
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring structural and electronic effects in three isomers of tris{bis(trifluoromethyl)phenyl}borane: towards the combined electrochemical-frustrated Lewis pair activation of H2†
Robin J.
Blagg
 *a,
Elliot J.
Lawrence
*a,
Katie
Resner
a,
Vasily S.
Oganesyan
a,
Thomas J.
Herrington
b,
Andrew E.
Ashley
b and
Gregory G.
Wildgoose
*a
*a,
Elliot J.
Lawrence
*a,
Katie
Resner
a,
Vasily S.
Oganesyan
a,
Thomas J.
Herrington
b,
Andrew E.
Ashley
b and
Gregory G.
Wildgoose
*a
aSchool of Chemistry, University of East Anglia, Norwich, NR4 7TJ, UK. E-mail: r.blagg@uea.ac.uk; elliot.lawrence@uea.ac.uk; g.wildgoose@uea.ac.uk
bDepartment of Chemistry, Imperial College London, South Kensington, London, SW7 2AZ, UK
First published on 28th July 2015
Abstract
Three structural isomers of tris{bis(trifluoromethyl)phenyl}borane have been studied as the acidic component of frustrated Lewis pairs. While the 3,5-substituted isomer is already known to heterolytically cleave H2 to generate a bridging-hydride; ortho-substituents in the 2,4- and 2,5-isomers quench such reactivity through electron donation into the vacant boron pz orbital and steric blocking of the boron centre; as shown by electrochemical, structural and computational studies. Electrochemical studies of the corresponding borohydrides identify that the two-electron oxidation of terminal-hydrides occurs at more positive potentials than observed for [HB(C6F5)3]−, while the bridging-hydride oxidizes at a higher potential still, comparable to that of free H2.
Introduction
Since the pioneering work of Stephan's group1 the field of frustrated Lewis pair (FLP) chemistry has grown rapidly.2–7 The archetypal FLP system combines the Lewis acid B(C6F5)3 with a sterically demanding Lewis base such as P(tBu)3, to effect the heterolytic cleavage of H2, resulting in the formation of the corresponding hydridic and protic products respectively.FLPs have found applications as catalysts or mediators for a variety of reactions such as the metal-free hydrogenation of imines and nitriles,8 alkynes,9 silyl enol ethers,10 and ketones.11,12 The activation of small molecules such as CO2,13,14 and alkynes by FLPs has also found applications in the synthesis of heterocycles and other aromatic systems.15
While much of the literature has focused on tris(pentafluorophenyl)borane, B(C6F5)3 as the Lewis acidic component in FLPs, other electron-deficient boranes have been used, including a range of halogenated triarylboranes,14,16–18 and borenium cations,19,20 Other examples of Lewis acids that have found use in FLPs include: the triaryl aluminium species, Al(C6F5)3, which generates a bridging-hydride following cleavage of H2;21 and the carbon based N-methylacridinium salts which activate H2 even in the presence of H2O.22 FLPs are not limited to the main group, with zirconocene–phosphane complexes pioneered by Wass and co-workers shown to act as intramolecular FLPs that exhibit unprecedented reactivity towards small molecules;23 similar chemistry has also been demonstrated with zirconocene-amines which act as hydrogenation catalysts with a wide range of substrates.24
In 2014 we introduced the concept of “combined electrochemical-frustrated Lewis pairs”,20,25,26 that couple the heterolytic cleavage of H2 by a conventional FLP with in situ electrochemical oxidation of the resultant borohydride and subsequent regeneration of the parent borane. The combined electrochemical-FLP systems were shown to be electrocatalytic for the oxidation of H2 (to form two protons and two electrons – a key reaction in many hydrogen-based energy technologies). These preliminary reports represent the first application of FLP chemistry other than to catalyse the hydrogenation of small molecules. For example, by combining the archetypal B(C6F5)3/P(tBu)3 FLP in the presence of H2 whilst at the same time oxidizing the resultant [HB(C6F5)3]− intermediate formed, the potential for oxidation (the energetic driving force) of the borohydride at a glassy carbon electrode was found to be reduced by 0.61 V, in comparison to the direct oxidation of H2 (observed at ca. +1.49 V vs. [FeCp2]0/+ under the same conditions), an energy saving equivalent to 117.7 kJ mol−1.25
Herein we report studies on three isomers of tris{bis(trifluoromethyl)phenyl}borane (Fig. 1) and their associated tris{bis(trifluoromethyl)phenyl}borohydrides. Electrochemical and computational investigations explain their varying ability to heterolytically cleave H2 as part of a FLP and allow us to extend our studies into combined electrochemical-FLPs.
 | ||
| Fig. 1 Isomers of tris{bis(trifluoromethyl)phenyl}borane. | ||
Results and discussion
Synthesis and characterization
The synthesis of tris{3,5-bis(trifluoromethyl)phenyl}borane 1 has been previously reported,18,27 and whilst a synthetic route to tris{2,4-bis(trifluoromethyl)phenyl}borane 2 has been reported by Cornet et al.28 their method led to a mixture of products requiring the isolation of 2 in low yield by fractional sublimations. The commercial availability of 2,4-bis(trifluoromethyl)bromobenzene and 2,5-bis(trifluoromethyl)bromobenzene allows us to report an improved synthetic route to 2 and also the synthesis of the novel tris{2,5-bis(trifluoromethyl)phenyl}borane 3.Boranes 2 and 3 were synthesized by lithium–halogen exchange of BrC6H3(CF3)2 with nBuLi at −77 °C generating the aryllithium species LiC6H3(CF3)2. Treatment of this with a third of an equivalent of BCl3, followed by warming to room temperature, subsequent removal of the volatiles and purification by sublimation or recrystallization allowed for the isolation of pure 2 and 3, in 90 and 77% yields respectively. It should be noted that applying the same synthetic route for the synthesis of 1, leads to a mixture of products with [Li(OEt2)n][B{3,5-(CF3)2C6H3}4] as the major component.
The crystal structures of 129 and 228 have been previously reported. Single crystals of 3 were obtained by slow diffusion of a saturated CH2Cl2 solution of 3 into n-hexane at −25 °C, from which the crystal structure was obtained in collaboration with the EPSRC UK National Crystallography Service30 (Fig. 2 and S1,† and Table 2).
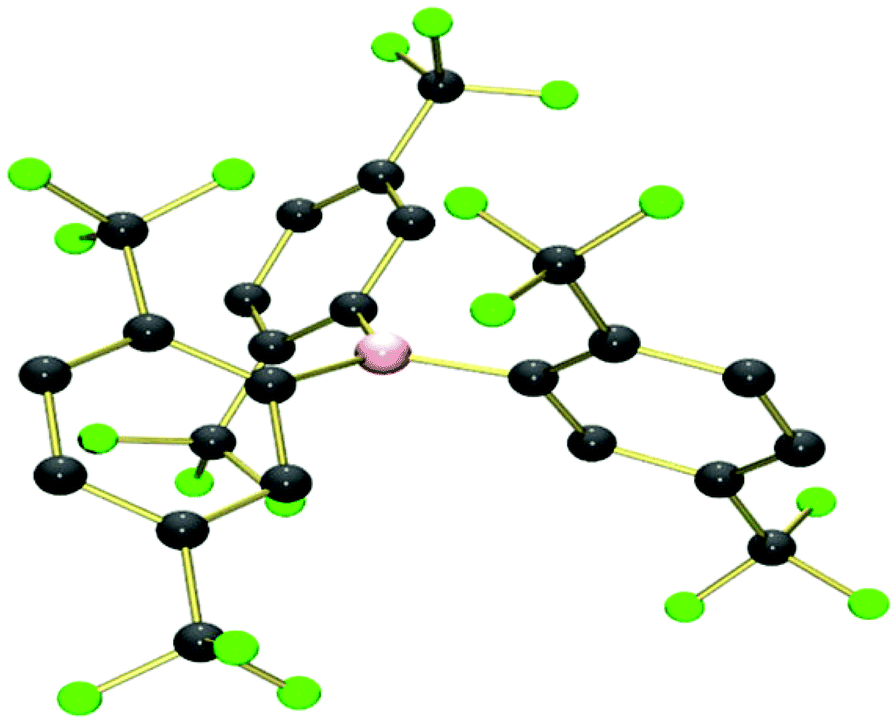 | ||
| Fig. 2 The crystallographic molecular structure of B{2,5-(CF3)2C6H3}33 (hydrogen atoms removed for clarity). | ||
The structures of 1–3 all show similar features, with a trigonal-planar boron centre and the three aryl rings twisted with respect to the BC3 plane to minimize steric interactions between the aryl rings. The degree of twist can be quantified by an appropriate choice of C–B–C–C torsion angles, which are both smaller and more consistent for 1 {mean 36(2)°, range 33.2–38.9°} than for 2 {mean 54(11)°, range 42.3–68.3°} or 3 {mean 53(9)°, range 40.9–61.2°}, due to the steric bulk of ortho-trifluoromethyl substituents. For 2 and 3 this results in the ortho-CF3 groups orientated above/below the boron centre resulting in B⋯F distances of 2.81(1), 2.80(7) Å for 2 and 3 respectively. The orientation of these groups suggests the possibility of electron donation from the fluorine atoms into the formally vacant pz orbital at boron; an effect together with the steric influence of the ortho-CF3 groups which would be expected to have a significant influence on the Lewis acidity/reactivity of these compounds. An example of the type of reactivity which could be expected from the ortho-CF3 groups, was observed by Cornet et al., who reported evidence of B–Cl/B–F exchange in mixtures containing the boranes B(ArF)2Cl where ArF = 2,4,6-(CF3)3C6H2, 2,4-(CF3)2C6H3 or 2,6-(CF3)2C6H3.28
The reactivity of 1 as the Lewis acidic component of an FLP has been previously studied,18 which showed that the reaction of 1 with H2 in the presence of 2,2,6,6-tetramethylpiperidine (tmp) leads to rapid formation of the bridging hydride species [tmpH][(μ-H)(1)2]. Analogous reactions of 2 and 3 with H2 in the presence of the Lewis bases tmp or P(tBu)3, result in neither Lewis acid–base adduct formation nor any evidence of H2 activation observable by NMR spectroscopy over a minimum period of 48 hours.
Direct synthesis of authentic terminal-hydride species [HB{C6H3(CF3)2}3]− proved to be possible for all three isomers 1–3 by direct reaction of the borane with sodium triethylborohydride in toluene solution, resulting in near quantitative conversion to the corresponding borohydrides. NMR spectra of the terminal-hydride species Na[1-H]–Na[3-H] show doublets in the 11B spectra (δB: −9.1, −15.3, −14.2 and 1JBH: 88, 93, 84 Hz respectively) and broad 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:1 quartets in the proton spectra (δH: +3.66, +4.06, +3.06 respectively). Additionally, for Na[2-H] and Na[3-H] clear evidence of through-space coupling between the hydride and ortho-CF3 groups is observed in the 19F spectra (JFH = 6.8, 7.6 Hz respectively); further, in the case of Na[3-H] this coupling is also observable in the proton spectrum with the hydride signal observable as a 1:1:1:1 quartet of broad 1:3:3:1 quartets (Fig. 3).
:1:1:1 quartets in the proton spectra (δH: +3.66, +4.06, +3.06 respectively). Additionally, for Na[2-H] and Na[3-H] clear evidence of through-space coupling between the hydride and ortho-CF3 groups is observed in the 19F spectra (JFH = 6.8, 7.6 Hz respectively); further, in the case of Na[3-H] this coupling is also observable in the proton spectrum with the hydride signal observable as a 1:1:1:1 quartet of broad 1:3:3:1 quartets (Fig. 3).
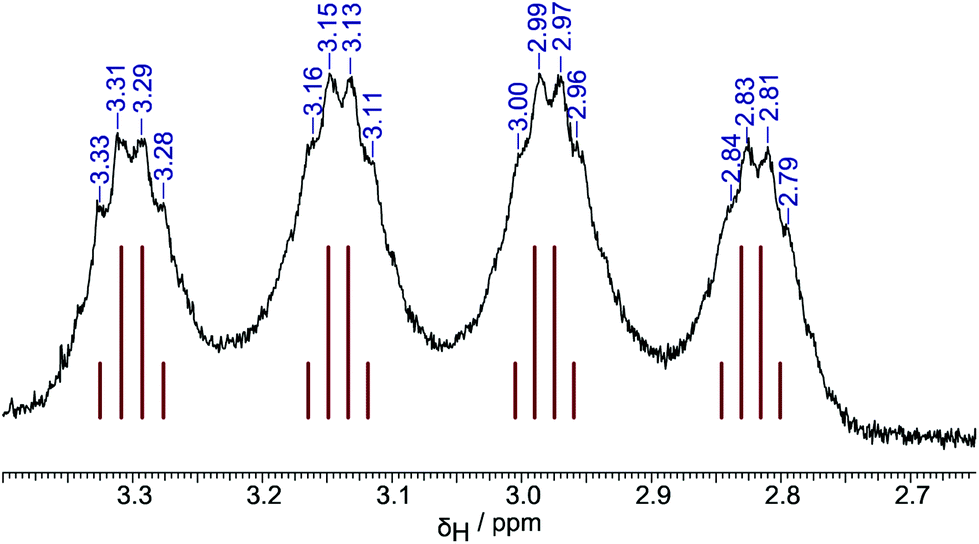 | ||
| Fig. 3 Hydride resonance in 1H NMR spectrum of Na[3-H], showing coupling to both 11B (I = 3/2) and an ortho-CF3 group {3× 19F, I = 1/2). | ||
Synthesis of Na[2-H] and Na[3-H] allows for the confirmation that the inability of the 2,3/P(tBu)3 FLPs to cleave H2 is not due to unfavourable thermodynamics, but due to the significant kinetic barrier resulting from the steric and electronic effects of the ortho-CF3 groups. Reaction with authentic [(tBu)3PH]Cl results in rapid metathesis (indicated by precipitation of NaCl) and formation of the salts [(tBu)3PH][2-H] and [(tBu)3PH][3-H]. NMR spectra of which, show no liberation of H2, regeneration of free borane/phosphine, or any other evidence of reaction over a 66 hour period.
Electrochemical studies
Cyclic voltammetric studies of 1–3 were performed in the non-coordinating solvent CH2Cl2 using [nBu4N][B(C6F5)4] as the added electrolyte at a glassy carbon electrode (GCE). In all three cases a one-electron, reduction process is observed with quasi-reversible (moderately fast) electron transfer kinetics. However for 1 (Fig. 4a) the process appears to be chemically irreversible (but not electrochemically irreversible) at scan rates up to 2.0 V s−1 due to fast chemical follow-up kinetics cf. the rate of electron transfer, while for both 2 (Fig. 4b) and 3 (Fig. 4c) an associated oxidation wave is observed indicating that the radical anions 2˙− and 3˙− are sufficiently stable on the electrochemical timescale for their subsequent (re-)oxidation to be observable.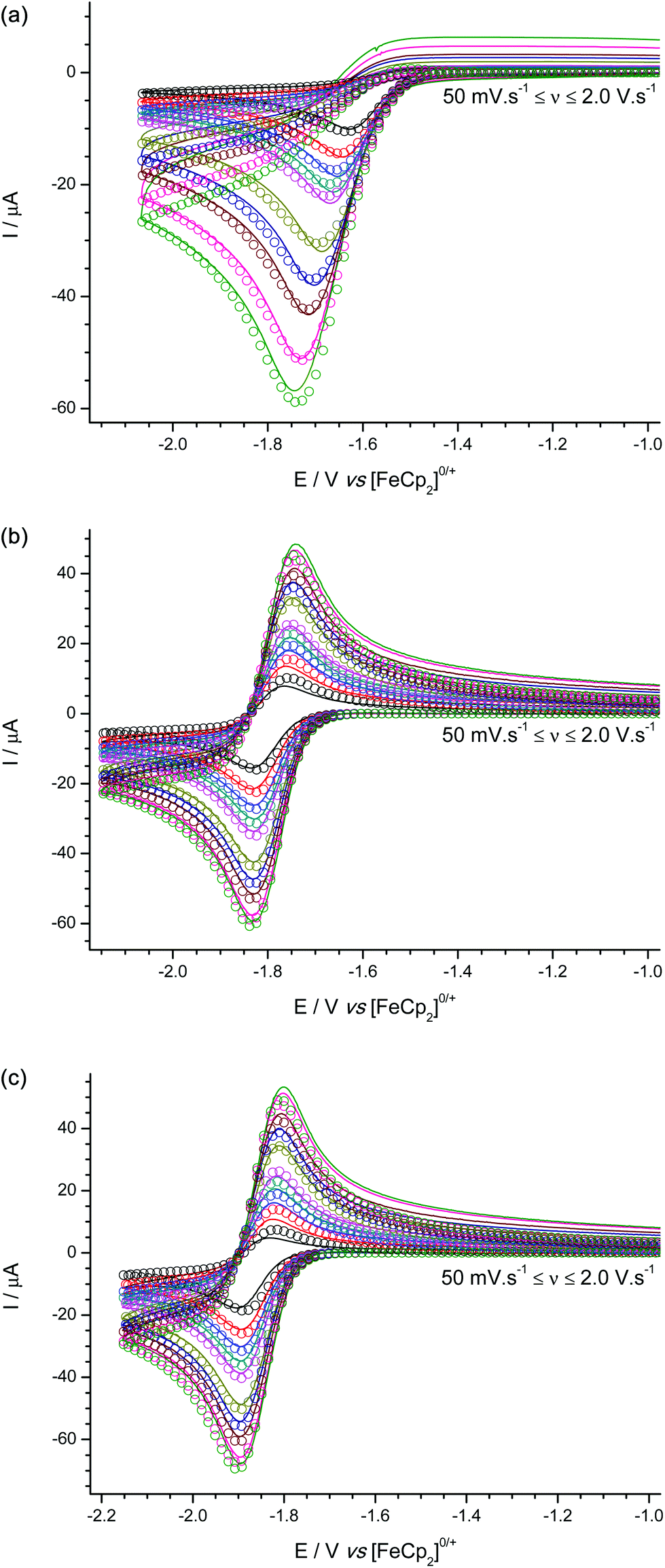 | ||
| Fig. 4 Experimental (line) and simulated (open circles) cyclic voltammograms for the reduction of (a) 1, (b) 2, and (c) 3. | ||
Based on our previous experience from an electrochemical study of B(C6F5)3,31 we performed digital simulations of the experimental voltammetric data modelled using an EC-mechanism (i.e. a reversible, heterogeneous electron transfer step followed by an irreversible, homogeneous chemical step which generates electro-inactive products. Other postulated mechanisms produced a poor fit to the data). These digital simulations allowed us to extract pertinent mechanistic parameters such as the formal redox potentials and charge transfer coefficients (E° and α respectively) and kinetic parameters for the electron transfer (k°) and follow-on chemical step (kf) as shown in Table 1.
| 1 | 2 | 3 | ||
|---|---|---|---|---|
| a Diffusion constants (D) obtained via1H and 19F DOSY NMR spectroscopy. b k f values are modelled as a pseudo first-order process. | ||||
| BArF18 + e− = BArF18˙− | E° vs. [FeCp2]0/+/V | −1.61 ± 0.01 | −1.79 ± 0.01 | −1.85 ± 0.01 |
| α | 0.419 | 0.498 | 0.468 | |
| k°/cm s−1 | 4.56 × 10−3 | 2.61 × 10−2 | 2.22 × 10−2 | |
| BArF18˙− ⇒ ‘decomposition’ |
k
f/s−1b |
≥25 | 1.92 × 10−2 | 8.08 × 10−2 |
|
D(BArF18) = D(BArF18˙−)/cm2 s−1a |
3.76 × 10−5 | 1.13 × 10−5 | 1.13 × 10−5 | |
| B{2,5-(CF3)2C6H3}33 | |
|---|---|
| Empirical formula | C24H9BF18 |
| Formula weight | 650.12 |
| Temperature/K | 100 |
| Crystal system | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a/Å | 7.2951(5) |
| b/Å | 10.6358(7) |
| c/Å | 15.9794(11) |
| α/° | 85.950(4) |
| β/° | 86.582(4) |
| γ/° | 74.846(4) |
| Volume/Å3 | 1192.64(14) |
| Z | 2 |
| ρ calc/mg mm−3 | 1.810 |
| μ/mm−1 | 0.204 |
| F(000) | 640.0 |
| Crystal size/mm3 | 0.30 × 0.06 × 0.02 |
| Radiation | Mo Kα (λ = 0.71075 Å) |
| 2Θ range for data collection | 3.974 to 54.96° |
| Index ranges | −9 ≤ h ≤ 8, −13 ≤ k ≤ 13, −20 ≤ l ≤ 20 |
| Reflections collected | 17457 |
| Independent reflections | 5459 [Rint = 0.0820, Rsigma = 0.0575] |
| Data/restraints/parameters | 5459/0/388 |
| Goodness-of-fit on F2 | 1.055 |
| Final R indexes [I ≥ 2σ(I)] | R 1 = 0.0555, wR2 = 0.1508 |
| Final R indexes [all data] | R 1 = 0.0712, wR2 = 0.1631 |
| Largest diff. peak/hole/e A−3 | 0.56/−0.39 |
The formal reduction potentials (E°) suggest that 1 is the most electrophilic of the three boranes, while 3 is the least. While the difference in electrophilicity between 2 and 3 is consistent with a simplified view of the inductive electron withdrawing effects of the different meta- and para-(CF3) group positions based on Hammett parameters {σmeta(CF3) = 0.43 vs. σpara(CF3) = 0.54}32 the relative electrophilicity of 1 cannot be similarly rationalized. This is in part because simple Hammett parameters for ortho-(CF3) substituents do not satisfactorily account for any additional steric and/or electronic effects. We can rationalize the reduced electrophilicity of 2 and 3 in comparison to 1 by considering the electronic effect of having ortho-CF3 groups present in 2 and 3. As noted above, the crystal structures of 2 and 3 show the ortho-(CF3) groups are positioned at sufficiently close distances above the central BC3 plane such that donation from the lone pairs on the fluorine atoms into the vacant boron pz orbital on boron could occur; such donation of electron density would be expected to reduce the electrophilicity of the boron centre, as is observed experimentally by our electrochemical measurements and indicated in electronic structure DFT calculations (vide infra).
Furthermore, the steric shielding of the boron centre by the ortho-(CF3) groups in 2 and 3 is qualitatively evident from the observation of more reversible redox processes in the cyclic voltammetric data. Quantitatively this is shown in the values of the rate constants (kf) obtained from voltammetric digital simulation for the radical anion decomposition step, which is assumed to proceed in a similar fashion as was previously ascertained for the analogous [B(C6F5)3]˙− intermediate via reaction between solvent molecules and the boron centre in the reduced radical anion intermediates, 1˙−, 2˙−, or 3˙−.31 The value of kf is at least three orders of magnitude greater for the decomposition of 1˙− than for 2˙− or 3˙−, where, in the latter two cases, the presence of ortho-CF3 groups provides significant steric shielding to the boron centre. It is worth noting here that these findings demonstrate the ease with which synthetic chemists working in this area can gain powerful insights into the chemistry of Lewis acidic species by the application of simple, rapid electrochemical characterization techniques in addition to the more ubiquitous crystallographic and spectroscopic characterization techniques. Simple examination of the shape and position of the voltammetry of each borane, obtained in a 20 minute experiment using inexpensive equipment can tell us qualitatively that in comparison to 1, boranes 2 and 3 are more sterically hindered, and less electrophilic, and therefore less Lewis acidic and less likely to be active FLP components for H2 activation. The voltammetry even allows us to infer why this is so, given that the only thing boranes 2 and 3 have that 1 does not have and that could simultaneously sterically shield the borane and reduce the electronic demand at the boron centre are the o-CF3 groups.
In comparison with the archetypal Lewis acid, B(C6F5)3 {which is observed under the same conditions as a quasi-reversible reduction at E° = −1.518 V vs. [FeCp2]0/+ (see Fig. S2†)}, boranes 1–3 are all less electrophilic; while [B(C6F5)3]˙− is more stable (smaller kf) with respect to follow-on decomposition reactions than 1˙−, but still considerably less stable than the ortho-(CF3) stabilized 2˙− and 3˙−.
For the terminal hydrides Na[1-H]–Na[3-H] (Fig. 5a–c) oxidations occurred with peak potentials of +1.08, +1.31, +1.13 V vs. [FeCp2]0/+ at a scan rate of 100 mV s−1 respectively; with no evidence of electroactive product species (such as the parent boranes) being regenerated in sufficient quantities to be observed.
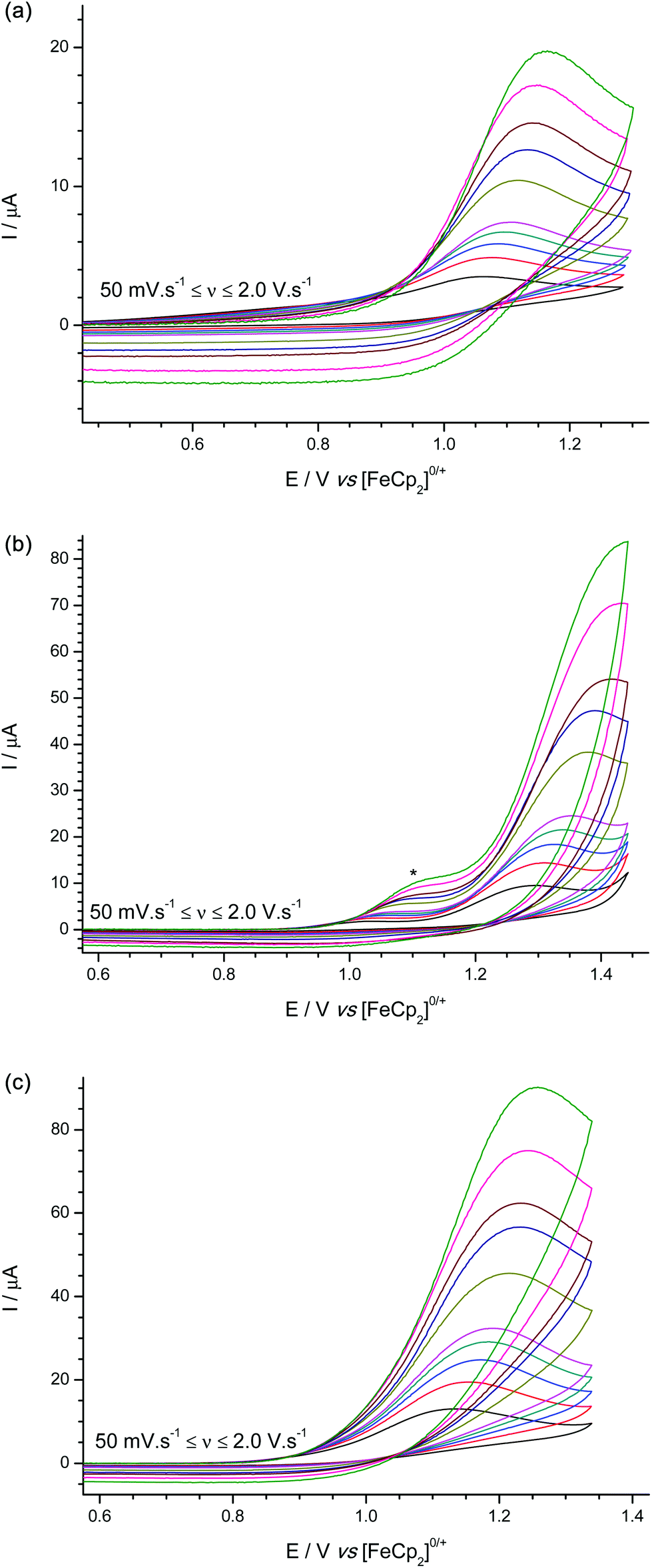 | ||
| Fig. 5 Experimental cyclic voltammograms for the oxidation of (a) Na[1-H], (b) Na[2-H], and (c) Na[3-H] shoulder (*) visible at the higher scan rates due to trace impurity in the solvent/electrolyte. | ||
The bridging hydride [tmpH][(μ-H)(1)2] (Fig. 6) oxidation is observed as a shoulder on the edge of the solvent window, at ca. +1.55 V vs. [FeCp2]0/+ at a scan rate of 100 mV s−1 (observable distinct to the solvent/electrolyte breakdown at scan rates below 1.0 V s−1), trace amounts of the terminal hydride species [1-H]− are also observed. Following oxidation of [(μ-H)(1)2]−, sweeping to negative potentials results in observation (at 100 mV s−1 scan rate and above) of an irreversible reduction wave at ca. −1.7 V vs. [FeCp2]0/+ characteristic of the reduction wave observed for the parent borane 1.
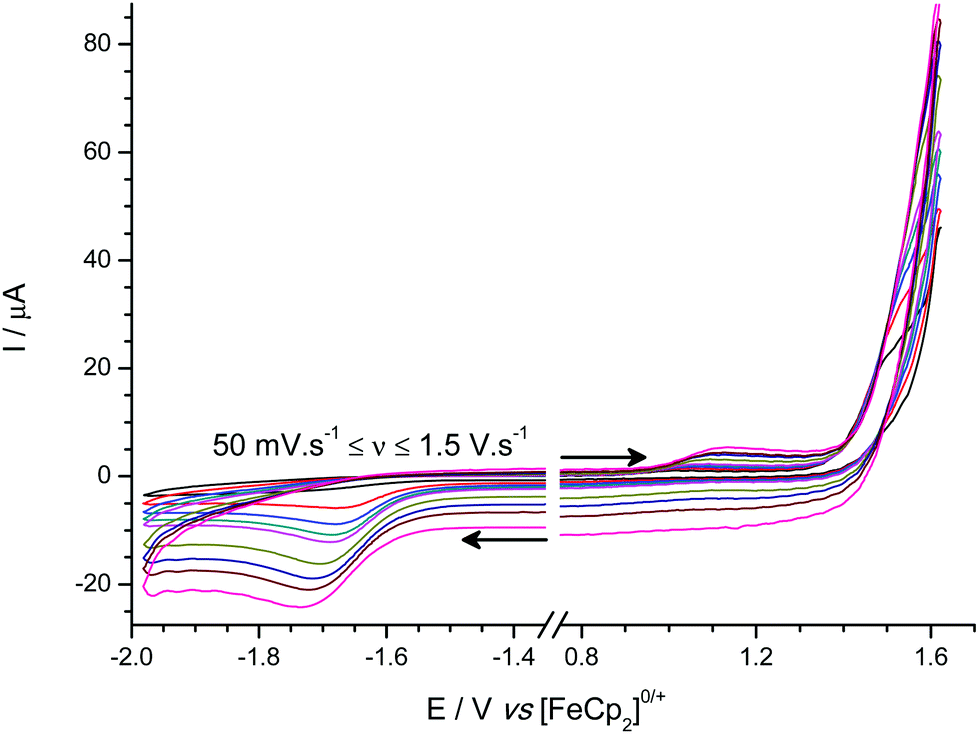 | ||
| Fig. 6 Experimental cyclic voltammogram(s) for the oxidation of [tmpH][(μ-H)(1)2]. | ||
In comparison to our previous studies on [HB(C6F5)3]−,25 all of the terminal-hydrides oxidize at more positive potentials, whilst the bridging-hydride species, [(μ-H)(1)2]−, is oxidized at an even more positive potential approaching that of the direct oxidation of H2 at a GCE (ca. +1.5 V vs. [FeCp2]0/+), and is effectively behaving as an electrolyte under these conditions.
Computational studies
To further our understanding of the tris{bis(trifluoromethyl)phenyl}borane isomers 1–3 we have investigated them and their associated radical-anions 1˙−–3˙− using density functional theory (DFT).For all the neutral boranes 1–3 the LUMOs (Fig. 7) showed a high degree of boron pz character (19, 22, 19% respectively, the majority of the remaining contributions being from the phenyl π-systems) as do the SOMOs (Fig. S4†) for the radical-anions 1˙−–3˙−.
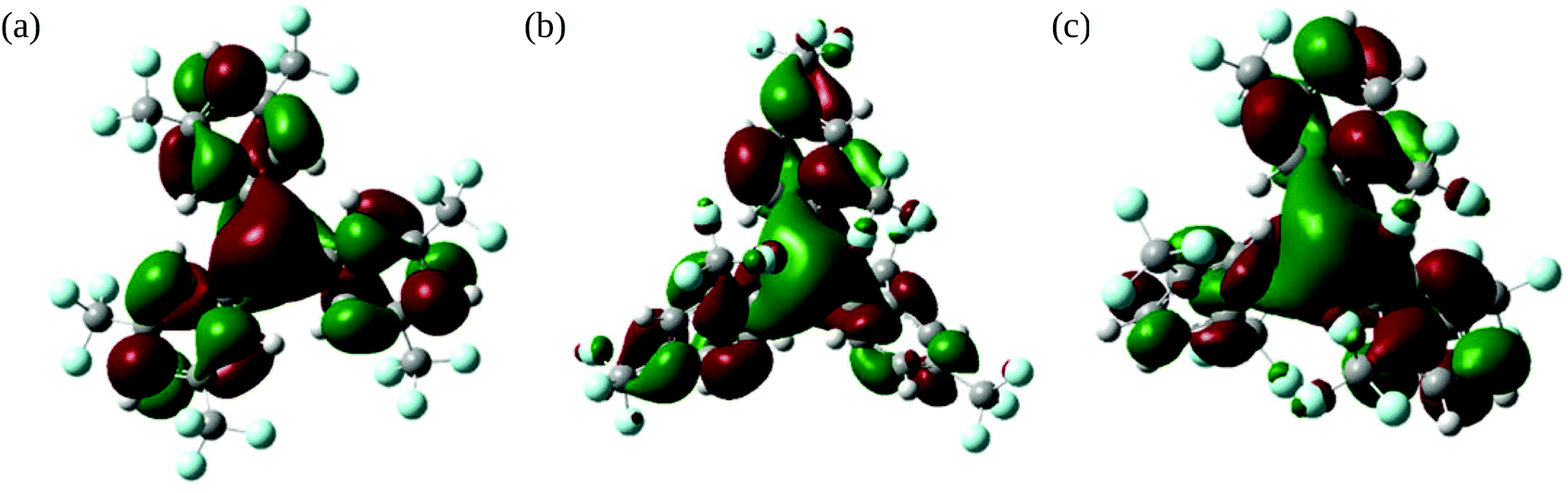 | ||
| Fig. 7 Lowest unoccupied molecular orbitals (LUMOs) for (a) 1, (b) 2, and (c) 3. | ||
Total molecular energy calculations indicate that the ground state of 1 is lower in energy than those of 2 and 3 by ca. 43 kJ mol−1. Analysis of calculated Mulliken atomic charges for 1–3, show that any stabilisation of 1 compared to 2 and 3 cannot be attributed to electron withdrawing effects of the aryl rings; and therefore may be attributed to the reduced steric hindrance caused by the lack of ortho-CF3 groups in 1.
As noted previously, the ortho-CF3 groups in 2 and 3 are orientated such that there is potential for B⋯F bonding interactions. Such interactions are clearly identified by the calculated bonding parameters: the B⋯F bonding parameter for 2 is 0.065, whilst for 3 it is 0.076 (in both cases averaged over all contributing B⋯F pairs). As expected, in 1, where no interaction occurs, the B⋯F bonding parameter to the meta-CF3 groups is <0.001.
Conclusions
We have investigated three structural isomers of tris{bis(trifluoromethyl)phenyl}borane in terms of both their electrochemical redox chemistries and also as the Lewis acidic components of an FLP for the heterolytic cleavage of H2.The two isomers of tris{bis(trifluoromethyl)phenyl}borane that incorporate ortho-CF3 groups were not found to be active as the Lewis acidic component of FLPs for H2 cleavage reactions. This lack of reactivity is due to a combination of, kinetic effects resulting from in part steric shielding of the boron centre; but also quenching of the boranes’ electrophilicity through B⋯F bonding interactions, which are quantified by DFT calculations and electrochemical measurements. Whilst electrochemical studies show that all three isomers are less electrophilic than the archetypal Lewis acid B(C6F5)3 the link between Lewis acidity, activity towards H2 in an FLP, and electrophilicity, as shown in our previous studies20,25,26 and those of other groups33–35 is complex and requires further study.
The direct synthesis of all three tris{bis(trifluoromethyl)phenyl}borohydride species under mild conditions allows for their reduction potentials to be measured, and their propensity for combined electrochemical-frustrated Lewis pair catalysis to be screened in a straightforward manner. The oxidation potentials of all three terminal borohydrides studied were found to be more positive than that of [HB(C6F5)3]− yet are still less than the potential required for the direct oxidation of H2 at a GCE under identical conditions.
Electrochemical studies of the bridging hydride formed when H2 is cleaved by the B{3,5-(CF3)2C6H3}3/tmp FLP, show that this species is oxidized at comparable potentials to that of the direct oxidation of H2. However, following oxidation, the regeneration of the parent borane species is clearly observed, which is not the case for any of the terminal borohydride Na[1-H]–Na[3-H] species studied.
What this report demonstrates is the utility of electrochemical characterization methods to enable synthetic chemists to rapidly screen prospective new Lewis acids using simple electrochemical techniques to gain insights into the chemical behaviour of new species. It also provides insights to guide the design of new Lewis acids for researchers wishing to employ the combined electrochemical-frustrated Lewis pair activation of H2, which is the focus of our ongoing studies.
Experimental
All reactions and manipulations were performed under an atmosphere of dry, oxygen-free N2, using either standard Schlenk techniques or in either a MBraun UNIlab or LABmaster glovebox. All solvents were dried prior to use by refluxing over an appropriate drying agent {Na/benzophenone for petroleum ether (b.p 40–60 °C) and diethyl ether; Na for toluene; CaH2 for dichloromethane}, collected by distillation under an inert N2 atmosphere and stored over 4 Å molecular sieves prior to use. All other reagents were obtained from commercial suppliers and used as received.NMR Spectra were obtained on either a Bruker Avance III 500 MHz or Bruker AV 400 MHz spectrometer, all deuterated solvents were dried over 4 Å molecular sieves prior to use. For 1H spectra residual protio-solvent was used as an internal standard; for 13C the solvent resonance(s) were used as an internal standard;36 for 19F spectra CFCl3 was used as an external standard; for 11B spectra BF3·Et2O was used as an external standard. 1H and 19F DOSY experiments were performed on a Bruker Avance III 500 MHz spectrometer equipped with a broadband multinuclear probe, using a longitudinal eddy current delay incorporating bipolar gradients for diffusion and spoil gradients (ledbpgp2s) pulse sequence.37
Mass spectrometry was performed by the EPSRC Mass Spectrometry Service at the University of Swansea for 3, or by Dr L. Haigh at Imperial College using a Micromass Autospec Premier spectrometer for 2. Elemental analyses were performed by Mr S. Boyer of the Elemental Analysis Service at London Metropolitan University.
Single crystals of 3 were grown by slow diffusion of a saturated CH2Cl2 solution of the compound into n-hexane; data collection and processing was performed at the UK National Crystallographic Service at the University of Southampton.30 Using Olex2,38 the structure was solved and space group assigned with SuperFlip/EDMA39 using charge flipping, and then refined with the ShelXL version 2014/740 refinement program using least squares minimization.
CCDC 1061234 contains the supplementary crystallographic data for this paper.
Electrochemical studies were carried out using a Metrohm Autolab PGSTAT302N potentiostat linked to a computer running Metrohm Autolab NOVA version 1.11 software, in conjunction with a three electrode cell comprising: a glassy carbon disc working electrode (Bioanalytical Systems Inc., ca. 7.0 mm2 area calibrated using the [FeCp2]0/+ redox couple), a platinum wire (99.99% purity) counter electrode, and a silver wire (99.99% purity) pseudo-reference electrode; all electrodes were polished with 0.3 μm α-alumina and dried prior to use. All electrochemical measurements were performed at ambient temperature under a dry N2 atmosphere, in CH2Cl2 containing 0.05 M [nBu4N][B(C6F5)4] as the supporting electrolyte and between 1.0 and 2.0 mM of the analyte species of interest. Cyclic voltammetric measurements were iR-compensated using positive-feedback to within 85 ± 5% of the uncompensated solution resistance. [nBu4N][B(C6F5)4] was synthesized according to published methods.41 All potentials were referenced to the [FeCp2]0/+ redox couple, which was added as an internal standard. Simulations of electrochemical processes were performed using ElchSoft DigiElch version 7.096 software.42
DFT calculations were performed using the Gaussian 09 computational package.43 Geometry optimization calculations have been carried out using the three-parameter exchange functional of Becke44 (B3) and the correlation functional of Lee, Yang, and Parr (LYP), B3LYP.45 The 6-311+G(d,p) basis set has been implemented for all atoms.46 Structures were geometry optimized in the gas phase with the default convergence criteria and confirmed as minima through frequency calculations. All optimized structures were confirmed as minima by frequency analysis with thermodynamic properties extracted for the gas phase at 298.15 K and 1 atm. Bonding parameters between B and F atoms were calculated as the absolute values of the associated non-diagonal elements of the condensed to atoms electron density matrix. Density matrixes were computed in a separate calculation taking into consideration basis set superposition error (BSSE) correction using an unrestricted Hartree–Fock (H–F) calculation with counterpoise (CP) correction approach as implemented in Gaussian suite.
B{3,5-(CF3)2C6H3}3118,27 and [tmpH][(μ-H)(B{3,5-(CF3)2C6H3}3)2] [tmpH][(μ-H)(1)2]18 were synthesized as previously reported.
B{2,4-(CF3)2C6H3}32
2,4-Bis(trifluoromethyl)bromobenzene (2.00 g, 1.16 cm3, 6.83 mmol) and Et2O (100 cm3) were combined and the solution cooled to −77 °C. With the aid of rapid stirring, nBuLi (2.87 cm3, 7.17 mmol, 2.5 M in hexanes) was added slowly by means of a syringe. Following one hour of stirring, BCl3 (2.28 cm3, 2.28 mmol, 1.0 M in heptane) was syringed into the amber solution and the mixture permitted to warm slowly to room temperature. The volatiles were removed in vacuo and the off white residue extracted with CH2Cl2 (3 × 25 cm3) and filtered through Celite. Volatiles were removed under vacuum, and following a high vacuum sublimation step (10−6 mbar) at 85 °C, a pure white solid was obtained. Yield 1.33 g (2.04 mmol, 90%).1H NMR (400.4 MHz, CD2Cl2, 25 °C, δ): +8.06 (s, 3H, 3-H), +7.87 (d, 3H, 3JHH = 8 Hz, 5-H), +7.46 (d, 3H, 3JHH = 8 Hz, 6-H); 11B NMR (128.4 MHz, CD2Cl2, 25 °C, δ): +74.0 (br.s); 13C{1H} NMR (100.6 MHz, CD2Cl2, 25 °C, δ): +144.2 (br, 1-C), +135.9 (s, 6-CH), +134.2 (q, 2JCF = 34 Hz, 2/4-C), +133.7 (q, 2JCF = 34 Hz, 2/4-C), +127.9 (q, 3JCF = 3 Hz, 5-CH), +123.9 (q, 1JCF = 273 Hz, 2/4-CF3), +123.6 (sept., 3JCF = 3 Hz, 3-CH), +123.6 (q, 1JCF = 273 Hz, 2/4-CF3). 19F NMR (376.8 MHz, CD2Cl2, 25 °C, δ): −56.6 (s, 9F, 2-CF3), −63.8 s, 9F, 4-CF3). HRMS-EI (m/z): [M]+ calc. for C24H9BF18, 650.0510; found, 650.0491. Elemental analysis (calc. for C24H9B1F18): C 44.34 (44.48), H 1.40 (1.47).
B{2,5-(CF3)2C6H3}33
2,5-Bis(trifluoromethyl)bromobenzene (3.1 cm3, 17.9 mmol) and Et2O (100 cm3) were combined and cooled to −77 °C. nBuLi (11 cm3, 17.6 mmol, 1.6 M in hexanes) was added to the stirred solution. After one hour BCl3 (5.8 cm3, 5.8 mmol, 1.0 M in heptane) was added to the orange solution and the mixture permitted to slowly warm to room temperature. The volatiles were removed in vacuo and the pale yellow residue extracted with CH2Cl2 (3 × 25 cm3) and filtered (via cannula). The product is then recrystallized from CH2Cl2/n-hexane and isolated as a white micro-crystalline solid. Yield 2.89 g (4.44 mmol, 77%).1H NMR (500.21 MHz, CD2Cl2, 25 °C, δ): +7.95 (s, 3H, 3/4-H), +7.95 (s, 3H, 3/4-H), +7.47 (s, 3H, 6-H); 11B NMR (160.49 MHz, CD2Cl2, 25 °C, δ): +70.7 (br.s); 13C{1H} NMR (125.78 MHz, CD2Cl2, 25 °C, δ): +141.5 (br.s, 1-C), +137.1 (q, 2JCF = 33 Hz, 2/5-C), +133.3 (q, 2JCF = 33 Hz, 2/5-C), +132.4 (q, 3JCF = 3.7 Hz, 3/4/6-C), +129.3 (q, 3JCF = 3.7 Hz, 3/4/6-C), +127.8 (br.m, 3/4/6-C), +124.1 (q, 1JCF = 275 Hz, 2/5-CF3), +123.9 (q, 1JCF 273 Hz, 2/5-CF3); 19F NMR (470.67 MHz, CD2Cl2, 25 °C, δ): −56.5 (s, 9F, 2-CF3), −63.7 (s, 9F, 5-CF3). HRMS-APCI (m/z): [M − F]+ calc. for C24H9BF17, 631.0525; found, 631.0519. Elemental analysis (calc. for C24H9B1F18): C 44.22 (44.48), H 1.38 (1.47).
Na[HB{3,5-(CF3)2C6H3}3] Na[1-H]
To a solution of 1 (0.40 g, 0.62 mmol) in toluene (10 cm3) was added Na[HBEt3] (0.6 cm3, 0.6 mmol, 1.0 M in toluene), the reaction mixture was stirred for 6 hours to give a colourless solution. All volatiles were removed in vacuo, to give a white residue which was washed with petroleum ether (2 × 5 cm3) and dried in vacuo to give a white solid. Yield 0.40 g (0.59 mmol, 95%).1H NMR (500.21 MHz, CD3CN, 25 °C, δ): +7.70 (s, 6H, 2,6-H), +7.58 (s, 3H, 4-H), +3.66 (br.q, 1JHB = 84 Hz, 1H, HB); 11B NMR (160.49 MHz, CD3CN, 25 °C, δ): −9.1 (d, 1JBH = 88 Hz); 13C{1H} NMR (125.78 MHz, CD3CN, 25 °C, δ): +165.0 (q, 1JCF = 49 Hz, 1-C), +135.7 (s, 2,6-C), +130.1 (q, 2JCF = 32 Hz, 3,5-C), +126.0 (q, 1JCF = 272 Hz, CF3), +118.5 (s, 4-C); 19F NMR (470.67 MHz, CD3CN, 25 °C, δ): −63.0 (s, 18F, CF3). Elemental analysis (calc. for C24H10B1F18Na): C 42.95 (42.76), H 1.61 (1.50).
Na[HB{2,4-(CF3)2C6H3}3] Na[2-H]
To a solution of 2 (0.275 g, 0.42 mmol) in toluene (8 cm3) was added Na[HBEt3] (0.43 cm3, 0.43 mmol, 1.0 M in toluene), the reaction mixture was stirred for 4 hours to give a cloudy white suspension. The reaction mixture was concentrated in vacuo reducing its volume to ca. 2 cm3, and the product precipitated by addition of petroleum ether and cooling to −25 °C. The white solid was isolated by filtration and dried in vacuo. Yield 0.231 g (0.34 mmol, 81%).1H NMR (500.21 MHz, CD3CN, 25 °C, δ): +7.76 (s, 3H, 3-H), +7.47 (d, 3JHH = 7.9 Hz, 3H, 5-H), +7.11 (d, 3JHH = 7.9 Hz, 3H, 6-H), +4.06 (br.q, 1JHB = 93 Hz, 1H, BH); 11B NMR (160.49 MHz, CD3CN, 25 °C, δ): −15.33 (d, 1JBH = 93 Hz); 13C{1H} NMR (125.78 MHz, CD3CN, 25 °C, δ): +139.1 (s, 6-CH), +135.3 (q, 2JCF = 29 Hz, 2/4-C), +126.7 (q, 3JCF = 3.7 Hz, 5-CH), +126.7 (q, 1JCF = 275 Hz, 2/4-CF3), +126.3 (q, 2JCF = 32 Hz, 2/4-C), +126.2 (q, 1JCF = 272 Hz, 2/4-CF3), +122.6 (sept., 3JCF = 3.7 Hz, 3-CH),; 19F NMR (470.67 MHz, CD3CN, 25 °C, δ): −59.5 (d, JFH = 6.8 Hz, 9F, 2-CF3), −62.6 (s, 9F, 4-CF3). Elemental analysis (calc. for C24H10B1F18Na): C 42.83 (42.76), H 1.57 (1.50).
Na[HB{2,5-(CF3)2C6H3 }3] Na[3-H]
To a solution of 3 (0.275 g, 0.42 mmol) in toluene (8 cm3) was added Na[HBEt3] (0.43 cm3, 0.43 mmol, 1.0 M in toluene), the reaction mixture was stirred for 4 hours to give a colourless solution. The reaction mixture was concentrated in vacuo reducing its volume to ca. 2 cm3, and the product crystallized by addition of petroleum ether and cooling to −25 °C. The white crystalline solid was isolated by filtration and dried in vacuo. Yield 0.207 g (0.31 mmol, 74%).1H NMR (500.21 MHz, CD2Cl2, 25 °C, δ): +7.70 (d, 3JHH = 8.2 Hz, 3H, 3/4-H), +7.45 (d, 3JHH = 8.5 Hz, 3H, 3/4-H), +7.23 (br.s, 3H, 6-H), +3.06 (br.qq, 1JHB = 82 Hz, JHF ≈ 9.1 Hz, 1H, BH); 11B NMR (160.49 MHz, CD2Cl2, 25 °C, δ): −14.23 (d, 1JBH = 84 Hz); 13C{1H} NMR (125.78 MHz, CD2Cl2, 25 °C, δ): +134.8 (q, 2JCF = 28.0 Hz, 2/5-C), +133.7 (q, 3JCF = 3.7 Hz, 3/4/6-C), +132.3 (q, 2JCF = 31.0 Hz, 2/5-C), +127.1 (q, 1JCF = 275 Hz, 2/5-CF3), +126.3 (br.m, 3/4/6-C), +124.9 (q, 1JCF = 273 Hz, 2/5-CF3), +121.6 (q, 3JCF 3.9 Hz, 3/4/6-C); 19F NMR (470.67 MHz, CD2Cl2, 25 °C, δ): −59.3 (d, JFH = 7.6 Hz, 9F, 2-CF3), −63.6 (s, 9F, 5-CF3). Elemental analysis (calc. for C24H10B1F18Na): C 42.93 (42.76), H 1.61 (1.50).
Acknowledgements
G. G. W. and A. E. A. thank the Royal Society for financial support by University Research Fellowships. E. J. L. thanks the EPSRC for financial support by a DTA studentship under grant number EP/J500409. The research leading to these results has received funding from the European Research Council under the ERC Grant Agreement no. 307061 (PiHOMER).We thank Dr C. MacDonald (University of East Anglia) for assistance with the 1H and 19F DOSY NMR experiments.
We acknowledge the use of the EPSRC funded National Chemical Database Service hosted by the Royal Society of Chemistry, and the EPSRC UK National Mass Spectrometry Facility (NMSF) at the University of Swansea. We thank the EPSRC UK National Crystallography Service at the University of Southampton for the collection of the crystallographic data.30 We thank the Research Computing Service at the University of East Anglia for access to the high performance computing cluster.
References
- G. C. Welch and D. W. Stephan, J. Am. Chem. Soc., 2007, 129, 1880–1881 CrossRef CAS PubMed.
- D. W. Stephan, Dalton Trans., 2009, 3129–3136 RSC.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed.
- D. W. Stephan, Comp. Inorg. Chem. II, 2013, 1, 1069–1103 CAS.
- D. W. Stephan and G. Erker, Chem. Sci., 2014, 5, 2625–2641 RSC.
- D. W. Stephan, Acc. Chem. Res., 2015, 48, 306–316 CrossRef CAS PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- P. A. Chase, G. C. Welch, T. Jurca and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 8050–8053 CrossRef CAS PubMed.
- K. Chernichenko, Á. Madarász, I. Pápai, M. Nieger, M. Leskelä and T. Repo, Nat. Chem., 2013, 5, 718–723 CrossRef CAS PubMed.
- H. Wang, R. Fröhlich, G. Kehra and G. Erker, Chem. Commun., 2008, 5966–5968 RSC.
- T. Mahdi and D. W. Stephan, J. Am. Chem. Soc., 2012, 136, 15809–15812 CrossRef PubMed.
- D. J. Scott, M. J. Fuchter and A. E. Ashley, J. Am. Chem. Soc., 2014, 136, 15813–15816 CrossRef CAS PubMed.
- G. Ménard, T. M. Gilbert, J. A. Hatnean, A. Kraft, I. Krossing and D. W. Stephan, Organometallics, 2013, 32, 4416–4422 CrossRef.
- Z. Lu, Y. Wang, J. Liu, Y. Lin, Z. H. Li and H. Wang, Organometallics, 2013, 32, 6753–6758 CrossRef CAS.
- R. L. Melen, Chem. Commun., 2014, 50, 1161–1174 RSC.
- S. C. Binding, H. Zaher, F. M. Chadwicka and D. O'Hare, Dalton Trans., 2012, 41, 9061–9066 RSC.
- D. J. Scott, M. J. Fuchter and A. E. Ashley, Angew. Chem., Int. Ed., 2014, 53, 10218–10222 CrossRef CAS PubMed.
- T. J. Herrington, A. J. W. Thom, A. J. P. White and A. E. Ashley, Dalton Trans., 2012, 41, 9019–9022 RSC.
- J. M. Farrell, J. A. Hatnean and D. W. Stephan, J. Am. Chem. Soc., 2012, 134, 15728–15731 CrossRef CAS PubMed.
- E. J. Lawrence, T. J. Herrington, A. E. Ashley and G. G. Wildgoose, Angew. Chem., Int. Ed., 2014, 53, 9922–9925 CrossRef CAS PubMed.
- G. Ménard and D. W. Stephan, Angew. Chem., Int. Ed., 2012, 53, 8272–8275 CrossRef PubMed.
- E. R. Clark and M. J. Ingleson, Angew. Chem., Int. Ed., 2014, 53, 11306–11309 CrossRef CAS PubMed.
- A. M. Chapman, M. F. Haddow and D. F. Wass, J. Am. Chem. Soc., 2011, 133, 18463–18478 CrossRef CAS PubMed.
- X. Xu, G. Kehr, C. G. Daniliuc and G. Erker, J. Am. Chem. Soc., 2015, 137, 4550–4557 CrossRef CAS PubMed.
- E. J. Lawrence, V. S. Oganesyan, D. L. Hughes, A. E. Ashley and G. G. Wildgoose, J. Am. Chem. Soc., 2014, 136, 6031–6036 CrossRef CAS PubMed.
- E. J. Lawrence, R. J. Blagg, D. L. Hughes, A. E. Ashley and G. G. Wildgoose, Chem. – Eur. J., 2015, 21, 900–906 CrossRef PubMed.
- E. L. Kolychev, T. Bannenberg, M. Freytag, C. G. Daniliuc, P. G. Jones and P. D. M. Tamm, Chem. – Eur. J., 2012, 18, 16938–16946 CrossRef CAS PubMed.
- S. M. Cornet, K. B. Dillon, C. D. Entwistle, M. A. Fox, A. E. Goeta, H. P. Goodwin, T. B. Marder and A. L. Thompson, Dalton Trans., 2003, 4395–4405 RSC.
- W. V. Konze, B. L. Scott and G. J. Kubas, Chem. Commun., 1999, 1807–1808 RSC.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683–689 RSC.
- E. J. Lawrence, V. S. Oganesyan, G. G. Wildgoose and A. E. Ashley, Dalton Trans., 2013, 42, 782–789 RSC.
- D. H. McDaniel and H. C. Brown, J. Org. Chem., 1958, 23, 420–427 CrossRef CAS.
- T. A. Rokob, A. Hamza and I. Pápai, J. Am. Chem. Soc., 2009, 131, 10701–10710 CrossRef CAS PubMed.
- I. B. Sivaev and V. I. Bregadze, Coord. Chem. Rev., 2014, 270–271, 75–88 CrossRef CAS PubMed.
- Z. M. Heiden and A. P. Lathem, Organometallics, 2015, 34, 1818–1827 CrossRef CAS.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- D. H. Wu, A. D. Chen and C. S. Johnson, J. Magn. Reson., Ser. A, 1995, 115, 260–264 CrossRef CAS.
- L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2015, 71, 59–75 CrossRef CAS PubMed; O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS; L. Palatinus and A. van der Lee, J. Appl. Crystallogr., 2008, 41, 975–984 CrossRef; L. Palatinus, S. J. Prathapab and S. van Smaalen, J. Appl. Crystallogr., 2012, 45, 575–580 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef CAS PubMed; G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef PubMed.
- S. Lancaster, ChemSpider Synthetic Pages, 2003, 10.1039/SP215, http://cssp.chemspider.com/215; T. E. Krattf, Boulder Scientific Company, Eur. Pat, 1186608, 2002 CrossRef CAS PubMed; R. J. LeSuer, C. Buttolph and W. E. Geiger, Anal. Chem., 2004, 76, 6395–6401 CrossRef CAS PubMed.
- DigiElch-Professional, ElechSoft, Kleinromstedt, Germany Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta Jr., F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Comprising: annotated ORTEP plot of the X-ray crystal structure of 3; cyclic voltammograms of B(C6F5)3 under corresponding conditions; further computational data. CCDC 1061234. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt01918d |
| This journal is © The Royal Society of Chemistry 2016 |