
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
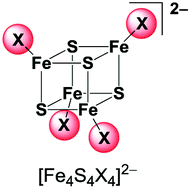
Halide coordinated homoleptic [Fe4S4X4]2− and heteroleptic [Fe4S4X2Y2]2− clusters (X, Y = Cl, Br, I)—alternative preparations, structural analogies and spectroscopic properties in solution and solid state†
Andreas O.
Schüren
ab,
Verena K.
Gramm
a,
Maximilian
Dürr
c,
Ana
Foi
b,
Ivana
Ivanović-Burmazović
c,
Fabio
Doctorovich
b,
Uwe
Ruschewitz
a and
Axel
Klein
*a
aDepartment für Chemie, Institut für Anorganische Chemie, Universität zu Köln, Greinstraße 6, 50939 Köln, Germany. E-mail: axel.klein@uni-koeln.de
bDepartamento de Química Inorgánica, Analítica, y Química Física, Facultad de Ciencias Exactas y Naturales, INQUIMAE-CONICET, Universidad de Buenos Aires, Ciudad Universitaria, Pabellón 2, Piso 3, C1428EHA Buenos Aires, Argentina
cDepartment Chemie und Pharmazie, Lehrstuhl für Bioanorgansiche Chemie, Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstraße 1, 91058 Erlangen, Germany
First published on 13th November 2015
Abstract
New facile methods to prepare iron sulphur halide clusters [Fe4S4X4]2− from [Fe(CO)5] and elemental sulphur were elaborated. Reactions of ferrous precursors like tetrahalidoferrates(II) or simple ferrous halides with [Fe(CO)5] and sulphur turned out to be efficient methods to prepare homoleptic [Fe4S4X4]2− (X = Cl, Br) and heteroleptic clusters [Fe4S4X4−nYn]2− (X = Cl, Br; Y = Br, I). Solid materials were obtained as salts of BTMA+ (= benzyltrimethylammonium); the new compounds containing [Fe4S4Br4]2− and [Fe4S4X2Y2]2− (X, Y = Cl, Br, I) were all isostructural to (BTMA)2[Fe4S4I4] (monoclinic, Cc) as inferred from synchrotron X-ray powder diffraction. While the solid materials contain defined heteroleptic clusters with a halide X![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Y ratio of 2:2, dissolving these compounds leads to rapid scrambling of the halide ligands forming mixtures of all five possible [Fe4S4X4−nYn]2− clusters as could be shown by UHR-ESI MS. The variation of X and Y allowed assignment of the absorption bands in the visible and NIR; the long-wavelength bands around 1100 nm were tentatively assigned to intervalence charge transfer (IVCT) transitions.
:Y ratio of 2:2, dissolving these compounds leads to rapid scrambling of the halide ligands forming mixtures of all five possible [Fe4S4X4−nYn]2− clusters as could be shown by UHR-ESI MS. The variation of X and Y allowed assignment of the absorption bands in the visible and NIR; the long-wavelength bands around 1100 nm were tentatively assigned to intervalence charge transfer (IVCT) transitions.
Introduction
Cubane-shaped [Fe4S4] clusters represent a very interesting class of cofactors in biology and are involved in various functions in a cell's life cycle.1–6 Due to their mixed valent character with two ferric (FeIII) and two ferrous (FeII) iron atoms these clusters show extraordinary electrochemical properties which are crucial for their biological functions in electron transfer and redox catalysis reactions.7 During the last 40 years numerous [Fe4S4]2+ clusters with terminal thiolate ligands have been prepared in order to model the structural, electronic and electrochemical properties as well as the reactivity of the naturally occurring clusters.8–16 Furthermore, several studies have recently been conducted to elucidate their availability as precursors for iron sulphur material synthesis as well as single molecule applications.17–24 Halide based [Fe4S4X4]2− with X = Cl, Br or I are known to be useful precursors because the halides can be easily exchanged with thiolate or other ligands by salt metathesis reactions. Homoleptic halide clusters have thus been studied previously and some preparation methods have been published (Scheme 1).25 | ||
| Scheme 1 Reported preparation pathways for [Fe4S4X4]2− clusters. (Method A: Henderson et al.;28 Method B: Holm et al.;26,27 Method C: Müller et al.;29 Method D: Kanatzidis et al.;20 Method E: Pohl et al.30,31). | ||
They can be either formed by ligand exchange from hydrogen sulfide or thiolate coordinated [Fe4S4]2+ clusters (see Scheme 1A and B) or directly starting from relatively cheap bulk chemicals.26–28 Müller and co-workers have been able to prepare [Fe4S4Cl4]2− and [Fe4S4Br4]2− clusters from ferric or ferrous iron halides and excess H2S in the presence of triethylamine as base (Scheme 1C).29 The chloride cluster can be also synthesised from iron(II) chloride and sulphur that has been reduced in situ with thiophenolate (Scheme 1D).20 A convenient preparation method for the iodide cluster using iron pentacarbonyl, sulphur, iodine and an iodide salt has been reported by Pohl and co-workers (Scheme 1E).30,31
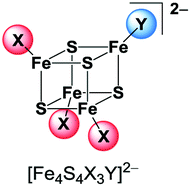
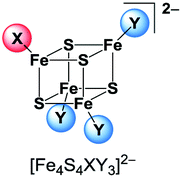
While many examples for [Fe4S4] clusters with a homoleptic coordination sphere of thiolate, halide, nitrogen or phosphorus ligands are known, the number of clusters with a heteroleptic ligand set is much smaller. Although several clusters [Fe4S4L2L′2]2− and [Fe4S4L3L′]2− have been published exhibiting a 2:2 or 3:1 coordination sphere of two different ligand types L and L′ (L, L′ = Cl−, Br−, I−, phenolates, thiophenolates and dithiocarbamates) they are very difficult to isolate. In solution the ligands are exchanged among the clusters leading to coordinative disproportionation (eqn (1)).9,10,32,33
 | (1) |
This exchange equilibrium is hampered if the cluster's charge is neutral due to one neutral ligand type L′′ like in [Fe4S4I2(Ph3PS)2]34 [Fe4S4(SPh)2(tBu3P)2]35 or [Fe4S4(SR)2(SC(NMe2))2]36 (R = 2,4,6-triisopropylphenyl). The products of ligand exchange on neutral [Fe4S4L2L′′2] are ionic and are less favourable in non-polar solvents.37,38 In order to completely avoid this exchange, tripodal thiolate ligands were established to stabilise clusters with 3:1 coordination.39–48
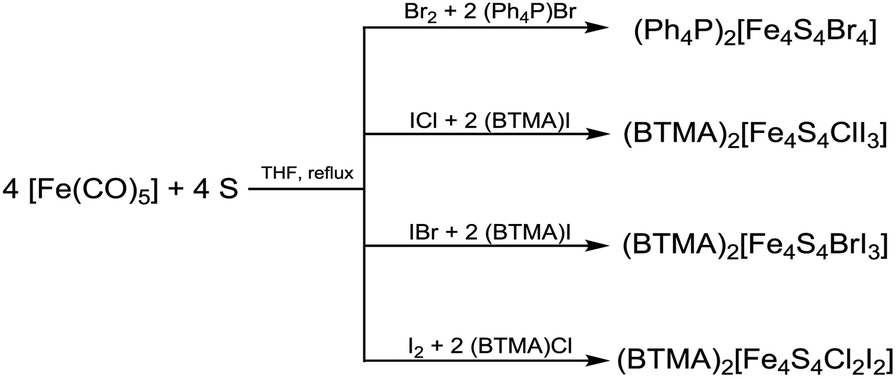
[Fe4S4Br2Cl2]2− is so far the only reported example of a heteroleptic halide [Fe4S4] cluster that has been isolated and characterised in solid state.29 The preparation of further clusters [Fe4S4X2Y2]2− and [Fe4S4X3Y]2− having a 2:2 or 3:1 coordination (Scheme 2) of two different halide ligands is desirable hence these clusters will be excellent precursors for selective ligand exchange reactions to form heteroleptic clusters with more intricate ligands. Furthermore, these clusters may provide deeper insight into the electronic structure of the [Fe4S4] core due to the different electronegativity of the halide ligands and the relatively small number of atoms.
 | ||
| Scheme 2 Homoleptic and heteroleptic halide [Fe4S4] cluster with 3:1 and 2:2 coordination. | ||
In this contribution we will report on new, remarkably improved preparation methods for homoleptic [Fe4S4X4]2− and heteroleptic [Fe4S4X2Y2]2− clusters (X = Cl, Br; Y = Br, I) modifying the above mentioned method by Pohl that was previously only reported for the preparation of the iodide cluster.30,31 The application of iron(II) halides and related precursors turned out to be the most efficient procedure to prepare those clusters in excellent yields. By this procedure we prepared e.g. the new clusters [Fe4S4Cl2I2]2− and [Fe4S4Br2I2]2−. Furthermore, we utilised different elemental halogens and interhalogens as well as halides to examine the scope of this procedure. The resulting materials, which were mainly salts of the BTMA+ (= benzyltrimethylammonium) cation were characterised in the solid by X-ray powder diffraction (XRPD) and IR spectroscopy and in solution by ESI MS and UV-vis-NIR absorption spectroscopy.
Results and discussion
Preparations and analytics
It turned out that homoleptic [Fe4S4X4]2− as well as heteroleptic [Fe4S4X2Y2]2− clusters can be obtained most efficiently if Pohl's method is modified by replacing one equivalent of [Fe(CO)5] and the halogen compound by simple iron(II) halide precursors (Scheme 3). | ||
| Scheme 3 Cluster preparations starting from [Fe(CO)5] and sulphur used in the present work. | ||
Heating [Fe(CO)5], sulphur, anhydrous ferrous halides and BTMA halides (BTMA+ = benzyltrimethylammonium) in MeCN for several days under an inert atmosphere allowed to obtain black, air and moisture sensitive solutions of [Fe4S4Cl4]2− and [Fe4S4Br4]2− (Scheme 3, eqn (2)) as confirmed by UV-vis-NIR absorption spectra (see later). The observed absorptions bands are in line with those reported for the cluster ions [Fe4S4Cl4]2− and [Fe4S4Br4]2−.25
High yields (87%) of black crystalline material were obtained by slow diffusion of diethyl ether into these solutions at −18 °C. The material obtained for X = Br is (BTMA)2[Fe4S4Br4]. In contrast to this, the crystalline material from [Fe4S4Cl4]2− solution was identified as the prismane shaped hexanuclear cluster (BTMA)3[Fe6S6Cl6] (eqn (2), Scheme 3) although in solution the tetranuclear, cubane species [Fe4S4Cl4]2− is stable and has been isolated previously from such solutions as salts of organic ammonium and phosphonium cations.20–31 We assume that the tetranuclear species is dominating a mixture of several possible cluster species (bi-, tetra-, hexanuclear) which are interconnected through rearrangements.49 The reason why crystallisation using the BTMA+ ions lead to the hexanuclear species probably lies in the better crystallisation of the latter over the tetranuclear species. To verify this assumption we will try to get single crystals of corresponding compounds in the future.
 | (2) |
We also utilised [FeCl4]2− and [FeBr4]2− for cluster formation. These precursors combine the iron(II) halide and the halide source. From reactions of (Et4N)2[FeCl4] or (Ph4P)2[FeBr4], respectively, with [Fe(CO)5] and sulphur in MeCN (Et4N)2[Fe4S4Cl4] and (Ph4P)2[Fe4S4Br4] were obtained as black solids in moderate to good yields of 69–92% (Scheme 3, eqn (3), Table 1). Obviously, both cluster formation methods have the same pathway since it is known that ferrous halides form tetrahalidoferrates(II) [FeX4]2− in presence of excess halide.50 Therefore, these [FeX4]2− are assumed to be intermediates for cluster assembly.
| Compound | Starting materials | Conditions | Yield | Reference |
|---|---|---|---|---|
| (BTMA)2[Fe4S4I4] | [Fe(CO)5] + S8 + I2 + (BTMA)I | THF, reflux | 95% | 30, 31 |
| (BTMA)2[Fe4S4I4] | [Fe(CO)5] + S8 + I2 + (BTMA)I | MeCN, reflux | 81% | This work |
| (BTMA)2[Fe4S4Br4] | [Fe(CO)5] + S8 + FeBr2 + (BTMA)Br | MeCN, reflux | 87% | This work |
| (BTMA)2[Fe4S4Br2Cl2] | [Fe(CO)5] + S8 + FeCl2 + (BTMA)Br | MeCN, reflux | 89% | This work |
| (BTMA)2[Fe4S4Cl2I2] | [Fe(CO)5] + S8 + FeCl2 + (BTMA)I | MeCN, reflux | 77% | This work |
| (BTMA)2[Fe4S4Br2I2] | [Fe(CO)5] + S8 + FeBr2 + (BTMA)I | MeCN, reflux | 85% | This work |
| (Et4N)2[Fe4S4Cl4] | [Fe(CO)5] + S8 + (Et4N)2[FeCl4] | MeCN, reflux | 69% | This work |
| (Ph4P)2[Fe4S4Br4] | [Fe(CO)5] + S8 + (Ph4P)2[FeBr4] | MeCN, reflux | 92% | This work |
| (Ph4P)2[Fe4S4Br4] | [Fe(CO)5] + S8 + Br2 + (BTMA)Br | THF, reflux | 71% | This work |
| (BTMA)2[Fe4S4Cl2I2] | [Fe(CO)5] + S8 + I2 + (BTMA)Cl | THF, reflux | 75% | This work |
| (BTMA)2[Fe4S4ClI3] | [Fe(CO)5] + S8 + ICl + (BTMA)I | THF, reflux | 94% | This work |
| (BTMA)2[Fe4S4BrI3] | [Fe(CO)5] + S8 + IBr + (BTMA)I | THF, reflux | 81% | This work |
| (Ph4P)2[Fe4S4Cl4] | FeCl3 + (Ph4P)Cl + H2S excess | DMF, MeOH, Et3N | 80% | 29 |
| (Ph4P)2[Fe4S4Br4] | FeBr3 + (Ph4P)Br + H2S excess | DMF, MeOH, Et3N | 78% | 29 |
| (Ph4P)2[Fe4S4Br2Cl2] | FeBr3 + (Ph4P)Cl + H2S excess | DMF, MeOH, Et3N | 68% | 29 |
| (Ph4P)2[Fe4S4Cl4] | FeCl2 + NaSPh excess + S8 + (Ph4P)Cl | MeCN | — | 20 |
| (R4N)2[Fe4S4Cl4] | (R4N)2[Fe4S4(tBuS)4] + benzoyl chloride | MeCN | 80–92% | 26, 27 |
| (Et4N)2[Fe4S4Br4] | (Et4N)2[Fe4S4(tBuS)4] + benzoyl bromide | MeCN | 50–65% | 26, 27 |
| (Et4N)2[Fe4S4I4] | (Et4N)2[Fe4S4Cl4] + NaI excess | MeCN | 75% | 26, 27 |
| (Pr4N)2[Fe4S4Cl4] | (Pr4N)2[Fe4S4(SH)4] + 1,2-dichloroethane | — | 85% | 28 |
Remarkably, no product formation was observed when using ferrous iodide precursors. For example, from our attempts to perform analogous reactions with [Fe(CO)4I2] no defined products were obtained (eqn (3)). Like with iron(II) chloride and bromide we assume a rapid formation of tetraiodidoferrate(II) in those solutions. In contrast to [FeCl4]2− and [FeBr4]2−, [FeI4]2− seems to be inert towards the desired cluster formation reactions. This is supported by the fact that crystalline (BTMA)2[FeI4] was isolated in quantitative yield from those solutions when being exposed to air and moisture.
 | (3) |
The iodide cluster [Fe4S4I4]2− was prepared as the BTMA+ salt in high yield (94%) by reproducing Pohl's original procedure using [Fe(CO)5], sulphur, iodine and iodide (pathway E in Schemes 1 and 3). Synchrotron XRPD (see also later) revealed a small amount of impurities in (BTMA)2[Fe4S4I4] which could not be identified so far. We tried to recrystallise the product from CH2Cl2 at elevated pressure as published.30,31 Depending on water and oxygen content of the solvent CH2Cl2 cluster degradation was observed by UV-vis-NIR spectroscopy and crystals of (BTMA)2[FeI4] were found in the resulting material.
 | (4) |
Reactions using ferrous halides FeY2 with (BTMA)X under similar conditions lead to formation of the heteroleptic clusters [Fe4S4Br2Cl2]2−, [Fe4S4Cl2I2]2− and [Fe4S4Br2I2]2− in MeCN (eqn (4) and Scheme 3). UV-vis-NIR absorption spectroscopy from reaction solutions shows both absorptions bands at 693–702 nm and 1088–1128 nm which are characteristic for [Fe4S4X4]2− clusters (see later in the UV-vis-NIR section).25 Black crystalline salts of BTMA+ were obtained for all these three cubane clusters in high yields of 77–89% upon ether diffusion. Elemental analysis showed a halide ratio X:Y of 2:2 for the bulk materials. This could either mean that the pure [Fe4S4X2Y2]2− clusters or a mixture of species [Fe4S4X4−nYn]2− with an averaged X:Y ratio of 2:2 are present in these solids. Synchrotron X-ray powder diffraction show that these crystalline materials represent indeed the pure heteroleptic clusters (BTMA)2[Fe4S4X2Y2] (see also later).
Remarkably, attempts to prepare heterobimetallic clusters [MFe3S4X4]2− (M = Mn, Co; X = Cl, Br, I) by this method result only in formation of monometallic clusters [Fe4S4X4]2−. Various experiments were carried out using (Et4N)2[CoCl4], (Ph4P)2[CoBr4], CoI2·nH2O, MnI2·4H2O as precursors. The reaction mixtures were analysed by ESI MS and UV-vis-NIR absorption spectroscopy, the materials obtained after precipitation using diethyl ether were characterised by elemental analysis and XRPD. Neither in the reaction mixtures nor in the isolated materials we found evidence for the heterobimetallic clusters. E.g. when using (Et4N)2[CoCl4] a MS signal at m/z = 624.1 is found in MeCN solutions which fits well to a [Et4N + Fe4S4Cl4]− species concerning peak position and isotopic pattern, whereas no signal at m/z = 626.7 corresponding to a [Et4N + CoFe3S4Cl4]− species is observed (Fig. S1, ESI†). Obviously, the transition metal halide only serves as halide source for the assembly of the monometallic cluster. In the case of (Ph4P)2[CoBr4], the precursor could be identified from XRPD analysis in the solid reaction product, the rest of the materials were largely amorphous. For the simple Co or Mn iodides we assume the formation of CoS and MnS which are virtually insoluble in MeCN.
We also attempted to modify Pohl's original method (Scheme 1E) by replacing iodine by bromine or the interhalogens ICl and IBr, respectively. Under similar conditions using [Fe(CO)5], sulphur, bromine and bromide [Fe4S4Br4]2− can be prepared in THF solution (eqn (5)).
 | (5) |
The tetraphenylphosphonium salt (Ph4P)2[Fe4S4Br4] was isolated as a black solid in good yields (Table 1). Its identity and purity was confirmed by XRPD. Analogous reactions using ICl and IBr for the preparation of [Fe4S4X3Y]2− clusters were conducted in THF. Black amorphous solids were isolated upon solvent evaporation that turned out to be mixture of all possible [Fe4S4X4−nYn]2− clusters. The amorphous character of these solids already indicates that these materials contain very probably a mixture of different clusters. This will be discussed later in detail with ESI MS results. Similar observations can be made from reactions using iodine and chloride in order to form [Fe4S4Cl2I2]2− clusters (eqn (5)).
Although [Fe4S4X4]2− can be prepared using the previously mentioned halogen/interhalogen pathway it turned out to be ineffective since the procedure is limited to THF as solvent. Unfortunately, solubility of the clusters is quite low in THF. Thus, they precipitate rather quickly and no crystalline material was obtained. Whereas [Fe4S4I4]2− can also be prepared from iodine in MeCN solutions, reactions between bromine or interhalogenes, respectively and the solvent prevent cluster formation.
Negative ESI MS
ESI mass spectra (negative mode) of (BTMA)2[Fe4S4I4] dissolved in THF is dominated by the molecular peak of the parent ion [Fe4S4I4]2− with m/z = 429.8 (Fig. S2, ESI†). The simulated isotopic pattern fits well to the two-fold negatively charged cluster ion. A further remarkable peak with m/z = 556.6 represents the species [Fe2S2I3]−. This is a product of oxidative cluster degradation. Like thiolate clusters, [Fe4S4X4]2− may decay in the presence of excess ligand with oxidation to binuclear clusters [Fe2S2X4]2− which are converted to [Fe2S2X3]− losing one ligand.9,10,25 Furthermore, small quantities of [FeI3]−m/z = 437.1 and [FeI4]−m/z = 563.2 are present that are formed from [FeI4]2− by loss of iodide or oxidation, respectively. [FeI4]2− that was also isolated as BTMA salt from decomposed [Fe4S4I4]2− seems to be a final product of cluster decay.From diluted solutions of (BTMA)2[Fe4S4Br4] in acetone a well resolved UHR-ESI (neg. mode) MS was obtained. Fig. 1 represents the molecular peak of [Fe4S4Br4]2−, the full MS is provided in the ESI.† The dianion [Fe4S4Br4]2− shows the most intense peak at m/z = 335.6497. Its mass value and its isotopic pattern fit very well to the calculated data (Fig. 1A). The distance between each peak inside the pattern of m/z = 0.5 confirms its dianionic character. A peak at m/z = 821.4260 with slightly lower intensity represents the singly charged [BTMA + Fe4S4Br4]− species. Extensive fragmentation seems not to be induced at this collision energy. Only weak peaks of [Fe4S4Br3]− (m/z = 590.3813) and [Fe4S4Br3O2]− (m/z = 622.3710) can be detected. They belong to an ion series starting with [Fe4S4Br4]2− which loses one bromide ligand followed by binding an oxygen molecule (Scheme 4). This may be a first step in cluster degradation upon oxidation. Similarly, [Fe4S4Cl3O2]− has been detected.51
 | ||
| Fig. 1 Molecular peaks of [Fe4S4Br4]2−, [Fe4S4Br2Cl2]2−, [Fe4S4 Cl2I2]2− and [Fe4S4Br2I2]2− ions in neg. UHR-ESI MS in acetone (above, collected data; below, simulated isotopic pattern). | ||
 | ||
| Scheme 4 Ion series of [Fe4S4Br4]2− decay in UHR-ESI source. | ||
Bromide ions were not detected. Product ions of oxidative splitting of cubane clusters in the presence of excess ligand can only be found in traces. Those are [Fe2S2Br4]2− (m/z = 247.7414) and [Fe2S2Br3]− (m/z = 414.5670) which belong to an analogous fragmentation series as we have observed for the iodide cluster and what is well-known for thiolate clusters.9,10,25 Additionally, further adduct species [M + Fe4S4Br4]− with M = alkali metals can be observed. So, the [Fe4S4Br4]2− cluster turned out to be relatively stable towards decay in the absence of oxygen.
UHR-ESI MS of diluted solutions of the heteroleptic clusters (BTMA)2[Fe4S4X2Y2] obtained from the FeY2/X− pathway (eqn (4)) in acetone exhibit the presence of all five possible dianionic cluster species represented by the formula [Fe4S4X4−nYn]2− with the [Fe4S4X2Y2]2− dianions being the most abundant in case of X = Cl, Y = Br and X = Br, Y = I (Table 2).
| m/z (intensity) | |||
|---|---|---|---|
| X = Cl | X = Cl | X = Br | |
| Y = Br | Y = I | Y = I | |
| a Characterised by m/z values and isotopic pattern. The given intensities were relative intensities in the experiment. | |||
|
245.7521 (43%) | 245.7523 (19%) | 335.6488 (15%) |
|
268.7258 (19%) | 292.7184 (3%) | 359.6419 (44%) |

|
291.6992 (100%) | 337.6883 (46%) | 382.6360 (100%) |
|
313.6743 (90%) | 383.6560 (100%) | 405.6303 (75%) |

|
335.6492 (44%) | 429.6239 (42%) | 429.6232 (29%) |
Apart of the dianionic [Fe4S4X4−nYn]2− species also their adducts [BTMA + Fe4S4X4−nYn]− are detected (see ESI†). The molecular peaks are well resolved and are in good to very good agreement with calculated isotopic patterns (Fig. 1B–D). For the combination X = Cl, Y = I, the BTMA+ adduct [BMTA + Fe4S4Cl2I2]− (m/z = 825.5033) is the most prevalent species, while the dianion [Fe4S4Cl2I2]2− (m/z = 337.6883) is not the most abundant. The signal for [Fe4S4ClI3]2− (m/z = 383.6560) is also quite intense while the signal for [Fe4S4Cl3I]2− (m/z = 291.7184) almost vanished. Measurements on (BTMA)2[Fe4S4Cl2I2] and (BTMA)2[Fe4S4ClI3] in THF show the same peculiar distribution. For reasons we do not understand so far the [Fe4S4ClI3]2− ion seems to be much more stable than [Fe4S4Cl2I2]2− under the MS conditions. In contrast to this, the solid sample of (BTMA)2[Fe4S4Cl2I2] seems to contain exclusively the [Fe4S4Cl2I2]2− cluster (see section on synchrotron X-ray powder diffraction).
As found for the [Fe4S4Br4]2− fragmentation series the mixed clusters decompose by loss of one halide ligand and binding of O2 (Scheme 4). The abundance of these species is quite similar to the distribution of their parent ions [Fe4S4X4−nYn]2−. Product ions of oxidative cluster splitting like [Fe2S2X4−nYn]2− and [Fe2S2X3−nYn]− were not detected (see ESI†).9,10,25
The presence of all five possible clusters [Fe4S4X4−nYn]2− is due to rapid scrambling of X and Y ligands and has been observed for other [Fe4S4] clusters in solution.10,51 This scrambling is usually very fast and is only suppressed if the cluster's total charge is zero (vide supra).9,10,34,36–38,52 When the stock solutions for MS were allowed to stand under inert atmosphere for a day no significant changes to the distributions of [Fe4S4X4−nYn]2− ions were observed (see ESI†) which means that immediately after dissolving the materials equilibria between the different species is obtained.
The ESI MS obtained from THF solutions of (BTMA)2[Fe4S4Cl2I2], (BTMA)2[Fe4S4ClI3] and (BTMA)2[Fe4S4BrI3] prepared from [Fe(CO)5] and ICl, IBr or I2 respectively (eqn (5)) also show distribution of all possible clusters. In contrast to the (BTMA)2[Fe4S4X2Y2] materials obtained by the iron(II) halide method (eqn (4)), their spectra exhibit much more different molecular ions containing iron, sulphur and halide. Although we cannot exclude that this difference is due to the solvent (THF vs. acetone), we assume that the material already contains cluster mixtures and by-products like prismane clusters which can be identified by their fragments [Fe6S6Cl2I3]2− and [Fe6S6I5]2− (see ESI†). The rather amorphous character of these materials compared to the crystalline (BTMA)2[Fe4S4X2Y2] materials supports this idea.
UV-vis-NIR absorption spectroscopy
UV-vis-NIR absorption spectra have been frequently used to identify [Fe4S4I4]2−, [Fe4S4Br4]2−, [Fe4S4Cl4]2− and other cluster ions in solution.22,25,53 Especially, the absorption bands in the visible and near infrared region are good tracers to identify [Fe4S4X4]2− clusters and were utilised by us for reaction control. Samples of [Fe4S4X4]2− and [Fe4S4X2Y2]2− from preparations with FeX2 and halide in MeCN show that halide [Fe4S4] clusters are present in all reaction mixtures. This is important since from solutions containing [Fe4S4Cl4]2− a solid cubane cluster could not be isolated. Instead, (BTMA)3[Fe6S6Cl6] was obtained. However, the recorded absorption bands in this case fit well to data reported by Holm and co-workers for [Fe4S4Cl4]2− (Table 3)25 showing that [Fe4S4Cl4]2− is dominant in the reaction solution. When dissolving the materials (BTMA)2[Fe4S4Br4] and (BTMA)2[Fe4S4I4] corresponding spectra were obtained (Fig. 2, Table 3). | ||
| Fig. 2 Long-wavelength part of the UV-vis-NIR absorption spectra of halide cubane clusters in MeCN. The spectra of the [Fe4S4X2Y2]2− represent rather the mixture of all possible [Fe4S4X4−nYn]2− species. | ||
| λ max [nm] (ελ [cm−1 M−1]) | ||||||
|---|---|---|---|---|---|---|
| All spectra recorded in MeCN solution.a From the reaction mixture.b In DMF from ref. 25.c The spectra of the compounds containing [Fe4S4X2Y2]2− represent rather the mixtures of all possible [Fe4S4X4−nYn]2− species. | ||||||
| (BTMA)2[Fe4S4Cl4]a | 260 | — | 507 | 690 | 1078 | 1202 |
| (Et4N)2[Fe4S4Cl4]b | 262 | 275 sh | 505 sh | 690 | 1080 | 1200 |
| (BTMA)2[Fe4S4Br4] | 289 (18528) |
303 sh (17766) |
471 sh (2588) | 698 (1900) | 1106 (624) | 1190 sh (566) |
| (BTMA)2[Fe4S4I4] | 360 | 390 sh | 490 sh | 712 sh | 1141 | 1230 sh |
| (BTMA)2[Fe4S4Br2Cl2]c | 278 sh (13330) |
301 sh (11151) |
504 sh (1990) | 693 (1591) | 1088 (521) | 1192 sh (461) |
| (BTMA)2[Fe4S4Cl2I2]c | 315 sh (6376) | 353 sh (5711) | 492 sh (2259) | 698 (1512) | 1110 (500) | 1174 sh (468) |
| (BTMA)2[Fe4S4Br2I2]c | 276 sh (21311) |
304 sh (19436) |
482 sh (2791) | 705 (1700) | 1128 (551) | 1205 sh (512) |
The UV-vis-NIR absorption spectra of the heteroleptic clusters [Fe4S4Br2Cl2]2−, [Fe4S4Cl2I2]2− and [Fe4S4Br2I2]2− in solution (reaction mixtures and dissolved isolated material) (Fig. 2, Table 3) exhibit very similar absorption bands as the homoleptic clusters [Fe4S4Br4]2− and [Fe4S4I4]2−.25 However, from the MS data we assume that these spectra represent all five possible species from ligand X scrambling (see Table 2). The spectra in the vis-NIR range were dominated by broad absorption bands with maxima around 1100 nm and intense bands around 700 nm (Fig. 2). Further strong absorptions for all compounds were found around 500 nm and the UV range is dominated by very strong absorptions probably due to the BTMA+ cations (Table 3, further spectra in the ESI†).25
The absorptions in the visible range have been frequently used to establish the formation or transformation of [FexSy]n− clusters (x = 2, 3, 4; y = 2 or 4)54–58 and have been assigned to ligand(X)-to-metal(Fe) charge transfer (LMCT) (at 700 nm), or μ-S/RS-to-metal(Fe) charge transfer (LMCT) (at 500 nm) transitions, respectively.25,49,54,59,60 In contrast to this, the broad near infrared absorptions with maxima ranging from 1080–1230 nm have been widely ignored. They are present in all [Fe4S4X4]2− clusters (X = halides or thiolates) but their origin remains to be elucidated.22,25,61 It has been proposed61 that they are the result of the mixed-valent oxidation state of the [Fe4S4]2+ core with two ferrous and two ferric centres (Scheme 5).
 | ||
| Scheme 5 Electron exchange between ferric and ferrous iron mediated by bridging sulfide ligands (left); magnetic structure of the [Fe4S4]2+ core (right).2,7 | ||
Multimetallic complexes of osmium,62 ruthenium63 and iron64,65 in mixed-valent oxidation states typically exhibit broad absorption bands in near infrared which were ascribed to so-called intervalence charge transfer (IVCT) or metal-to-metal charge transfer (MMCT).65–70 These transitions are established for mixed valent compounds as a good measure of the electron delocalisation.63,71–73 While for [Fe2S2] clusters74,75 and related materials,76–80 the IVCT character of such low energy absorptions in the NIR range was established by spectroscopy and calculations, for the [Fe4S4] clusters this remains to be done, although some mixed valent oligoiron compounds have been studied in this respect.54,81–83 This goes along with quantum chemical calculations84,85 finding the binuclear [Fe2S2] cluster easier to treat, due to their rather localised character. In contrast to this, higher nuclearity clusters are considered to be partially delocalised, lying between class II and class III according to the Robin and Day classification.63,71–73,84 Based on this we tentatively assign the near infrared absorption of the [Fe4S4X4]2− and [Fe4S4X2Y2]2− clusters to IVCT transitions. The electron exchange is mediated by the bridging sulphur ligands in the [Fe4S4] core, presumably through the so-called hole transfer mechanism.62,71–73 Due to the fast electron (or hole) transfer the oxidation state of the iron atoms is averaged to 2.5 (Scheme 5), in line with EPR and Mössbauer experiments on such clusters.2,7,29,56,61,86 Both the IVCT character of these long-wavelength absorptions and the hole transport mechanism needs to be proven by a quantum mechanical model for the [Fe4S4X4]2− clusters, which is not yet available.
For the X-to-Fe LMCT at around 700 nm a slight red-shift with decreasing electronegativity of X (Fig. 2, Table 3) is observed for the homoleptic clusters.25 For the [Fe4S4X2Y2]2− clusters this shift is observed as well in a [Fe4S4Br2Cl2]2− > [Fe4S4Cl2I2]2− > [Fe4S4Br2I2]2− sequence, confirming the assignment. From MS experiments we assume that these spectra represent very probably all possible species [Fe4S4X4−nYn]2− from ligand scrambling, however MS also showed that the species [Fe4S4X2Y2]2− are dominant in solution. Comparable studies on [Fe2S2X4−nYn]2− show a similar trend for LMCT absorption bands upon gradual replacement of chloride by bromide.87
Also the near infrared IVCT bands show red-shifts with increasing ligand electronegativity. This strongly suggests an influence of the halide ligands to the accepting and donating Fe centred orbitals in these transitions. Like the X-to-Fe LMCT bands the IVCT energies lie also between the associated homoleptic clusters. Unfortunately, the S-to-Fe LMCT absorptions around 500 nm are not well resolved and no correlation of absorption energies and X electronegativity can be made.
Synchrotron X-ray powder diffraction
The structure of (BTMA)2[Fe4S4I4] (monoclinic, space group Cc, Z = 4) has been reported by Pohl and co-workers.31 X-ray powder diffraction (XRPD) should allow to detect whether the new materials were isostructural to this. Phase analysis using synchrotron radiation is preferred over laboratory sources as narrow half-widths and consequent reduction in overlap can be obtained, greatly simplifying the identification of individual reflections.The XRPD of (BTMA)2[Fe4S4I4] is in line with simulated data from the published crystal structure (see Fig. S24, ESI†).31 Closer inspection reveals some additional reflections due to a minor amount of a microcrystalline, so far unknown, by-product. The XRPD data of (BTMA)2[Fe4S4Br4] shows that this material is indeed isostructural to (BTMA)2[Fe4S4I4] (Fig. S25, ESI†) and here, no additional reflections of a second phase or impurities are observed.
The XRPDs recorded from (BTMA)2[Fe4S4Br2Cl2], (BTMA)2[Fe4S4Cl2I2] and (BTMA)2[Fe4S4Br2I2] samples are in good agreement with the (BTMA)2[Fe4S4Br4] pattern (Fig. 3). Slight variations in reflection positions and intensities are due to different lattice parameters and atom types. The reflections do not exhibit a significant broadening (no increased full width at half maximum). It is apparent from this that the sample is homogenous without any phase width. The halide ratio X:Y of 2:2 found for these materials by elemental analysis could also represent mixtures of clusters with an average ratio X:Y of 2:2. However, XRPD clearly shows that only the defined [Fe4S4X2Y2]2− clusters are present in the bulk crystalline material.
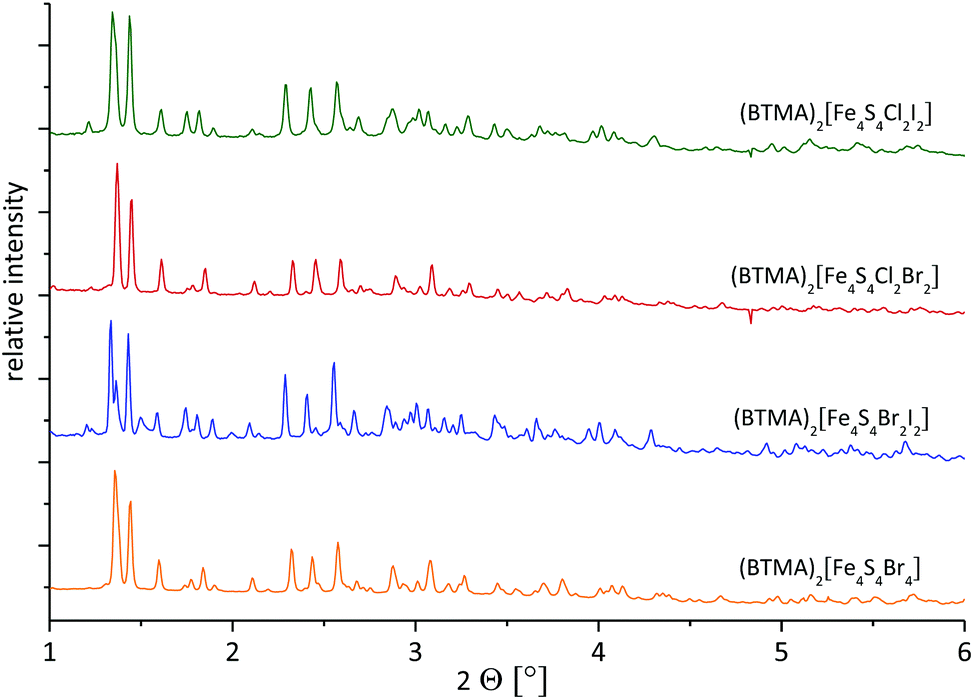 | ||
| Fig. 3 Synchrotron XRPD of homoleptic (BTMA)2[Fe4S4Br4] and heteroleptic (BTMA)2[Fe4S4Br2Cl2], (BTMA)2[Fe4S4Cl2I2] and (BTMA)2[Fe4S4Br2I2] clusters (experimental data with λ = 0.207203 Å; recorded at approx. 293 K). | ||
As can be seen from MS and also UV-vis-NIR spectroscopy, in solutions of (BTMA)2[Fe4S4X2Y2] all five possible species [Fe4S4X4−nYn]2− are present in equilibria with the [Fe4S4X2Y2]2− clusters being the most abundant. We assume, that upon adding diethyl ether to the reaction mixture (to solutions in THF or MeCN) the most abundant clusters [Fe4S4X2Y2]2− start to crystallise and the other species [Fe4S4X4−nYn]2− are consumed through fast exchange equilibria.
Also for (BTMA)2[Fe4S4Br2Cl2], (BTMA)2[Fe4S4Cl2I2] and (BTMA)2[Fe4S4Br2I2], the monoclinic space group Cc can be assumed. As reported for (BTMA)[Fe4S4I4]2−,31 a significant distortion of the [Fe4S4] core due to intermolecular halide–halide interactions can be expected in all our clusters with the BTMA cation. This distortion should be stronger in comparison to [Fe4S4] clusters in (Et4N)6[(Fe4S4I4)2(Fe2S2I4)] (tetragonal, P42bc),30 (Ph4P)2[Fe4S4I4] (tetragonal, I41/a)31 or in the Ph4P+ salts of [Fe4S4Cl4]2−, [Fe4S4Br2Cl2]2− and [Fe4S4Br4]2− (monoclinic, C2/c).29
The XRPD of (Ph4P)2[Fe4S4Br4] (eqn (5)) collected at λ = 0.551155 Å with synchrotron radiation fits well to theoretical data calculated from the published structure with respect to reflection positions (Fig. S26, ESI†).29 However, the measured intensities show significant deviations from the calculated values, which might be attributed to an inhomogeneous particle shape (preferred orientation). Furthermore, the experimental data exhibit a high background, which might be due to amorphous phases. This is in line with the generally more pronounced amorphous character of the materials produced by this method (eqn (5)) (see MS section).
Vibrational spectroscopy
Solid samples (as polyethylene pellets) of (BTMA)2[Fe4S4Br4] exhibit four dominant vibrations bands in far IR. From comparison with [Fe4S4X4]2− salts (X = Cl, Br, I) of Ph4P+ and Bu4N+ the 377 cm−1 frequency can be assigned to stretching νas(Fe–S) modes of the [Fe4S4] core and is invariant for all examined clusters in the form of BTMA+ salts (Fig. 4 and Table 4).29,88–91 A significant blue-shift to higher energies is observed for the corresponding Ph4P+ and Bu4N+ salts. This can be ascribed to specific interactions in the crystal structures of (cation)2[Fe4S4X4] compounds. | ||
| Fig. 4 FIR spectra of BTMA halide [Fe4S4] cluster salts. | ||
a
| ν as (Fe–S) core | ν as (Fe–X) terminal | ν(Fe–S) core (I) | ν(Fe–S) core (II) | |
|---|---|---|---|---|
| a From measurement of microcrystalline samples as polyethylene pellets, in [cm−1]. b From ref. 29. c From ref. 88. | ||||
| (Ph4P)2[Fe4S4Cl4]b | 382 (w) | 353 (s) | — | — |
| (Bu4N)2[Fe4S4Cl4]c | 389 | 360 | 278 | 253 |
| (BTMA)2[Fe4S4Br4] | 377 (m) | 307 (s) | 269 (w) 260 (sh) | 227 (w) |
| (Ph4P)2[Fe4S4Br4]b | 382 (w) | 305 (s) | — | — |
| (Bu4N)2[Fe4S4Br4]c | 384 | 310 | 279 | 230/242 |
| (BTMA)2[Fe4S4I4] | 377 (s) | 289 (s) | 273 (s) 258 (m) | 229 (m) |
| (Bu4N)2[Fe4S4I4]c | 381 | 296 | 264 | 232 |
| (Ph4P)2[Fe4S4Br2Cl2]b | 380 (m) | 354 (s) 305 (s) | — | — |
| (BTMA)2[Fe4S4Cl2I2] | 378 (m) | 356 (s) 290 (s) | 272 (m) 260 (m) | 229 (w) |
| (BTMA)2[Fe4S4Br2Cl2] | 378 (m) | 356 (s) 306 (s) | 270 (m) 260 (sh) | 232 (w) |
| (BTMA)2[Fe4S4Br2I2] | 376 (m) | 306 (s) 291 (s) | 272 (sh) 260 (m) | 231 (w) |
In the structure of (BTMA)2[Fe4S4I4] (monoclinic, Cc) an attractive intermolecular interaction to two individual iodide ligands of each cluster lead to a one-dimensional network of [Fe4S4X4]2− cluster across the structure.31 Thus, the symmetry of the [Fe4S4] core is significantly reduced in the BTMA+ salts compared to the corresponding structures of Ph4P+ and Bu4N+ salts.31 It is reasonable to assume that lower cluster symmetry goes along with lower vibration energies of typical cluster core vibrations. This is roughly the case when comparing typical vibrations in Table 4.
The stretching ν(Fe–Br) modes of the terminal bromido ligands were recorded for (BTMA)2[Fe4S4Br4] at 307 cm−1 following the trend of published data.29,88 In addition, bands at 269, 260 and 227 cm−1 can be assigned to vibrational modes of bridging Fe–S bonds similar to those reported for the Bu4N+ salt.89–91 Like the νas(Fe–S) vibrations they are blue-shifted but also split due to symmetry reduction in the BTMA+ structure.
The frequencies found for the ν(Fe–S) stretching modes in heteroleptic clusters [Fe4S4Br2Cl2]2−, [Fe4S4Cl2I2]2− and [Fe4S4Br2I2]2− are consistent with those of the homoleptic bromide and iodine clusters. For the ν(Fe–X) modes it seems that in each case two individual bands were observed for the heteroleptic clusters with about 355 cm−1 for Fe–Cl, 306 cm−1 for Fe–Br and 290 cm−1 for Fe–I groups. Those frequencies do not differ from those found in homoleptic clusters.
This is remarkable since the idealised symmetry of the 2:2 coordinated clusters is already reduced from Td to C2v (Scheme 6) which should have an effect on the vibrational modes. The shifts and splitting of the bands that might be expected upon symmetry reduction seem to be merged within the broad bands. Thus only the broadening of the bands indicates the loss of symmetry in the structures. On the other hand, orientation effects in the crystal structure probably overrule the molecular symmetry of the clusters.
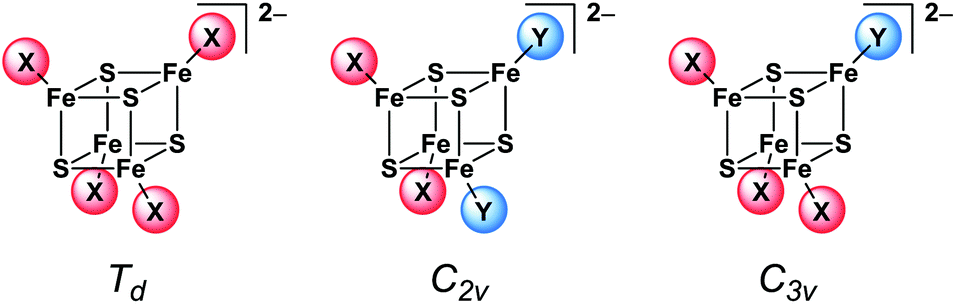 | ||
| Scheme 6 Idealised cluster symmetry. | ||
Conclusions
By evaluating the scope of a previously reported method to prepare [Fe4S4I4]2− from [Fe(CO)5], sulphur and iodide sources a couple of new improved preparations for [Fe4S4X4]2− clusters with various combinations of chloride, bromide and iodide were developed. The utilisation of simple iron(II) halide precursors like iron(II) halide and tetrahalidoferrates(II) has turned out to be the most efficient way to produce these clusters in high yields and purities (see Table 1). But even more valuable is its convenience as an one-pot, nearly atom-economic procedure which offers broad applicability of these clusters towards utilisation as precursors for the preparation of functionalised [Fe4S4] containing materials. Further variations of the reaction conditions and efforts to extend this method also to the synthesis of heterobimetallic clusters [MFe3S4X4]2− (M = Mn, Co; X = Cl, Br, I) have been less successful but have provided some insight into the mechanisms of formation of such clusters.Besides the reproduction of the established homoleptic [Fe4S4Cl4]2−, [Fe4S4Br4]2− and heteroleptic [Fe4S4Br2Cl2]2− clusters, the new clusters [Fe4S4Cl2I2]2− and [Fe4S4Br2I2]2− were isolated and characterised for the first time. The application of synchrotron XRPD revealed the isostructural analogy between the clusters in solid state as salts of BTMA+ (= benzyltrimethylammonium). Thus, a similar high distortion of the cluster ions in these structures like in the previously reported (BTMA)2[Fe4S4I4] (monoclinic, space group Cc) can be assumed. This is confirmed by NIR vibrational spectroscopy through significant differences of typical [Fe4S4] core vibrations when comparing the BTMA+ salts with Ph4P+ or Bu4N+ derivatives.
In the solid the (BTMA)2[Fe4S4X2Y2] materials are homogenous and consist only of [Fe4S4X2Y2]2− clusters, which is evident from XRPD. When dissolved a rapid halide ligand exchange produces mixtures of [Fe4S4X4−nYn]2− clusters as revealed by negative UHR-ESI MS. All these species are also observed in the reaction mixtures. However, upon crystallisation, the main component of such a mixture, the desired [Fe4S4X2Y2]2− cluster, seems to crystallise selectively giving the pure materials (BTMA)2[Fe4S4X2Y2]. In contrast to this, during the crystallisation of solutions containing mainly [Fe4S4Cl4]2− (MS and UV-vis-NIR) (BTMA)3[Fe6S6Cl6] is obtained, containing the hexanuclear prismane-shaped cluster [Fe6S6Cl6]3−. We assume that although the tetranuclear species is dominating the reaction mixture, further cluster species (hexanuclear, binuclear, …) are present, interconnected through rearrangement reactions. Proper choice of the cations is in both cases a good option to obtain pure compounds out of the mixtures of cluster species we observed in all of the investigated preparation methods. Another is surely the use of polydentate ligands.
UV-vis-NIR spectroscopy reveals characteristic near infrared absorptions for homoleptic [Fe4S4X4]2− and heteroleptic clusters [Fe4S4X2Y2]2− and for the latter very probably a mixture of [Fe4S4X4−nYn]2− species is observed. Due to the variation of X and Y the bands around 700 nm can clearly be assigned to X-to-Fe LMCT transitions The long-wavelength absorptions around 1100 nm are also slightly influenced by the variation of X and Y and were assigned to IVCT transitions within the mixed valent [Fe4S4]2+ core. The details of the electronic structure of the heteroleptic clusters are of certain interest and will be focus of our future efforts.
Experimental section
Materials and procedures
All manipulations and sample preparations were performed under anaerobic and dry argon atmosphere (>99996%, Airproducts) using standard Schlenk and high-vacuum techniques or a MBraun labmaster 130 dry box. Glassware was flame-dried immediately before use. THF was dried using a MBRAUN MB SPS-800 solvent purification system and degassed by pump-freeze–thaw techniques. Diethyl ether was distilled under argon from sodium/benzophenone and MeCN from calcium hydride prior to use. Commercially available [Fe(CO)5] was filtered through a pad of sea sand and silica and stored under argon atmosphere. Further chemicals were used as purchased. (Et4N)2[FeCl4] and (Ph4P)2[FeBr4] have been prepared similar to published procedures.50
Instrumentation
Elemental analysis was obtained using a Hekatech CHNS EuroEA 3000 analyzer. To complete combustion samples have been analysed with additional V2O5 in an atmosphere with 20 ml of oxygen. UV-vis-NIR absorption spectra were recorded in solution on a PerkinElmer Lambda 950 UV/Vis Scan spectrophotometer. IR spectra were collected with a Bruker IFS66νS spectrometer. Samples have been prepared as polyethylene pellet (abbreviations s = strong, m = medium, w = weak, sh = shoulder). ESI mass spectra with THF and MeCN solutions have been recorded with an APPLIED BIOSYSTEMS LC/MS system API 5000 at a collision energy of 10 eV and a potential of 5000 V. Sample solutions in THF were diluted to concentrations of 10−5 to 10−6 M and introduced to the machine with a 1–5 μL flow rate. Tubes and the machine have been purged with pure argon immediately before each use. For data analysis APPLIED BIOSYSTEMS software ANALYST 1.4.2 was utilised. UHR-ESI MS measurements of acetone solutions were performed on a UHR-TOF Bruker Daltonik (Bremen, Germany) maXis 5G, an ESI-TOF MS capable of resolution of at least 60000 FWHM. Detection was in negative-ion mode and the source voltage was 3.2 kV. The flow rates were 250 μL per hour. The machine was calibrated prior to every experiment via direct infusion of the Agilent ESI-TOF low concentration tuning mixture, which provided an m/z range of singly charged peaks up to 2700 Da in both ion modes. Preliminary X-ray powder diffraction data collection under laboratory conditions was carried out with a Huber G670 diffractometer using Guinier geometry and germanium-monochromatised Mo-K(α)1 radiation. For reflection detection image-plate techniques were used. Synchrotron data collection was performed at DESY (Hamburg, beamline P02.1, storage ring Petra III with λ = 0.207203 Å and a PerkinElmer XRD 1621 flat panel detector) or at DELTA (Dortmund, beamline 9 with λ = 0.551155 Å, silicon (311) double crystal monochromator and a MARRESEARCH MAR 345 flat panel detector). Samples have been measured in capillaries sealed in an argon atmosphere (0.5 mm diameter). The collected data was transformed with Fit2d92 and analysed with WinXPow.93
Cluster preparations with halogens and interhalogens
(BTMA) 2 [Fe 4 S 4 I 4 ] (Method A in THF, see Scheme 1) 1.27 g (5.0 mmol) iodine, 0.64 g (20.0 mmol) sulphur and 2.77 g (10.0 mmol) (BTMA)I were suspended in 100 ml THF. To the stirred slurry 2.70 ml (20.5 mmol) [Fe(CO)5] was added in small portions. Upon heating the mixture under reflux for 19 h its colour changed to black. After cooling the solvent was evaporated to dryness under reduced pressure and the remaining black microcrystalline solid was dried in vacuum at 50 °C for 20 min. Yield: 5.46 g (94%). (−)ESI-MS (THF): m/z (%) = 429.8 (95) [Fe4S4I4]2−, 556.6 (28) [Fe2S2I3]−. Elemental analysis for C20H32I4Fe4N2S4 (1159.74): calcd C 20.71, H 2.78, N 2.42, S 11.06; found C 20.65, H 2.77, N 2.25, S 11.22. FIR (PE pellet): νmax [cm−1]: 610 (w), 513 (w), 452 (m), 416 (w), 377 (s), 289 (s), 273 (sh), 258 (m), 227 (m), 198 (m), 183 (sh). UV-vis-NIR:λmax (MeCN) [nm]: 292, 319 (sh), 361, 392 (sh), 493 (sh), 708.(Method B in MeCN) 1.27 g (5.0 mmol) iodine, 0.64 g (20.0 mmol) sulphur and 2.77 g (10.0 mmol) (BTMA)I were suspended in 40 ml MeCN. To the stirred slurry 2.70 ml (20.5 mmol) [Fe(CO)5] was added. The mixture was heated under reflux for 16 h and a colour change to black could be observed. After cooling the reaction mixture was filtered through a frit. The filtrate was layered with 200 ml diethyl ether and stored for 3 days at −18 °C. Black needles precipitated from the solution were filtered off and dried in vacuum. Yield: 4.66 g (81%). Elemental analysis for C20H32I4Fe4N2S4 (1159.74): calcd C 20.71, H 2.78, N 2.42, S 11.06; found C 20.71, H 2.63, N 2.55, S 10.89. UV-vis-NIR:λmax (MeCN) [nm]: 247, 291, 320 (sh), 360, 390 (sh), 490 (sh), 709, 1141, 1230 (sh).
(Ph 4 P) 2 [Fe 4 S 4 Br 4 ] 4.19 g (10.0 mmol) (Ph4P)Br and 0.65 g (20.0 mmol) sulphur were suspended in 100 ml THF and cooled to 0 °C. After addition of 0.50 ml (9.8 mmol) bromine 2.70 ml (20.0 mmol) [Fe(CO)5] was added drop wise within 2 min. The reaction mixture was stirred for 5 min and then heated under reflux for 39 h. Upon heating a black solid precipitated from the dark red solution. The black microcrystalline solid was collected by filtration, washed with 3 × 10 ml THF and dried in vacuum for 10 min. By layering the combined filtrates with 260 ml diethyl ether a second crop of black microcrystals could be obtained. Yield over all fractions: 4.78 g (71%).
(BTMA) 2 [Fe 4 S 4 BrI 3 ] 2.77 g (10.0 mmol) (BTMA)I, 0.64 g (20.0 mmol) sulphur and 1.03 g (5.0 mmol) iodine monobromide were suspended in 100 ml THF and cooled to −30 °C. 2.7 ml (20.0 mmol) [Fe(CO)5] was added in portions of 0.5 ml within 2 min causing a strong evolution of gas. Gradually, the reaction mixture turned black. After removing the cooling bath the mixture was allowed to warm up and was finally heated under reflux for 21 h. The solvent was removed under reduced pressure at 40 °C. A sticky black mass was obtained by drying the remaining residue for 1 h in vacuum at 50 °C. Yield: 4.51 g (81%). (−)ESI-MS (THF): m/z (%) = 429.7 (95) [Fe4S4I4]2−, 405.8 (58) [Fe4S4BrI3]2−, 382.8 (23) [Fe4S4Br2I2]2−, 358.8 (8) [Fe4S4Br3I]2−, 335.8 (10) [Fe4S4Br4]2−.
(BTMA) 2 [Fe 4 S 4 ClI 3 ] A slurry of 2.77 g (10.0 mmol) (BTMA)I and 0.64 g (20.0 mmol) sulphur in 100 ml THF was cooled to −60 °C. After addition of 0.25 ml (5.0 mmol) iodine monochloride 2.7 ml (20.0 mmol) [Fe(CO)5] was added in small portions to the orange coloured suspension. While warming up to room temperature a strong evolution of gas could be observed from the mixture. Upon heating under reflux for 23 h the mixture's colour changed slowly to black. The solvent was removed under reduced pressure at 35 °C. A sticky black mass was obtained by drying the remaining residue for 2 h in vacuum at 50 °C. Yield: 5.00 g (94%). (−)ESI-MS (THF): m/z (%) = 580.4 (60) [Fe6S6I5]2−, 548.6 (64) [Fe6S4I5]2−, 429.7 (87) [Fe4S4I4]2−, 383.7 (35) [Fe4S4ClI3]2−, 337.8 (29) [Fe4S4Cl2I2]2−, 291.9 (5) [Fe4S4Cl3I]2−, 246.1 (18) [Fe4S4Cl4]2−.
(BTMA) 2 [Fe 4 S 4 Cl 2 I 2 ] To a slurry of 1.86 g (10.0 mmol) (BTMA)Cl, 0.64 g (10.0 mmol) sulphur and 1.27 g (5.0 mmol) iodine in 150 ml THF 2.7 ml (20.0 mmol) [Fe(CO)5] was added in small portions of 0.5 ml within 2 min. An intensive evolution of gas could be observed from the reddish-brown suspension turning slowly to black upon heating under reflux for 89 h. After cooling the solvent was evaporated to dryness under reduced pressure. The remaining residue was dried for 4 h at 70 °C in vacuum yielding a black sticky mass. Yield: 3.78 g (75%). (−)ESI-MS (THF): m/z (%) = 580.4 (94) [Fe6S6I5]2−, 548.6 (100) [Fe6S4I5]2−, 488.7 (40) [Fe6S6Cl2I3]2−, 456.5 (50) [Fe6S4Cl2I3]2−, 429.7 (30) [Fe4S4I4]2−, 383.7 (20) [Fe4S4ClI3]2−, 337.8 (18) [Fe4S4Cl2I2]2−, 291.9 (3) [Fe4S4Cl3I]2−, 246.1 (47) [Fe4S4Cl4]2−.
Cluster preparations with ferrous tetrahalidoferrates [FeX4]2−
(Et 4 N) 2 [Fe 4 S 4 Cl 4 ] To a slurry of 1.74 g (3.8 mmol) (Et4N)2[FeCl4] and 0.49 g (15.2 mmol) sulphur in 120 ml MeCN 1.50 ml (11.4 mmol) [Fe(CO)5] were added via syringe. The yellow reaction mixture was heated under reflux for 17 h. A colour change to black could be observed upon heating. The solvent was evaporated to dryness under reduced pressure. The remaining residue was dried in vacuum at 50 °C for 15 min yielding a black microcrystalline powder. Yield: 1.99 g (69%). (−)ESI-MS (MeCN): m/z (%) = 624.0 (26) [Et4N + Fe4S4Cl4]−, 492.9 (55) [Fe4S4Cl4]−, 365.5 (45) [Fe3S4Cl2]−, 282.9 (60) [Fe2S2Cl3]−, 245.6 (100) [Fe4S4Cl4]2−.(Ph 4 P) 2 [Fe 4 S 4 Br 4 ] To a slurry of 1.00 g (1.0 mmol) (Ph4P)2[FeBr4] and 0.12 g (3.8 mmol) sulphur in 30 ml MeCN 0.38 ml (2.9 mmol) [Fe(CO)5] were added. The reaction mixture was heated under reflux for 1 day. Upon heating the reaction mixture turned black quickly. The solvent was evaporated to dryness under reduced pressure. The remaining residue was dried in vacuum at 50 °C for 1 h yielding a black microcrystalline powder. Yield: 1.18 g (92%).
Cluster preparations with ferrous halides
(BTMA) 2 [Fe 4 S 4 Cl 4 ] respectively (BTMA) 3 [Fe 6 S 6 Cl 6 ] 1.28 g (10.1 mmol) FeCl2, 1.28 g (40.0 mmol) sulphur and 3.71 g (20.0 mmol) (BTMA)Cl were suspended in 40 ml MeCN. After addition of 3.94 ml (30.0 mmol) [Fe(CO)5] the resulting orange mixture was heated under reflux for 3 days. A colour change to black was observed immediately upon heating. The mixture was allowed to cool and filtered through a frit. UV-vis reaction control confirms [Fe4S4Cl4]2− in solution. UV-vis-NIR:λmax (MeCN) [nm]: 260, 507, 690, 1078, 1202 (sh).The black filtrate was layered with 160 ml of diethyl ether and stored for 3 weeks at −18 °C. Black column-shaped crystals were filtered off and dried under vacuum. Yield: 7.18 g (91%). Elemental analysis for C30H48Cl6Fe6N3S6 (1190.35): calcd C 30.26, H 4.06, N 3.53, S 16.16; found C 30.38, H 4.07, N 3.71, S 16.09. FIR (PE pellet): νmax [cm−1]: 612 (w), 513 (w), 454 (m), 419 (w), 381 (sh), 351 (s), 300 (m), 283 (sh), 250 (sh), 200 (w), 214 (w).
(BTMA) 2 [Fe 4 S 4 Br 4 ] 1.11 g (5.1 mmol) FeBr2, 0.64 g (20.0 mmol) sulphur and 2.30 g (10.0 mmol) (BTMA)Br were suspended in 40 ml MeCN. After addition of 1.97 ml (15.0 mmol) [Fe(CO)5] the resulting orange mixture was heated under reflux for 1 day. The suspension turned black quickly upon heating. The mixture was allowed to cool down and filtered through a frit. The black filtrate was layered with 180 ml of diethyl ether and stored for 3 weeks at −18 °C. Black needle-shaped crystals were filtered off and dried under vacuum. Yield: 4.20 g (87%). (−)ESI-MS (acetone): m/z (%) = 335.6479 (100) [Fe4S4Br4]2−, 821.4260 (60) [BTMA + Fe4S4Br4]−, 590.3813 (4) [Fe4S4Br3]−, 622.3710 (4) [Fe4S4Br3O2]−, 671.2970 (4) [Fe4S4Br4]−, 694.2866 (2) [Na + Fe4S4Br4]−, 678.3131 (7) [Li + Fe4S4Br4]−. Elemental analysis for C20H32Br4Fe4N2S4 (971.37): calcd C 24.72, H 3.32, N 2.88, S 13.20; found C 24.53, H 3.46, N 2.99, S 13.25. FIR (PE pellet): νmax [cm−1]: 610 (w), 515 (w), 453 (m), 418 (w), 377 (s), 307 (s), 269 (w), 260 (sh), 227 (m), 214 (w). UV-vis:λmax (MeCN) [nm]: 240, 261 (sh), 269, 289, 303 (sh), 469 (sh), 507 (sh), 697, 1106, 1190 (sh).
(BTMA) 2 [Fe 4 S 4 Cl 2 I 2 ] 1.27 g (10.0 mmol) FeCl2, 1.28 g (40.0 mmol) sulphur and 5.54 g (20.0 mmol) (BTMA)I were suspended in 40 ml MeCN. After addition of 4.00 ml (30.3 mmol) [Fe(CO)5] the resulting orange mixture was heated under reflux for 6 h and then stirred overnight. A colour change to black was observed immediately upon heating. The mixture was allowed to cool and filtered through a frit. The black filtrate was layered with 150 ml of diethyl ether and stored for 3 days at −18 °C. Black needle-shaped crystals were filtered off and dried under vacuum. A second crystalline crop was obtained from the filtrate upon storing it for additional 16 weeks at −18 °C. Yield over all fractions: 7.48 g (77%). (−)ESI-MS (acetone): m/z (%) = 429.6239 (42) [Fe4S4I4]2−, 383.6560 (100) [Fe4S4ClI3]2−, 337.6883 (46) [Fe4S4Cl2I2]2−, 292.7184 (3) [Fe4S4Cl3I]2−, 245.7523 (19) [Fe4S4Cl4]2−, 1009.3728 (2) [BTMA + Fe4S4I4]−, 917.4389 (9) [BTMA + Fe4S4ClI3]−, 825.5033 (15) [BTMA + Fe4S4Cl2I2]−, 733.5675 (9) [BTMA + Fe4S4Cl3I]−, 643.6284 (3) [BTMA + Fe4S4Cl4]−, 732.3404 (<1) [Fe4S4I3]−, 640.4057 (7) [Fe4S4ClI2]−, 548.4708 (21) [Fe4S4Cl2I]−, 456.5353 (16) [Fe4S4Cl3]−, 764.3302 (<1) [Fe4S4I3O2]−, 672.3954 (5) [Fe4S4ClI2O2]−, 580.4606 (17) [Fe4S4Cl2IO2]−, 488.5251 (12) [Fe4S4Cl3O2]−. Elemental analysis for C20H32Cl2I2Fe4N2S4 (976.47): calcd C 24.59, H 3.30, N 2.87, S 13.13; found C 24.45, H 3.25, N 2.77, S 12.95. FIR (PE pellet): νmax [cm−1]: 609 (w), 513 (w), 452 (m), 416 (w), 378 (s), 356 (s), 346 (sh), 290 (s), 272 (m), 260 (m), 228 (m), 212 (m). UV-vis-NIR:λmax (MeCN) [nm]: 315 (sh), 353 (sh), 492 (sh), 695, 1110, 1174 (sh).
(BTMA) 2 [Fe 4 S 4 Br 2 Cl 2 ] 1.27 g (10.0 mmol) FeCl2, 1.28 g (40.0 mmol) sulphur and 4.60 g (20.0 mmol) (BTMA)Br were suspended in 40 ml MeCN. After addition of 4.00 ml (30.3 mmol) iron [Fe(CO)5] the resulting orange mixture was heated under reflux for 17 h. A colour change to black was observed immediately upon heating. The mixture was left to stand for 3 days and was then filtered through a frit. The black filtrate was layered with 200 ml of diethyl ether and stored for 3 days at −18 °C. Black needle-shaped crystals were filtered off and dried under vacuum. Yield: 7.81 g (89%). (−)ESI-MS (acetone): m/z (%) = 335.6492 (44) [Fe4S4Br4]2−, 313.6743 (90) [Fe4S4Br3Cl]2−, 291.6992 (100) [Fe4S4Br2Cl2]2−, 268.7258 (19) [Fe4S4BrCl3]2−, 245.7521 (43) [Fe4S4Cl4]2−, 821.4248 (11) [BTMA + Fe4S4Br4]−, 777.4755 (36) [BTMA + Fe4S4Br3Cl]−, 731.5284 (47) [BTMA + Fe4S4Br2Cl2]−, 687.5786 (27) [BTMA + Fe4S4BrCl3]−, 643.6280 (6) [BTMA + Fe4S4Cl4]−, 590.3805 (2) [Fe4S4Br3]−, 546.4314 (13) [Fe4S4Br2Cl]−, 502.4818 (24) [Fe4S4BrCl2]−, 456.5348 (11) [Fe4S4Cl3]−, 622.3699 (1) [Fe4S4Br3O2]−, 578.4210 (10) [Fe4S4Br2ClO2]−, 534.4714 (17) [Fe4S4BrCl2O2]−, 488.5246 (8) [Fe4S4Cl3O2]−. Elemental analysis for C20H32Cl2Br2Fe4N2S4 (882.47): calcd C 27.21, H 3.17, N 3.65, S 14.53; found C 27.23, H 3.08, N 3.66, S 14.69. FIR (PE pellet): νmax [cm−1]: 609 (w), 515 (w), 453 (m), 419 (w), 378 (m), 356 (s), 347 (sh), 306 (s), 270 (m), 260 (sh), 233 (m). UV-vis-NIR:λmax (MeCN) [nm]: 261, 278 (sh), 301 (sh), 504 (sh), 689, 1088, 1192 (sh).
(BTMA) 2 [Fe 4 S 4 Br 2 I 2 ] 2.16 g (10.0 mmol) FeBr2, 1.28 g (40.0 mmol) sulphur and 5.54 g (20.0 mmol) (BTMA)I were suspended in 40 ml MeCN. After addition of 4.00 ml (30.3 mmol) [Fe(CO)5] the resulting orange mixture was heated to reflux for 16 h. The suspension turned black quickly upon heating. The mixture was allowed to cool and filtered through a frit. The black filtrate was layered with 195 ml of diethyl ether and stored for 3 days at −18 °C. Black needle-shaped crystals were filtered off and dried under vacuum. Yield: 9.03 g (85%). (−)ESI-MS (acetone): m/z (%) = 429.6232 (28) [Fe4S4I4]2−, 405.6303 (72) [Fe4S4BrI3]2−, 382.6360 (96) [Fe4S4Br2I2]2−, 359.6419 (42) [Fe4S4Br3I]2−, 335.6488 (14) [Fe4S4Br4]2−, 1009.3717 (2) [BTMA + Fe4S4I4]−, 961.3868 (7) [BTMA + Fe4S4BrI3]−, 915.3993 (14) [BTMA + Fe4S4Br2I2]−, 867.4132 (9) [BTMA + Fe4S4Br3I]−, 821.4241 (2) [BTMA + Fe4S4Br4]−, 732.3395 (<1) [Fe4S4I3]−, 684.3543 (2) [Fe4S4BrI2]−, 638.3662 (5) [Fe4S4Br2I]−, 590.3805 (6) [Fe4S4Br3]−, 764.3295 (<1) [Fe4S4I3O2]−, 716.3439 (1) [Fe4S4BrI2O2]−, 670.3558 (4) [Fe4S4Br2IO2]−, 622.3701 (4) [Fe4S4Br3O2]−. Elemental analysis for C20H32Br2I2Fe4N2S4 (1065.37): calcd C 22.54, H 3.03, N 2.63, S 12.03; found C 22.48, H 2.99, N 2.51, S 12.16. FIR (PE pellet): νmax [cm−1]: 609 (w), 514 (w), 452 (m), 416 (w), 376 (s), 306 (s), 291 (s), 272 (m), 260 (m), 232 (m), 210 (m). UV-vis-NIR:λmax (MeCN) [nm]: 276 (sh), 304 (sh), 482 (sh), 702, 1128, 1205 (sh).
Acknowledgements
The authors are grateful to Dr Stefan Liebig, Ireneus Grzesiak and Dr Stefanie Busch, Institute for Inorganic Chemistry, Cologne, Germany, for fruitful discussions and help on X-ray powder diffraction. We are indebted to HASYLAB at DESY, Hamburg, Germany and DELTA, Dortmund, Germany for the kind support of synchrotron radiation. Dr Dennis Rokitta, Hospital of the University of Cologne, Department of Pharmacology, Clinical Pharmacology Unit, is thanked for providing the opportunity for ESI mass spectrometry measurements. The work was financially supported for AOS by DAAD and CONICET; for AK by DFG KL1194/13-1. AF, AOS and FD are members of CONICET. I.I.-B. and M.D. gratefully acknowledge support through the “Solar Technologies Go Hybrid” initiative of the State of Bavaria.Notes and references
- M. C. Kennedy, T. A. Kent, M. Emptage, H. Merkle, H. Beinert and E. Münck, J. Biol. Chem., 1984, 259, 14463–14471 Search PubMed.
- H. Beinert, R. H. Holm and E. Münck, Science, 1997, 277, 653–659 CrossRef CAS PubMed.
- D. C. Johnson, D. R. Dean, A. D. Smith and M. K. Johnson, Annu. Rev. Biochem., 2005, 74, 247–281 CrossRef CAS PubMed.
- M. K. Johnson and A. D. Smith, Iron-Sulfur Proteins, in Encyclopedia of Inorganic and Bioinorganic Chemistry, John Wiley & Sons, Ltd, 2011, pp. 2589–2619 Search PubMed.
- C. Greco, M. Bruschi, P. Fantucci, U. Ryde and L. De Gioia, J. Am. Chem. Soc., 2011, 133, 18742–18749 CrossRef CAS PubMed.
- I. Span, K. Wang, W. Wang, J. Jauch, W. Eisenreich, A. Bacher, E. Oldfield and M. Groll, Angew. Chem., Int. Ed., 2013, 125, 2172–2175 CrossRef.
- H. Beinert, J. Biol. Inorg. Chem., 2000, 5, 2–15 CrossRef CAS PubMed.
- R. H. Holm, Acc. Chem. Res., 1977, 10, 427–434 CrossRef CAS.
- R. H. Holm, in Comprehensive Coordination Chemistry II, Pergamon, Oxford, 2003, ch. 8.3, pp. 61–90 Search PubMed.
- P. Venkateswara Rao and R. H. Holm, Chem. Rev., 2004, 104, 527–559 CrossRef CAS PubMed.
- S. C. Lee, W. Lo and R. H. Holm, Chem. Rev., 2014, 114, 3579–3600 CrossRef CAS PubMed.
- K. Tanifuji, N. Yamada, T. Tajima, T. Sasamori, N. Tokitoh, T. Matsuo, K. Tamao, Y. Ohki and K. Tatsumi, Inorg. Chem., 2014, 53, 4000–4009 CrossRef CAS PubMed.
- A. Alwaaly, I. Dance and R. A. Henderson, Chem. Commun., 2014, 50, 4799–4802 RSC.
- I. Dance and R. A. Henderson, Dalton Trans., 2014, 43, 16213–16226 RSC.
- I. Dance, Dalton Trans., 2015, 44, 4707–4717 RSC.
- C. T. Saouma, W. D. Morris, J. W. Darcy and J. M. Mayer, Chem. – Eur. J., 2015, 21, 9256–9260 CrossRef CAS PubMed.
- C. J. Pickett, S. K. Ibrahim and D. L. Hughes, Faraday Discuss., 2000, 116, 235–244 RSC.
- P. V. Vanitha and P. O'Brien, J. Am. Chem. Soc., 2008, 130, 17256–17257 CrossRef CAS PubMed.
- M. Akhtar, J. Akhter, M. A. Malik, P. O'Brien, F. Tuna, J. Raftery and M. Helliwell, J. Mater. Chem., 2011, 21, 9737 RSC.
- B. D. Yuhas, A. L. Smeigh, A. P. Samuel, Y. Shim, S. Bag, A. P. Douvalis, M. R. Wasielewski and M. G. Kanatzidis, J. Am. Chem. Soc., 2011, 133, 7252–7255 CrossRef CAS PubMed.
- V. C.-C. Wang, M. Can, E. Pierce, S. W. Ragsdale and F. A. Armstrong, J. Am. Chem. Soc., 2013, 135, 2198–2206 CrossRef CAS PubMed.
- Y. Shim, B. D. Yuhas, S. M. Dyar, A. L. Smeigh, A. P. Douvalis, M. R. Wasielewski and M. G. Kanatzidis, J. Am. Chem. Soc., 2013, 135, 2330–2337 CrossRef CAS PubMed.
- Y. Shim, R. M. Young, A. P. Douvalis, S. M. Dyar, B. D. Yuhas, T. Bakas, M. R. Wasielewski and M. G. Kanatzidis, J. Am. Chem. Soc., 2014, 136, 13371–13380 CrossRef CAS PubMed.
- M. Schwarz and C. Röhr, Inorg. Chem., 2015, 54, 1038–1048 CrossRef CAS PubMed.
- G. B. Wong, M. A. Bobrik and R. H. Holm, Inorg. Chem., 1978, 17, 578–584 CrossRef CAS.
- B. M. Segal, H. R. Hoveyda and R. H. Holm, Inorg. Chem., 1998, 37, 3440–3443 CrossRef CAS.
- M. A. Bobrik, K. O. Hodgson and R. H. Holm, Inorg. Chem., 1977, 16, 1851–1858 CrossRef CAS.
- K. Bates, M. Wouldhave and R. A. Henderson, Dalton Trans., 2008, 6527–6529 RSC.
- A. Müller, N. H. Schladerbeck, E. Krickemeyer, H. Bögge, K. Schmitz, E. Bill and A. X. Trautwein, Z. Anorg. Allg. Chem., 1989, 570, 7–36 CrossRef.
- W. Saak and S. Pohl, Z. Naturforsch., B: Anorg. Chem. Org. Chem., 1985, 40, 1105–1112 Search PubMed.
- S. Pohl and W. Saak, Z. Naturforsch., B: Chem. Sci., 1988, 43, 457–462 CAS.
- M. G. Kanatzidis, N. C. Baenziger, D. Coucouvanis, A. Simopoulos and A. Kostikas, J. Am. Chem. Soc., 1984, 106, 4500–4511 CrossRef CAS.
- D. Coucouvanis, M. G. Kanatzidis, E. Simhon and N. C. Baenziger, J. Am. Chem. Soc., 1982, 104, 1874–1882 CrossRef CAS.
- W. Saak and S. Pohl, Z. Naturforsch., B: Chem. Sci., 1988, 43, 813–817 CAS.
- M. A. Tyson, K. D. Demadis and D. Coucouvanis, Inorg. Chem., 1995, 34, 4519–4520 CrossRef CAS.
- U. Bierbach, W. Saak, D. Haase and S. Pohl, Z. Naturforsch., B: Chem. Sci., 1991, 46, 1629–1634 CrossRef CAS.
- S. Pohl and U. Bierbach, Z. Naturforsch., B: Chem. Sci., 1991, 46, 68–74 CrossRef CAS.
- F. Osterloh, W. Saak and S. Pohl, J. Am. Chem. Soc., 1997, 119, 5648–5656 CrossRef CAS.
- R. H. Holm, Pure Appl. Chem., 1998, 70, 931–938 CrossRef CAS.
- T. Terada, K. Hirabayashi, D. Liu, T. Nakamura, T. Wakimoto, T. Matsumoto and K. Tatsumi, Inorg. Chem., 2013, 52, 11997–12004 CrossRef CAS PubMed.
- D. L. Gerlach, D. Coucouvanis and N. Lehnert, Eur. J. Inorg. Chem., 2013, 3883–3890 CrossRef CAS.
- E. P. L. van der Geer, Q. Li, G. v. Koten, R. J. M. Klein Gebbink and B. Hessen, Inorg. Chim. Acta, 2008, 361, 1811–1818 CrossRef CAS.
- C. Walsdorff, W. Saak and S. Pohl, J. Chem. Soc., Dalton Trans., 1997, 1857–1861 RSC.
- C. Walsdorff, W. Saak, D. Haase and S. Pohl, Chem. Commun., 1997, 1931–1932 RSC.
- J. Zhou, Z. Hu, E. Münck and R. H. Holm, J. Am. Chem. Soc., 1996, 118, 1966–1980 CrossRef CAS.
- T. D. P. Stack, J. A. Weigel and R. H. Holm, Inorg. Chem., 1990, 29, 3745–3760 CrossRef CAS.
- T. D. P. Stack and R. H. Holm, J. Am. Chem. Soc., 1987, 109, 2546–2547 CrossRef CAS.
- E. Victor and S. J. Lippard, Inorg. Chem., 2014, 53, 5311–5320 CrossRef CAS PubMed.
- M. G. Kanatzidis, W. R. Hagen, W. R. Dunham, R. K. Lester and D. Coucouvanis, J. Am. Chem. Soc., 1985, 107, 953–961 CrossRef CAS.
- N. S. Gill, F. B. Taylor, W. E. Hatfield, W. E. Parker, C. S. Fountain and F. L. Bunger, Inorg. Synth., 2007, 9, 136–142 CrossRef.
- Y.-J. Fu, J. Laskin and L.-S. Wang, Int. J. Mass Spectrom., 2006, 255–256, 102–110 CrossRef CAS.
- M. Harmjanz, C. Junghans, U.-A. Optiz, B. Bahlmann and S. Pohl, Z. Naturforsch., B: Chem. Sci., 1996, 51, 1040–1048 CrossRef CAS.
- S. Rutchik, S. Kim and M. A. Walters, Inorg. Chem., 1988, 27, 1513–1515 CrossRef.
- J.-F. You, B. S. Snyder, G. C. Papaefthymiou and R. H. Holm, J. Am. Chem. Soc., 1990, 112, 1067–1076 CrossRef CAS.
- H. Gao, S. Subramanian, J. Couturier, S. G. Naik, S.-K. Kim, T. Leustek, D. B. Knaff, H.-C. Wu, F. Vignols, B. H. Huynh, N. Rouhier and M. K. Johnson, Biochemistry, 2013, 52, 6633–6645 CrossRef CAS PubMed.
- B. Zhang, S. Bandyopadhyay, P. Shakamuri, S. G. Naik, B. H. Huynh, J. Couturier, N. Rouhier and M. K. Johnson, J. Am. Chem. Soc., 2013, 135, 15153–15164 CrossRef CAS PubMed.
- M. M.-J. Couture, V. J. J. Martin, W. W. Mohn and L. D. Eltis, Biochim. Biophys. Acta, 2006, 1764, 1462–1469 CrossRef CAS PubMed.
- A. D. Smith, G. N. L. Jameson, P. C. Dos Santos, J. N. Agar, S. Naik, C. Krebs, J. Frazzon, D. R. Dean, B. H. Huynh and M. K. Johnson, Biochemistry, 2005, 44, 12955–12969 CrossRef CAS PubMed.
- Y.-J. Kim and J.-H. Han, Bull. Korean Chem. Soc., 2012, 33, 48–54 CrossRef CAS.
- B. V. DePamphilis, B. A. Averill, T. Herskovitz, L. Que Jr. and R. H. Holm, J. Am. Chem. Soc., 1974, 96, 4159–4167 CrossRef CAS PubMed.
- L. M. Lawson Daku, J. Pécaut, A. Lenormand-Foucaut, B. Vieux-Melchior, P. Iveson and J. Jordanov, Inorg. Chem., 2003, 42, 6824–6850 CrossRef PubMed.
- W. Kaim and G. K. Lahiri, Angew. Chem., Int. Ed., 2007, 46, 1778–1796 CrossRef CAS PubMed.
- W. Kaim and B. Sarkar, Coord. Chem. Rev., 2007, 251, 584–594 CrossRef CAS.
- M. I. Belinsky, J. Magn. Magn. Mater., 2007, 310, e486–e488 CrossRef CAS.
- M. I. Belinsky, Chem. Phys., 1999, 240, 303–311 CrossRef CAS.
- W. Kaim, Coord. Chem. Rev., 2011, 255, 2503–2513 CrossRef CAS.
- C. Díaz and A. Arancibia, Polyhedron, 2000, 19, 2679–2687 CrossRef.
- W.-Q. Chen, Y.-M. Chen, T. Lei, W. Liu and Y. Li, Inorg. Chem. Commun., 2012, 19, 4–9 CrossRef CAS.
- W. Kaim and B. Schwederski, Coord. Chem. Rev., 2010, 254, 1580–1588 CrossRef CAS.
- W. Kaim, Eur. J. Inorg. Chem., 2012, 343–348 CrossRef CAS.
- W. Kaim, A. Klein and M. Glöckle, Acc. Chem. Res., 2000, 33, 755–763 CrossRef CAS PubMed.
- D. M. D'Alessandro and F. R. Keene, Chem. Rev., 2006, 106, 2270–2298 CrossRef PubMed.
- D. M. D'Alessandro and F. R. Keene, Chem. Soc. Rev., 2006, 35, 424–440 Search PubMed.
- A. Albers, S. Demeshko, S. Dechert, E. Bill, E. Bothe and F. Meyer, Angew. Chem., Int. Ed., 2011, 50, 9191–9194 CrossRef CAS PubMed.
- J. M. McCormick and E. I. Solomon, J. Am. Chem. Soc., 1990, 112, 2005–2007 CrossRef CAS.
- F. Li, M. Chakrabarti, Y. Dong, K. Kauffmann, E. L. Bominaar, E. Münck and L. Que Jr., Inorg. Chem., 2012, 51, 2917–2929 CrossRef CAS PubMed.
- M. Glöckle, N. E. Katz, M. Ketterle and W. Kaim, Inorg. Chim. Acta, 2002, 336, 55–60 CrossRef.
- M. Glöckle, W. Kaim, A. Klein, E. Roduner, G. Hübner, S. Zalis, J. van Slageren, F. Renz and P. Gütlich, Inorg. Chem., 2001, 40, 2256–2262 CrossRef.
- U. Pfaff, A. Hildebrandt, D. Schaarschmidt, T. Hahn, S. Liebing, J. Kortus and H. Lang, Organometallics, 2012, 31, 6761–6771 CrossRef CAS.
- R. Warratz, H. Aboulfadl, T. Bally and F. Tuczek, Chem. – Eur. J., 2009, 15, 1604–1617 CrossRef CAS PubMed.
- K. L. Taft, G. C. Papaefthymiou and S. J. Lippard, Inorg. Chem., 1994, 33, 1510–1520 CrossRef CAS.
- K. L. Taft, A. Caneschi, L. E. Pence, C. D. Delfs, G. C. Papaefthymiou and S. J. Lippard, J. Am. Chem. Soc., 1993, 115, 11753–11766 CrossRef CAS.
- P. Albores and E. Rentschler, Inorg. Chem., 2010, 49, 8953–8961 CrossRef CAS PubMed.
- E. I. Solomon, X. Xie and A. Dey, Chem. Soc. Rev., 2008, 37, 623–638 RSC.
- M. Parthey and M. Kaupp, Chem. Soc. Rev., 2014, 43, 5067–5088 RSC.
- E. Bill, Hyperfine Interact., 2011, 205, 139–147 CrossRef.
- C. Tonon, H. Iernoa, J. Jordanov, J. Laugier and J.-M. Greneche, Chem. Ber., 1997, 130, 235–239 CrossRef CAS.
- A. Kern, C. Näther and F. Tuczek, Inorg. Chem., 2004, 43, 5011–5020 CrossRef CAS PubMed.
- K. Nakamoto, in Infrared and Raman Spectra of Inorganic and Coordination Compounds: Applications in Coordination, Organometallic, and Bioinorganic Chemistry, John Wiley & Sons, 5th edn, 1997, vol. B Search PubMed.
- Y. Xiao, M. Koutmos, D. A. Case, D. Coucouvanis, H. Wang and S. P. Cramer, J. Chem. Soc., Dalton Trans., 2006, 2192–2201 RSC.
- D. Mitra, S. J. George, Y. Guo, S. Kamali, S. Keable, J. W. Peters, V. Pelmenschikov, D. A. Case and S. P. Cramer, J. Am. Chem. Soc., 2013, 135, 2530–2543 CrossRef CAS PubMed.
- A. P. Hammersley, FIT2D, V10.3, ESRF, Grenoble, France, 1998 Search PubMed.
- STOE WinXPow, version 3.01.03, STOE and Cie GmbH, Darmstadt, Germany, 2010 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Negative mode ESI mass and UV-vis-NIR spectra as well as synchrotron XRPD as mentioned in the text. See DOI: 10.1039/c5dt02769a |
| This journal is © The Royal Society of Chemistry 2016 |