
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceResolution of P-stereogenic P-heterocycles via the formation of diastereomeric molecular and coordination complexes (a review)
Péter
Bagi
*a,
Viktória
Ujj
b,
Mátyás
Czugler
a,
Elemér
Fogassy
a and
György
Keglevich
*a
aDepartment of Organic Chemistry and Technology, Budapest University of Technology and Economics, 1521 Budapest, Hungary. E-mail: gkeglevich@mail.bme.hu; Fax: +36 1 4633648; Tel: +36 1 4631111/5883
bComInnex Inc., Záhony u. 1031 Budapest, Hungary
First published on 29th October 2015
Abstract
TADDOL derivatives and the Ca2+-salts of tartaric acid derivatives were found to be versatile and generally applicable resolving agents for the preparation of the enantiomers of P-stereogenic heterocyclic phosphine oxides and phosphinates via the formation of the corresponding diastereomeric molecular and coordination complexes. A few of the diastereomeric intermediates were characterized by single crystal X-ray crystallography to gain insights into the binding mode of the corresponding heterocyclic phosphine oxide (“guest”) and the resolving agent (“host”) and to study the underlying phenomenon of enantiomeric recognition.
 Péter Bagi | Péter Bagi was born in Nyíregyháza (Hungary) in 1986. He graduated at the Budapest University of Technology and Economics in 2011 as a chemical engineer and earned his PhD in 2014. Currently, he is a postdoctoral fellow at the Budapest University of Technology and Economics and, besides educational activity, he has been working on the preparation and application of optically active P-stereogenic compounds. His research field includes optical resolution and the development of new catalysts. |
 Viktória Ujj | Viktória Ujj was born in Vác (Hungary) in 1983. She graduated as a chemical engineer at the Budapest University of Technology and Economics in 2006, and obtained her PhD with her thesis on the resolution of P-chiral P-heterocycles in 2010. Then, she started working in the field of pharmaceutical research. Since 2012, she has been working as a senior research scientist at ComInnex Inc., focusing on the syntheses of heterocyclic organic compounds. |
 Mátyás Czugler | Mátyás Czugler was born in Tárnok (Hungary) in 1948, and graduated at the Technical University of Budapest as a chemical engineer in 1972. His research in crystallography involves mainly solid state description of organic supramolecular ensembles, often with biological importance. His DSc was defended in 2006. He led the Crystallography Department (Hungarian Academy) as academic research professor in the period of 2005–2013. In 2007, he was appointed as Adjunct Professor at TUB. His track record shows over 150 publications and experience in crystallizing and solving non-routine crystal structures. |
 Elemér Fogassy | Elemér Fogassy was born in 1934 in Budapest, and graduated at the Technical University of Budapest (Hungary) as a Chemical Engineer in 1957. He earned his PhD in 1974, and defended his DSc degree in 1986. He was working for pharmaceutical factories in the period of 1957–64, then he joined the Department of Organic Chemical Technology, TUB, and was appointed as Professor in 1987. From 2010, he is an Emeritus Professor. He is the author or co-author of 315 papers/patents in the subject of optical resolution. He received the Academic Patent Award and the Price for Pharmaceutical Research. |
 György Keglevich | György Keglevich was born in 1957 in Budapest, and became a chemical engineer in 1981. He obtained PhD and DSc in 1990 and 1994, respectively, and was appointed to Professor in 1996. He has been the Head of Organic Chemistry and Technology Department, Budapest University of Technology and Economics since 1999. His research interests include organophosphorus and environmentally-friendly (“green”) chemistry, catalytic and microwave-assisted reactions, and industrial organophosphorus syntheses. He has published 460 articles, and he serves as an editor for Curr. Org Chem., Curr Org Synth., Curr Green Chem., Lett. Org. Chem. and Lett. Drug Design Disc. |
1. Introduction
Optically active organophosphorus compounds are of great importance in organic syntheses.1 An important application is the use of chiral P(III) derivatives (phosphorous, phosphonous and phosphinous esters/amides and especially phosphines) as P-ligands in transition metal complexes which are potential catalysts in enantioselective homogeneous catalytic transformations, such as hydrogenation or hydroformylation.2–5 Among chiral organophosphorus compounds, the species bearing P-stereogenic center(s) are of special interest.6–10P-stereogenic organophosphorus compounds cannot be found in the natural pool of chirality in an optically active form, thus resolution and asymmetric synthesis remain the primary sources for the preparation of such compounds.9,11 Despite the wide variety of asymmetric syntheses,12–21 the resolution of racemic P-compounds is a well-established method for the preparation of the corresponding optically active organophosphorus compounds.9,11,21,22 In the following few paragraphs, selected examples are shown for the preparation of optically active P-stereogenic heterocycles via the formation of diastereomers. The systematization of the optical resolution of the acyclic derivatives was not our aim, as this was covered by the review of Pietrusiewicz and Zablocka, as well as by the book of Grabulosa.9,11
One of the first examples for the preparation of optically active P-heterocycles via the formation of covalent diastereomers was described by Campbell and Way. They prepared diastereomeric amides by the reaction of an optically active dibenzophosphole oxide bearing a carboxyl functional group with 1-phenylethylamine.23 Mathey et al. reported the synthesis of 1-phosphanorbornadiene-2-carboxaldehydes which were resolved via the corresponding diastereomeric acetals formed with (S,S)-1,2-diphenylethane-1,2-diol.24
Tang and his co-workers elaborated the resolution of t-butyl-hydroxy-benzodihydrooxaphosphole oxide by reacting the racemate with menthyl chloroformate. The separation of the covalent diastereomers followed by hydrolysis afforded the enantiomerically pure t-butyl-hydroxy-benzodihydrooxaphosphole oxide.25,26 Pietrusiewicz et al. reported the oxidative resolution of phenylhexahydrocyclopenta[b]phosphole with menthyl bromoacetate. The corresponding enantiopure phenylhexahydrocyclopenta[b]phosphole oxide was obtained after purification and hydrolysis of the corresponding diastereomeric phosphonium salt.11,27
The resolution of P-stereogenic compounds may be elaborated via the formation of diastereomeric salts if the corresponding organophosphorus compound is a phosphonium salt or it contains an acidic or basic functional group. Ostrogovich and Kerek elaborated the resolution of 1-(4-diethylaminophenyl)-2-phospholene oxide by forming diastereomeric salts with camphorsulfonic acid.28 Kobayashi et al. described the optically active borane complex of 1-phenylphospholane-2-carboxylic acid prepared via optical resolution by applying quinine.29 For the preparation of optically active phosphonium salts, the silver salt of dibenzoyl-tartaric acid, camphorsulfonic acid or menthoxy-acetic acid finds wide-spread application. Using these resolving agents, the resolution of a few P-stereogenic phosphonium salts bearing a heterocyclic moiety is elaborated (Fig. 1).30–35
 | ||
| Fig. 1 Resolution of P-heterocyclic phosphonium salts using the silver salt of camphorsulfonic acid. | ||
Organophosphorus compounds having neither acidic nor basic functional groups may be resolved via the formation of diastereomeric molecular complexes. O,O′-Dibenzoyl-(2R,3R)-tartaric acid was suitable for the preparation of optically active phosphine oxides,36–43 among which a few P-heterocyclic and P-stereogenic examples can be found, such as 1-phenyl-2-phospholene oxide,11 DuanPhos oxide44 and a bis-benzophosphetane oxide (Fig. 2).45
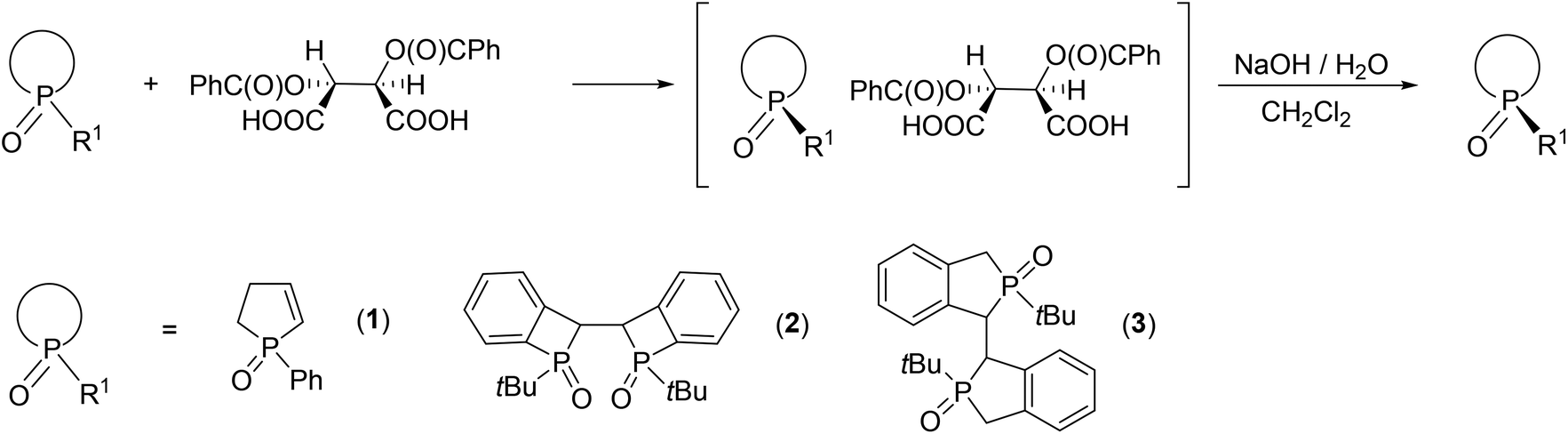 | ||
| Fig. 2 Resolution of P-heterocyclic phosphine oxides using O,O′-dibenzoyl-(2R,3R)-tartaric acid. | ||
Exploiting the coordination ability of phosphines to transition metals, the palladium complex of phenylethylamine or naphthylethylamine is a generally applicable resolving agent for P-stereogenic phosphines via the formation of the corresponding diastereomeric coordination complex. In this manner several mono- or bis-P-heterocyclic phosphines could be separated into their enantiomers (Fig. 3).46–54
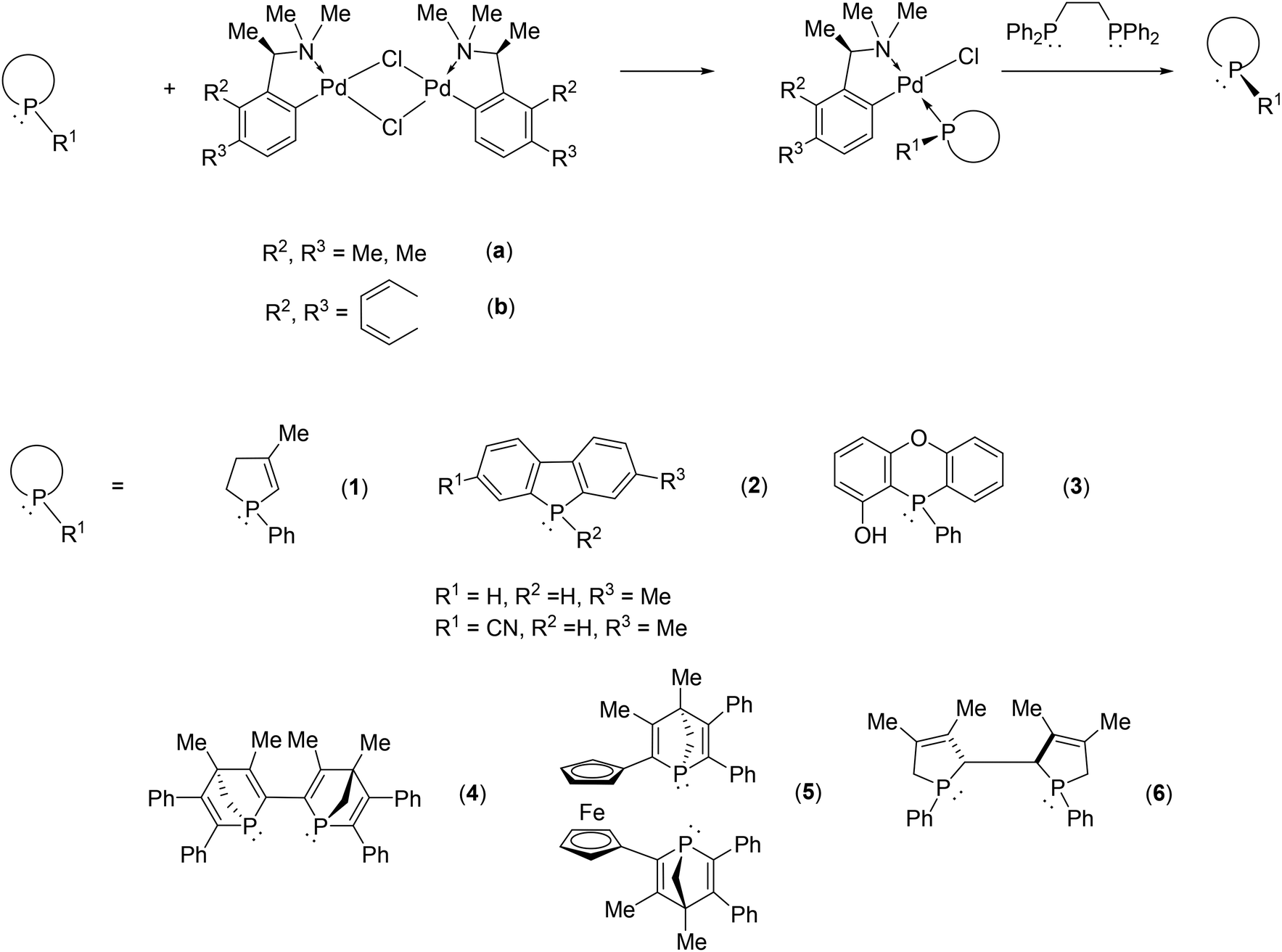 | ||
| Fig. 3 Resolution of P-heterocyclic phosphines using the palladium complex of phenylethylamine or naphthylethylamine. | ||
Herein, our aim was to summarize the results obtained in the field of the resolution methods developed by our research group for the preparation of optically active five- and six-membered P-heterocyclic phosphine oxides, phosphinates and a few other derivatives (1–8) by applying TADDOL derivatives [(−)-9 and (−)-10] or the Ca2+-salts of tartaric acid derivatives [(−)-11 and (−)-12] as the resolving agents (Fig. 4).
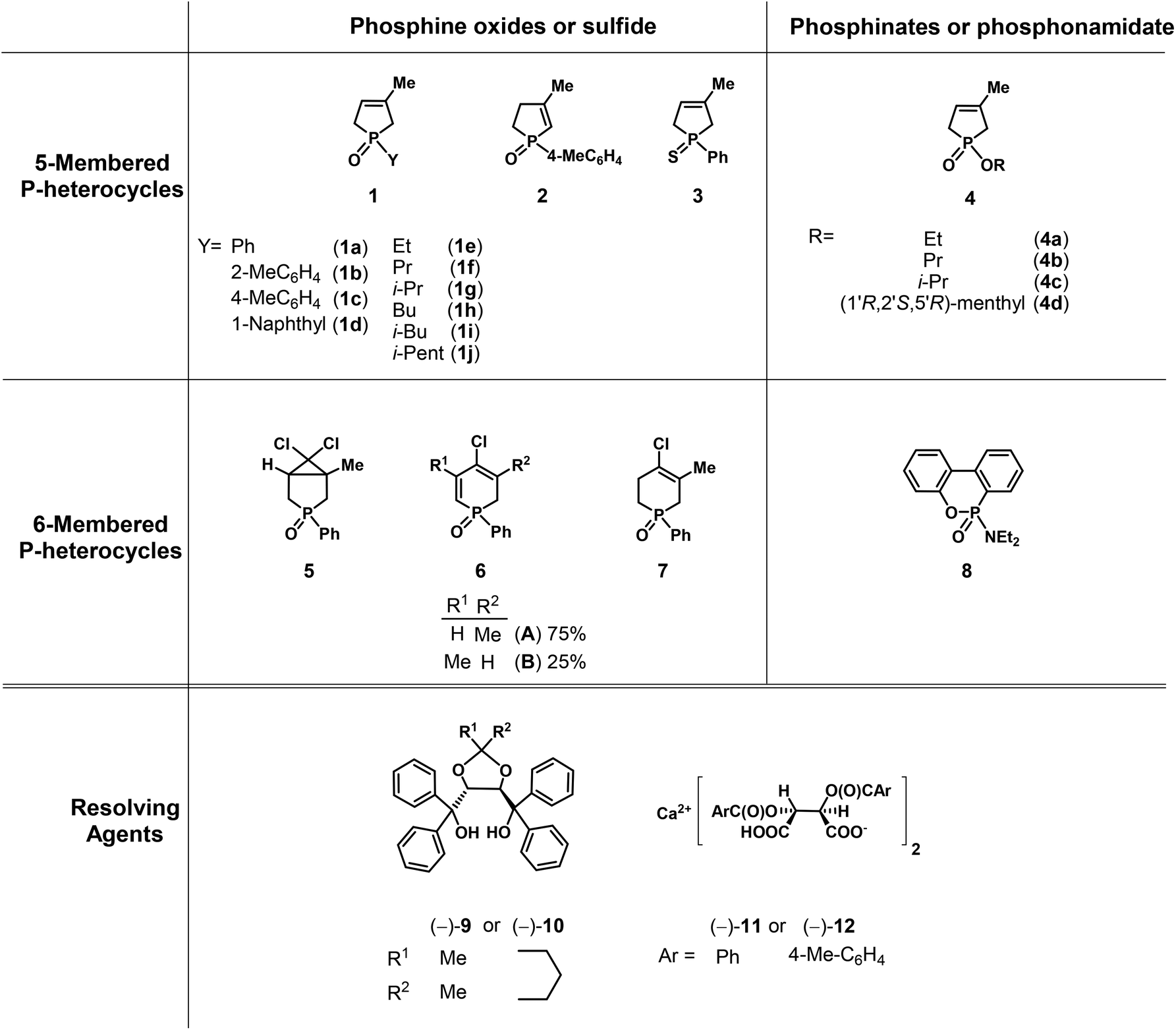 | ||
| Fig. 4 The racemic compounds (1–8) and the resolving agents [(−)-9 and (−)-12] included in this review. | ||
2. The optical resolution of P-heterocycles
2.1. Optical resolution of five- and six-membered P-heterocycles (1–8) with TADDOL derivatives [(−)-9 and (−)-10]
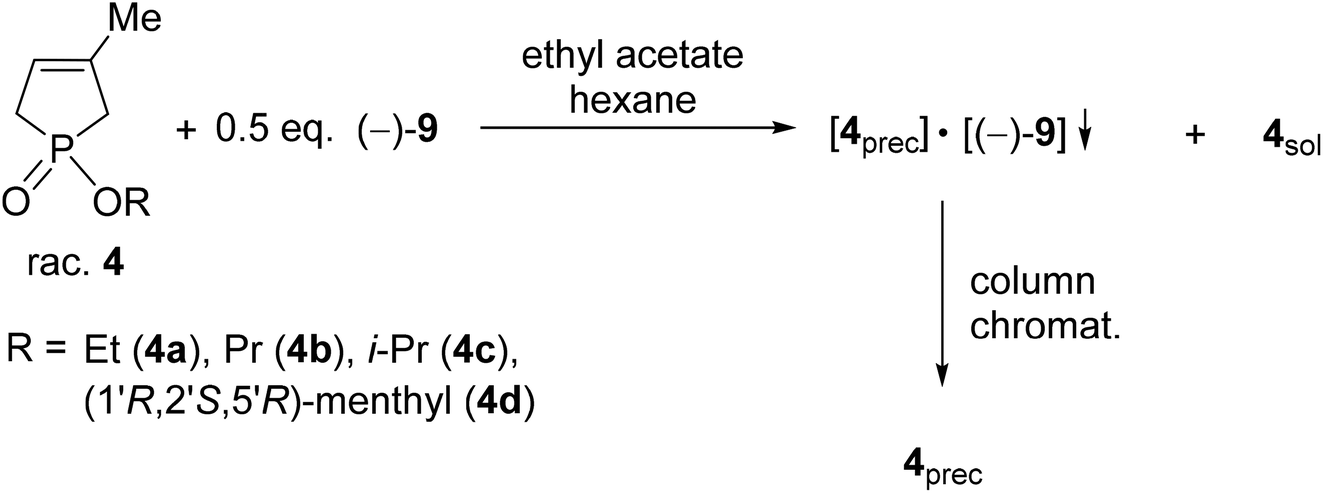
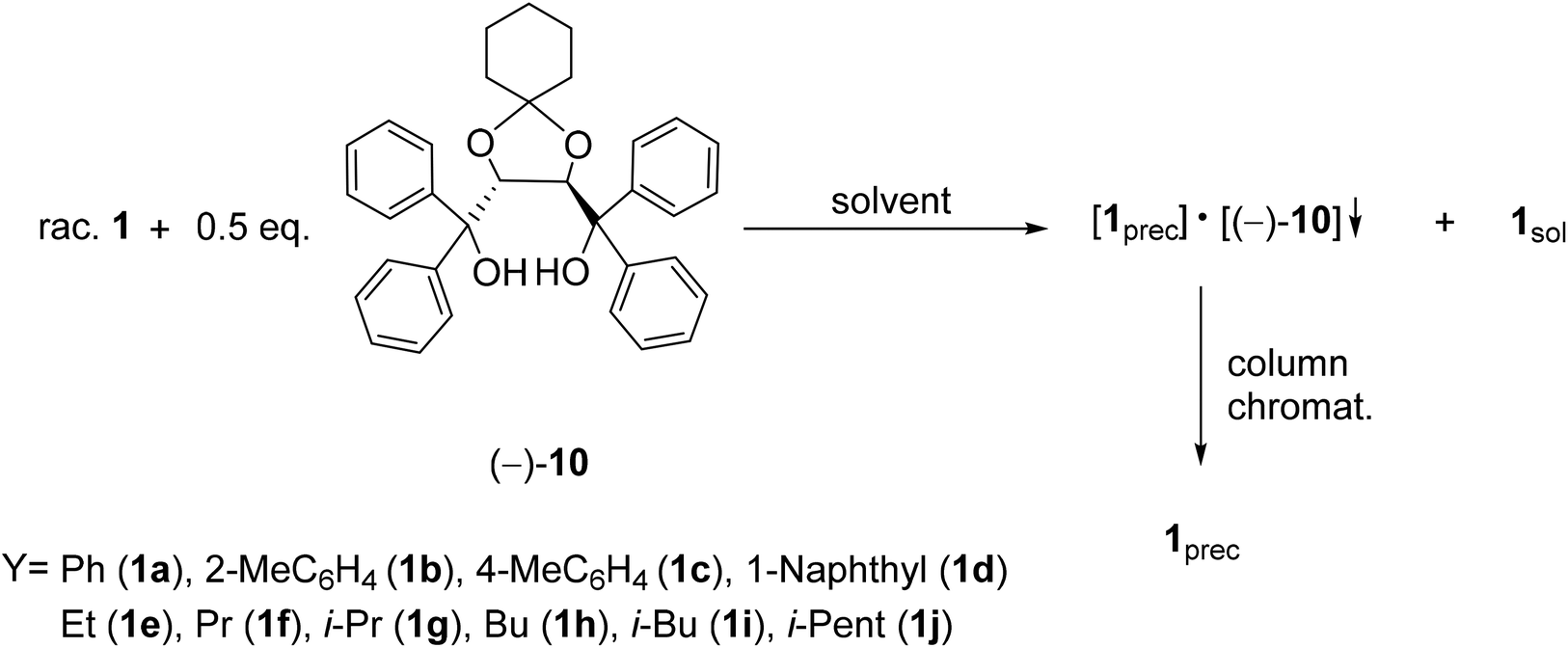
It was the first breakthrough, when TADDOL (−)-9 was applied in the resolution of 1-aryl- and alkyl-3-methyl-3-phospholene 1-oxide (1a–j).56 According to our resolution method (Scheme 1), the racemic phospholene oxide (1a–j) and half equivalent of TADDOL [(−)-9] were dissolved in ethyl acetate. Hexane was then added to the solution to promote the precipitation of the corresponding diastereomeric molecular associates. It was found that the maximum resolving capability could be obtained by using the “half equivalent method”.57–60 The diastereomeric complexes formed were further purified by two recrystallizations, and the 3-phospholene oxide (1a–j) enantiomers were then recovered from the corresponding diastereomers by flash column chromatography (Scheme 1). In all instances, the enantiomeric purity of the given enantiomeric mixtures was determined by HPLC or GC on chiral stationary phases. The selected results are summarized in Table 1.
 | ||
| Scheme 1 General protocol for the resolution of 1-aryl- and 1-alkyl-3-methyl-3-phospholene 1-oxides (1a–j) with TADDOL [(−)-9]. | ||
| Entry | Y | Diastereomeric complex | eea (%) | Yielda,b (%) | S , (−) | Abs. config.d | Ref. |
|---|---|---|---|---|---|---|---|
| a Enantiomeric excess, yield and resolving capability obtained after recrystallizations. b Based on the half of the racemate that is regarded to be 100% for each antipode. c Resolving capability, also known as the Fogassy parameter [S = (Yield/100) × (ee/100)].66 d Determined by X-ray crystallography or CD spectroscopy. e 1 equiv. of TADDOL [(−)-9] was used. f 0.33 equiv. of TADDOL [(−)-9] was used. g The composition of the diastereomer was different from that of the predominant associate shown in Scheme 1. | |||||||
| 1 | Ph (1a) | (1a)·[(−)-9] | 97 | 44 | 0.43 | (S) | 56, 61 |
| 2 | 2-MeC6H4 (1b) | (1b)·[(−)-9] | 57 | 49 | 0.28 | (S) | 61 |
| 3 | 4-MeC6H4 (1c) | (1c)·[(−)-9] | 69 | 42 | 0.29 | (S) | 61 |
| 4 | 1-Naphthyl (1d) | (1d)·[(−)-9] | 70 | 42 | 0.29 | (S) | 61 |
| 5 | Et (1e) | (1e)·[(−)-9] | 24 | 36 | 0.09 | (R) | 61 |
| 6 | Pr (1f) | (1f)·[(−)-9] | 95 | 35 | 0.33 | (R) | 61 |
| 7 | i-Pr (1g) | (1g)·[(−)-9] | 6 | 81 | 0.05 | (R) | 62 |
| 8e | Bu (1h) | (1h)·[(−)-9]2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) g g |
74 | 23 | 0.17 | (S) | 63 |
| 9 | i-Bu (1i) | (1i)·[(−)-9] | 35 | 14 | 0.05 | (R) | 64 |
| 10f | i-Pent (1j) | (1j)3·[(−)-9]2g |
18 | 38 | 0.07 | (R) | 65 |
Applying TADDOL [(−)-9] as the resolving agent for the enantiomeric separation of 3-phospholene oxides (1), the corresponding enantiomeric excess values fell in the range of 6–97%, whereas the resolving capability values (S) were 0.05–0.43 (Table 1).
It was observed that the ethyl derivative (1e) could be resolved with a lower efficiency (S) than the propyl derivative (1f) (compare Table 1, entries 5 and 6). It was also found that the enantiomers of 3-phospholene oxides bearing a branched substituent (1g, 1i and 1j) could be prepared with lower resolving capability (S) than the derivatives having the normal alkyl chain (1e, 1f and 1h) (compare Table 1, entries 7, 9 and 10 with 5, 6 and 8). On the one hand, these results reveal that an adequate length of the alkyl chain is necessary for the interactions being responsible for the enantiomeric recognition between the host [(−)-9] and the guest molecules (1e–j). On the other hand, the results also show that the increasing steric bulk of the alkyl chain may hinder or weaken the non-bonding interactions between the corresponding 3-phospholene oxide (1e–j) and the TADDOL [(−)-9] molecule that leads to less efficient enantiomeric separation.
In the case of phenyl-3-phospholene oxide (1a), it was observed that kinetic or thermodynamic effects did not affect significantly the resolving capability values obtained.61 It was found that (S)-aryl-3-phospholene oxides (1a–d) could be prepared with TADDOL [(−)-9] (Table 1, entries 1–4). However, among the alkyl derivatives (1e–j), the TADDOL [(−)-9] resolving agent preferred diastereomeric complex formation with the (R)-enantiomers in all but one instance, and the only exception was the butyl-3-phospholene oxide (1h) (Table 1, entries 5–10). The 1H NMR spectra of the corresponding diastereomers revealed that the ratio of the corresponding 3-phospholene oxide (1a–g and 1i) and TADDOL [(−)-9] was 1:1 in most instances (Table 1, entries 1–7 and 9).
 | ||
| Scheme 2 General procedure for the resolution of 1-alkoxy-3-methyl-3-phospholene 1-oxides (4a–d) with TADDOL [(−)-9]. | ||
| Entry | R | Diastereomeric complex | eea (%) | Yielda,b (%) | S , (−) | Abs. config.d | Ref. |
|---|---|---|---|---|---|---|---|
| a Enantiomeric excess, yield and resolving capability obtained after recrystallizations. b Based on the half of the racemate that is regarded to be 100% for each antipode. c Resolving capability, also known as the Fogassy parameter [S = (Yield/100) × (ee/100)].66 d Determined by X-ray crystallography or CD spectroscopy. e The sign of optical rotation. The absolute P-configuration was not determined. | |||||||
| 1 | Et (4a) | (4a)·[(−)-9] | 44 | 25 | 0.11 | (S) | 61 |
| 2 | Pr (4b) | (4b)·[(−)-9] | >99 | 38 | 0.38 | (R) | 67 |
| 3 | i-Pr (4c) | (4c)·[(−)-9] | >99 | 5 | 0.05 | (R) | 61 |
| 4 | Men (4d) | (4d)·[(−)-9] | >99 | 25 | 0.25 | (−)e | 68 |
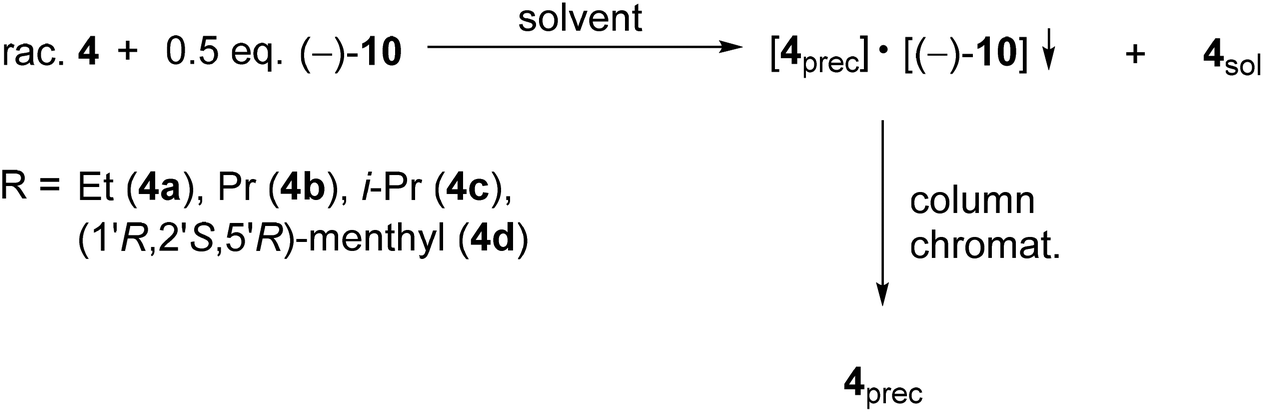
The corresponding optically active alkoxy-3-phospholene oxides (4a–d) were prepared with an ee above 99% in three out of four instances (Table 2, entries 2–4). Similarly to the results obtained in the case of the alkyl derivatives (1e–j), it was observed that the length and the bulkiness of the alkoxy chain influenced the overall efficiency of the resolution (S). The enantiomeric excess and the resolving capability (S) values were lower in the case of the ethoxy derivative (4a) than in that of the propoxy derivative (4b) (Table 2, entries 1 and 2). The (R)-i-propoxy-3-methyl-3-phospholene oxide [(R)-4c] was prepared with lower resolving capability than the propoxy derivative (4b) despite the fact that the corresponding enantiomeric excess values were identical (Table 2, entries 2 and 3).
Besides the phosphinates (4a–c) bearing only one asymmetric center, the 1:1 diastereomeric mixture of 1-[(1′R,2′S,5′R)-(−)-menthyloxy]-3-methyl-3-phospholene 1-oxide (4d) could also be separated using TADDOL [(−)-9], and the (−)-4d diastereomer was obtained with a diastereomeric purity above 99% (Table 2, entry 4). Interestingly, the separation of the two diastereomers of 4d was not successful under achiral conditions (e.g. column chromatography or crystallization).
 | ||
| Scheme 3 General route for the resolution of 1-aryl- and 1-alkyl-3-methyl-3-phospholene 1-oxides (1a–j) with spiro-TADDOL [(−)-10]. | ||
| Entry | Y | Diastereomeric complex | eea (%) | Yielda,b (%) | S , (−) | Abs. config.d | Ref. |
|---|---|---|---|---|---|---|---|
| a Enantiomeric excess, yield and resolving capability obtained after recrystallizations. b Based on the half of the racemate that is regarded to be 100% for each antipode. c Resolving capability, also known as the Fogassy parameter [S = (Yield/100) × (ee/100)].66 d Determined by X-ray crystallography or CD spectroscopy. e 1 equiv. of spiro-TADDOL [(−)-10] was used. f The composition of the diastereomer was different from that of the predominant associate shown in Scheme 3. | |||||||
| 1 | Ph (1a) | (1a)·[(−)-10] | >99 | 29 | 0.29 | (S) | 56, 61 |
| 2 | 2-MeC6H4 (1b) | (1b)·[(−)-10] | >99 | 41 | 0.41 | (S) | 61 |
| 3 | 4-MeC6H4 (1c) | (1c)·[(−)-10] | >99 | 30 | 0.30 | (S) | 61 |
| 4 | 1-Naphthyl (1d) | (1d)·[(−)-10] | >99 | 55 | 0.55 | (S) | 61 |
| 5 | Et (1e) | (1e)·[(−)-10] | 58 | 45 | 0.26 | (R) | 61 |
| 6e | Pr (1f) | (1f)·[(−)-10]2f |
89 | 30 | 0.27 | (S) | 61 |
| 7 | i-Pr (1g) | (1g)·[(−)-10] | 64 | 60 | 0.39 | (R) | 62 |
| 8e | Bu (1h) | (1h)·[(−)-10]2f |
95 | 52 | 0.49 | (S) | 63 |
| 9 | i-Bu (1i) | (1i)·[(−)-10] | 25 | 39 | 0.10 | (S) | 64 |
| 10 | i-Pent (1j) | (1j)2·[(−)-10] | Rac. | — | — | — | 65 |
In all but one instance, spiro-TADDOL [(−)-10] was also found to be an applicable resolving agent for the preparation of optically active aryl- and alkyl-3-phospholene oxides (1a–i) with an ee of 25–99%, and resolving capability values of 0.10–0.55 (Table 3, entries 1–9). The i-pentyl-3-phospholene oxide (1j) was the only five-membered P-heterocycle, where no enantiomeric discrimination could be observed (Table 3, entry 10).
The direct comparison of the results obtained with TADDOL [(−)-9] or spiro-TADDOL [(−)-10] in a mixture of ethyl acetate and hexane shows that in almost all instances the application of spiro-TADDOL [(−)-10] resulted in better enantiomeric separation of the corresponding 3-phospholene oxide (1a–i) leading to higher resolving capability values (compare Tables 1 and 3). Considering the resolving capability values, the phenyl- and propyl-3-phospholene oxides (1a and 1f) were the exceptions, where the application of TADDOL [(−)-9] was more beneficial than that of spiro-TADDOL [(−)-10] (compare Table 1, entry 1 with Table 3, entry 1 and Table 1, entry 6 with Table 3, entry 6).
In all but three instances, the composition of the diastereomers was 1:1 [(1)·(−)-10] similarly to the results obtained with TADDOL [(−)-9] (compare Tables 1 and 3). In the case of the propyl-, butyl- and the i-pentyl-3-phospholene oxides (1f, 1h and 1j) the composition of the diastereomer was different, as the given complex contained the corresponding 3-phospholene oxide (1f, 1h or 1j) and spiro-TADDOL [(−)-10] in a ratio of 1:2 or 2:1 (Table 3, entries 6, 8 and 10).
According to our expectation, in most instances both TADDOL derivatives [(−)-9 or (−)-10] preferred diastereomeric complex formation with the same enantiomer of the corresponding aryl- or alkyl-3-phospholene oxide (1a–e, 1g, 1h and 1j). However, in the case of the propyl- and i-butyl derivatives (1f and 1i), despite the same absolute configuration and chiral environment of the resolving agents [(−)-9 and (−)-10], different 3-phospholene oxide enantiomers could be prepared with TADDOL [(−)-9] or spiro-TADDOL [(−)-10] (compare Tables 1 and 3).
In this series of experiments, it was also observed that the sterically demanding alkyl chains on the 3-phospholene moiety (1i or 1j) decreased the overall efficiency of the resolution with TADDOL derivatives [(−)-9 or (−)-10] (Table 3, entries 9 and 10).
First, the resolution of phenyl-3-phospholene oxide (1a) with spiro-TADDOL [(−)-10] was carried out in ethyl acetate and hexane as described above, but 2 equivalents of a given solvent (i.e. acetone, DMSO, DMF, acetonitrile, acetic acid, water, ethanol, methyl ethyl ketone or methyl isobutyl ketone) were also added to the reaction mixture. It was found that additives with a dielectric constant between 5 and 40 improved the resolution efficiency. Whereas, the resolving capability values decreased significantly, when solvents having a dielectric constant above 40 (e.g. DMSO or water) were added to the reaction mixture.69
This correlation observed between the dielectric constant of the solvent and the efficiency of the resolution led us to the conclusion that the application of H-acceptor or H-donor solvents could be beneficial for the optical resolution of 1-aryl- and 1-alkyl-3-phospholene oxides (1a–j) with TADDOL derivatives [(−)-9 or (−)-10], as the solvents may influence the coordination sphere of the TADDOL derivatives [(−)-9 or (−)-10] which subsequently affect the enantiomeric recognition and resolving capability. This assumption was also supported by the fact that in a few instances acetone was incorporated into the molecular complex, when crystals were grown using acetone.56
Motivated by these results, the resolution of a few 3-phospholene oxides (1a–f) was carried out with spiro-TADDOL [(−)-10] in acetone or in a mixture of acetone and hexane. In other experiments (1g–j), alcohols were used as solvents (Scheme 3). The selected results are summarized in Table 4.
| Entry | Y | Solvent | Diastereomeric complex | eea (%) | Yielda,b (%) | S , (−) | Abs. config.d | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Enantiomeric excess, yield and resolving capability obtained after recrystallizations. b Based on the half of the racemate that is regarded to be 100% for each antipode. c Resolving capability, also known as the Fogassy parameter [S = (Yield/100) × (ee/100)].66 d Determined by X-ray crystallography or CD spectroscopy. e 1 mol of acetone was incorporated into the diastereomer. f 1 equiv. of spiro-TADDOL [(−)-10] was used. g The composition of the diastereomer was different from that of the predominant associate shown in Scheme 3. | ||||||||
| 1 | Ph (1a) | Acetone | (1a)·[(−)-10] | 97 | 83 | 0.81 | (S) | 69 |
| 2 | 2-MeC6H4 (1b) | Acetone | (1b)·[(−)-10] | 88 | 48 | 0.42 | (S) | 69 |
| 3 | 4-MeC6H4 (1c) | Acetone–hexane | (1c)·[(−)-10] | 97 | 18 | 0.18 | (S) | 70 |
| 4 | 1-Naphthyl (1d) | Acetone | (1d)·[(−)-10] | 71 | 32 | 0.23 | (S) | 69 |
| 5e | Et (1e) | Acetone | (1e)·[(−)-10]2g |
55 | 41 | 0.23 | (S) | 70 |
| 6e,f | Pr (1f) | Acetone | (1f)·[(−)-10]2g |
91 | 66 | 0.60 | (S) | 70 |
| 7 | i-Pr (1g) | EtOH | (1g)·[(−)-10]2g |
95 | 50 | 0.47 | (S) | 62 |
| 8f | Bu (1h) | EtOH | (1h)·[(−)-10]2g |
70 | 17 | 0.12 | (R) | 63 |
| 9f | Bu (1h) | i-PrOH | (1h)·[(−)-10]2g |
78 | 43 | 0.33 | (S) | 63 |
| 10f | i-Bu (1i) | EtOH | (1i)·[(−)-10]2g |
96 | 26 | 0.25 | (R) | 64 |
| 11f | i-Pent (1j) | i-PrOH | (1j)·[(−)-10]2g |
>99 | 16 | 0.16 | (R) | 65 |
In 7 out of 11 cases (Table 4, entries 1, 2, 5, 6, 7, 10 and 11), the application of the above mentioned H-acceptor or H-donor solvents was beneficial as the resolving capability values were higher or showed parity with the results obtained in a mixture of ethyl acetate and hexane (compare Tables 3 and 4). One of the most remarkable examples was the phenyl-3-phospholene oxide (1a), where the resolving capability values changed from 0.29 to 0.81 with the solvent change (compare Table 3, entry 1 and Table 4, entry 1). Moreover, the i-pentyl-3-phospholene oxide (1j) could be prepared with spiro-TADDOL [(−)-10] in i-propanol with an ee above 99%, whereas no enantiomeric separation was observed in a mixture of ethyl acetate and hexane (compare Table 3, entry 10 and Table 4, entry 11). Another intriguing observation was that in the case of the ethyl-, i-propyl-, butyl- and i-butyl derivatives (1e and 1g–i) the solvent influenced which 3-phospholene oxide enantiomer was incorporated into the corresponding diastereomeric associate. In this manner, both antipodes of the corresponding 3-phospholene oxide (1e and 1g–i) could be prepared with the same resolving agent that was spiro-TADDOL [(−)-10] simply by changing the solvent (compare Table 3, entries 5, 7, 8, and 9 with Table 4, entries 5, 7, 8, 9 and 10). The underlying phenomena of this solvent dependent enantioselection may be that the solubility of the corresponding diastereomeric complexes is different in various solvents, or the corresponding solvents incorporated into the diastereomeric associates change the mode of binding of the host [(−)-10] and guest molecules (1e and 1g–i).
In a few cases (1e–j), a change in the solvent influenced the composition of the diastereomers, and diastereomeric complexes having a composition of (1)·[(−)-10]2 were formed in all, but one instance contrary to the previously prevalent diastereomeric composition of (1)·[(−)-10] (compare Table 3, entries 5–10 and Table 4, entries 5–11).
 | ||
| Scheme 4 General method for the resolution of 1-alkoxy-3-methyl-3-phospholene 1-oxides (4a–d) with spiro-TADDOL [(−)-10]. | ||
| Entry | R | Solvent | Diastereomeric complex | eea (%) | Yielda,b (%) | S , (−) | Abs. config.d | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Enantiomeric excess, yield and resolving capability obtained after recrystallizations. b Based on the half of the racemate that is regarded to be 100% for each antipode. c Resolving capability, also known as the Fogassy parameter [S = (Yield/100) × (ee/100)].66 d Determined by X-ray crystallography or CD spectroscopy. e 1 equiv. of spiro-TADDOL [(−)-10] was used. f The sign of optical rotation. The absolute P-configuration was not determined. g The composition of the diastereomer was different from that of the predominant associate shown in Scheme 4. | ||||||||
| 1 | Et (4a) | EtOAc–hexane | (4a)·[(−)-10] | 95 | 50 | 0.48 | (R) | 61 |
| 2 | Et (4a) | acetone | (4a)·[(−)-10]2g |
58 | 38 | 0.22 | (R) | 70 |
| 3 | Pr (4b) | EtOAc–hexane | (4b)·[(−)-10] | 93 | 43 | 0.40 | (R) | 67 |
| 4e | Pr (4b) | EtOH | (4b)·[(−)-10]2g |
93 | 15 | 0.14 | (S) | 67 |
| 5 | i-Pr (4c) | EtOAc–hexane | (4c)·[(−)-10] | >99 | 37 | 0.37 | (R) | 61 |
| 6 | i-Pr (4c) | acetone–hexane | (4c)·[(−)-10] | >99 | 56 | 0.56 | (R) | 69 |
| 7 | Men (4d) | EtOAc–hexane | (4d)·[(−)-10] | 90 | 45 | 0.41 | (−)f | 68 |
In the case of the alkoxy-3-phospholene oxides (4a–d), the enantiomeric excess values were in the range of 58–99%, whereas the corresponding resolving capability values were 0.14–0.56 (Table 5). The direct comparison of the result obtained with either spiro-TADDOL [(−)-10] or TADDOL [(−)-9] in a mixture of ethyl acetate and hexane revealed that the application of spiro-TADDOL [(−)-10] was more advantageous considering the resolving capability values (compare Table 2, entries 1–4 and Table 5, entries 1, 3, 5 and 7). Moreover, the TADDOL derivatives [(−)-9 or (−)-10] formed diastereomeric molecular complexes with the same antipodes of the corresponding alkoxy-3-phospholene oxides (4b–d) in all but one instance. The exception was the ethoxy derivative (4a), in which case different antipodes of 4a could be prepared with TADDOL [(−)-9 or spiro-TADDOL [(−)-10] (compare Table 2, entry 1 and Table 5, entry 1) similarly to the case described for the two alkyl-3-phospholene oxides (1f and 1i).
In two out of three cases, the solvents other than the mixture of ethyl acetate and hexane lowered the overall efficiency (S) of the resolution of the alkoxy-3-phospholene oxides (4a and 4b) (compare Table 5, entry 1 with 2 and entry 3 with 4). Moreover, in the case of the propoxy derivative (4b), the solvent influenced the enantiopreference of spiro-TADDOL [(−)-10], and both antipodes of 4b could be prepared with the same resolving agent in different solvents (Table 5, entries 3 and 4).
 | ||
| Scheme 5 The resolution of 1-(4-methylphenyl)-3-methyl-2-phospholene 1-oxide (2) with spiro-TADDOL [(−)-10]. | ||
 | ||
| Scheme 6 The resolution of 1-phenyl-3-methyl-3-phospholene sulfide (3) with spiro-TADDOL [(−)-10]. | ||
 | ||
| Scheme 7 The resolution of 6,6-dichloro-1-methyl-3-phenyl-3-phosphabicyclo[3.1.0]hexane 3-oxide (5) with spiro-TADDOL [(−)-10]. | ||
 | ||
| Scheme 8 The resolution of 4-chloro-1-phenyl-1,2-dihydrophosphinine 1-oxide (6) with spiro-TADDOL [(−)-10]. | ||
 | ||
| Scheme 9 The resolution of 4-chloro-5-methyl-1-phenyl-1,2,3,6-tetrahydrophosphinine 1-oxide (7) with TADDOL [(−)-9]. | ||
 | ||
| Scheme 10 The resolution of 6-diethylamino-dibenzo[c,e][5,6]oxaphosphorine 6-oxide (8) with spiro-TADDOL [(−)-10]. | ||
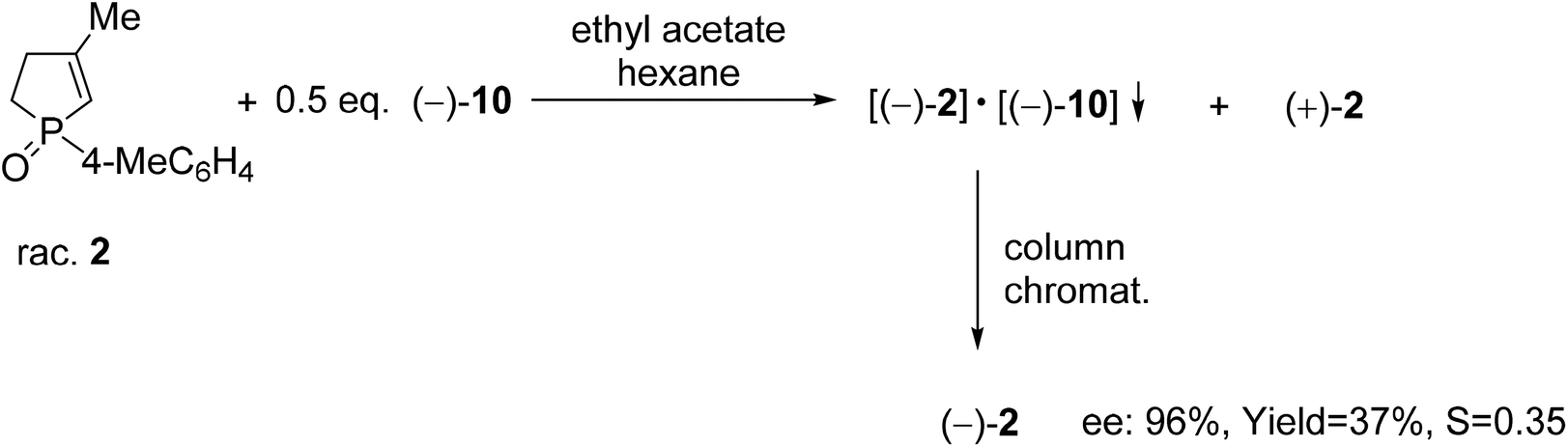
The resolution procedure applying spiro-TADDOL [(−)-10] as the resolving agent was also suitable for the enantiomeric separation of 1-(4-methylphenyl)-3-methyl-2-phospholene 1-oxide (2). The (−)-2 enantiomer was obtained with an ee of 96% and a resolving capability of 0.35 which is in good agreement with the results obtained in the case of the corresponding 3-phospholene oxide analogue (1c) (compare Scheme 5 and Table 3, entry 3). This result suggests that the position of the double-bond in the phospholene moiety does not affect significantly the overall efficiency of resolution with spiro-TADDOL [(−)-10].70
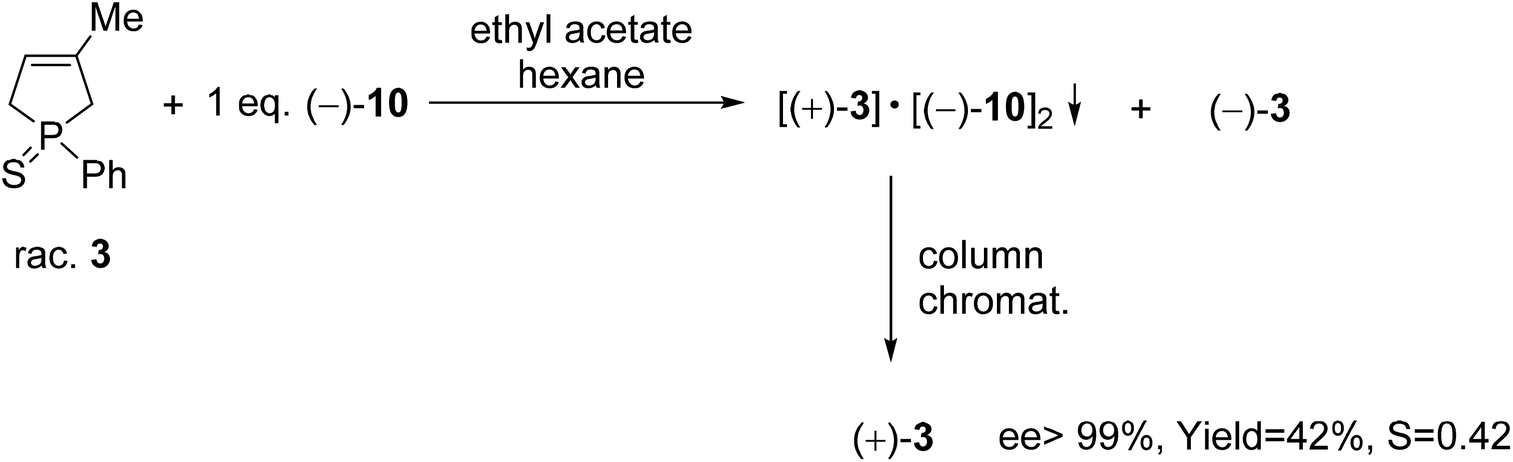
The (+)-1-phenyl-3-methyl-3-phospholene sulfide [(+)-3] was also prepared by resolution with spiro-TADDOL [(−)-10], the ee was above 99% in this case (Scheme 6). The crude product of the phospholene sulfide (3) was used as the starting material in this resolution experiment. Interestingly, less efficient enantioseparation of phosphine sulfide 3 was achieved, when purified phenyl-3-phospholene sulfide (3) was used as the racemic starting material.61
The wide applicability of TADDOL derivatives [(−)-9 and (−)-10] for the preparation of optically active five-membered phosphine oxides (1 and 2), a sulfide (3) and phosphinates (4) motivated us to extend this method to six-membered derivatives as well, such as phosphine oxides (5–7) and a phosphonic ester-amide (8). Among the six-membered P-heterocyclic phosphine oxides, the (+)-6,6-dichloro-1-methyl-3-phenyl-3-phosphabicyclo[3.1.0]hexane 3-oxide [(+)-5] bearing the dichlorocyclopropane ring and the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O function in the position trans disposition could be prepared with an ee of 55% using spiro-TADDOL [(−)-10] as the resolving agent (Scheme 7).71
O function in the position trans disposition could be prepared with an ee of 55% using spiro-TADDOL [(−)-10] as the resolving agent (Scheme 7).71
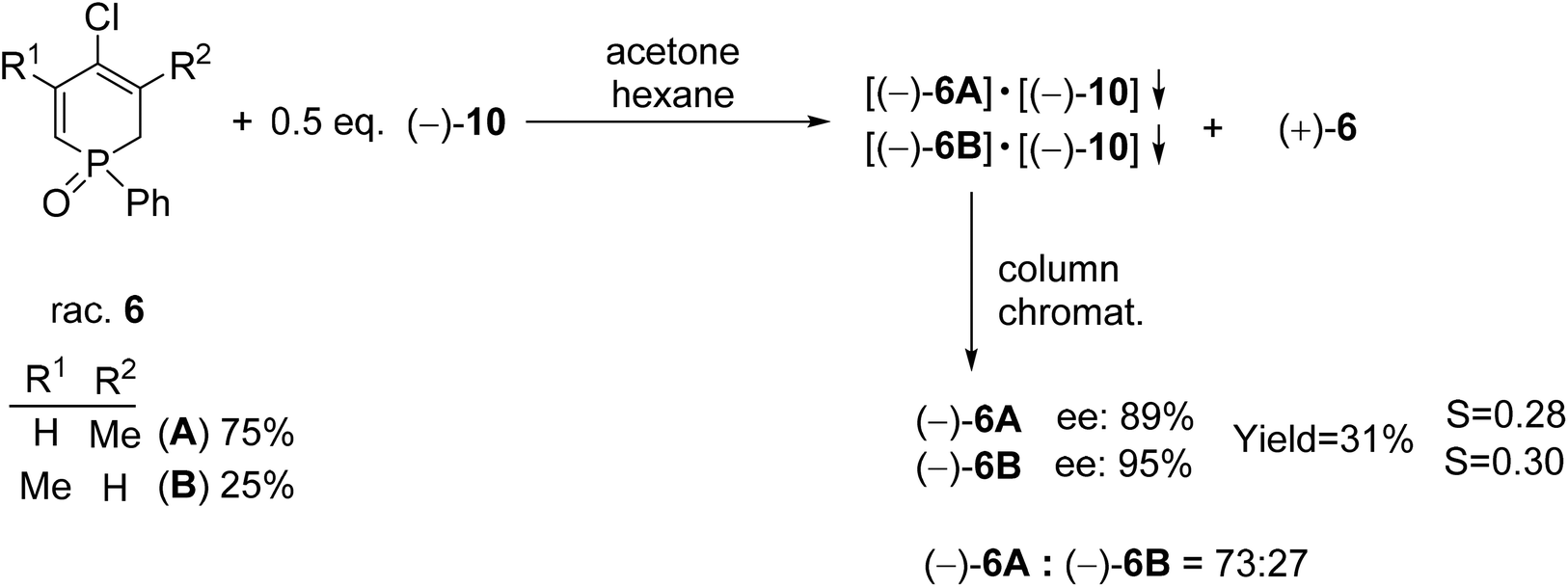
Racemic 4-chloro-1-phenyl-1,2-dihydrophosphinine 1-oxide (6) comprised a 3:1 mixture of two double-bond isomers (6A and 6B) that could not be separated. Therefore, the mixture of dihydrophosphinine oxides 6A and 6B was resolved using spiro-TADDOL [(−)-10] in a mixture of acetone–hexane. The ratio of the double-bond isomers (6A and 6B) did not change significantly during the resolution procedure. However, the enantiomeric excess of 6A and 6B was slightly different (89% and 95%, respectively) (Scheme 8).72
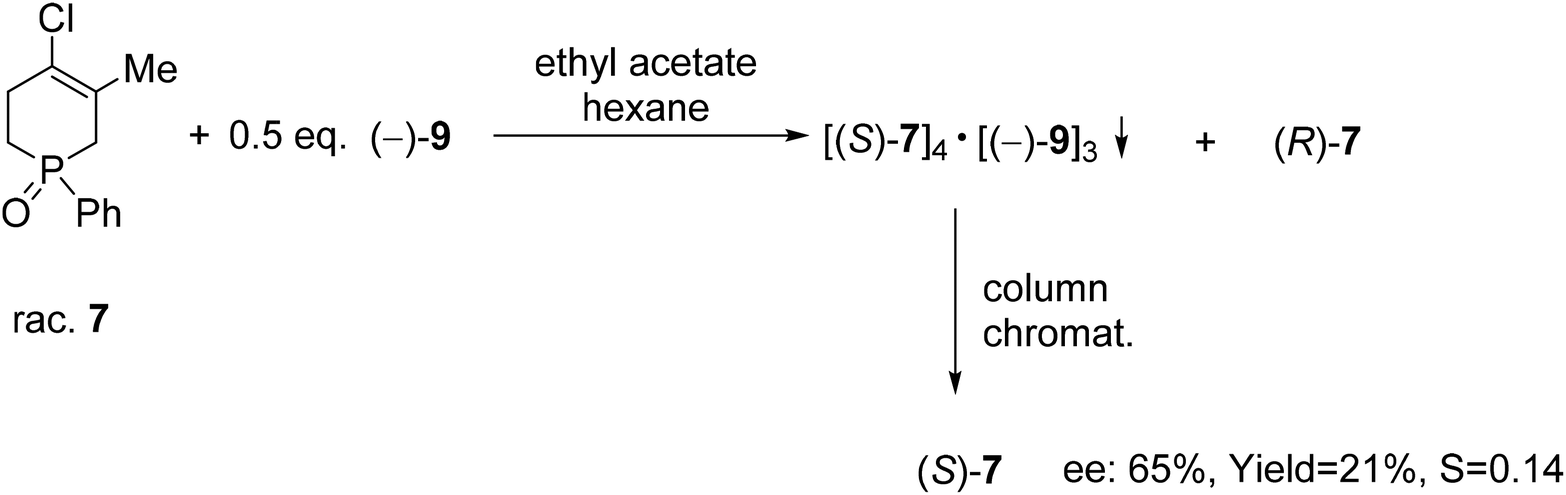
TADDOL [(−)-9] was also suitable for the partial resolution of 4-chloro-5-methyl-1-phenyl-1,2,3,6-tetrahydrophosphinine 1-oxide (7), whereupon (S)-7 was obtained with an ee of 65% (Scheme 9).71 Recently, X-ray crystallographic and CD spectroscopic studies have allowed us to assign (S) absolute configuration to the (−)-4-chloro-5-methyl-1-phenyl-1,2,3,6-tetrahydrophosphinine 1-oxide enantiomer [(−)-7].73
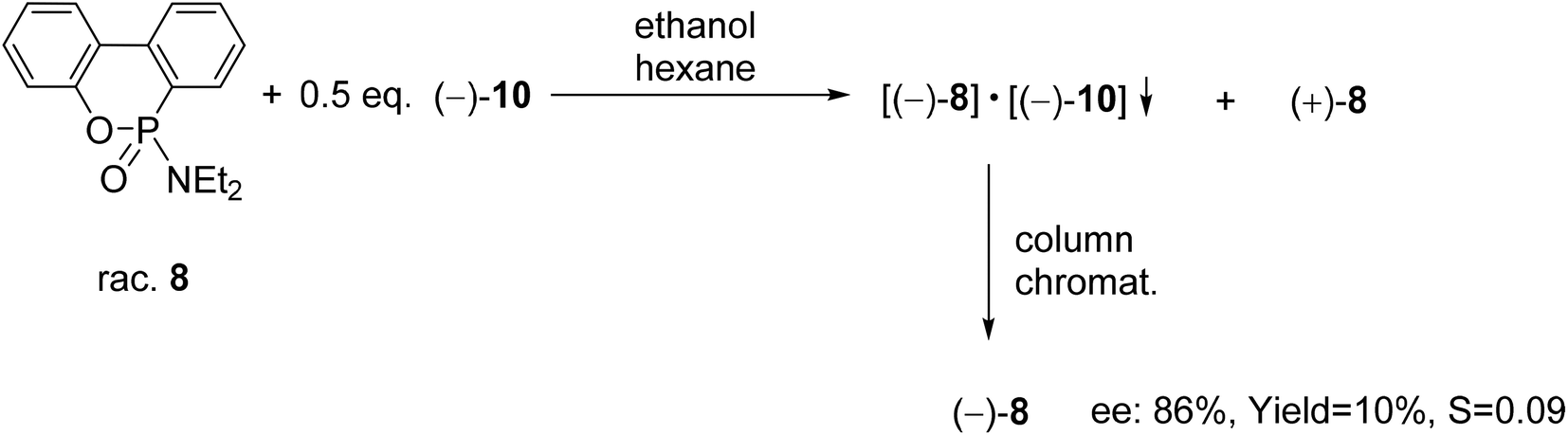
A six-membered phosphonic ester-amide, the 6-diethylamino-dibenzo[c,e][5,6]oxaphosphorine 6-oxide (8) was partially resolved with spiro-TADDOL [(−)-10], and as a result, (−)-8 was obtained with an ee of 86% (Scheme 10).70
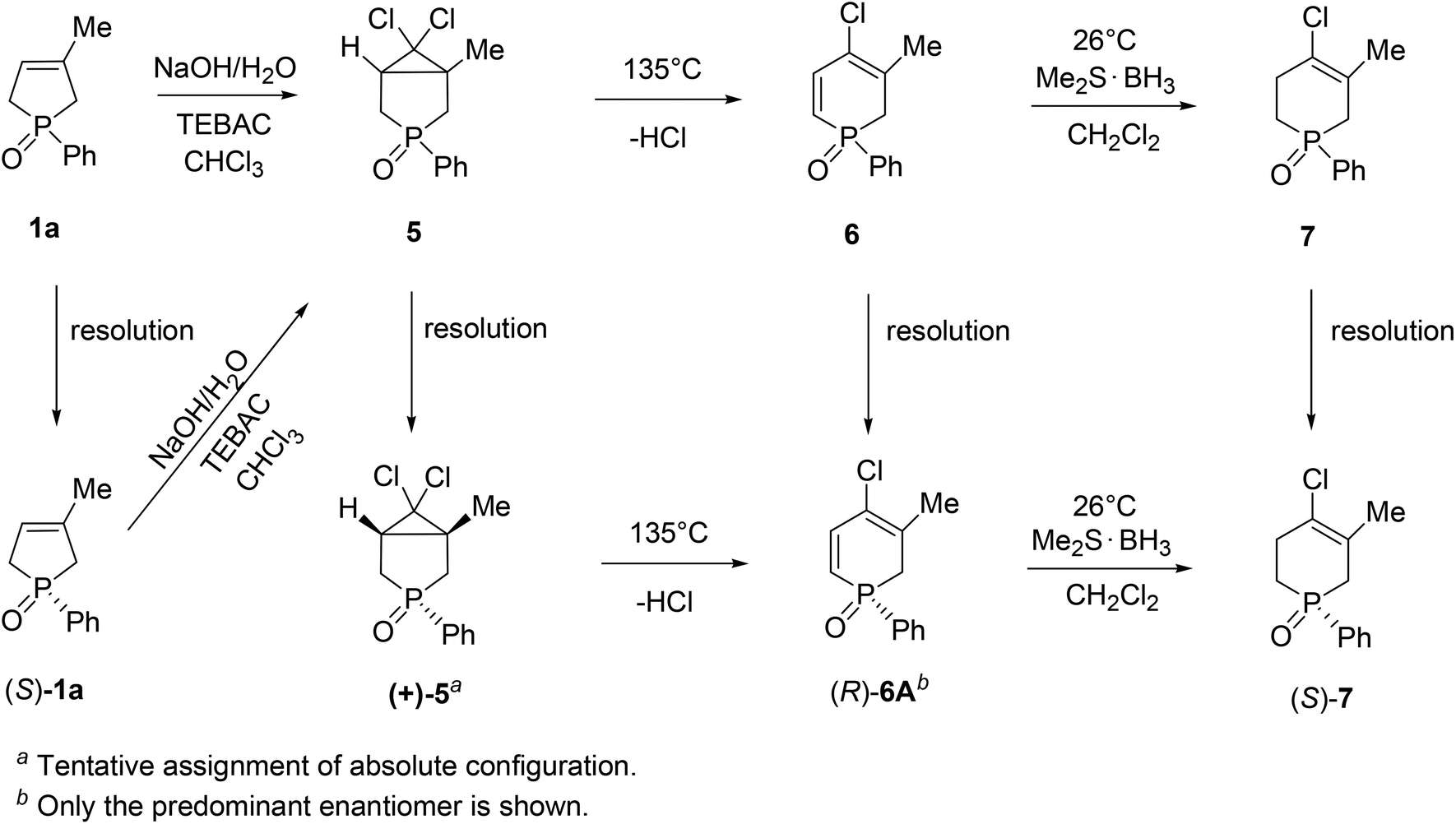
Besides the systematic resolution experiments of the six-membered heterocyclic phosphine oxides (5–7) with TADDOL derivatives (−)-9 or (−)-10, we investigated the stereochemical outcome of the 3-methyl-1-phenyl-3-phospholene 1-oxide (1a) → 6,6-dichloro-1-methyl-3-phenyl-3-phosphabicyclo[3.1.0]hexane 3-oxide (5) → 4-chloro-1-phenyl-1,2-dihydrophosphinine 1-oxide (6) → 4-chloro-5-methyl-1-phenyl-1,2,3,6-tetrahydrophosphinine 1-oxide (7) reaction sequence described earlier by our research group.74,75 It was proven that the dichlorocarbene addition on the double-bond of phenyl-3-phospholene oxide (1a) involved racemization on the P-stereogenic center. However, the thermolytic ring opening of 3-phosphabicyclo[3.1.0]hexane oxide (5), and the selective reduction of the α,β-double bond of the 4-chloro-3-methyl-1-phenyl-1,2-dihydrophosphinine 1-oxide (6A) could be accomplished without the loss of optical activity.
Knowing the stereochemical outcome of the 6A → 7 transformation, and considering the absolute configuration of the phenyl-1,2,3,6-tetrahydrophosphinine oxide (7) enabled us to make an indirect assignment of (R) absolute configuration to the P atom of (−)-4-chloro-3-methyl-1-phenyl-1,2-dihydrophosphinine 1-oxide (6A) enantiomer.71 In Scheme 11, the possible ways of preparing optically active six-membered P-heterocyclic phosphine oxides (5–7) are summarized.
 | ||
| Scheme 11 Preparation of optically active six-membered P-heterocyclic phosphine oxides (5–7). | ||
2.2. Optical resolution of five- and six-membered P-heterocycles with the calcium salts of tartaric acid derivatives
To overcome these disadvantages deriving from the application of TADDOL derivatives (−)-9 or (−)-10, we attempted the application of cheap and readily available tartaric acid and its derivatives. Unfortunately, none of the tartaric acid derivatives tried out could form a diastereomeric molecular complex with 3-phospholene oxides (1 or 4). Then, we focused on the acidic calcium salts of O,O′-dibenzoyl- and O,O′-di-p-toluoyl-(2R,3R)-tartaric acids [Ca(H-DBTA)2 (−)-11 and Ca(H-DPTTA)2 (−)-12, respectively]. These resolving agents combine the advantage of the chiral recognition ability of the tartaric acid scaffold and the coordination ability of the calcium ion which is beneficial for the enantiomeric separation of α-alkoxyalcohols, α-alkoxycarboxylic acids, as well as α- and β-hydroxycarboxylic acid esters.76–78
The first proof of this concept was the successful resolution of 1-phenyl- and 1-(1-naphthyl)-3-methyl-3-phospholene oxides (1a and 1d) with Ca(H-DBTA)2 (−)-11 or Ca(H-DPTTA)2 (−)-12.79 Then, these resolution methods were extended to other 1-aryl- and 1-alkyl-3-phospholene oxides (1b, 1c and 1e–j). According to our general procedure, the solutions of the given 3-phospholene oxides (1a–j) were added to the solution of the resolving agent (−)-11. The crystalline diastereomeric coordination complexes obtained were purified by digestion in the suitable solvent. Then the phospholene oxides (1a–j) were recovered by the treatment of the dichloromethane solution of the diastereomeric complexes with aqueous ammonia (Scheme 12). The best results obtained after optimization are summarized in Table 6.
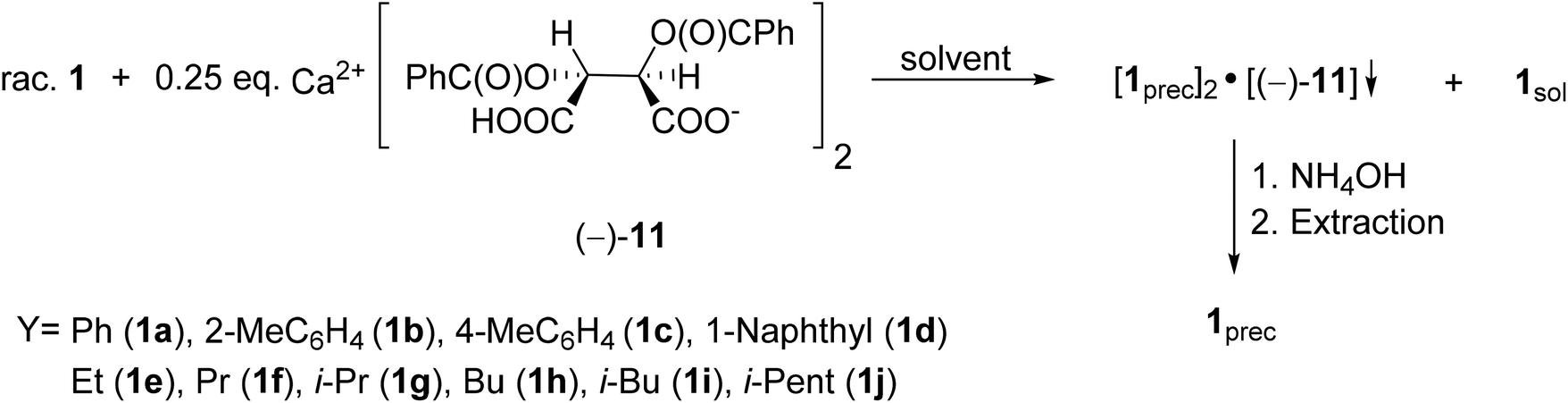 | ||
| Scheme 12 General route for the resolution of 1-aryl- and 1-alkyl-3-methyl-3-phospholene 1-oxides (1a–j) with Ca(H-DBTA)2 [(−)-11]. | ||
| Entry | Y | Solvent | Diastereomeric complex | eea (%) | Yielda,b (%) | S , (−) | Abs. config. | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Enantiomeric excess, yield and resolving capability obtained after digestions. b Based on the half of the racemate that is regarded to be 100% for each antipode. c Resolving capability, also known as the Fogassy parameter [S = (Yield/100) × (ee/100)].66 d Solvent used for crystallization. e Solvent used for digestions. f 0.5 equiv. of Ca(H-DBTA)2 [(−)-11] was used. g Water was incorporated into the diastereomer. h The composition of the diastereomer was different from that of the predominant associate shown in Scheme 12. | ||||||||
| 1 | Ph (1a) | EtOAc–EtOHd | (1a)2·(−)-11g | 96 | 52 | 0.50 | (R) | 80 |
| EtOH–watere | ||||||||
| 2 | 2-MeC6H4 (1b) | MeCN–EtOHd,e | (1b)2·(−)-11 | 93 | 33 | 0.31 | (R) | 80 |
| 3 | 4-MeC6H4 (1c) | MeCN–EtOHd,e | (1c)2·(−)-11 | 44 | 28 | 0.12 | (S) | 80 |
| 4 | 1-Naphthyl (1d) | EtOAc–EtOHd | (1d)2·(−)-11 | 99 | 42 | 0.42 | (R) | 80 |
| EtOH–watere | ||||||||
| 5 | Et (1e) | EtOAc–EtOHd,e | (1e)2·(−)-11 | 49 | 25 | 0.12 | (R) | 80 |
| 6f | Pr (1f) | EtOH–waterd,e | (1f)·(−)-11g,h | 96 | 18 | 0.17 | (S) | 80 |
| 7 | i-Pr (1g) | EtOHd,e | (1g)2·(−)-11 | 43 | 8 | 0.03 | (R) | 62 |
| 8 | Bu (1h) | MeCN–EtOHd,e | (1h)2·(−)-11 | 76 | 21 | 0.16 | (S) | 63 |
| 9 | Bu (1h) | EtOAc–EtOHd,e | (1h)2·(−)-11 | 13 | 14 | 0.02 | (R) | 63 |
| 10 | i-Bu (1i) | EtOAc–EtOHd,e | (1i)2·(−)-11 | 29 | 7 | 0.02 | (R) | 64 |
| 11 | i-Pent (1j) | MeCN–EtOHd,e | (1j)2·(−)-11 | 99 | 17 | 0.17 | (S) | 65 |
Upon applying Ca(H-DBTA)2 [(−)-11] as the resolving agent, the corresponding enantiomeric excess values of the aryl- and alkyl-3-phospholene oxides (1a–j) fell in the range of 13–99%, whereas the resolving capability values (S) were 0.02–0.50 (Table 6). In 5 out of 10 cases, the 3-phospholene oxides (1a, 1b, 1d, 1f and 1j) could be prepared with an ee above 90% (Table 6, entries 1, 2, 4, 6 and 11). It is noteworthy that the resolving capability values (S) obtained in the instance of the aryl-3-phospholene oxides (1a–d) (S = 0.12–0.50) were significantly higher than in the case of the corresponding alkyl derivatives (1e–j) (S = 0.02–0.17) (compare Table 6, entries 1–4 and 5–11). The underlying reason for this difference may be that the strength and the nature of the non-bonding interactions between the resolving agent [(−)-11] and the given 3-phospholene oxide are presumably different in the case of the aryl- and alkyl derivatives (1a–d or 1e–j).
Generally, the enantiomeric excess values obtained with the acidic Ca2+-salt of the O,O′-dibenzoyl-(2R,3R)-tartaric acid [(−)-11] were comparable with the ee values obtained with TADDOL derivatives [(−)-9 or (−)-10]. However, Ca(H-DBTA)2 [(−)-11] underperformed TADDOL and spiro-TADDOL [(−)-9 and (−)-10] in terms of the resolving capability values (S) (compare Table 6 with Tables 1, 3 and 4).
In this series of resolution experiments, the solvent did not influence in which an antipode of the 3-phospholene oxide (1a–g, 1i and 1j) was incorporated into the diastereomer. The only exception was the 1-butyl-3-phospholene oxide (1h) (Table 6, entries 8 and 9). However, the significance of this solvent dependent enantioselection was not as pronounced, as in the case of spiro-TADDOL [(−)-10] (Tables 3 and 4).
The 1H NMR studies revealed that the predominant composition of the diastereomers may be described by the formula Ca(1)2(H-DBTA)2 (Table 6). The only exception was the propyl-3-phospholene oxide (1f), where one 3-phospholene oxide molecule was incorporated into the diastereomer (Table 6, entry 6).
 | ||
| Scheme 13 General protocol for the resolution of 1-aryl- and 1-alkyl-3-methyl-3-phospholene 1-oxides (1a–j) with Ca(H-DPTTA)2 [(−)-12]. | ||
| Entry | Y | Solvent | Diastereomeric complex | eea (%) | Yielda,b (%) | S , (−) | Abs. config. | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Enantiomeric excess, yield and resolving capability obtained after digestions. b Based on the half of the racemate that is regarded to be 100% for each antipode. c Resolving capability, also known as the Fogassy parameter [S = (Yield/100) × (ee/100)].66 d 0.5 equiv. of Ca(H-DPTTA)2 [(−)-12] was used. e Water was incorporated into the diastereomer. f The composition of the diastereomer was different from that of the predominant associate shown in Scheme 13. | ||||||||
| 1d | Ph (1a) | EtOH–water | (1a)·(−)-12e,f | 93 | 55 | 0.51 | (S) | 80 |
| 2 | 2-MeC6H4 (1b) | EtOH–water | (1b)2·(−)-12 | 45 | 13 | 0.06 | (R) | 80 |
| 3 | 4-MeC6H4 (1c) | EtOH–water | (1c)2·(−)-12 | 32 | 15 | 0.05 | (S) | 80 |
| 4 | 1-Naphthyl (1d) | EtOH–water | (1d)2·(−)-12e | 69 | 29 | 0.20 | (R) | 80 |
| 5 | Et (1e) | EtOH–water | (1e)2·(−)-12e | 73 | 34 | 0.25 | (S) | 80 |
| 6 | Pr (1f) | EtOH–water | (1f)2·(−)-12 | 41 | 42 | 0.17 | (S) | 80 |
| 7 | i-Pr (1g) | EtOAc–EtOH–water | (1g)2·(−)-12 | 92 | 10 | 0.09 | (S) | 62 |
| 8 | Bu (1h) | EtOH–water | (1h)2·(−)-12 | 77 | 27 | 0.21 | (S) | 63 |
| 9 | i-Bu (1i) | EtOAc–EtOH–water | (1i)2·(−)-12 | 73 | 23 | 0.17 | (R) | 64 |
| 10 | i-Pent (1j) | EtOH–water | (1j)4·[(−)-12]5f |
95 | 32 | 0.30 | (S) | 81 |
Regarding the aryl-3-phospholene oxides (1a–d), lower resolving capability values (S) could be obtained with Ca(H-DPTTA)2 [(−)-12] than with Ca(H-DBTA)2 [(−)-11] (compare Table 6, entries 2–4 and Table 7, entries 2–4). The only exception was the 1-phenyl-3-phospholene oxide (1a), in which case the results showed parity (compare Table 6, entry 1 and Table 7, entry 1). Interestingly, this tendency reversed for the series of the alkyl-3-phospholene oxides (1e–j), where, in 5 out of 6 instances, the application of Ca(H-DPTTA)2 [(−)-12] was more advantageous (compare Table 6, entries 5 and 7–11 with Table 7, entries 5 and 7–10). Both Ca(H-DBTA)2 and Ca(H-DPTTA)2 [(−)-11 and (−)-12] were equally applicable for the enantiomeric separation of 1-propyl-3-phospholene oxide (1f) in terms of resolving capability (S), but a higher enantiomeric excess could be obtained with Ca(H-DBTA)2 [(−)-11] (compare Table 6, entry 6 and Table 7, entry 6). In this series of experiments, the formation of diastereomers with a composition of Ca(1)2(H-DPTTA)2 was observed in most of the cases (Table 7) similarly to the instances described for the use of Ca(H-DBTA)2 [(−)-11] (Table 6).
The impact of the similar chiral environment of the O,O′-dibenzoyl- or O,O′-di-p-toluoyl-(2R,3R)-tartaric acid was that the resolving agents (−)-11 and (−)-12 preferred diastereomeric complex formation with the same enantiomers of a given 3-phospholene oxide (1b–d, 1f and 1h–1j) in the majority of the instances (compare Table 6, entries 2–4, 6 and 8–11 with Table 7, entries 2–4, 6 and 8–10). However, in the case of the phenyl-, ethyl or i-propyl-derivatives (1a, 1e and 1g) different antipodes could be prepared with Ca(H-DBTA)2 and Ca(H-DPTTA)2 [(−)-11 or (−)-12] (compare Table 6, entries 1, 5 and 7 with Table 7, entries 1, 5 and 7). It is worth mentioning that no solvent dependent enantiopreference could be observed, when Ca(H-DPTTA)2 [(−)-12] was used as the resolving agent.
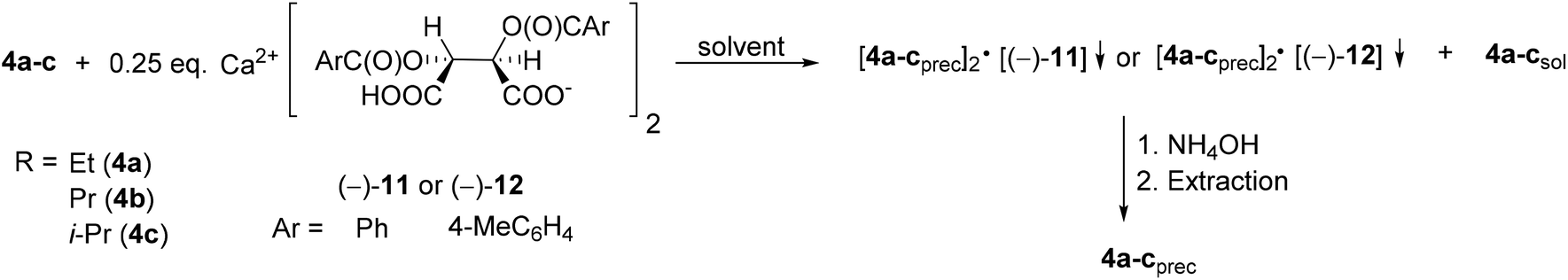 | ||
| Scheme 14 General method for the resolution of 1-alkoxy-3-methyl-3-phospholene 1-oxides (4a–c) with Ca(H-DBTA)2 or Ca(H-DPTTA)2 [(−)-11 or (−)-12]. | ||
| Entry | R | Resolving agent | Solvent | Diastereomeric complex | eea (%) | Yielda,b (%) | S , (−) | Abs. config. | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Enantiomeric excess, yield and resolving capability obtained after digestions. b Based on the half of the racemate that is regarded to be 100% for each antipode. c Resolving capability, also known as the Fogassy parameter [S = (Yield/100) × (ee/100)].66 | |||||||||
| 1 | Et (4a) | (−)-11 | MeCN–EtOH | (4a)2·(−)-11 | 91 | 29 | 0.26 | (R) | 80 |
| 2 | Pr (4b) | (−)-11 | MeCN–EtOH | (4b)2·(−)-11 | 59 | 18 | 0.11 | (R) | 67 |
| 3 | Pr (4b) | (−)-11 | EtOAc–EtOH | (4b)2·(−)-11 | 17 | 34 | 0.06 | (S) | 67 |
| 4 | i-Pr (4c) | (−)-11 | MeCN–EtOH | (4c)2·(−)-11 | 92 | 36 | 0.33 | (S) | 80 |
| 5 | Et (4a) | (−)-12 | EtOH–water | (4a)2·(−)-12 | 75 | 44 | 0.33 | (R) | 80 |
| 6 | Pr (4b) | (−)-12 | EtOAc–EtOH–water | (4b)2·(−)-12 | 55 | 36 | 0.20 | (R) | 67 |
| 7 | i-Pr (4c) | (−)-12 | EtOH–water | (4c)2·(−)-12 | Rac. | — | — | — | 80 |
When Ca(H-DBTA)2 [(−)-11] was the resolving agent, the alkoxy-3-phospholene oxides (4a–c) were prepared with an ee of 17–92%, whereas the corresponding resolving capability values fell in the range of 0.06–0.33 (Table 8, entries 1–4). By applying Ca(H-DPTTA)2 [(−)-12], the (R)-enantiomer of ethoxy- and propoxy-3-phospholene oxide could be prepared with an ee of 75% and 55%, respectively, and with a resolving capability value of 0.33 and 0.20, respectively (Table 8, entries 5 and 6). It is noteworthy that Ca(H-DPTTA)2 [(−)-12] could not differentiate between the two antipodes of i-propoxy-3-phospholene oxide (4c) (Table 8, entry 7).
The results in Table 8 indicate that the longer alkoxy chain may influence the non-bonding interactions with the resolving agent in a negative manner (compare Table 8, entry 1 with 2 and 3 and entry 5 with 6).
The 1-propoxy-3-phospholene oxide (4b) was the only alkoxy derivative whose (R)- and (S)-enantiomers could be prepared with the same resolving agent, Ca(H-DBTA)2 [(−)-11] using different solvents (ee for (R)-4b: 59% and ee for (S)-4b: 17%) (Table 8, entries 2 and 3).
The diastereomers formed on resolution contained the corresponding 1-alkoxy-3-phospholene oxides (4a–c) and Ca(H-DBTA)2 [(−)-11] or Ca(H-DPTTA)2 [(−)-12] in a ratio of 2:1 similarly to the cases described for the resolution of most of the aryl- and alkyl-3-phospholene oxides (compare Table 8 with Tables 6 and 7).
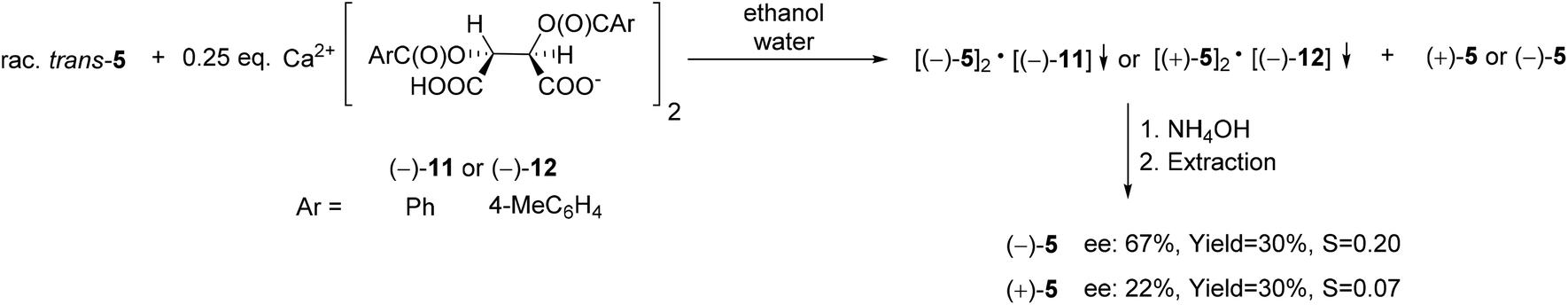 | ||
| Scheme 15 The resolution of 6,6-dichloro-1-methyl-3-phenyl-3-phosphabicyclo[3.1.0]hexane 3-oxide (5) with Ca(H-DBTA)2 or Ca(H-DPTTA)2 [(−)-11 or (−)-12]. | ||
 | ||
| Scheme 16 The resolution of 4-chloro-5-methyl-1-phenyl-1,2,3,6-tetrahydrophosphinine 1-oxide (7) with Ca(H-DPTTA)2 [(−)-12]. | ||
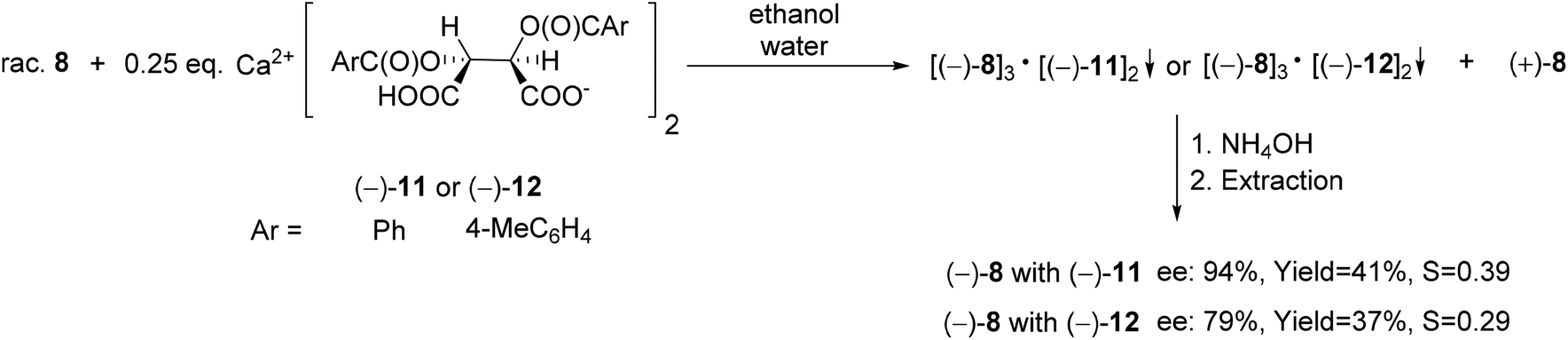 | ||
| Scheme 17 The resolution of 6-diethylamino-dibenzo[c,e][5,6]oxaphosphorine 6-oxide (8) with Ca(H-DBTA)2 or Ca(H-DPTTA)2 [(−)-11 or (−)-12]. | ||
Both Ca(H-DBTA)2 [(−)-11] and Ca(H-DPTTA)2 [(−)-12] were applicable for the partial enantiomeric separation of phenyl-phosphabicyclo[3.1.0]hexane oxide (5). Interestingly, resolving agents (−)-11 or (−)-12 formed diastereomeric coordination complexes with the different enantiomers of phosphabicyclo[3.1.0]hexane oxide 5. In this manner, (−)-5 and (+)-5 could be prepared with an ee of 67% and 22%, respectively (Scheme 15).71
Using Ca(H-DPTTA)2 [(−)-12] as the resolving agent, enantiopure (R)-1-phenyl-1,2,3,6-tetrahydrophosphinine 1-oxide [(R)-7] could be prepared (Scheme 16). It is noted that no crystalline diastereomers were formed, when Ca(H-DBTA)2 [(−)-11] was used.79
The acidic calcium salts O,O′-dibenzoyl- or O,O′-di-p-toluoyl-(2R,3R)-tartaric acid [(−)-11 or (−)-12] were also suitable resolving agents for the preparation of the (−)-6-diethylamino-dibenzo[c,e][5,6]oxaphosphorine 6-oxide [(−)-8]. Among all of the resolving agents [(−)-9–(−)-12] used for the enantioseparation of dibenzooxaphosphorine oxide 8, the highest ee and resolving capability values were obtained with Ca(H-DBTA)2 [(−)-11] (Scheme 17).70
2.3. X-Ray analysis of the diastereomeric complexes
The crystallization process plays an important role in these investigations not only by forming well defined solid precipitates, but also through the manipulation possibilities through solubility and insolubility properties, as well as affecting e.g. the dielectric properties by a designed engineering approach (fine tuning by crystal engineering). Thus, crystal structure determinations56–64,67,73,79 of these complex associate molecular systems may be of use in responding to queries regarding the structural background of the physical processes taking place during the experiments.Crystallographic data shed light not only on the solvent56,64 or co-solvent by identifying not only the presence of these species but also their role in the structure of the respective crystals. An interesting example was provided by the crystal structure of the quaternary (R)-i-butyl-3-phospholene oxide [(R)-1i]–spiro-TADDOL [(−)-10]–isopropanol 1:2:1 molecular assembly.64 In this case, a molecule of the applied solvent was also included in the crystal. The competitive binding of the solvent to the two host molecules and also to the (R)-i-butyl-3-phospholene oxide [(R)-1i] resolution target could be traced back to the slightly stronger binding of the solvent than that of the respective (R)-i-butyl-3-phospholene oxide guest [(R)-1i].
In another neutral complex case, the key feature of the resolution process was the formation of the 2:1 associate of spiro-TADDOL [(−)-10] with the (S)-(−)-1-isopropyl-3-methyl-3-phospholene oxide [(S)-1g] guest.65 Apart from the well-defined classical H-bridges, a large number of short C–H⋯O contacts were also present. Further support for the quantification of binding forces was obtained from force field calculations and from a semi-empirical computational analysis. These provided nearly same and cohesive binding energies in the range of −135 to −125 kJ mol−1.62 Thus, the major binding effects were successfully recognized in the structure discussed (Fig. 5).
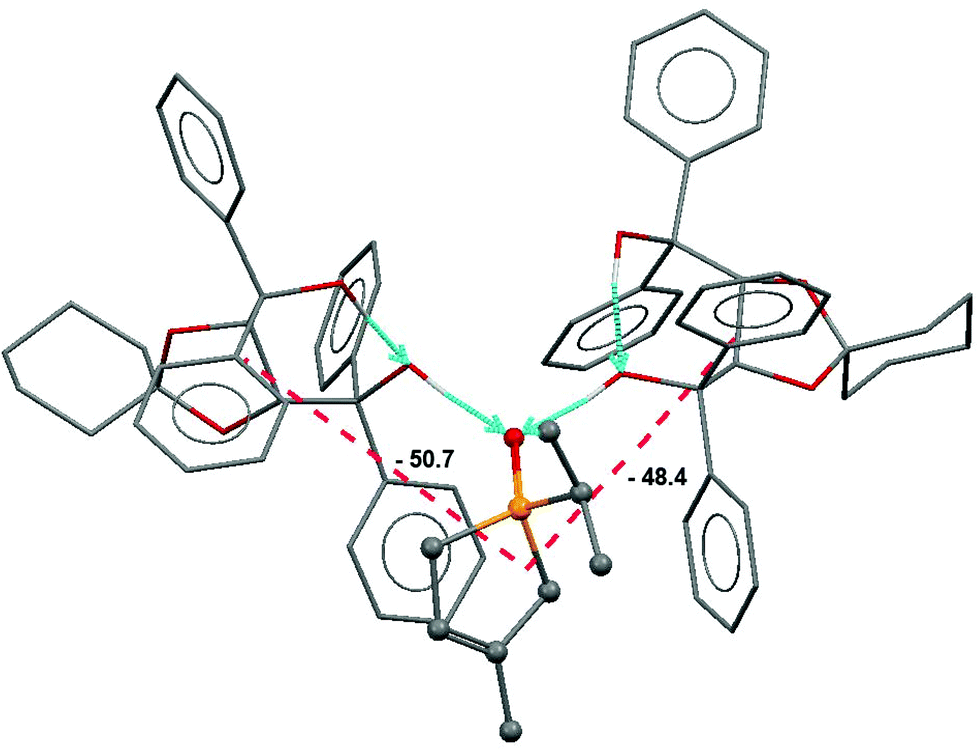 | ||
| Fig. 5 The two largest attractive interactions (in kJ mol−1) indicated also by red broken lines between the respective molecular centres in the crystal of the (S)-(−)-1-isopropyl-3-methyl-3-phospholene oxide guest [(S)-1g] with two independent spiro-TADDOL [(−)-10] hosts. Arrows indicate the donor–acceptor directions of the H-bond vectors. | ||
This identifies the major cohesive forces in the solid state that possibly also operate under the conditions of resolution.
The crystal structures of the acidic Ca-tartrate salt complexes67,73,79 disclosed a more alike pattern both in the composition, and in the way of binding the target phospholene oxides (1). The common feature in these crystals is that two acidic tartrate anions and two phospholene oxides contributed to a square-pyramid coordination sphere around the Ca2+ cation and the formation of a catena-type endless chain using crystallographic translations. The acidic proton forms a H-bridge with the anion, so that the coordination from the carboxylic/carboxylate groups provide a partial negative charge distributed evenly. A further feature of the coordination sphere is that either a perfect or an approximate twofold symmetry or pseudo-symmetry rules their global shape.
As it turned out clearly from this series of crystal structure determinations,56–64,67,73,79 the obvious and common binding of the P-oxides is realized through the PO oxygen atom. As it is described, the binding is primarily established through H-bonding in the neutral type assemblies incorporating TADDOL derivatives [(−)-9 and (−)-10]. The acidic Ca salt matrices involved the binding of the PO function into the sixfold coordination sphere of the metal ion. These observations may be explained by the enhanced negative charge on the PO oxygen atom.
3. Conclusions
In this paper, our versatile resolution methods applying TADDOL derivatives [(−)-9 and (−)-10] or the Ca2+-salts of tartaric acid derivatives [(−)-11 and (−)-12] are summarized, and the methods are widely applicable for the preparation of five- and six-membered P-heterocyclic phosphine oxides, phosphinates, a phosphine sulfide and a phosphonic ester-amide (1–8) in an optically active form (Fig. 6). The corresponding enantiomers (1–8) were obtained with an ee above 95% in the majority of the instances. The P-heterocycles (1–8) discussed are unknown in the literature in an optically active form. The absolute configuration of the P-stereogenic centers was also determined in most of the instances. In a few cases, diastereomeric molecular or coordination complexes were characterized by X-ray crystallography and the characteristic non-bonding interactions between the host and the molecules were identified. The optically active P-heterocyclic phosphine oxides are precursors of the corresponding P(III)-compounds which could be applied as ligands in platinum complexes (14).62,81–85 | ||
| Fig. 6 The best results for the resolution of five- and six-membered P-heterocyclic phosphine oxides, phosphinates, a phosphine sulfide and a phosphonic ester-amide (1–8). | ||
Acknowledgements
The authors are grateful to the Hungarian Research Fund for financial support (Grant No. K83118, K104769 and PD116096).References
- L. D. Quin, A guide to organophosphorus chemistry, John Wiley & Sons, New York, 2000 Search PubMed.
- T. Imamoto, in Handbook of Organophosphorus Chemistry, ed. R. Engel, Marcel Dekker, New York, 1992, ch. 1, pp. 1–54 Search PubMed.
- R. Noyori, Asymmetric Catalysis in Organic Synthesis, John Wiley & Sons, New York, 1994 Search PubMed.
- Phosphorus Ligands in Asymmetric Catalysis, ed. A. Börner, Wiley-VCH, Weinheim, 2008 Search PubMed.
- Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley, Weinheim, 2010 Search PubMed.
- H. Brunner and W. Zettlmeier, Handbook of enantioselective catalysis with transition metal compounds, VCH, Weinheim, 1993 Search PubMed.
- Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer-Verlag, Heidelberg, 2000 Search PubMed.
- W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029–3070 CrossRef CAS PubMed.
- A. Grabulosa, P-Stereogenic Ligands in Enantioselective Catalysis, The Royal Society of Chemistry, Cambridge, 2010 Search PubMed.
- S. Lühr, J. Holz and A. Börner, ChemCatChem, 2011, 3, 1708–1730 CrossRef.
- K. M. Pietrusiewicz and M. Zablocka, Chem. Rev., 1994, 94, 1375–1411 CrossRef CAS.
- O. Korpiun and K. Mislow, J. Am. Chem. Soc., 1967, 89, 4784–4786 CrossRef CAS.
- S. Juge, M. Stephan, J. A. Laffitte and J. P. Genet, Tetrahedron Lett., 1990, 31, 6357–6360 CrossRef CAS.
- J. V. Carey, M. D. Barker, J. M. Brown and M. J. H. Russell, J. Chem. Soc., Perkin Trans. 1, 1993, 831–839 RSC.
- E. J. Corey, Z. Chen and G. J. Tanoury, J. Am. Chem. Soc., 1993, 115, 11000–11001 CrossRef CAS.
- M. J. Johansson and N. Kann, Mini-Rev. Org. Chem., 2004, 1, 233–247 CrossRef CAS.
- A. Grabulosa, J. Granell and G. Muller, Coord. Chem. Rev., 2007, 251, 25–90 CrossRef CAS.
- D. S. Glueck, Synlett, 2007, 2627–2634 CrossRef CAS.
- O. I. Kolodiazhnyi, Tetrahedron: Asymmetry, 2012, 23, 1–46 CrossRef CAS.
- O. I. Kolodiazhnyi, V. P. Kukhar and A. O. Kolodiazhna, Tetrahedron: Asymmetry, 2014, 25, 865–922 CrossRef CAS.
- E. Bergin, C. T. O'Connor, S. B. Robinson, E. M. McGarrigle, C. P. O'Mahony and D. G. Gilheany, J. Am. Chem. Soc., 2007, 129, 9566–9567 CrossRef CAS PubMed.
- K. Nikitin, K. V. Rajendran, H. Müller-Bunz and D. G. Gilheany, Angew. Chem., Int. Ed., 2014, 53, 1906–1909 CrossRef CAS PubMed.
- I. G. M. Campbell and J. K. Way, J. Chem. Soc., 1961, 2133–2141 RSC.
- S. Lelièvre, F. Mercier, L. Ricard and F. Mathey, Tetrahedron: Asymmetry, 2000, 11, 4601–4608 CrossRef.
- W. Tang, A. G. Capacci, A. White, S. Ma, S. Rodriguez, B. Qu, J. Savoie, N. D. Patel, X. Wei, N. Haddad, N. Grinberg, N. K. Yee, D. Krishnamurthy and C. H. Senanayake, Org. Lett., 2010, 12, 1104–1107 CrossRef CAS PubMed.
- W. Tang, B. Qu, A. G. Capacci, S. Rodriguez, X. Wei, N. Haddad, B. Narayanan, S. Ma, N. Grinberg, N. K. Yee, D. Krishnamurthy and C. H. Senanayake, Org. Lett., 2010, 12, 176–179 CrossRef CAS PubMed.
- Z. Pakulski, O. M. Demchuk, J. Frelek, R. Luboradzki and K. M. Pietrusiewicz, Eur. J. Org. Chem., 2004, 3913–3918 CrossRef CAS.
- G. Ostrogovich and F. Kerek, Angew. Chem., Int. Ed. Engl., 1971, 83, 496–497 Search PubMed.
- X.-M. Sun, K. Manabe, W. W. L. Lam, N. Shiraishi, J. Kobayashi, M. Shiro, H. Utsumi and S. Kobayashi, Chem. – Eur. J., 2005, 11, 361–368 CrossRef PubMed.
- F. G. Holliman and F. G. Mann, J. Chem. Soc., 1947, 1634–1642 RSC.
- F. A. Hart and F. G. Mann, J. Chem. Soc., 1955, 4107–4114 RSC.
- J. R. Corfield, J. R. Shutt and S. Trippett, J. Chem. Soc., Chem. Commun., 1969, 789–790 RSC.
- K. D. Berlin and C.-H. Chen, J. Org. Chem., 1971, 36, 2791–2796 CrossRef CAS.
- K. L. Marsi and H. Tuinstra, J. Org. Chem., 1975, 40, 1843–1844 CrossRef CAS.
- N. Gurusamy and K. D. Berlin, J. Am. Chem. Soc., 1982, 104, 3114–3119 CrossRef CAS.
- H. Takaya, K. Mashima, K. Koyano, M. Yagi, H. Kumobayashi, T. Taketomi, S. Akutagawa and R. Noyori, J. Org. Chem., 1986, 51, 629–635 CrossRef CAS.
- Y. Hamada, F. Matsuura, M. Oku, K. Hatano and T. Shioiri, Tetrahedron Lett., 1997, 38, 8961–8964 CrossRef CAS.
- U. Matteoli, V. Beghetto, C. Schiavon, A. Scrivanti and G. Menchi, Tetrahedron: Asymmetry, 1997, 8, 1403–1409 CrossRef CAS.
- T. Miura and T. Imamoto, Tetrahedron Lett., 1999, 40, 4833–4836 CrossRef CAS.
- T. Benincori, S. Gladiali, S. Rizzo and F. Sannicolo, J. Org. Chem., 2001, 66, 5940–5942 CrossRef CAS PubMed.
- S. D. de Paule, S. Jeulin, V. Ratovelomanana-Vidal, J. P. Genet, N. Champion and P. Dellis, Tetrahedron Lett., 2003, 44, 823–826 CrossRef.
- J. Omelanczuk, A. Karaçar, M. Freytag, P. G. Jones, R. Bartsch, M. Mikolajczyk and R. Schmutzler, Inorg. Chim. Acta, 2003, 350, 583–591 CrossRef CAS.
- J. Holt, A. M. Maj, E. P. Schudde, K. M. Pietrusiewicz, L. Sieroń, W. Wieczorek, T. Jerphagnon, I. W. C. E. Arends, U. Hanefeld and A. J. Minnaard, Synthesis, 2009, 2061–2065 CAS.
- D. Liu and X. Zhang, Eur. J. Org. Chem., 2005, 646–649 CrossRef CAS.
- T. Imamoto, K. V. L. Crépy and K. Katagiri, Tetrahedron: Asymmetry, 2004, 15, 2213–2218 CrossRef CAS.
- K. Tani, L. D. Brown, J. Ahmed, J. A. Ibers, M. Yokota, A. Nakamura and S. Otsuka, J. Am. Chem. Soc., 1977, 99, 7876–7886 CrossRef CAS.
- E. Duran, E. Gordo, J. Granell, M. Font-Bardia, X. Solans, D. Velasco and F. Lopez-Calahorra, Tetrahedron: Asymmetry, 2001, 12, 1987–1997 CrossRef CAS.
- E. Duran, E. Gordo, J. Granell, D. Velasco and F. Lopez-Calahorra, Tetrahedron Lett., 2001, 42, 7791–7793 CrossRef CAS.
- F. Doro, M. Lutz, J. N. H. Reek, A. L. Spek and P. van Leeuwen, Eur. J. Inorg. Chem., 2008, 1309–1317 CrossRef CAS.
- F. Robin, F. Mercier, L. Ricard, F. Mathey and M. Spagnol, Chem. – Eur. J., 1997, 3, 1365–1369 CrossRef CAS.
- F. Mathey, F. Mercier, F. Robin and L. Ricard, J. Organomet. Chem., 1998, 557, 117–120 CrossRef CAS.
- G. Frison, F. Brebion, R. Dupont, F. Mercier, L. Ricard and F. Mathey, C. R. Chim., 2002, 5, 245–249 CrossRef CAS.
- F. Bienewald, L. Ricard, F. Mercier and F. Mathey, Tetrahedron: Asymmetry, 1999, 10, 4701–4707 CrossRef CAS.
- G. He, K. F. Mok and P.-H. Leung, Organometallics, 1999, 18, 4027–4031 CrossRef CAS.
- D. Seebach, A. K. Beck and A. Heckel, Angew. Chem., Int. Ed., 2001, 40, 92–138 CrossRef CAS.
- T. Novák, J. Schindler, V. Ujj, M. Czugler, E. Fogassy and G. Keglevich, Tetrahedron: Asymmetry, 2006, 17, 2599–2602 CrossRef.
- W. J. Pope and S. J. Peachey, J. Chem. Soc., Dalton Trans., 1899, 1066–1093 RSC.
- M. Ács, E. Fogassy, C. Kassai, D. Kozma and M. Nógrádi, CRC Handbook of Optical Resolutions via Diastereoisomeric Salt Formation, CRC Press, Boca Raton, 2002 Search PubMed.
- E. Fogassy, M. Nógrádi, D. Kozma, G. Egri, E. Pálovics and V. Kiss, Org. Biomol. Chem., 2006, 4, 3011–3030 CAS.
- F. Faigl, E. Fogassy, M. Nógrádi, E. Pálovics and J. Schindler, Tetrahedron: Asymmetry, 2008, 19, 519–536 CrossRef CAS.
- T. Novák, V. Ujj, J. Schindler, M. Czugler, M. Kubinyi, Z. A. Mayer, E. Fogassy and G. Keglevich, Tetrahedron: Asymmetry, 2007, 18, 2965–2972 CrossRef.
- P. Bagi, K. Juhász, I. Timári, K. E. Kövér, D. Mester, M. Kállay, M. Kubinyi, T. Szilvási, P. Pongrácz, L. Kollár, K. Karaghiosoff, M. Czugler, L. Drahos, E. Fogassy and G. Keglevich, J. Organomet. Chem., 2015, 797, 140–152 CrossRef CAS.
- P. Bagi, A. Fekete, M. Kállay, D. Hessz, M. Kubinyi, T. Holczbauer, M. Czugler, E. Fogassy and G. Keglevich, Chirality, 2014, 26, 174–182 CrossRef CAS PubMed.
- P. Bagi, A. Fekete, M. Kállay, D. Hessz, M. Kubinyi, T. Holczbauer, M. Czugler, E. Fogassy and G. Keglevich, Heteroatom Chem., 2015, 26, 79–90 CrossRef CAS.
- P. Bagi, K. Juhász, D. Hessz, M. Kállay, M. Kubinyi, K. Karaghiosoff, M. Czugler, E. Fogassy and G. Keglevich, 2015, unpublished work.
- R. A. Sheldon, Chirotechnology: Industrial Synthesis of Optically Active Compounds, Marcel Dekker, New York, 1993 Search PubMed.
- P. Bagi, M. Kállay, D. Hessz, M. Kubinyi, T. Holczbauer, M. Czugler, E. Fogassy and G. Keglevich, Tetrahedron: Asymmetry, 2014, 25, 318–326 CrossRef CAS.
- G. Keglevich, J. Dvorszki, V. Ujj and K. Ludányi, Heteroatom Chem., 2010, 21, 271–277 CrossRef CAS.
- V. Ujj, T. Szuhánszki, J. Schinder, M. Czugler, E. Fogassy and G. Keglevich, Hung. J. Ind. Chem., 2008, 36, 131–136 Search PubMed.
- V. Ujj, Ph.D Thesis, 2010.
- P. Bagi, A. Laki and G. Keglevich, Heteroatom Chem., 2013, 24, 179–186 CrossRef CAS.
- V. Ujj, A. Kerényi, A. Laki, E. Fogassy and G. Keglevich, Lett. Org. Chem., 2010, 7, 110–113 CrossRef CAS.
- P. Bagi, K. Karaghiosoff, M. Czugler, D. Hessz, M. Kállay, M. Kubinyi, T. Szilvási, P. Pongrácz, L. Kollár, I. Timári, K. E. Kövér, L. Drahos, E. Fogassy and G. Keglevich, Heteroatom Chem., 2015 Search PubMed , submitted.
- G. Keglevich, Synthesis, 1993, 931–942 CrossRef CAS.
- G. Keglevich, Curr. Org. Chem., 2006, 10, 93–111 CrossRef CAS.
- A. Mravik, Z. Böcskei, Z. Katona, I. Markovits and E. Fogassy, Angew. Chem., Int. Ed. Engl., 1997, 36, 1534–1536 CrossRef CAS.
- A. Mravik, Z. Bocskei, K. Simon, F. Elekes and Z. Izsaki, Chem. – Eur. J., 1998, 4, 1621–1627 CrossRef CAS.
- A. Mravik, Z. Böcskei, Z. Katona, I. Markovits, G. Pokol, D. K. Menyhárd and E. Fogassy, J. Chem. Soc., Chem. Commun., 1996, 1983–1984 RSC.
- V. Ujj, J. Schindler, T. Novák, M. Czugler, E. Fogassy and G. Keglevich, Tetrahedron: Asymmetry, 2008, 19, 1973–1977 CrossRef CAS.
- V. Ujj, P. Bagi, J. Schindler, J. Madarász, E. Fogassy and G. Keglevich, Chirality, 2010, 22, 699–705 CAS.
- P. Bagi, T. Kovács, T. Szilvási, P. Pongrácz, L. Kollár, L. Drahos, E. Fogassy and G. Keglevich, J. Organomet. Chem., 2014, 751, 306–313 CrossRef CAS.
- A. Kerényi, V. Kovács, T. Körtvélyesi, K. Ludányi, L. Drahos and G. Keglevich, Heteroatom Chem., 2010, 21, 63–70 CrossRef.
- P. Pongrácz, L. Kollár, A. Kerényi, V. Kovács, V. Ujj and G. Keglevich, J. Organomet. Chem., 2011, 696, 2234–2237 CrossRef.
- G. Keglevich, P. Bagi, Á. Szöllősy, T. Körtvélyesi, P. Pongrácz, L. Kollár and L. Drahos, J. Organomet. Chem., 2011, 696, 3557–3563 CAS.
- P. Bagi, T. Szilvási, P. Pongrácz, L. Kollár, L. Drahos and G. Keglevich, Curr. Org. Chem., 2014, 18, 1529–1538 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |