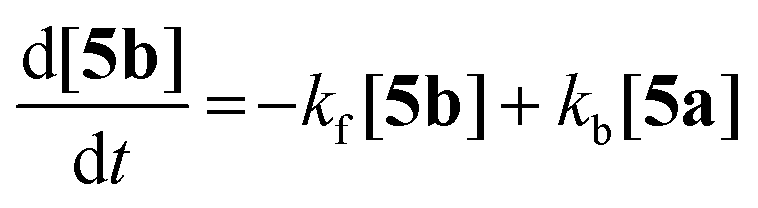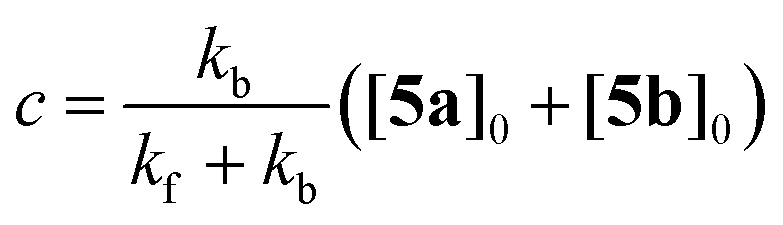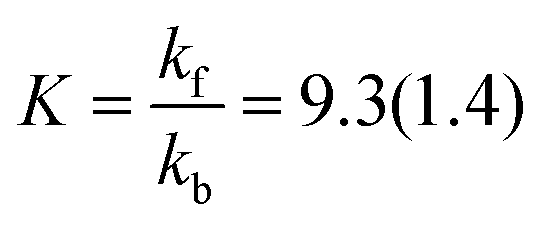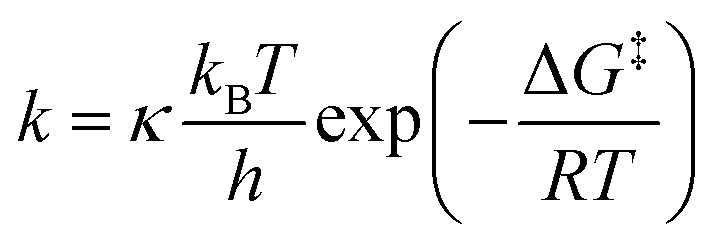DOI:
10.1039/C5DT02757H
(Paper)
Dalton Trans., 2016,
45, 1998-2007
Synthetic strategies to bicyclic tetraphosphanes using P1, P2 and P4 building blocks†
Received
20th July 2015
, Accepted 28th August 2015
First published on 28th August 2015
Abstract
Different reactions of Mes* substituted phosphanes (Mes* = 2,4,6-tri-tert-butylphenyl) led to the formation of the bicyclic tetraphosphane Mes*P4Mes* (5) and its unknown Lewis acid adduct 5·GaCl3. In this context, the endo–exo isomer of 5 was fully characterized for the first time. The synthesis was achieved by reactions involving “self-assembly” of the P4 scaffold from P1 building blocks (i.e. primary phosphanes) or by reactions starting from P2 or P4 scaffolds (i.e. a diphosphene or cyclic tetraphosphane). Furthermore, interconversion between the exo–exo and endo–exo isomer were studied by 31P NMR spectroscopy. All compounds were fully characterized by experimental as well as computational methods.
Introduction
Ring systems composed of group 15 elements (pnictogens, Pn) are an intriguing and widely investigated aspect of main group chemistry.1–3 Within this field of research, the chemistry of phosphorus based ring systems has become an important branch of inorganic chemistry,4,5 especially in view of the fact that various phosphorus ring systems can be obtained by direct activation of white phosphorus.6–8 A lot of work has been carried out to improve the selectivity of these activation processes, involving functionalization of P4 by Lewis acids and bases, (transition) metals, radicals, or singlet carbenes such as N-heterocyclic carbenes (NHCs) or cyclic alkylaminocarbenes (CAACs).9–12
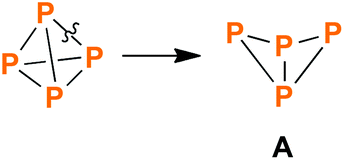
Among a plethora of structural motifs found in phosphorus ring systems, the simple tetraphosphabicyclo[1.1.0]butane scaffold (A, Scheme 1) is of special interest. Firstly, it can formally be derived from tetrahedral P4 by “simple” cleavage of one PP bond. Secondly, and more importantly, tetraphosphabicyclo[1.1.0]butanes were indeed obtained by P4 activation, thus representing worthwhile target molecules in phosphorus chemistry.
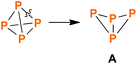 |
| | Scheme 1 Breaking one of the PP bonds in tetrahedral P4 formally yields the tetraphosphabicylco[1.1.0]butane scaffold (A). | |
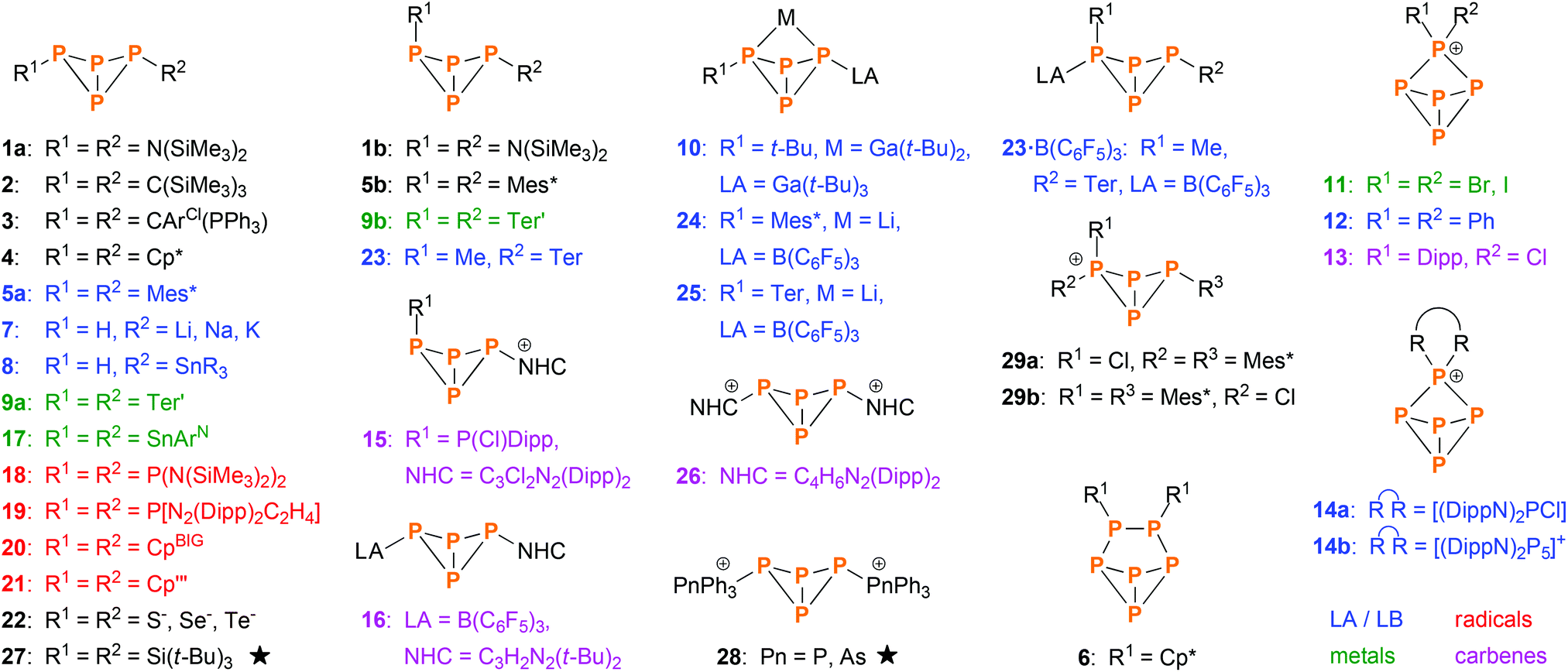
The first tetraphosphabicyclo[1.1.0]butane, (Me3Si)2N–P4–N(SiMe3)2 (1a, 1b, Scheme 2), was synthesized by the group of Niecke in 1982.13 It was derived from P2 building blocks utilizing PP coupling reactions. In the following years, some more examples emerged that were derived from P2 or P3 units, as reported by Cowley (2),14 Schmidpeter (3),15 Jutzi (4–6),16–19 Weber (5a),20 and Romanenko (5a).21 However, the majority of bicyclic tetraphosphanes was synthesized by P4 activation, the first example being exo–exo-Mes*P4Mes* (5a, Mes* = 2,4,6-tri-tert-butylphenyl), which was reported by Fluck and co-workers in 1985.22,23 Improvement of P4 activation methods led to various other bicyclic P4 species, which can be categorized by the aforementioned types of P4 activation (Scheme 2); important contributions were made by the groups of Baudler (7, 8),24,25 Power (9–10),26,27 Krossing (11),28 Weigand (12–15)29,30 Tamm (16),31 Roesky and Stalke (17),32 Lappert (18),33 Masuda (19),34 Scheer (20, 21),10 Karaghiosoff (22),35 Lammertsma (23–25),12 and Jones (26).36 Additionally, various examples of transition metal complexes that incorporate bicyclic P4 scaffolds were reported.11,37–42 In contrast, very few examples of self-assembly reactions exist (i.e. the bicyclic P4 scaffold is built from P1 building blocks in a single reaction); literature reports include Wiberg's (t-Bu)3Si–P4–Si(t-Bu)3 (27)43 and Weigand's [Ph3Pn–P4–PnPh3]2+ (28, Pn = P, As).44 Just recently, we reported on the cation [Mes*P4(Cl)Mes*]+ (29),45 which in itself was not derived by a self-assembly reaction, but its cyclic P4 precursor was.46
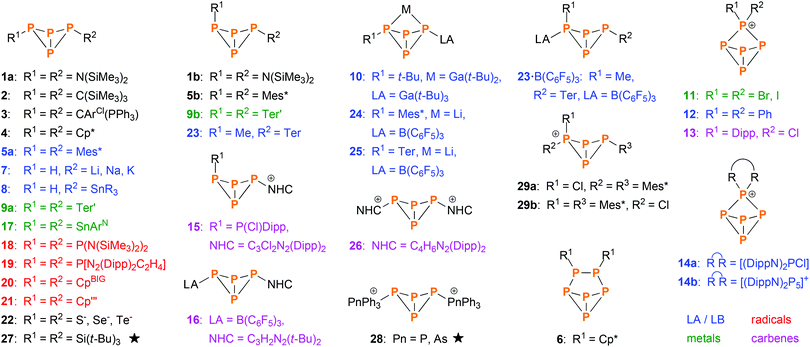 |
| | Scheme 2 Overview of known bicyclic tetraphosphane species (not including transition metal complexes). Most were synthesized by direct activation of P4 using Lewis acids and bases (blue), metals or metal fragments (green), radical species (red) or singlet carbenes (violet). The rest was derived from other precursors, mainly P2 and P4 building blocks. Species indicated by a star were derived directly from P1 building blocks in a self-assembly reaction. ArCl = 2,6-dichlorophenyl; ArN = C6H3-2,6-(C(NDipp)CH3)2; Cp = cyclopentadienyl; Cp* = pentamethyl-Cp; Cp′′′ = 2,3,5-tri-tert-butyl-Cp; CpBIG = pentakis(4-n-butylphenyl)-Cp; Dipp = 2,6-diisopropylphenyl; SnR3 = SnMe3, SnPh3, Sn(c-Hex)3 or Sn(o-Tol)3; Ter = 2,6-dimesitylphenyl; Ter′ = 2,6-bis(diisopropylphenyl)phenyl; LA = Lewis acid, LB = Lewis Base, NHC = N-heterocyclic carbene. | |
Interestingly, exo–exo-substituted tetraphosphabicyclo[1.1.0]butanes are considerably more common than their endo–exo-substituted counterparts, indicating that the latter are energetically less favoured. We therefore took interest in the synthesis and characterization of rare endo–exo-substituted derivatives, which could be synthesized from P1, P2 and P4 building blocks and were analysed by experimental and computational methods.
Results and discussion
Synthesis and characterization of endo–exo-Mes*P4Mes* (5b)
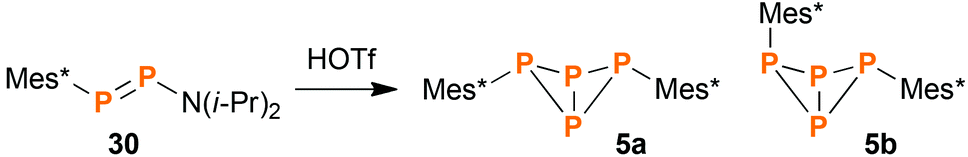
During our research in functionalized cyclo-tetraphosphanes,46 we came across a publication of Romanenko et al.,21 wherein the authors described the synthesis of exo–exo-Mes*P4Mes* (5a) and the asymmetrically substituted bicyclic system Mes*P4N(i-Pr)2 by reacting the diphosphene Mes*PPN(i-Pr)2 (30) with equimolar amounts of HOTf (Tf = SO2CF3). Intrigued by this curious method to synthesize bicyclic tetraphosphanes, we tried to reproduce the experiment described in the publication. However, fractional crystallization of the product mixture did not yield the reported compound Mes*P4N(i-Pr)2, but rather endo–exo-Mes*P4Mes* (5b, Scheme 3), alongside the known product 5a and minor amounts of the diphosphene Mes*PPMes* (31).47,48 Probably the available NMR and MS data were misinterpreted in the original publication. Compound 5b was now fully characterized for the first time, including NMR, Raman and IR spectroscopy, mass spectrometry, and single crystal X-ray diffraction.
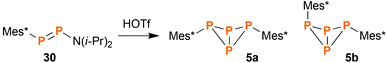 |
| | Scheme 3 Reaction of diphosphene 30 with HOTf, yielding bicyclic phosphanes 5a and 5b as main products. | |
Spectroscopic characterization
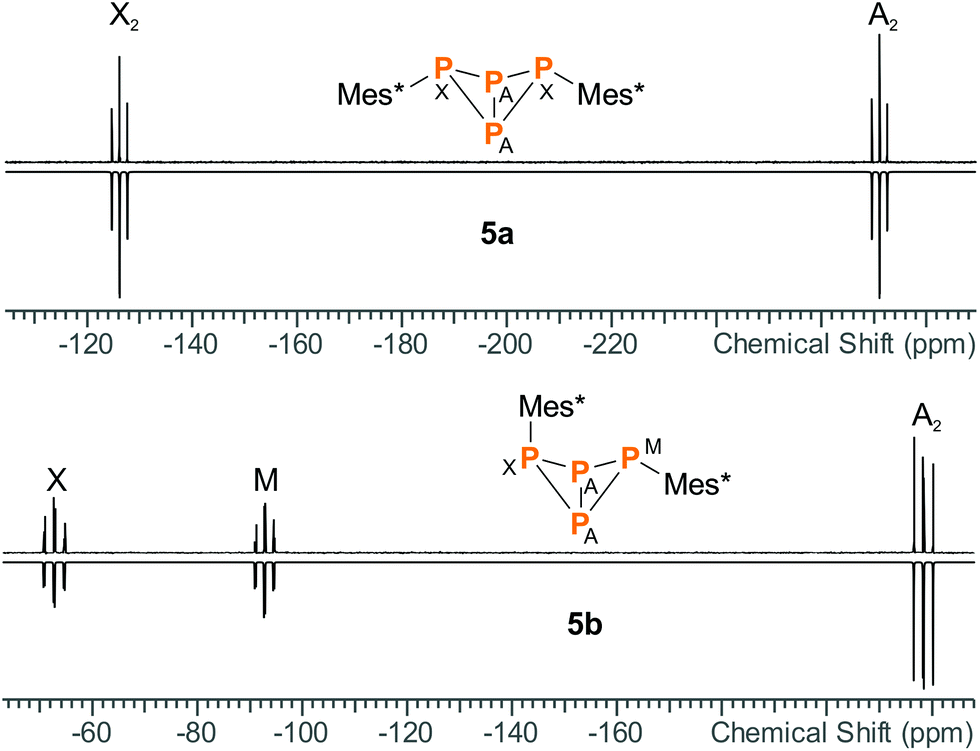
Apart from minor impurities, the 31P NMR spectrum of the reaction mixture showed an A2X2 (−273.2, −128.3 ppm) and an A2MX spin system (−220.4, −94.8, −54.7 ppm) in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4. The former set of signals was assigned to 5a (17% yield based on 31P NMR integrals), the latter to 5b (67%); hence, the endo–exo-isomer was actually formed in significant excess. The same NMR data were obtained for pure 5a and 5b (Fig. 1), which agree well with calculated NMR shifts and coupling constants (ESI†) as well as previously reported NMR data.17,22 Moreover, both isomers could be nicely distinguished in the Raman spectrum due to different excitation energies of the vibrational “breathing” mode of the P4 scaffold (Table 1). The corresponding Raman bands were easily identified as the most intense signals in both spectra (5a: 592 cm−1, 5b: 568 cm−1). The wavenumbers compare well to the symmetrical A1 mode of tetrahedral P4 in the gas phase (600 cm−1).49 In case of 5b, two distinct P–C valence modes were identified at 584 (exo) and 591 cm−1 (endo). In 5a, the single P–C valence band was superimposed by the “breathing” mode of the bicyclic scaffold and could not be discerned.
:4. The former set of signals was assigned to 5a (17% yield based on 31P NMR integrals), the latter to 5b (67%); hence, the endo–exo-isomer was actually formed in significant excess. The same NMR data were obtained for pure 5a and 5b (Fig. 1), which agree well with calculated NMR shifts and coupling constants (ESI†) as well as previously reported NMR data.17,22 Moreover, both isomers could be nicely distinguished in the Raman spectrum due to different excitation energies of the vibrational “breathing” mode of the P4 scaffold (Table 1). The corresponding Raman bands were easily identified as the most intense signals in both spectra (5a: 592 cm−1, 5b: 568 cm−1). The wavenumbers compare well to the symmetrical A1 mode of tetrahedral P4 in the gas phase (600 cm−1).49 In case of 5b, two distinct P–C valence modes were identified at 584 (exo) and 591 cm−1 (endo). In 5a, the single P–C valence band was superimposed by the “breathing” mode of the bicyclic scaffold and could not be discerned.
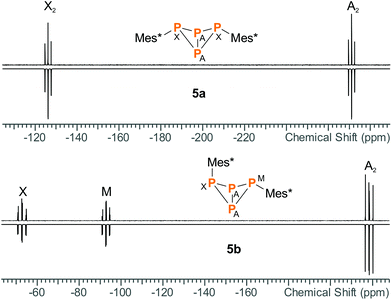 |
| | Fig. 1 Experimental (up) and simulated (down) 31P NMR spectra of 5a and 5b. | |
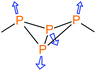
Table 1 Main vibrational modes of bicyclic tetraphosphanes in the Raman spectrum. Assignment of the symmetries based on approximate C2v symmetry of the P4 scaffold
| Vibration mode |
Description |
Frequency [cm–1] |
|
Symmetrical valence “breathing mode” (A1 mode, in phase) |
5a: 592 |
|
5b: 568 |
|
5a·GaCl3: 597 |
|
5b·GaCl3: 567 |
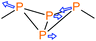
|
Peripheral bond stretch (B1 mode) |
5a: 447 |
|
5b: 500 |
|
5a·GaCl3: 497 |
|
5b·GaCl3: 518 |
|
Transannular bond stretch (A1 mode, out of phase) |
5a: 412 |
|
5b: — |
|
5a·GaCl3: 436 |
|
5b·GaCl3: — |
|
Peripheral bond stretch (B2 mode) |
5a: 412 |
|
5b: 412 |
|
5a·GaCl3: 451 |
|
5b·GaCl3: 438 |
Molecular structure
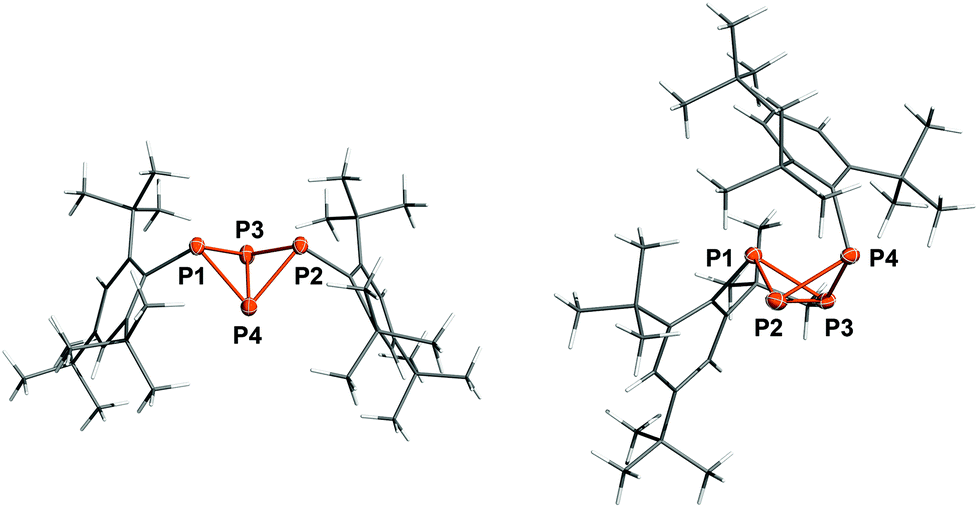
Crystallization from n-hexane yielded single crystals of 5b in the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) while crystallization from CH2Cl2 afforded crystals in the space group P21/n (Fig. 2, right). The P–P and P–C bond lengths are similar in both modifications of 5b: the P1–P2 and P1–P3 bonds (av. 2.227, 2.233 Å) are close to the sum of the covalent radii (∑rcov = 2.22 Å),50 whereas the P2–P4 and P3–P4 bonds (av. 2.213, 2.210 Å) as well as the transannular bond P2–P3 (av. 2.177 Å) are slightly shorter. This is in contrast to the structure of the previously reported exo–exo-isomer 5a (Fig. 2, left),22 where all four peripheral P–P bonds exhibit similar lengths (av. 2.225 Å); the transannular bond, however, is likewise only 2.1634(8) Å. The bond angles at the P atoms are all close to 60° and thus compare to tetrahedral P4. Interestingly, the Mes* substituent in 5b is bent backwards above the P4 scaffold, so the p-tert-butyl group rests on top of the exo-Mes* substituent, effectively shielding the top side of the bicyclic ring system. Hence, the fold angle of the butterfly-shaped P4 scaffold is quite different in all three cases; it varies from 95.66(3)° (5a) across 105.75(5)° (triclinic 5b) to 107.78(3)° (monoclinic 5b), which can be attributed to Pauli repulsion between the endo-substituent and the opposite bridging atom (P1) in case of 5b as well as packing effects to account for the difference between the two modifications. A similar effect was observed in case of Ter′P4Ter′ (9a, 9b, Ter′ = 2,6-bis(diisopropylphenyl)phenyl), where the difference between exo–exo (92.9°) and endo–exo-isomer (108.1°) is even larger.27
while crystallization from CH2Cl2 afforded crystals in the space group P21/n (Fig. 2, right). The P–P and P–C bond lengths are similar in both modifications of 5b: the P1–P2 and P1–P3 bonds (av. 2.227, 2.233 Å) are close to the sum of the covalent radii (∑rcov = 2.22 Å),50 whereas the P2–P4 and P3–P4 bonds (av. 2.213, 2.210 Å) as well as the transannular bond P2–P3 (av. 2.177 Å) are slightly shorter. This is in contrast to the structure of the previously reported exo–exo-isomer 5a (Fig. 2, left),22 where all four peripheral P–P bonds exhibit similar lengths (av. 2.225 Å); the transannular bond, however, is likewise only 2.1634(8) Å. The bond angles at the P atoms are all close to 60° and thus compare to tetrahedral P4. Interestingly, the Mes* substituent in 5b is bent backwards above the P4 scaffold, so the p-tert-butyl group rests on top of the exo-Mes* substituent, effectively shielding the top side of the bicyclic ring system. Hence, the fold angle of the butterfly-shaped P4 scaffold is quite different in all three cases; it varies from 95.66(3)° (5a) across 105.75(5)° (triclinic 5b) to 107.78(3)° (monoclinic 5b), which can be attributed to Pauli repulsion between the endo-substituent and the opposite bridging atom (P1) in case of 5b as well as packing effects to account for the difference between the two modifications. A similar effect was observed in case of Ter′P4Ter′ (9a, 9b, Ter′ = 2,6-bis(diisopropylphenyl)phenyl), where the difference between exo–exo (92.9°) and endo–exo-isomer (108.1°) is even larger.27
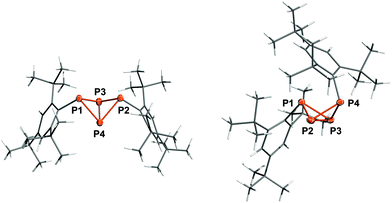 |
| | Fig. 2 Molecular structure of 5a (left) and 5b (monoclinic, right). Ellipsoids are set at 50% probability (173 K). Selected bond lengths [Å] and angles [°]: 5a P1–P3 2.2310(7), P1–P4 2.2171(7), P2–P3 2.2294(7), P2–P4 2.2236(7), P3–P4 2.1634(8); P1–P3–P4–P2 −95.66(3); 5b P1–P2 2.2244(7), P1–P3 2.2326(8), P2–P4 2.2131(8), P3–P4 2.2101(8), P2–P3 2.1773(8); P1–P2–P3–P4 107.78(3). | |
Computational study
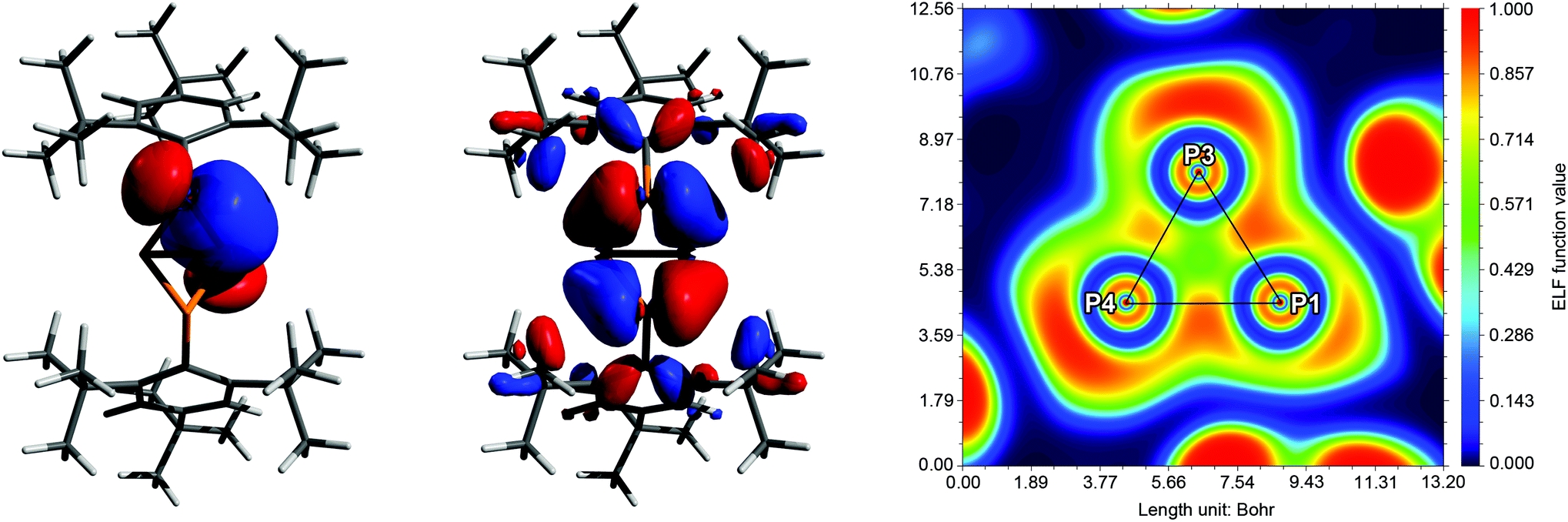
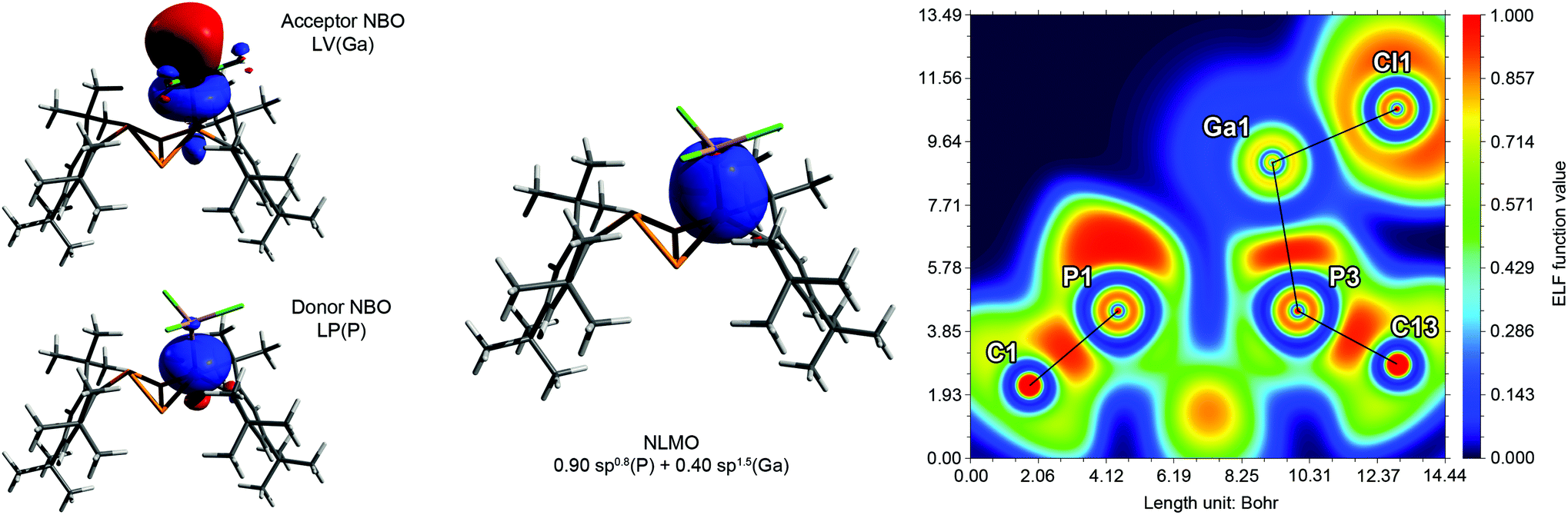
Comprehensive DFT calculations were carried out to compare the isomers 5a and 5b. According to NBO analysis,51 the bonding situation is similar in both cases. The Wiberg and NLMO bond indices are near unity for all P–P bonds. However, due to the small bond angles, the electron density is not distributed symmetrically around the P–P bond axes, but rather bent outwards, resulting in banana-shaped bonds. This is illustrated by the electron localization function (ELF, Fig. 3 right), where the local maxima between the P atoms are found outside the lines of nuclear centres. Additionally, the symmetry and shape of the natural bond orbitals (NBOs) and molecular orbitals (MOs) support this picture.
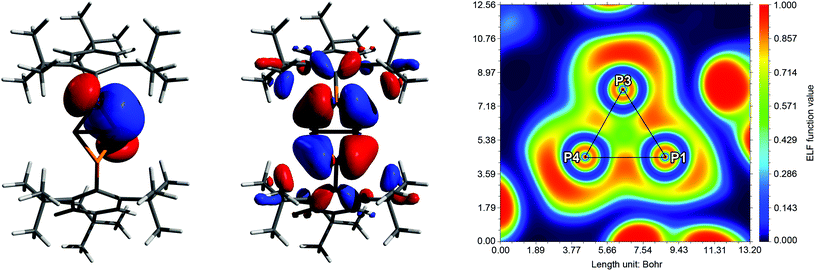 |
| | Fig. 3 Exemplary description of the bonding in 5a. Left: NBO/NLMO depiction of one PP bond, illustrating the “banana” bond character. Middle: HOMO−6 with large coefficients at the central P4 scaffold. Right: Electron localization function (ELF) depicted in a plane through atoms P1, P3 and P4. The maximum densities between the P atoms are found outside the lines of nuclear centres (black). The maxima at the corners of the triangle show the location of the lone pairs (LPs). | |
Isomerization of 5b to 5a
At the PBE0/aug-cc-pVDZ level of theory, the exo–exo-isomer 5a is energetically favoured with respect to 5b by 10.4 kJ mol−1 (ΔH298) or 8.80 kJ mol−1 (ΔG298), respectively. These findings prompted us to investigate the thermodynamic equilibrium between both isomers: Indeed, when heating a THF solution of 5a and 5b (1:4 ratio) to 75 °C over a period of 50 days, slow conversion of 5b to 5a was observed in the 31P NMR spectrum. The forward and backward reaction were modelled as first order kinetics according to eqn (1).| | 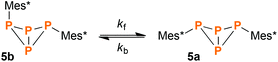 | (1) |
Hence, eqn (2) defines the reaction rate, [5a] and [5b] being the partial concentrations of 5a and 5b, respectively.
| | 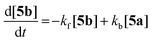 | (2) |
Let [5a]0 and [5b]0 denominate the initial concentrations at t = 0. Due to the conservation of mass, [5b]0–[5b] equals [5a]–[5a]0. Furthermore, the reaction rate becomes zero at equilibrium, when the system reaches a steady state with constant concentrations of both isomers. Using these boundary conditions to integrate eqn (2), we obtain
| | | [5b] = c + ([5b]0 − c)e−(kf+kb)t | (3) |
| | 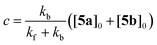 | (4) |
Least-squares fitting of eqn (3) against the experimentally determined concentrations gave kf = 2.77(11) × 10−6 s−1 and kb = 0.30(3) × 10−6 s−1. Accordingly, the experimental equilibrium constant is:
| | 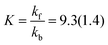 | (5) |
Thus, the experimental Gibbs energy (at 75 °C) can be calculated:
| | | ΔRG = −RTln K = −6.4(3) kJ mol−1 | (6) |
This value compares well to the calculated value of ΔRG348 = −8.54 kJ mol−1 for the gas phase reaction.
Intriguingly, an equilibrium between endo–exo and exo–exo-isomers was also described for Ter′P4Ter′ (9a, 9b); however, the system was found to be highly dynamic and both isomers afforded identical NMR spectra even at low temperature. Crystallization of either one of the isomers depended solely on the solvent used.27
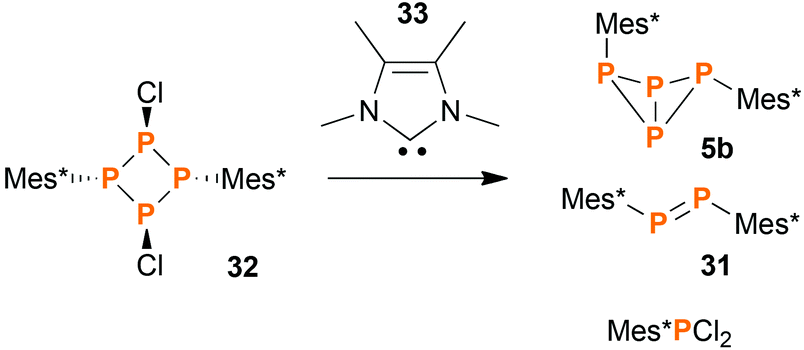
Synthesis of endo–exo-Mes*P4Mes* (5b) by carbene promoted degradation of [ClP(μ-PMes*)]2 (32)
Using the synthetic protocol described above, 5a and 5b were always obtained in mixture. Separation was difficult and could only be achieved for small amounts of substance by repeated re-crystallization. Designing a reaction that would yield 5b exclusively proved to be difficult: obviously, all reactions involving elevated temperatures led to (at least) partial formation of the thermodynamically favoured exo–exo-isomer 5a. Still, we found that the reaction of the cyclo-tetraphosphane [ClP(μ-PMes*)]2 (32)46 with tetramethylimidazolylidene (33) yielded bicyclic tetraphosphane 5 in a surprisingly good isomeric ratio (5a:5b) of 1:12 (Scheme 4). The isolated yield of pure 5b was low (14% based on 32), since the degradation of 32 did not proceed cleanly but rather gave a mixture of different products. Anyway, 5b could be crystallized from the reaction mixture in large, block-shaped crystals, making isolation comparatively easy.
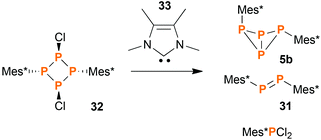 |
| | Scheme 4 The carbene promoted degradation of 32 yields a mixture of products, of which some were identified. | |
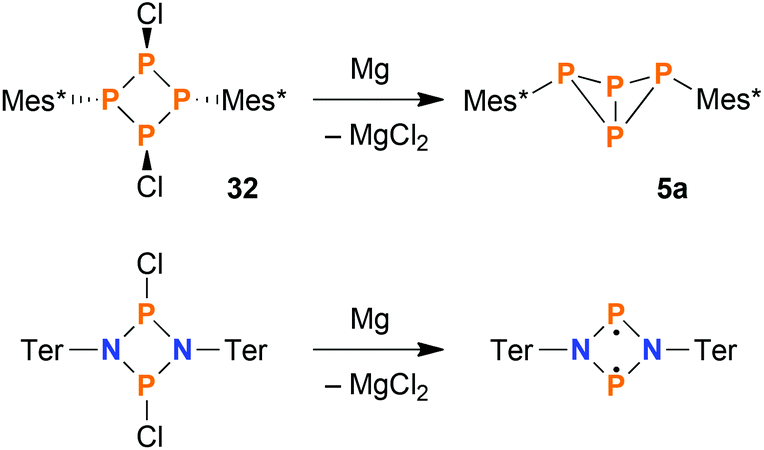
Selective synthesis of exo–exo-Mes*P4Mes* (5a) by reduction
Concerning the synthesis of pure 5a, the isomerisation at elevated temperatures certainly offered a viable possibility to increase the isomeric ratio in favour of the exo–exo isomer. Nevertheless, we found a much more straightforward way to synthesize pure 5a: starting from 32, reduction with stoichiometric amounts of Mg afforded 5a in high yields (based on 31P NMR integrals: 95%; isolated substance: 73%) in a clean reaction (Scheme 5, top). For comparison, the original synthesis of 5a published by Fluck et al. afforded the bicyclic phosphane in a yield of only 5.2%.22 Formally, the reduction of 32 with Mg can be compared to the reduction of dichloro-cyclo-diphosphadiazanes, [ClP(μ-NR)]2, which results in the formation of cyclo-diphosphadiazanediyls, [P(μ-NR)]2, provided that the substituent R is bulky enough to prevent dimerization (Scheme 5, bottom).52 Yet, in contrast to the bicyclic tetraphosphane, the NP species comprise a planar ring system with singlet biradical character, as there is no classical bonding interaction between the transannular P atoms in this case.
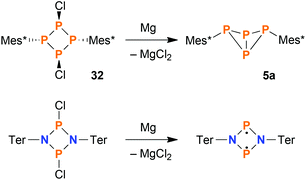 |
| | Scheme 5 Top: Reduction of cyclo-tetraphosphane 32 yields bicyclic phosphane 5a selectively. Bottom: Reduction of homologous dichloro-cyclo-diphosphadiazanes results in singlet open-shell biradicals with a drastically different bonding situation. | |
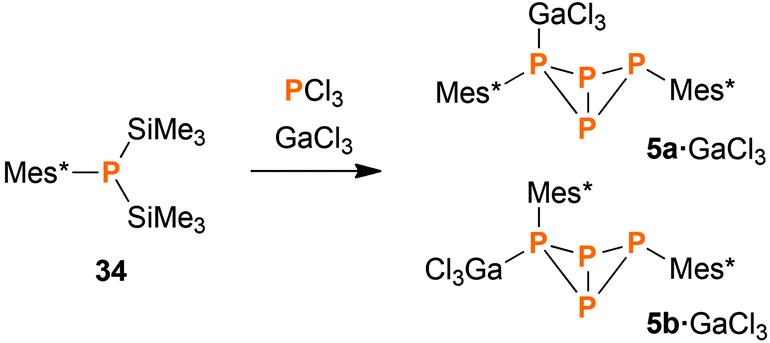
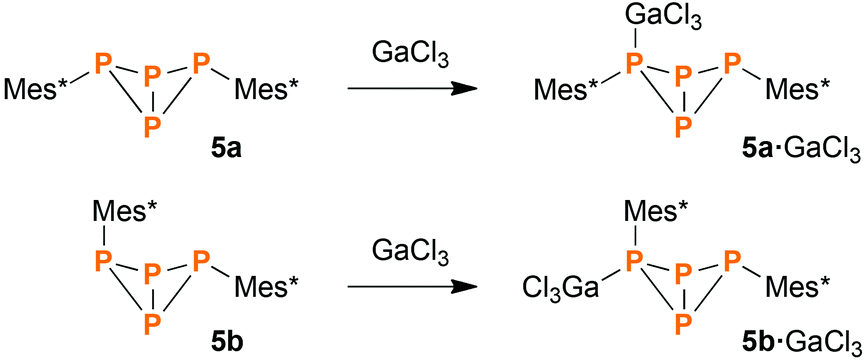
Synthesis of Mes*P4Mes*·GaCl3 (5·GaCl3) from P1 units
When heating a solution of Mes*P(SiMe3)2 (34) in the presence of PCl3, a mixture of various products was obtained. Interestingly, the exo–exo-isomer 5a was found to be one of the major products. Other species that could be identified were Mes*PPMes* (31), Mes*PCl2, P4, endo–exo-isomer 5b (minor amounts), and some higher aggregates of uncertain composition. However, the reaction was rather slow, so we decided to add a Lewis acid for activation purposes. Indeed, in the presence of GaCl3, complete conversion was detected after one hour even at low temperatures (Scheme 6). Upon slow warming to ambient temperature, colourless crystals of 5a·GaCl3 or 5b·GaCl3 were obtained. Depending on the solvent, either one could be crystallized selectively, even though the ratio of both isomers in solution (7:5) remained unaffected by the choice of solvent.
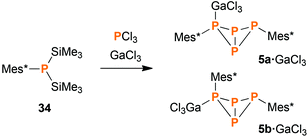 |
| | Scheme 6 Reacting 34 with PCl3 and GaCl3 leads to GaCl3 adducts of the bicyclic tetraphosphane 5, among other products. | |
Spectroscopic characterization
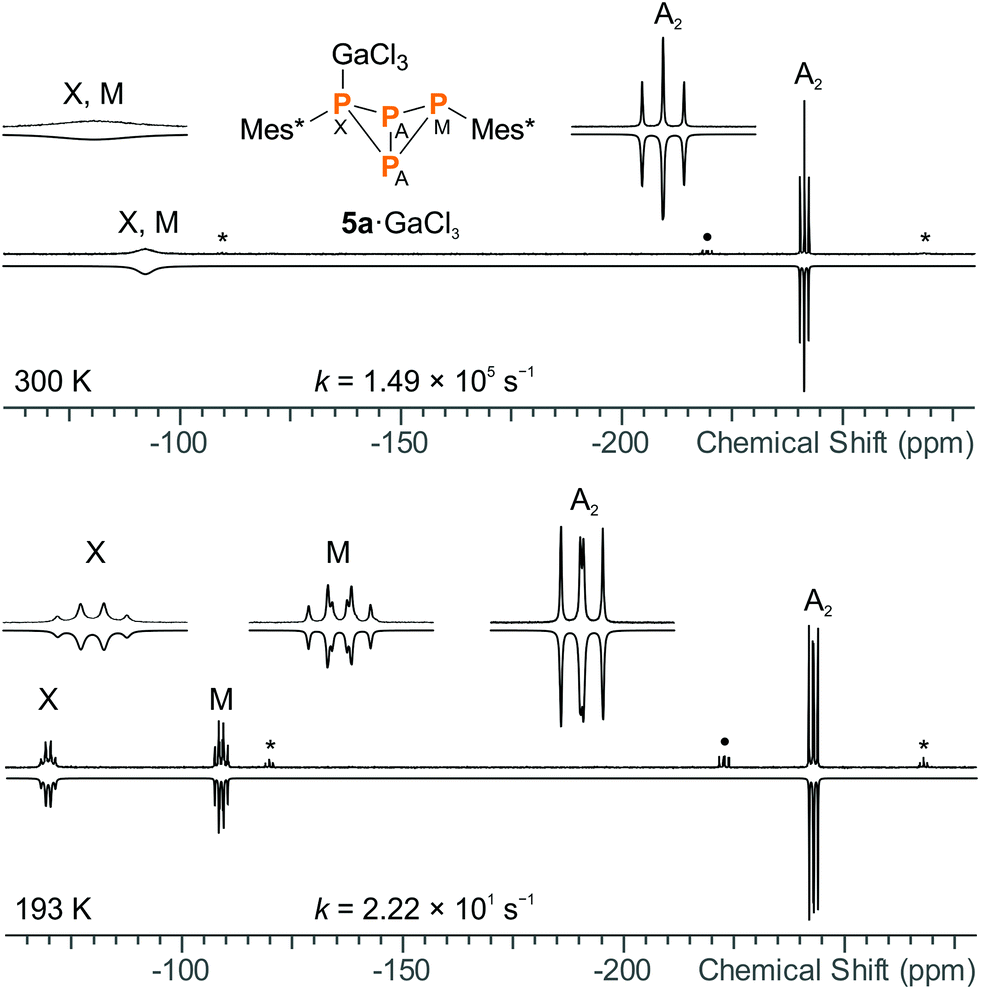
For both 5a·GaCl3 and 5b·GaCl3, an A2MX spin system was expected due to unsymmetrical substitution of the bicyclic scaffold. Nonetheless, the room temperature 31P NMR spectrum of 5a·GaCl3 only displayed two resonances (formal A2X2 pattern; −246.4, −97.1 ppm; Fig. 4), which were shifted downfield by ca. 30 ppm with respect to non-coordinating 5a. At −80 °C, though, the actual A2MX spin system was resolved (−248.1, −114.0, −74.8 ppm), with a rather large 2JMX coupling constant of +225 Hz (cf.5b: 2JMX = −27 Hz). To investigate the nature of this dynamic effect, temperature dependent NMR spectra were recorded. Basically, either an intramolecular or an intermolecular exchange (i.e. dissociation or bimolecular exchange) of the GaCl3 unit could be responsible for the observed line shapes, given that the concentration of free 5a is much lower than the concentration of the adduct 5a·GaCl3 (approx. ratio of 10−2 or less). However, at such low concentrations, the free phosphane could well be below the detection limit and it might not be discernible in the spectrum even at slow exchange. Accordingly, we added an excess of 5a, so the appearance of the NMR spectrum would change drastically if the free phosphane (i.e. dissociation) was involved in the observed exchange.53 However, the signals of the excess phosphane could be detected independently of those of the adduct and the signal pattern of 5a·GaCl3 remained unchanged, regardless of the excess of phosphane, proving an intramolecular mechanism (Scheme 7). Still, at higher temperatures above the coalescence of the M and X signal of 5a·GaCl3, slight broadening of the signals of free 5a was detected, hinting at an independent intermolecular exchange. Full line-shape analysis of the NMR signals (Fig. 4, ESI†) facilitated derivation of the rate constants k at different temperatures. Using the Eyring eqn (7), the Gibbs energy of activation of the intramolecular exchange could be determined.| | 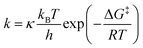 | (7) |
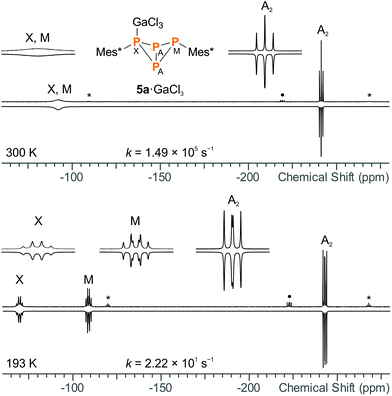 |
| | Fig. 4 Experimental (up) and simulated (down) 31P NMR spectra of 5a·GaCl3 at 27 °C (top) and −80 °C (bottom), demonstrating the dynamic exchange of the GaCl3 moiety in solution (* = 5a, • = impurity of 5b·GaCl3). GaCl3 was added to 5a in slightly sub-stoichiometric amounts (0.95 eq.) so the non-coordinating bicyclic phosphane was detectable in the NMR spectrum. | |
 |
| | Scheme 7 In case of 5a·GaCl3, a fast intramolecular exchange of the GaCl3 unit was observed. The Gibbs energy of activation for this process was determined to be 39.5 kJ mol−1. | |
Least-squares fitting gave a mean Gibbs energy of activation ΔG‡ = 39.5(4) kJ mol−1, which compares, for example, to the activation barrier of the exchange of NH3 in H3N·GaMe3 (35.6 kJ mol−1).54 According to the linearized Eyring plot, the enthalpy of activation ΔH‡ was found to be 39.5(4) kJ mol−1 and the entropy of activation ΔS‡ = −0.20(2) J mol−1 K−1, indicating a monomolecular transition state in agreement with the discussed intramolecular exchange reaction. The NMR data at slow exchange (−80 °C) correspond well with calculated NMR shifts and coupling constants (ESI†).
The endo–exo isomer 5b·GaCl3 showed an A2MX spin system (−224.5, −114.5, −50.0 ppm), which resembled the NMR spectrum of free 5b. Like in 5a·GaCl3, the A part was broadened due to coupling with Ga. Owing to the arrangement of the Mes* substituents, no intramolecular exchange was observed; the linewidths did not change significantly upon cooling to −80 °C. Nonetheless, upon addition of an excess of 5b the 31P NMR spectrum revealed a dynamic exchange between free phosphane and adduct, which is most likely caused by a bimolecular exchange of the type 5·GaCl3 + 5′ ⇌ 5′·GaCl3 + 5 (ESI†).
Both isomers were nicely distinguishable in the solid state Raman spectra, due to the different positions of the “breathing mode” bands, similar to the non-coordinating tetraphosphabicyclo[1.1.0]butanes 5a and 5b. In contrast to the latter, the most intense Raman signal was caused by the Ga–Cl stretching at 343 (5a·GaCl3) or 348 cm−1 (5b·GaCl3). P–C valence modes could be identified at 613 (Mes* at coordinating P, 5a·GaCl3) and 618 cm−1 (endo-Mes*, 5b·GaCl3), the other ones were superimposed by the intense “breathing mode” signals. Important vibrational modes of the P4 scaffold are summarized in Table 1.
Molecular structure
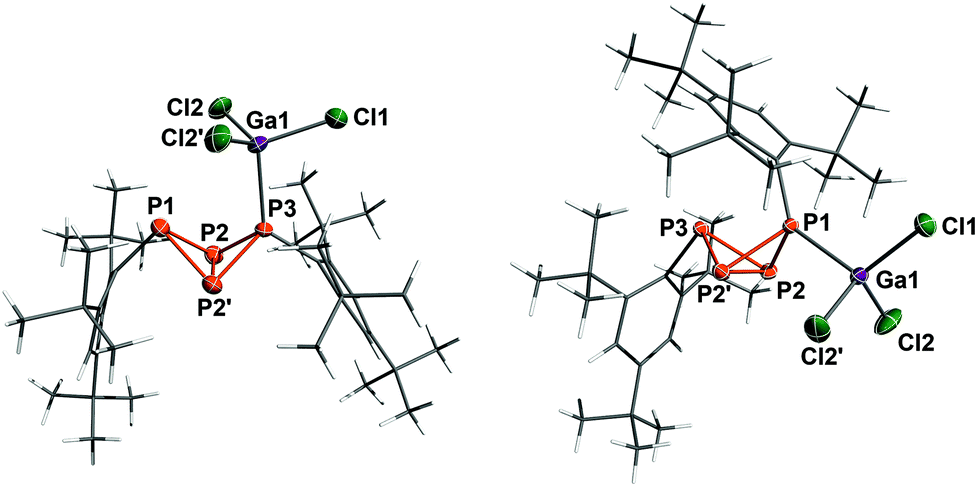
Lewis acid adduct 5a·GaCl3 crystallized in the orthorhombic space group Pnma as n-hexane solvate (Fig. 5). The PP bond lengths lie within the range of typical single bonds, however the P3–P2 (or P3–P2′) bond is slightly shortened (2.1805(9) Å); therefore, the bicyclic structure with two longer and two shorter bonds actually resembles the bicyclic scaffold in 5b. The transannular bond (P2–P2′: 2.200(1) Å), on the other hand, is noticeably longer than in 5a or 5b. In comparison with the sum of covalent radii (2.35 Å),50 the P3–Ga1 bond is somewhat elongated (2.4206(8) Å), most likely due to Pauli repulsion between the ortho-tert-butyl groups of the neighbouring Mes* moiety and the GaCl3 unit. The bonding angles at the coordinating P atom (P3) are considerably flattened, so one of the Mes* substituents is bent further outwards. Moreover, the fold angle of the bicyclic system amounts to 98.69(3)° and is therefore 3° larger than in 5a.
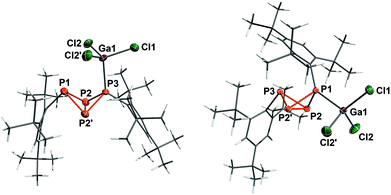 |
| | Fig. 5 Molecular structure of 5a·GaCl3 (left) and 5b·GaCl3 (right). Ellipsoids are set at 50% probability (173 K). Selected bond lengths [Å] and angles [°]: 5a·GaCl3 P1–P2 2.228(1), P2–P3 2.1805(9), P2–P2′ 2.200(1), Ga1–P3 2.4206(8); P1–P2–P2′–P3 98.96(3); 5b·GaCl3 P1–P2 2.1854(5), P2–P3 2.2365(6), P2–P2′ 2.2074(8), P1–Ga1 2.3966(5); P1–P2–P2′–P3 102.64(2). | |
Compound 5b·GaCl3 crystallized in the monoclinic space group P21/m either as toluene or CH2Cl2 solvate with similar cell parameters and molecular structure. Hence only the toluene solvate shall be discussed in the following: again, the bonds between the bridgehead atoms and the coordinating P atom (P2–P1 and P2′–P1: 2.1854(5) Å) are shortened in comparison with 5b, whereas the transannular bond is somewhat widened (P2–P2′: 2.2074(8) Å). The P–Ga bond length (2.3699(5) Å) is shorter than in 5a·GaCl3 and compares well to the sum of covalent radii. The angle of the P3–P2–P2′ plane to the P3–C13 bond axis (96.92(6)°) is about 8° smaller than in 5b, so the Mes* substituent moves closer to the bicyclic scaffold. In contrast to 5a·GaCl3, the fold angle of 5b·GaCl3 lessens with respect to the free bicyclic tetraphosphane by 3° (102.64(2)°).
In both isomers, the substitution pattern at the bicyclic P4 scaffold is comparable to the borane adduct TerP4Me·B(C6F5)3 (23·B(C6F5)3) or the bicyclic phosphino-phosphonium cation [Mes*P4(Cl)Mes*]+ (29).12,45
Computational study
NBO analysis revealed that the partial charges of the P atoms in 5a·GaCl3 and 5b·GaCl3 (bridgehead P: +0.10e, coordinating P: +0.24e, non-coordinating P: +0.31e) changed only slightly in comparison with 5a and 5b (bridgehead P: 0.00e, P(Mes*): +0.27e), implying a similar distribution of the electron density. Inspection of the molecular orbitals (MOs) showed that the principal bonding orbitals of the P4 scaffold remained intact. According to the electron localization function (ELF), the P–Ga bond is strongly polarized towards phosphorus (Fig. 6). The natural Lewis description actually suggests a non-bonding situation with a lone pair (LP) at phosphorus and an empty p-type orbital at Ga, with low P–Ga bond indices (NLMO: 0.34, Wiberg: 0.50). However, a second order perturbation analysis revealed a strong donor–acceptor interaction between the LP at P and the empty p-type orbital at Ga (5a·GaCl3: 530.5 kJ mol−1, 5b·GaCl3: 555.2 kJ mol−1); thus, the P–Ga bonding can be described as a classical dative bond. At the PBE0/aug-cc-pVDZ level of theory, the exo–exo isomer 5a·GaCl3 was calculated to be energetically favoured by 8.03 kJ mol−1 (ΔG298); therefore the energetic difference between exo–exo and endo–exo-isomer slightly decreased in comparison to non-coordinating 5a and 5b. This is reflected in the calculated Gibbs energies (ΔRG298) for the association of 5a or 5b and GaCl3, which amount to −46.7 kJ mol−1 or −47.4 kJ mol−1, respectively.
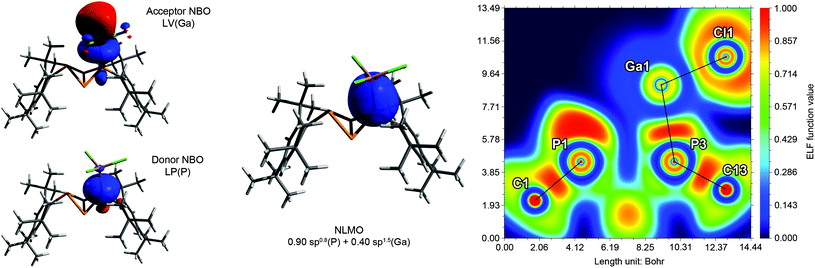 |
| | Fig. 6 Exemplary description of the bonding in 5a·GaCl3. Left: Donor and acceptor NBOs describing the dative bond from P to Ga. Middle: P–Ga bond in the natural localized MO (NLMO) picture. Right: Electron localization function (ELF) depicted in a plane through atoms P1, P3 and Ga1. The P–Ga and Ga–Cl bonds are strongly polarized and exhibit partly ionic character. The deformation of the LP at P1 is due to Pauli repulsion with Cl2 and Cl2′ (out of plane). | |
Equilibrium between 5a·GaCl3 and 5b·GaCl3
Similar to the free phosphanes, the GaCl3 adducts 5a·GaCl3 and 5b·GaCl3 were found to interconvert slowly in solution at ambient temperature. Over a period of several weeks, the establishment of equilibrium was monitored by 31P NMR spectroscopy. Interestingly, the equilibrium constant was found to be 1.6(2), thus the amount of endo–exo isomer at equilibrium was significantly higher than in case of the free phosphanes. This is in agreement with the calculations, although the actual effect is even more pronounced. According to the experiment, the energetic difference between both isomers is just 1.2(4) kJ mol−1 (ΔG).
Direct synthesis of Mes*P4Mes*·GaCl3 (5·GaCl3)
As expected, 5a·GaCl3 and 5b·GaCl3 could be synthesized from pure 5a and 5b by treatment with equimolar amounts of GaCl3. Mixing solutions of both reactants and subsequent evaporation of the solvent led to quantitative yield of the respective GaCl3 adduct (Scheme 8). Due to the short reaction time (ca. 5 min), isomerization could be avoided.
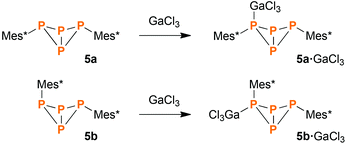 |
| | Scheme 8 Rational synthesis of 5a·GaCl3 and 5b·GaCl3. | |
Conclusions
We present new insights into the chemistry of tetraphosphabicyclo[1.1.0]butanes: various synthetic approaches were investigated, involving precursor molecules with one, two or four phosphorus atoms. Thereby, the endo–exo isomer of Mes*P4Mes* (5b) could be fully characterized for the first time and the interconversion of both isomers could by studied in detail. Furthermore, the hitherto unknown GaCl3 adducts of both exo–exo and endo–exo isomer (5a·GaCl3, 5b·GaCl3) were thoroughly investigated, including experimental assessment of dynamic behaviour in solution and computational studies of the bonding situation.
Due to isomerization in solution, it is difficult to obtain either isomer purely; ideally, synthetic strategies should be designed to (a) minimize the reaction time and (b) take place at low temperatures to avoid thermodynamic equilibrium.
Experimental
All manipulations were carried out under oxygen- and moisture-free conditions under argon using standard Schlenk or Drybox techniques. All starting materials containing the Mes* moiety were synthesized according to modified literature procedures; other reactants and solvents were obtained from commercial sources and thoroughly dried. Detailed information concerning experimental procedures, data acquisition and processing, as well as purification of chemicals can be found in the ESI.†
Synthesis of 5a
A solution of [ClP(μ-PMes*)]2 (206 mg, 0.30 mmol) in THF (3 mL) is added to magnesium turnings (10 mg, 0.42 mmol) and stirred at ambient temperature overnight. Subsequently, the solvent is removed in vacuo and the solid residue is extracted with n-hexane (5 mL). After filtration, the filtrate is concentrated and stored at 5 °C, resulting in crystallization of colourless exo–exo-Mes*P4Mes*. Yield: 135 mg (0.22 mmol, 73%). CHN calc. (found) in %: C 70.34 (70.32), H 9.51 (9.35). 31P{1H} NMR (CD2Cl2, 121.5 MHz): δ = −273.2 (t, 1JAX(31P,31P) = −177 Hz, 2 P, Pbridgehead), −128.3 (t, 1JAX(31P,31P) = −177 Hz, 2 P, PMes*). 1H NMR (CD2Cl2, 300.1 MHz): δ = 1.19 (s, 18 H, p-t-Bu), 1.63 (s, 36 H, o-t-Bu), 7.07 (m, 4 H, m-H). Raman (633 nm, 15 s, 10 scans, cm−1): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 3075 (1), 2963 (2), 2924 (2), 2903 (3), 2863 (1), 2777 (1), 2709 (1), 1588 (1), 1527 (1), 1473 (1), 1461 (1), 1442 (1), 1396 (1), 1365 (1), 1292 (1), 1281 (1), 1241 (1), 1208 (1), 1201 (1), 1175 (1), 1152 (1), 1128 (1), 1033 (2), 1020 (1), 934 (1), 921 (1), 891 (1), 822 (2), 775 (1), 637 (1), 592 (10), 563 (1), 490 (1), 475 (1), 463 (1), 447 (1), 432 (1), 412 (1), 387 (1), 351 (4), 294 (1), 259 (2), 211 (1), 191 (1), 177 (1), 136 (1), 125 (2), 107 (3), 86 (3).
= 3075 (1), 2963 (2), 2924 (2), 2903 (3), 2863 (1), 2777 (1), 2709 (1), 1588 (1), 1527 (1), 1473 (1), 1461 (1), 1442 (1), 1396 (1), 1365 (1), 1292 (1), 1281 (1), 1241 (1), 1208 (1), 1201 (1), 1175 (1), 1152 (1), 1128 (1), 1033 (2), 1020 (1), 934 (1), 921 (1), 891 (1), 822 (2), 775 (1), 637 (1), 592 (10), 563 (1), 490 (1), 475 (1), 463 (1), 447 (1), 432 (1), 412 (1), 387 (1), 351 (4), 294 (1), 259 (2), 211 (1), 191 (1), 177 (1), 136 (1), 125 (2), 107 (3), 86 (3).
Synthesis of 5b
Method 1: HOTf (180 mg, 1.12 mmol) is condensed onto a degassed solution of Mes*PPN(i-Pr)2 (489 mg, 1.12 mmol) in CH2Cl2 (8 mL) at −196 °C. The reaction mixture is slowly warmed to ambient temperature overnight. The solvent is removed in vacuo and the residue is extracted with n-hexane (5 mL). Insoluble solids are filtered off. The clear orange filtrate is concentrated, resulting in crystallization of a mixture of exo–exo and endo–exo-Mes*P4Mes* (1:4 ratio). Yield: 120 mg (0.20 mmol, 35%). Re-crystallization yields pure endo–exo-Mes*P4Mes*. Method 2: A mixture of [ClP(μ-PMes*)]2 (835 mg, 1.22 mmol) and Me4C3N2 (302 mg, 2.44 mmol) is dissolved in CH2Cl2 (10 mL) at −80 °C, resulting in a dark red solution. The reaction vessel is warmed to ambient temperature over a period of one hour, whereupon the solution is concentrated and stored at 5 °C, resulting in the crystallization of orange, block shaped crystals that were identified as Mes*PPMes*. The supernatant is separated and concentrated. Storage at 5 °C affords large colourless crystals of endo–exo-Mes*P4Mes*. Yield: 105 mg (0.17 mmol, 14%). CHN calc. (found) in %: C 70.34 (70.04), H 9.51 (9.47). 31P{1H} NMR (CD2Cl2, 121.5 MHz): δ = −220.4 (dd, 1JAX(31P,31P) = −234 Hz, 1JAM(31P,31P) = −213 Hz, 2 P, Pbh), −94.8 (td, 1JAM(31P,31P) = −213 Hz, 2JMX(31P,31P) = −27 Hz, 1 P, Pexo), −54.7 (td, 1JAX(31P,31P) = −234 Hz, 2JMX(31P,31P = −27 Hz, 1 P, Pendo). 1H NMR (CD2Cl2, 300.1 MHz): δ = 1.19 (s, 9 H, exo-Mes*, p-t-Bu), 1.30 (s, 9 H, endo-Mes*, p-t-Bu), 1.49 (s, 18 H, exo-Mes*, o-t-Bu), 1.66 (m 18 H, endo-Mes*, o-t-Bu), 7.02 (m, 2 H, exo-Mes*, m-H), 7.05 (m, 2 H, endo-Mes*, m-H). Raman (633 nm, 15 s, 20 scans, cm−1): = 3168 (1), 3074 (1), 3055 (1), 2959 (4), 2924 (4), 2902 (5), 2865 (2), 2778 (1), 2712 (1), 1584 (3), 1520 (1), 1475 (1), 1466 (2), 1444 (2), 1399 (1), 1392 (1), 1360 (1), 1283 (2), 1251 (1), 1203 (1), 1186 (1), 1182 (1), 1173 (1), 1148 (1), 1132 (2), 1033 (3), 1017 (2), 932 (1), 919 (2), 897 (1), 877 (1), 822 (4), 772 (1), 744 (1), 740 (1), 647 (1), 638 (1), 591 (2), 584 (2), 568 (10), 500 (2), 490 (1), 473 (1), 435 (1), 419 (2), 412 (2), 381 (4), 363 (2), 330 (1), 309 (1), 295 (1), 256 (2), 200 (1), 177 (1), 143 (4), 139 (3), 103 (7), 78 (6).
Synthesis of 5a·GaCl3
Method 1: Solutions of PCl3 (130 mg, 0.95 mmol) and GaCl3 (168 mg, 0.95 mmol) in n-hexane (2 mL each) are added consecutively to a stirred solution of Mes*P(SiMe3)2 (403 mg, 0.95 mmol) in n-hexane (5 mL) at −80 °C. The mixture is warmed to ambient temperature, resulting in the deposition of an intensively red oil. Overnight, large colourless crystals grow at the phase interface at ambient temperature. Yield: 45 mg (0.07 mmol, 8%). Method 2: A solution of GaCl3 (10 mg, 0.057 mmol) in CH2Cl2 (1 mL) is added to a solution of exo–exo-Mes*P4Mes* (35 mg, 0.057 mmol) in CH2Cl2 (2 mL). The mixture is stirred for ten minutes and the solvent is removed in vacuo, yielding an analytically pure powder of exo–exo-Mes*P4Mes*·GaCl3. Yield: 30 mg (0.038 mmol, 67%). CHN calc. (found) in %: C 54.68 (53.97), H 7.39 (6.89). 31P{1H} NMR (CD2Cl2, 121.5 MHz): δ = −246.4 (t, 1JAX(31P,31P) = −198 Hz, 2 P, Pbridgehead), −97.1 (broad, 2 P, PMes*). 31P{1H} NMR (CD2Cl2, 121.5 MHz, −80 °C): δ = −248.1 (dd, 1JAM(31P,31P) = −182 Hz, 1JAX(31P,31P) = −216 Hz, 2 P, Pbridgehead), −114.0 (dt, 1JAM(31P,31P) = −182 Hz, 2JMX(31P,31P) = +225 Hz, 1 P, PMes*), −74.8 (dt, 1JAX(31P,31P) = −216 Hz, 2JMX(31P,31P) = +225 Hz, 1 P, P(Ga)Mes*). 1H NMR (CD2Cl2, 300.1 MHz): δ = 1.20 (s, 18 H, Mes*, p-t-Bu), 1.68 (s, 36 H, o-t-Bu), 7.24 (s, 4 H, m-H). Raman (785 nm, 30 s, 4 scans, cm−1): = 3064 (1), 2976 (1), 2960 (2), 2928 (2), 2904 (3), 2869 (1), 2786 (1), 2717 (1), 1593 (3), 1582 (2), 1536 (1), 1477 (2), 1465 (3), 1443 (2), 1393 (2), 1362 (1), 1284 (3), 1250 (2), 1208 (2), 1176 (3), 1031 (3), 1025 (3), 1016 (2), 920 (2), 891 (1), 819 (5), 772 (1), 747 (1), 638 (1), 613 (3), 597 (9), 561 (4), 502 (1), 497 (1), 480 (2), 470 (1), 451 (2), 436 (2), 406 (3), 395 (4), 385 (3), 366 (4), 343 (10), 294 (2), 260 (4).
Synthesis of 5b·GaCl3
Method 1: A solution of PCl3 (131 mg, 0.95 mmol) in CH2Cl2 (1 mL) and a solution of GaCl3 (168 mg, 0.95 mmol) in CH2Cl2/toluene (1:1, 2 mL) are added consecutively to a stirred solution of Mes*P(SiMe3)2 (403 mg, 0.95 mmol) in CH2Cl2 (5 mL) at −80 °C. The mixture is warmed to −50 °C, whereupon the solution turns red. After further stirring at −50 °C for two hours, the solution is concentrated and subsequently warmed to ambient temperature overnight, resulting in crystallization of endo–exo-Mes*P4Mes*·GaCl3 (CH2Cl2 solvate) in colourless, block-shaped crystals. The solvent is removed by drying in vacuo. Yield: 50 mg (0.08 mmol, 9%). Method 2: A solution of GaCl3 (15 mg, 0.085 mmol) in CH2Cl2 (1 mL) is added to a solution of endo–exo-Mes*P4Mes* (52 mg, 0.085 mmol) in CH2Cl2 (2 mL). The mixture is stirred for ten minutes and the solvent is removed in vacuo, yielding an analytically pure powder of endo–exo-Mes*P4Mes*·GaCl3. Yield: 51 mg (0.064 mmol, 75%). CHN calc. (found) in %: C 54.68 (54.15), H 7.39 (7.15). 31P{1H} NMR (CD2Cl2, 121.5 MHz): δ = −224.5 (dd, 1JAM(31P,31P) = −192 Hz, 1JAX(31P,31P) = −249 Hz, 2 P, Pbridgehead), −114.5 (td, 1JAM(31P,31P) = −192 Hz, 1JMX(31P,31P) = +24 Hz, 1 P, Pexo), −50.0 (broad, 1 P, Pendo). 1H NMR (CD2Cl2, 300.1 MHz): δ = 1.21 (s, 9 H, exo-Mes*, p-t-Bu), 1.33 (s, 9 H, endo-Mes*, p-t-Bu), 1.49 (s, 18 H, exo-Mes*, o-t-Bu), 1.70 (m 18 H, endo-Mes*, o-t-Bu), 7.11 (d, J(1H,31P) = 1.9 Hz, 2 H, exo-Mes*, m-H), 7.32 (d, J(1H,31P) = 5.0 Hz, 2 H, endo-Mes*, m-H). Raman (633 nm, 10 s, 4 scans, cm−1): = 3055 (2), 3035 (1), 2973 (5), 2966 (6), 2905 (7), 2866 (3), 2782 (1), 2714 (1), 1603 (1), 1582 (4), 1525 (1), 1463 (2), 1441 (2), 1394 (1), 1362 (1), 1284 (2), 1208 (2), 1173 (1), 1133 (2), 1029 (3), 1011 (2), 1002 (3), 924 (1), 818 (3), 784 (2), 741 (2), 618 (3), 567 (7), 518 (1), 508 (1), 438 (3), 407 (2), 391 (2), 372 (2), 348 (10), 302 (1), 258 (3), 213 (1).
Detailed analytical data for all compounds, including low temperature NMR data, can be found in the ESI.†
Acknowledgements
We thank Dr Dirk Michalik for the measurement of low-temperature NMR spectra, Dr Alexander Hinz for a gift of tetramethylimidazolylidene, and the Fonds der Chemischen Industrie (JB) as well as the Deutsche Forschungsgemeinschaft (SCHU 1170/11-1) for financial support.
Notes and references
- L. Stahl, Coord. Chem. Rev., 2000, 210, 203–250 CrossRef CAS.
- M. S. Balakrishna, D. J. Eisler and T. Chivers, Chem. Soc. Rev., 2007, 36, 650–664 RSC.
- G. He, O. Shynkaruk, M. W. Lui and E. Rivard, Chem. Rev., 2014, 114, 7815–7880 CrossRef CAS PubMed.
- O. J. Scherer, Comments Inorg. Chem., 1987, 6, 1–22 CrossRef CAS.
- M. Baudler and K. Glinka, Chem. Rev., 1993, 93, 1623–1667 CrossRef CAS.
- M. Scheer, G. Balázs and A. Seitz, Chem. Rev., 2010, 110, 4236–4256 CrossRef CAS PubMed.
- B. M. Cossairt, N. A. Piro and C. C. Cummins, Chem. Rev., 2010, 110, 4164–4177 CrossRef CAS PubMed.
- N. A. Giffin and J. D. Masuda, Coord. Chem. Rev., 2011, 255, 1342–1359 CrossRef CAS.
- J. D. Masuda, W. W. Schoeller, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2007, 46, 7052–7055 CrossRef CAS PubMed.
- S. Heinl, S. Reisinger, C. Schwarzmaier, M. Bodensteiner and M. Scheer, Angew. Chem., Int. Ed., 2014, 53, 7639–7642 CrossRef CAS PubMed.
- S. Heinl and M. Scheer, Chem. Sci., 2014, 5, 3221–3225 RSC.
- J. E. Borger, A. W. Ehlers, M. Lutz, J. C. Slootweg and K. Lammertsma, Angew. Chem., Int. Ed., 2014, 53, 12836–12839 CrossRef CAS PubMed.
- E. Niecke, R. Rüger and B. Krebs, Angew. Chem., Int. Ed. Engl., 1982, 21, 544–545 CrossRef.
- A. H. Cowley, P. C. Knueppel and C. M. Nunn, Organometallics, 1989, 8, 2490–2492 CrossRef CAS.
- H.-P. Schrödel, H. Nöth, M. Schmidt-Amelunxen, W. W. Schoeller and A. Schmidpeter, Chem. Ber., 1997, 130, 1801–1805 CrossRef.
- P. Jutzi and T. Wippermann, J. Organomet. Chem., 1985, 287, C5–C7 CrossRef CAS.
- P. Jutzi and U. Meyer, J. Organomet. Chem., 1987, 333, C18–C20 CrossRef CAS.
- P. Jutzi, R. Kroos, A. Müller and M. Penk, Angew. Chem., 1989, 101, 628–629 CrossRef CAS.
- P. Jutzi and N. Brusdeilins, Z. Anorg. Allg. Chem., 1994, 620, 1375–1380 CrossRef CAS.
- L. Weber, G. Meine, R. Boese and N. Niederprün, Z. Naturforsch., B: Chem. Sci., 1988, 43, 415–721 CrossRef.
- V. D. Romanenko, V. L. Rudzevich, E. B. Rusanov, A. N. Chernega, A. Senio, J.-M. Sotiropoulos, G. Pfister-Guillouzo and M. Sanchez, J. Chem. Soc., Chem. Commun., 1995, 1383–1385 RSC.
- R. Riedel, H.-D. Hausen and E. Fluck, Angew. Chem., Int. Ed. Engl., 1985, 24, 1056–1057 CrossRef.
- E. Fluck, R. Riedel, H.-D. Hausen and G. Heckmann, Z. Anorg. Allg. Chem., 1987, 551, 85–94 CrossRef CAS.
- M. Baudler, C. Adamek, S. Opiela, H. Budzikiewicz and D. Ouzounis, Angew. Chem., Int. Ed. Engl., 1988, 27, 1059–1061 CrossRef.
- M. Baudler and B. Wingert, Z. Anorg. Allg. Chem., 1992, 611, 50–55 CrossRef CAS.
- M. B. Power and A. R. Barron, Angew. Chem., Int. Ed. Engl., 1991, 30, 1353–1354 CrossRef.
- A. R. Fox, R. J. Wright, E. Rivard and P. P. Power, Angew. Chem., Int. Ed., 2005, 44, 7729–7733 CrossRef CAS PubMed.
- I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2001, 40, 4406–4409 CrossRef CAS.
- M. H. Holthausen and J. J. Weigand, J. Am. Chem. Soc., 2009, 131, 14210–14211 CrossRef CAS PubMed.
- J. J. Weigand, M. H. Holthausen and R. Fröhlich, Angew. Chem., Int. Ed., 2009, 48, 295–298 CrossRef CAS PubMed.
- D. Holschumacher, T. Bannenberg, K. Ibrom, C. G. Daniliuc, P. G. Jones and M. Tamm, Dalton Trans., 2010, 39, 10590–10592 RSC.
- S. Khan, R. Michel, J. M. Dieterich, R. A. Mata, H. W. Roesky, J. P. Demers, A. Lange and D. Stalke, J. Am. Chem. Soc., 2011, 133, 17889–17894 CrossRef CAS PubMed.
- J.-P. Bezombes, P. B. Hitchcock, M. F. Lappert and J. E. Nycz, Dalton Trans., 2004, 499–501 RSC.
- N. A. Giffin, A. D. Hendsbee, T. L. Roemmele, M. D. Lumsden, C. C. Pye and J. D. Masuda, Inorg. Chem., 2012, 51, 11837–11850 CrossRef CAS PubMed.
- C. Rotter, M. Schuster and K. Karaghiosoff, Inorg. Chem., 2009, 48, 7531–7533 CrossRef CAS PubMed.
- A. Sidiropoulos, B. Osborne, A. N. Simonov, D. Dange, A. M. Bond, A. Stasch and C. Jones, Dalton Trans., 2014, 43, 14858–14864 RSC.
- M. Baudler and B. Wingert, Z. Anorg. Allg. Chem., 1993, 619, 1977–1983 CrossRef CAS.
- P. Jutzi, N. Brusdeilins, H.-G. Stammler and B. Neumann, Chem. Ber., 1994, 127, 997–1001 CrossRef CAS.
- O. J. Scherer, G. Schwarz and G. Wolmershäuser, Z. Anorg. Allg. Chem., 1996, 622, 951–957 CrossRef CAS.
- O. J. Scherer, T. Hilt and G. Wolmershäuser, Organometallics, 1998, 17, 4110–4112 CrossRef CAS.
- P. Barbaro, C. Bazzicalupi, M. Peruzzini, S. Seniori Costantini and P. Stoppioni, Angew. Chem., Int. Ed., 2012, 51, 8628–8631 CrossRef CAS PubMed.
- S. Pelties, D. Herrmann, B. de Bruin, F. Hartl and R. Wolf, Chem. Commun., 2014, 7014–7016 RSC.
- N. Wiberg, A. Wörner, H.-W. Lerner and K. Karaghiosoff, Z. Naturforsch., B: Chem. Sci., 2002, 57, 1027–1035 CAS.
- M. Donath, E. Conrad, P. Jerabek, G. Frenking, R. Fröhlich, N. Burford and J. J. Weigand, Angew. Chem., Int. Ed., 2012, 51, 2964–2967 CrossRef CAS PubMed.
- J. Bresien, K. Faust, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2015, 54, 6926–6930 CrossRef CAS PubMed.
- J. Bresien, C. Hering, A. Schulz and A. Villinger, Chem. – Eur. J., 2014, 20, 12607–12615 CrossRef CAS PubMed.
- M. Yoshifuji, I. Shima, N. Inamoto, K. Hirotsu and T. Higuchi, J. Am. Chem. Soc., 1981, 103, 4587–4589 CrossRef CAS.
- M. Yoshifuji, I. Shima, N. Inamoto, K. Hirotsu and T. Higuchi, J. Am. Chem. Soc., 1982, 104, 6167 CrossRef.
- Y. M. Bosworth, R. J. H. Clark and D. M. Rippon, J. Mol. Spectrosc., 1973, 46, 240–255 CrossRef CAS.
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 12770–12779 CrossRef PubMed.
-
E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis and F. Weinhold, NBO 6.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2013 Search PubMed.
- T. Beweries, R. Kuzora, U. Rosenthal, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2011, 50, 8974–8978 CrossRef CAS PubMed.
- H. Schmidbaur, A. Shiotani and H. F. Klein, J. Am. Chem. Soc., 1971, 93, 1555–1557 CrossRef CAS.
- J. B. DeRoos and J. P. Oliver, J. Am. Chem. Soc., 1967, 89, 3970–3977 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details, crystallographic and spectroscopic data. CCDC 1413820–1413827. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt02757h |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a