation which emanates from the substantial influences of the much higher levels of cation vacancies and oxygen interstitials upon the local SiO4 environment. These observations mirror previously reported 29Si MAS NMR studies.15,26 Similar to La8Sr2Si6O26, the La9.6Si6O26.4 system is characterised by a dominant resonance at δiso −78.4 ppm, however the perturbations to the SiO4 framework are observed by the additional upfield resonances at δiso −80.5, −82.3, −84.6 and −86.0 ppm. This trend of upfield shifted resonances is consistent with the increased electronegativity influencing the immediate Si environment induced by La3+ cation vacancies immediately surrounding the SiO4 monomeric unit, coupled with an additional O coordination/proximity to the SiO4 site via the presence of O interstitials.
 |
| | Fig. 10 The (a) 29Si MAS and (b) 29Si CPMAS NMR data (B0 = 9.40 T, ν0 = 59.61 MHz, νr = 10 kHz), (c) 17O MAS NMR data (B0 = 14.1 T, ν0 = 81.30 MHz, νr = 30 kHz), and (d) 1H MAS NMR data (B0 = 14.1 T, ν0 = 600.13 MHz, νr = 30 kHz) data from the hydrated La8Sr2Si6O26, dehydrated La8Sr2Si6O26, hydrated La9.6Si6O26.4 and dehydrated La9.6Si6O26.4 systems. The asterisk on each spectrum in (c) denotes a background signal from the ZrO2 rotor material. | |
The 29Si CPMAS NMR data shown in Fig. 10(b) contrast markedly with the corresponding MAS NMR data discussed above. It is immediately evident that the signal-to-noise of these CPMAS data is greatly inferior thus suggesting that, despite there being an abundance of H species (i.e. OH and H2O) to facilitate 1H–29Si cross-polarisation, the motion of the intercalated OH/H2O modulates the 1H–29Si dipolar interaction and greatly diminishes the efficiency of the Hartmann-Hahn condition. For both of the hydrated La8Sr2Si6O26 and La9.6Si6O26.4 systems only the monomeric SiO4 positions exhibit some observable intensity above the noise level, and this is probably facilitated by proximity to a component of immobile OH interstitial species. However, the dehydration of these systems shows that both the mobile and immobile OH and H2O species are largely removed, as evidenced by the absence of a 29Si signal above the baseline noise.
From Fig. 10(c) the 17O MAS NMR data shows that similar O speciation from both systems is observed. Each spectrum is dominated by 17O resonances at an apparent shift of δ ∼180–200 ppm that are attributed to O species associated with the framework SiO4 element. In addition, there are two additional signals attributed to interstitial species observed from these data; a clearly resolved 17O resonance at δ ∼40–50 ppm, which may represent a Si-based (Si–O–Si or Si–OH) interstitial O species bridging between SiO4 framework moieties, and a partially resolved shoulder at δ ∼260–270 ppm which is assigned to the less prevalent Si–O–La interstitial species. The latter assignment is consistent with the observation of interstitial O species in previously reported silicate- and germanate-based rare earth apatite SOFC materials.47,49 It can be observed that upon dehydration of these systems both of these interstitial species are essentially removed, with the more complete removal of the Si–O–Si/Si–OH interstitial species being observed from the La8Sr2Si6O26 system, suggesting that this component is mostly related to Si–OH (see Fig. 10(b)). Previous studies on the La8Y2Ge6O27 rare earth apatite phase has clearly shown that a resonance observed at δ ∼ 580 ppm is associated with O channel species;49 this channel species is not clearly evident in the silicate-based La8Sr2Si6O26 and La9.6Si6O26.4 systems under study here. In addition, the resonance at δ ∼380–390 ppm (marked with an asterisk) in Fig. 10(c) is identified as a 17O background signal arising from the ZrO2 MAS rotor material.
Although nominal O interstitial species within the hydrated and dehydrated La8Sr2Si6O26 and La9.6Si6O26.4 phases are indicated by 17O resonances at δ ∼40–50 ppm, a more precise identification of these species is afforded by the accompanying 1H MAS NMR data shown in Fig. 10(d). A substantial proportion of these interstitials are hydroxylated or –Si–OH species as suggested by the narrow 1H resonance at δ 1.0 ppm. The near-complete elimination of these species upon dehydration of the La8Sr2Si6O26 system, coupled with their partial removal upon dehydration of the La9.6Si6O26.4 system is demonstrated by the concomitant reduction of the δ ∼40–50 ppm resonance(s) in the 17O MAS NMR data (see Fig. 9(c)) and the δ 1.0 ppm resonance in the 1H MAS NMR data (see Fig. 10(d)). The resonance at δ ∼8 ppm represents H-bonded OH species residing in more occluded and sterically crowded environments; these are characterized by the larger downfield shift caused by the deshielding of the H-bonding arrangement and a broader residual linewidth induced by a much larger homonuclear 1H–1H dipolar interaction. In addition, the smaller resonances at δ 3.9 and 4.9 ppm represent different H2O environments within each unit cell. These resonances are also affected by the dehydration process.
These results demonstrate that the 17O enrichment via heterogeneous gas phase exchange under autoclave conditions facilitates the exchange of the O positions comprising the SiO4 groups in both the as-synthesized La8Sr2Si6O26 and La9.6Si6O26.4 systems. These O exchange characteristics are similar to those reported for the analogous germanate systems.49 This work therefore suggests that while a direct interstitial oxide ion conduction mechanism may occur down the channels, as proposed by Bechade et al.,38 there is likely to be significant contribution from exchange processes throughout the SiO4 tetrahedra network. In particular, such processes would explain the observation of significant conductivity perpendicular to the channel direction from single crystal studies.51
In comparison to the germanate (La10−xGe6O27−3x/2) apatite systems, there appears, however, to be less association of the interstitial oxide ion defects with the MO4 tetrahedra (for the germanate systems, the 17O NMR data indicated that the presence of interstitial oxide ions led to the formation of GeO5 units49). This closer association may explain the higher activation energies for oxide ion conduction for the germanate compared to the silicate apatites, with a degree of trapping of the oxide ion interstitials due to this association. This trapping contribution is supported by recent high temperature Raman studies on germanate apatites.43
4. Conclusions
In this work, we have provided a detailed account of the structures of both as prepared and hydrated La9.6Si6O26.4 and La8Sr2Si6O26. The results show that higher hydration levels are possible for the La9.6Si6O26.4 system, and the study provides an important clarification of the effect of oxide ion content on the interstitial oxide ion position. In particular, the structural studies show that for low oxide ion excess, the interstitial oxide ion position appears to lie close to the channel centre, while on increasing the interstitial oxide ion levels, there is progressive displacement of the oxide ions towards the channel periphery. This can be explained by the need for additional displacement to prevent short O–O interactions, and is allowed by the flexibility of the apatite framework: in particular a decrease in the LaO6 metaprism twist angle allows the expansion of the apatite channels to accommodate more interstitial oxide ions. The NMR studies show that the oxide ions of the SiO4 are readily exchangeable, similar to reports for Ge based apatites. These results suggest that oxide ion exchange processes with the SiO4 are likely to have a contribution to the oxide ion conductivity, and this would help to explain the observation of significant oxide ion conduction perpendicular to the channels from single crystal studies.
Acknowledgements
We would like to thank EPSRC and the University of Birmingham for DTG financial support of studentships for BJC and JFS, and we thank EPSRC for the funding of EP/I003932, EP/G009929 and EP/I004114/1. We would like to express thanks to SINQ for neutron diffraction time and Vladimir Pomjakushin for help with the neutron diffraction experiments on the La9.6Si6O26.4 system, and to ISIS at the Rutherford Appleton Laboratory for the provision of neutron diffraction beam time for the neutron diffraction experiments on the La8Sr2Si6O26 system. JVH and PRS thank Dr Thomas Kemp for his assistance with the acquisition of some of the solid state MAS NMR data. JVH thanks EPSRC and the University of Warwick for partial funding of the solid state NMR infrastructure at Warwick. PRS and JVH thank the University of Birmingham and the Birmingham Science City for the Bruker D8 diffractometer, Netzsch STA 449 F1 Jupiter Thermal Analyser and some solid state MAS NMR instrumentation used in this research which were obtained through the Birmingham Science City Advanced Materials Project 1: Creating and Characterising Next generation Advanced Materials project, with support from Advantage West Midlands (AWM) and partial funding from the European Regional Development Fund (ERDF).
References
- A. Orera and P. R. Slater, Chem. Mater., 2010, 22, 675 CrossRef CAS.
- A. J. Jacobson, Chem. Mater., 2010, 22, 660 CrossRef CAS.
- S. Nakayama, H. Aono and Y. Sadaoka, Chem. Lett., 1995, 431 CrossRef CAS.
- S. Nakayama, M. Sakamoto, M. Higuchi and K. Kodaira, J. Mater. Sci. Lett., 2000, 19, 91 CrossRef CAS.
- H. Arikawa, H. Nishiguchi, T. Ishihara and Y. Takita, Solid State Ionics, 2000, 136–137, 31 CrossRef CAS.
- S. Tao and J. T. S. Irvine, Mater. Res. Bull., 2001, 36, 1245 CrossRef CAS.
- J. E. H. Sansom, D. Richings and P. R. Slater, Solid State Ionics, 2001, 139, 205 CrossRef CAS.
- L. Leon-Reina, M. E. Martin-Sedeno, E. R. Losilla, A. Caberza, M. Martinez-Lara, S. Bruque, F. M. B. Marques, D. V. Sheptvakov and M. A. G. Aranda, Chem. Mater., 2003, 15, 2099 CrossRef CAS.
- E. J. Abram, C. A. Kirk, D. C. Sinclair and A. R. West, Solid State Ionics, 2005, 176, 1941 CrossRef CAS.
- J. R. Tolchard, P. R. Slater and M. S. Islam, Adv. Funct. Mater., 2007, 17, 2564 CrossRef CAS.
- J. R. Tolchard, M. S. Islam and P. R. Slater, J. Mater. Chem., 2003, 13, 1956 RSC.
- L. Leon-Reina, E. R. Losilla, M. Martinez-Lara, M. C. Martin-Sedeno, S. Bruque, P. Nunez, D. V. Sheptyakov and M. A. G. Aranda, Chem. Mater., 2005, 17, 596 CrossRef CAS.
- L. Leon-Reina, E. R. Losilla, M. Martinez-Lara, S. Bruque, A. Llobet, D. V. Sheptyakov and M. A. G. Aranda, J. Mater. Chem., 2005, 15, 2489 RSC.
- V. V. Kharton, A. L. Shaula, M. V. Patrakeev, J. C. Waerenborgh, D. P. Rojas, N. P. Vyshatko, E. V. Tsipis, A. A. Yaremchenko and F. M. B. Marques, J. Electrochem. Soc., 2004, 151, A1236 CrossRef CAS.
- J. E. H. Sansom, J. R. Tolchard, D. Apperley, M. S. Islam and P. R. Slater, J. Mater. Chem., 2006, 16, 1410 RSC.
- E. Kendrick, M. S. Islam and P. R. Slater, J. Mater. Chem., 2007, 17, 3104 RSC.
- Y. Masubuchi, M. Higuchi, S. Kikkawa, K. Kodaira and S. Nakayama, Solid State Ionics, 2004, 175, 357 CrossRef CAS.
- S. Celerier, C. Laberty-Robert, J. W. Long, K. A. Pettigrew, R. M. Stroud, D. R. Rolison, F. Ansart and P. Stevens, Adv. Mater., 2006, 18, 615 CrossRef CAS.
- L. Leon-Reina, J. M. Porras-Vasquez, E. R. Losilla and M. A. G. Aranda, J. Solid State Chem., 2007, 180, 1250 CrossRef CAS.
- E. Kendrick, J. R. Tolchard, J. E. H. Sansom, M. S. Islam and P. R. Slater, Faraday Discuss., 2007, 134, 181 RSC.
- E. Kendrick and P. R. Slater, Mater. Res. Bull., 2008, 43, 2509 CrossRef CAS.
- E. Kendrick and P. R. Slater, Mater. Res. Bull., 2008, 43, 3627 CrossRef CAS.
- S. S. Pramana, W. T. Klooster and T. J. White, Acta Crystallogr., Sect. B:
Struct. Sci., 2007, 63, 597 CAS.
- S. S. Pramana, W. T. Klooster and T. J. White, J. Solid State Chem., 2008, 181, 1717 CrossRef CAS.
- E. Kendrick, M. S. Islam and P. R. Slater, Chem. Commun., 2008, 715 RSC.
- A. Orera, E. Kendrick, D. C. Apperley, V. M. Orera and P. R. Slater, Dalton Trans., 2008, 5296 RSC.
- E. Kendrick and P. R. Slater, Solid State Ionics, 2008, 179, 981 CrossRef CAS.
- P. J. Panteix, I. Julien, P. Abelard and D. Bernache-Assolant, Ceram. Int., 2008, 34, 1579 CrossRef CAS.
- T. Iwata, K. Fukuda, E. Bechade, O. Masson, I. Julien, E. Champion and P. Thomas, Solid State Ionics, 2008, 178, 1523 CrossRef.
- R. Ali, M. Yashima, Y. Matsushita, H. Yoshioka, K. Okoyama and F. Izumi, Chem. Mater., 2008, 20, 5203 CrossRef CAS.
- J. R. Tolchard and P. R. Slater, J. Phys. Chem. Solids, 2008, 69, 2433 CrossRef CAS.
- E. Kendrick and P. R. Slater, Mater. Res. Bull., 2008, 43, 3627 CrossRef CAS.
- A. Orera and P. R. Slater, Solid State Ionics, 2010, 181, 110 CrossRef CAS.
- E. Kendrick, K. S. Knight and P. R. Slater, Mater. Res. Bull., 2009, 44, 1806 CrossRef CAS.
- J. M. Porras-Vazquez, E. R. Losilla, L. Leon-Reina, D. Marrero-Lopez and M. A. G. Aranda, J. Am. Ceram. Soc., 2009, 92, 1062 CrossRef CAS.
- C. Bonhomme, S. Beaudet-Savignat, T. Chartier, P.-M. Geffroy and A.-L. Sauvet, J. Eur. Ceram. Soc., 2009, 29, 1781 CrossRef CAS.
- A. Al-Yasari, A. Jones, D. C. Apperley, D. Driscoll, M. S. Islam and P. R. Slater, J. Mater. Chem., 2009, 19, 5003 RSC.
- E. Bechade, O. Masson, T. Iwata, I. Julien, K. Fukuda, P. Thomas and E. Champion, Chem. Mater., 2009, 21, 2508 CrossRef CAS.
- S. Guillot, S. Beaudet-Savignat, S. Lambert, R.-N. Vannier, P. Roussel and F. Porcher, J. Solid State Chem., 2009, 182, 3358 CrossRef CAS.
- E. Kendrick, A. Orera and P. R. Slater, J. Mater. Chem., 2009, 19, 7955 RSC.
- A. Orera, D. Headspith, D. C. Apperley, M. G. Francesconi and P. R. Slater, J. Solid State Chem., 2009, 182, 3294 CrossRef CAS.
- T. J. White and Z. L. Dong, Acta Crystallogr., Sect. B: Struct. Sci., 2003, 59, 1 CrossRef CAS.
- A. Orera, M. L. Sanjuán, E. Kendrick, V. M. Orera and P. R. Slater, J. Mater. Chem., 2010, 20, 2170 RSC.
- K. Fukuda, T. Asaka, M. Oyabu, D. Urushihara, A. Berghout, E. Bechade, O. Masson, I. Julien and P. Thomas, Chem. Mater., 2012, 24, 4623 CrossRef CAS.
- K. Fukuda, T. Asaka, N. Ishizawa, H. Mino, D. Urushihara, A. Berghout, E. Bechade, O. Masson, I. Julien and P. Thomas, Chem. Mater., 2012, 24, 2611 CrossRef CAS.
- K. Matsunaga and K. Toyoura, J. Mater. Chem., 2012, 22, 7265 RSC.
- H. Kiyono, Y. Matsuda, T. Shimada, M. Ando, I. Oikawa, H. Maekawa, S. Nakayama, S. Ohki, M. Tansho, T. Shimizu, P. Florian and D. Massiot, Solid State Ionics, 2012, 228, 64 CrossRef CAS.
- K. Fukuda, T. Asaka, R. Hamaguchi, T. Suzuki, H. Oka, A. Berghout, E. Bechade, O. Masson, I. Julien, E. Champion and P. Thomas, Chem. Mater., 2011, 23, 5474 CrossRef CAS.
- P. M. Panchmatia, A. Orera, G. J. Rees, M. E. Smith, J. V. Hanna, P. R. Slater and M. S. Islam, Angew. Chem., Int. Ed., 2011, 50, 9328 CrossRef CAS PubMed.
- A. Orera, T. Baikie, E. Kendrick, J. F. Shin, S. Pramana, R. Smith, T. J. White, M. L. Sanjuán and P. R. Slater, Dalton Trans., 2011, 40, 3903 RSC.
- T. An, T. Baikie, F. Wei, S. S. Pramana, M. K. Schreyer, R. O. Piltz, J. F. Shin, J. Wei, P. R. Slater and T. J. White, Chem. Mater., 2013, 25, 1109 CrossRef CAS.
- S. Nakayama, Y. Higuchi, M. Sugawara, A. Makiya, K. Uematsu and M. Sakamoto, Ceram. Int., 2014, 40, 1221 CrossRef CAS.
- G. Ou, X. R. Ren, L. Yao, H. Nishijima and W. Pan, J. Mater. Chem. A, 2014, 2, 13817 CAS.
- H. Yoshioka, H. Mieda, T. Funahashi, A. Mineshige, T. Yazawa and R. Mori, J. Eur. Ceram. Soc., 2014, 34, 373 CrossRef CAS.
- W. Liu, T. Tsuchiya, S. Miyoshi, S. Yamaguchi, K. Kobayashi and W. Pan, J. Power Sources, 2014, 248, 685 CrossRef CAS.
- C. Argirusis, E. Jothinathan, G. Sourkouni, O. Van der Biest and F. Jomard, Solid State Ionics, 2014, 257, 53 CrossRef CAS.
- M. M. Vieira, J. C. Oliveira, A. L. Shaula, B. Trindade and A. Cavaleiro, Surf. Coat. Technol., 2014, 247, 14 CrossRef CAS.
- A. Pons, J. Jouin, E. Bechade, I. Juilien, O. Masson, P. M. Geffroy, R. Mayet, P. Thomas, K. Fukuda and I. Kagomiya, Solid State Sci., 2014, 38, 150 CrossRef CAS.
- T. An, T. Baikie, M. Weyland, J. Shin, P. Slater, J. Wei and T. White, Chem. Mater., 2015, 27, 1217 CrossRef CAS.
- T. An, A. Orera, T. Baikie, J. S. Herrin, R. O. Piltz, P. R. Slater, T. J. White and M. L. Sanjuán, Inorg. Chem., 2014, 53, 9416 CrossRef CAS PubMed.
-
A. C. Larson and R. B. Von Dreele, Los Alamos National Laboratory, Report. No LA-UR-86-748, 1987.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a 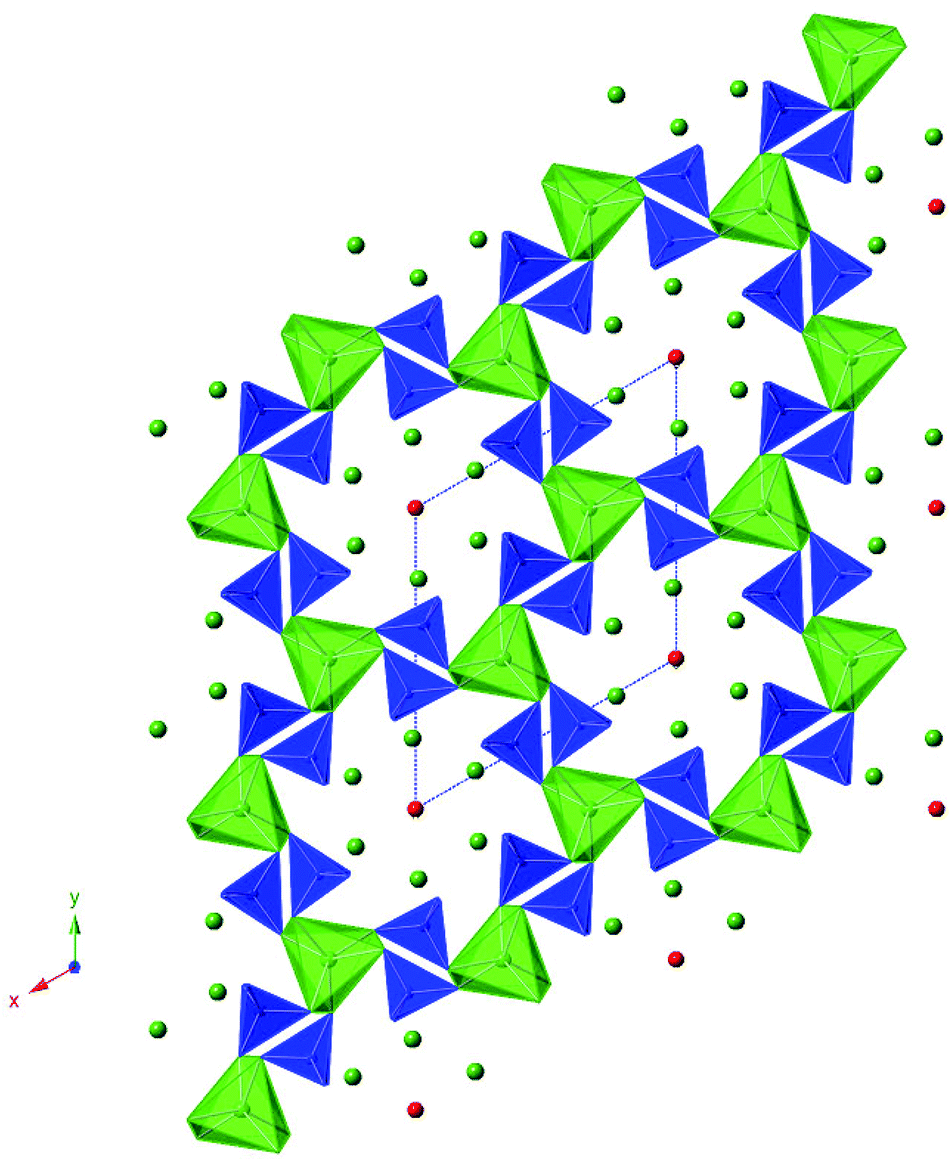
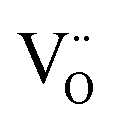 –O′′i (Kröger–Vink notation, O′′i = interstitial oxide ion with a double effective negative charge,
–O′′i (Kröger–Vink notation, O′′i = interstitial oxide ion with a double effective negative charge, 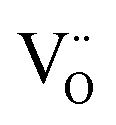 = oxide ion vacancy with a double effective positive charge)), and in the present study, a similar complex can be proposed. The observed length scale in this study for this complex is 2.27 Å which is smaller than the predicted value of 2.93 Å from the modelling work.10 However, it should be noted that the atomic displacement parameter for O(6) is very high perpendicular to the channel (100 × U22 = 34 Å2). This suggests significant local displacement from the refined position, which may hence allow an increase in the O′′i–
= oxide ion vacancy with a double effective positive charge)), and in the present study, a similar complex can be proposed. The observed length scale in this study for this complex is 2.27 Å which is smaller than the predicted value of 2.93 Å from the modelling work.10 However, it should be noted that the atomic displacement parameter for O(6) is very high perpendicular to the channel (100 × U22 = 34 Å2). This suggests significant local displacement from the refined position, which may hence allow an increase in the O′′i– 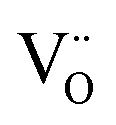 length. Overall it would suggest the presence of various interstitial oxide ion sites with differing displacements from the channel centre. In particular, the anisotropic thermal ellipsoids for the O(6) sites are directed towards channel lanthanum, La(3) (Fig. 5), which is not unexpected as O(6) is highly underbonded (bond valence sum calculations for this oxygen site give a value of only −1.08), and so some local displacement might be expected to aid the stability of this oxide ion site.
length. Overall it would suggest the presence of various interstitial oxide ion sites with differing displacements from the channel centre. In particular, the anisotropic thermal ellipsoids for the O(6) sites are directed towards channel lanthanum, La(3) (Fig. 5), which is not unexpected as O(6) is highly underbonded (bond valence sum calculations for this oxygen site give a value of only −1.08), and so some local displacement might be expected to aid the stability of this oxide ion site.