
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Restructuring of supported Pd by green solvents: an operando quick EXAFS (QEXAFS) study and implications for the derivation of structure–function relationships in Pd catalysis
Mark A.
Newton
*a,
John B.
Brazier
b,
Elena M.
Barreiro
b,
Hermann
Emerich
c,
Luis A.
Adrio
b,
Christopher J.
Mulligan
b,
Klaus
Hellgardt
d and
King Kuok (Mimi)
Hii
 *b
*b
aDepartment of Physics, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK. E-mail: m.newton2@warwick.ac.uk
bDepartment of Chemistry, Imperial College London, South Kensington, London SW7 2AZ, UK. E-mail: mimi.hii@imperial.ac.uk
cSwiss-Norwegian Beamline (SNBL), European Synchrotron Radiation Facility (ESRF), 6 rue Jules Horowitz, 38000, Grenoble, France
dDepartment of Chemical Engineering, Imperial College London, South Kensington, London SW7 2AZ, UK
First published on 14th November 2016
Abstract
Transmission electron microscopy (TEM) is commonly used as an ex situ technique to determine structural changes by comparing images of catalyst particles before and after a reaction. This requires the use of an alcoholic solvent to disperse the particles on a grid. In this work, we will show that Pd catalysts can be transformed during the procedure, by using EXAFS to determine the structure of Pd catalysts in different environments (as dry or wet samples). Supported palladium nanoparticles exposed to aqueous ethanolic solution (50% w/v) are transformed to a common, reduced, and hydrogen-contaminated state, irrespective of their initial habit or support. Catalysts comprised of nanosize PdO are reduced at ca. 350 K, whereas samples comprised of very small (ca. ≤10 atoms) Pd particles react with the solvent at just above room temperature and agglomerate with considerable loss of dispersion. As such any potential benefits to catalysis sought through the synthesis of very highly dispersed metallic Pd supported upon a range of inorganic dispersants will be rapidly erased through the action of such solvents.
Introduction
The use of solvents is unquestionably an important issue for the chemical industry. Being the dominant component in many chemical processes, their removal, recycling, and destruction often incur significant operational costs. In recent years, the demand for ‘greener’ solvents has been further escalated by increasing environmental and regulatory pressures. The pharmaceutical industry, in particular, has made substantial investment in defining the green credentials of solvents in their search for more sustainable solutions,1–6 resulting in the emergence of several solvent selection guides in recent years. Without exception, primary alcohols and water score highly in these assessments.Within the diverse range of reactions enabled by palladium catalysts, mixtures of ethanol and water are often employed as reaction media, including: hydrogenation,7–9 oxidation of alcohols,10,11 Suzuki–Miyaura,12,13 Sonogashira coupling,14 and Heck reaction,15,16 among others. We have recently shown that an aqueous solution of ethanol (50% w/v) can be effective in the reduction of Al2O3 supported nano-size PdO under relatively mild conditions,17 although this process is subject to a considerable size dependence: PdO nanoparticles of less than ca. 3–4 nm diameter are considerably more resistant to reduction than larger particles. Moreover, the reduced Pd particles are significantly contaminated with interstitial hydrogen, which is known to interfere with catalytic processes and adsorption of reactants to surfaces.18–21
Recently, it has been shown that solvent molecules can interact with the surface of colloidal ZnO nanoparticles into organised structures.22 Indeed, it is well known that the choice of solvent used in the preparation of metal nanoparticles can have a big effect on their structure, morphology and resultant physical properties.23,24 Similarly, metal surfaces can rearrange during catalytic turnover, which may ultimately affect the morphology of the nanoparticle.
It has long been recognised the right support is critical for catalyst stability, reactivity and selectivity, through metal–support interactions.25–27 In this study, we employed (quick scanning) extended X-ray absorption fine structure QEXAFS spectroscopy to examine four Pd catalysts on different supports as they are subjected to a flowing ethanol/water solvent mixture (50% w/v). We are able to show that dynamic structural changes in their compositions and morphologies occur under relatively mild conditions.
Results and discussion
Four different types of 5 wt% Pd catalysts were examined, including 3 commercial samples procured from Johnson Matthey PLC, supported on γ-alumina (Pd/Al2O3, JM325), anatase titania (Pd/TiO2) and charcoal (Pd/C, JM490), as well as a sample of Pd/Al2O3 prepared at Imperial College (IC) London. The samples were initially examined by TEM (Fig. 1). Analysis of the images revealed that the metal-oxide (Al2O3 and TiO2) supported Pd catalysts have similar mean particle sizes centred around 4 nm diameter, with varying degrees of distribution. In contrast, a much broader distribution was found in the sample of Pd/C (JM490), consisting largely of smaller particle sizes of ca. 2.5–3 nm and a considerable tail of larger particles, extending to ca. 8–9 nm.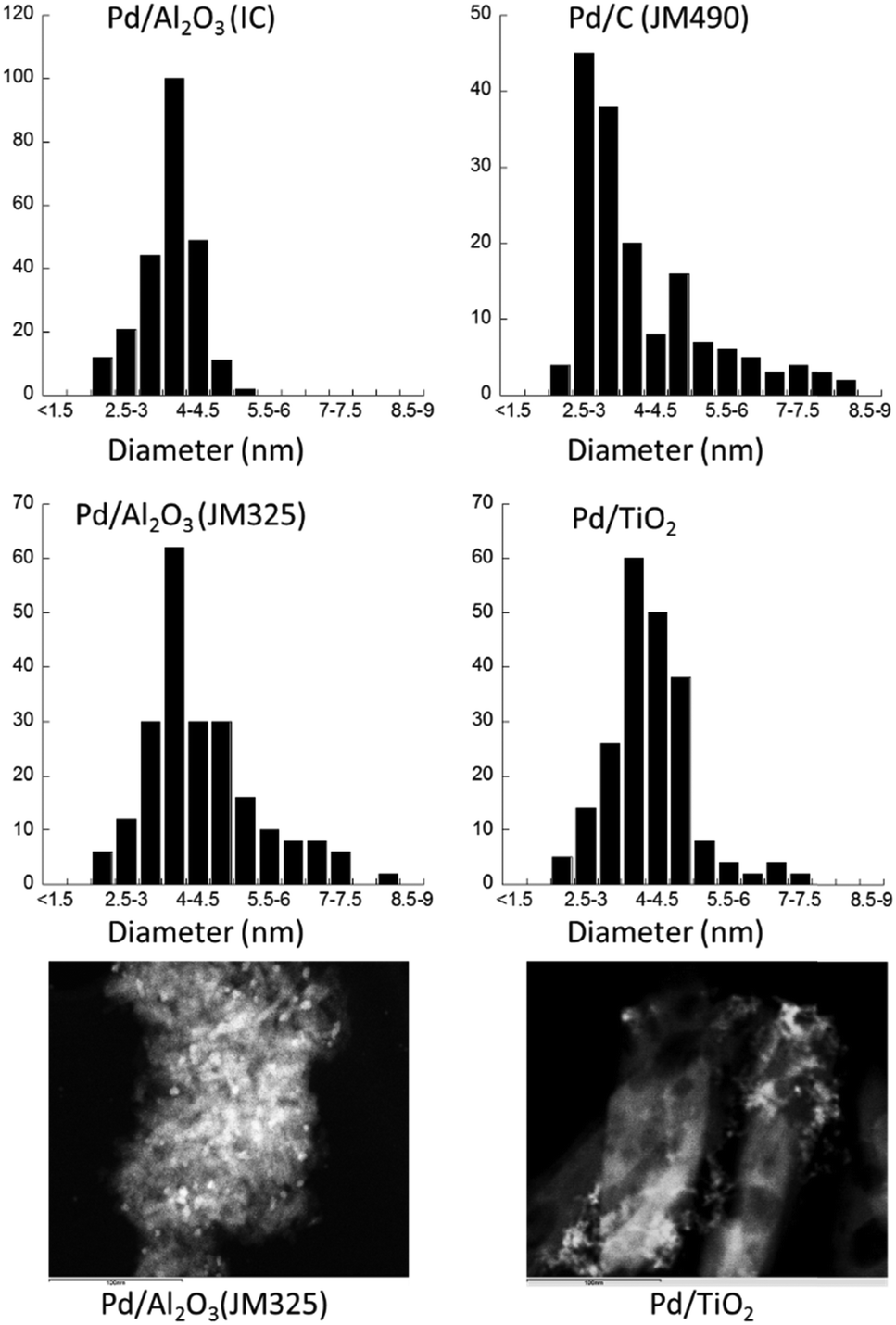 | ||
| Fig. 1 Particle size distributions obtained by TEM for each of the four supported Pd catalysts (all 5 wt% loading). Representative images for the Pd/Al2O3 (JM325) and Pd/TiO2 samples are also given. | ||
Carbon has the highest surface area among these supports (BET: 790 m2 g−1versus ca. 150–200 m2 g−1 for γ-alumina, and ca. 25 m2 g−1 for the anatase TiO2) and significant porosity, with average pore sizes of ca. 5 nm in diameter. Thus, the observed size distribution in Pd/C (JM490) is attributed to smaller and more populace Pd particles constrained within the pores of the support, whilst the large particles are those Pd that reside on the surface of the carbon particles.
The same samples were subsequently measured using Pd K-edge EXAFS as dry powders, which were compared with spectra of Pd foil and PdO, serving as references for Pd in the 0 and +2 oxidation states, respectively. The global oxidation state of each catalyst can thus be determined by their unique XAFS features near the absorption edge (Fig. 2); the spectra of Pd/C (JM490) and Pd/Al2O3 (IC) catalyst are very similar to the PdO reference sample, denoting that they are highly oxidised. In comparison, Pd/TiO2 and Pd/Al2O3 (JM325), were less oxidised, but also clearly not entirely reduced.
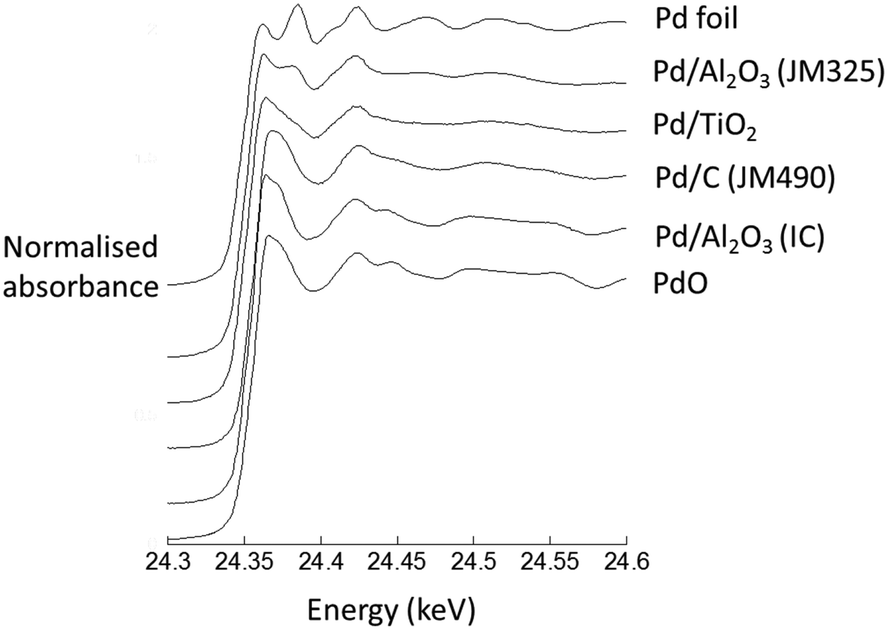 | ||
| Fig. 2 Normalised Pd K-edge XAFS for four different 5 wt% Pd catalysts in their native “dry” states. Spectra from a bulk PdO and a Pd foil are also given. | ||
Structural information can be derived by transforming the spectra into the k3-weighted Pd K-edge EXAFS (Fig. 3A) and their Fourier transformed (Fig. 3B) representations; data-fitting was achieved using EXCURV and indicated by the red lines.28 Structural and statistical information arising from these analyses are given in Table 1.
 | ||
| Fig. 3 (a) k3-Weighted Pd K-edge EXAFS derived from dry samples. (b) Fourier transforms of the k3-weighted data. Fits (from EXCURV28) are shown in red. | ||
| Sample | Element | CNa | R (Å) | DW (2σ2)c | R% | E F |
|---|---|---|---|---|---|---|
| a Co-ordination number (±10% of stated value). b Distance of the scattering atom from central atom (±1.5% of stated value). c Debye–Waller factor = 2σ2, where σ is the root mean square inter-nuclear separation (Å). EF = the edge position relative to the vacuum zero (Fermi energy, eV). R% = 33.37 = (∫[χT − χE]k3dk/[χE]k3dk) × 100 where χT and χE are the theoretical and experimental EXAFS and k is the photoelectron wave vector. Other parameters: AFAC, related to the proportion of electrons performing an EXAFS type scatter on absorption, is 1. The fitting range used was: k = 2.5–13 Å−1. | ||||||
| Pd/Al2O3 (IC) | O | 4.4 | 2.03 | 0.008 | 31.45 | 0.40 |
| Pd | 4.4 | 3.04 | 0.012 | |||
| Pd | 4.7 | 3.45 | 0.013 | |||
| Pd/C (JM 490) | O | 3.8 | 2.01 | 0.007 | 30.70 | 2.71 |
| Pd | 2.8 | 3.01 | 0.013 | |||
| Pd | 3.3 | 3.42 | 0.014 | |||
| Pd | 1.0 | 2.76 | 0.011 | |||
| Pd/TiO2 | O | 2.5 | 2.01 | 0.006 | 45.87 | 0.31 |
| Pd | 2.6 | 2.73 | 0.015 | |||
| Ti | 1.9 | 3.63 | 0.010 | |||
| Pd | 0.7 | 3.94 | 0.017 | |||
| Pd/Al2O3 (JM325) | O | 1.6 | 2.01 | 0.004 | 26.38 | 2.76 |
| Pd | 4.5 | 2.74 | 0.015 | |||
| Al | 2.3 | 3.43 | 0.010 | |||
The results afforded by the EXAFS data are striking. In contrast to the TEM analysis, the EXAFS characterisation divides the catalyst samples into two distinct groups: Pd/Al2O3 (IC) and Pd/C (JM 490) samples, that are essentially comprised of nanosize PdO; and the Pd/TiO2 and Pd/Al2O3 (JM325) that contain extremely small particles, with implied average atomicities of <10 atoms,29,30 and PdPd bond distances associated with a metallic Pd(0) state. Further, EXAFS analysis of the Pd/TiO2 sample is consistent with a structure similar to that previously determined by Bennett et al.,31 of Pd adsorbed upon a (1 × 2) reconstructed TiO2 (110) surface, i.e. six-atom Pd (111) rafts interacting with both lattice oxygen and Ti atoms. The Pd/Al2O3 (JM325) sample is best described as containing very small Pd clusters of ca. 10 atoms, interacting with oxygen from the support.
The clear contradiction between results derived from TEM and EXAFS for JM325 and Pd/TiO2 may be attributed to the sample preparation. EXAFS can be recorded directly using the dry samples (in this case, as a fixed plug bed loaded in a quartz tube). In contrast, the samples have to be suspended in isopropanol or ethanol before they are deposited on the grid before the TEM measurement can be made. As such the contradictory results point to the suspension process and/or the subsequent exposure of the wet sample to high energy electrons as capable of inducing considerable sintering of the Pd phase before TEM images can be obtained. This has significant implications for catalytic research, where TEM is routinely used as a characterisation technique.
The dry catalyst powders, mounted within the plug flow reactor designed for operando EXAFS studies,32 were exposed to 50% w/v EtOH/H2O at a flow rate of 0.1 mL min−1, whilst the temperature was increased at a rate of 1 K min−1. During this time, changes in the Pd catalyst were recorded spatially and temporally using transmission Pd K-edge quick EXAFS (QEXAFS): A 0.35 mm (vertical) × 3 mm (horizontal) X-ray beam was used to axially sample through the catalyst bed (5 mm) packed in a quartz tube (Fig. 4). Bi-directional QEXAFS was employed with a time per (uni-directional) spectrum (24 to 25.5 keV) of between 6 to 20 seconds to collect spectra sequentially along the catalyst bed at intervals of 0.5 mm, starting at the reactor inlet and ending at the outlet. This process was then repeated throughout the remainder of the experiment as the sample was heated from ambient temperature (298 K) to 350 K.
 | ||
| Fig. 4 Schematic representation of the relative sizes of the catalyst bed (5 mm), X-ray beam (0.35 mm) and the general method of experimentation employed to yield both spatial and temporal information regarding the development of the Pd catalysts within a single pass, plug-flow, environment. | ||
Fig. 5 shows the ends points of this experiment, in Fourier transform representation of the k3-weighted Pd K-edge EXAFS, measured at three axial positions within the catalyst bed (the inlet, the middle, and the outlet) at 350 K under the flowing solvent mixture. These can be compared with spectra of the pristine catalyst samples (Fig. 3). At the very inlet of the catalyst bed three of the supported catalyst systems – Pd/Al2O3 (IC), Pd/C (JM 490) and Pd/TiO2 – remain in a very similar state to that measured for the dry catalysts. In contrast, the highly dispersed Pd/Al2O3 (JM325) sample has been considerably changed; the FT spectrum indicating the presence of much larger and reduced Pd nanoparticles.
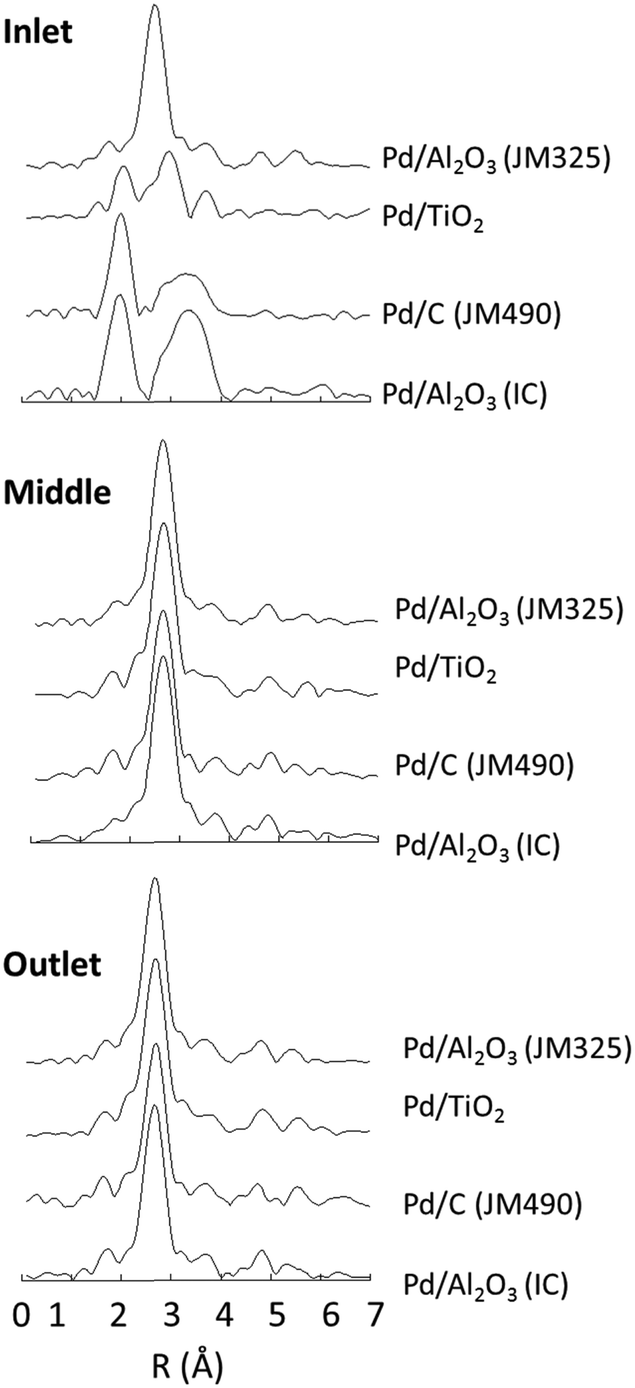 | ||
| Fig. 5 Axial variation of the final states of each catalyst after heating under 50% w/v EtOH/H2O, measured using Pd K-edge EXAFS (Fourier transform of the k3 weighted EXAFS data). | ||
At both middle and end of the catalyst beds the FT spectra recorded after exposure to the solvent flow are all found to be essentially identical; all the samples were transformed to a reduced, metallic state. Importantly, these results show that very small and highly dispersed Pd nanoparticles, such as that found in Pd/Al2O3 (JM325) and Pd/TiO2, facilely agglomerate into considerably larger Pd nanoparticles, such as those observed in the TEM images (Fig. 1), solely through the action of the solvent. As such, it may be concluded that the ethanol–water mixture is not only able to reduce the PdO phase, but is also able to rearrange the highly dispersed Pd to form more extended structures, irrespective of the support, and any Pd–support interactions that may exist.
The relative ease, and the extent to which each Pd sample is transformed by the action of the solvent is revealed by analysing the QEXAFS data collected during the spatio-temporal mapping of the catalyst beds as they are heated under the solvent flow. Fig. 6 shows an increase in the first shell coordination number (N1PdPd) due to fcc Pd(0) (Fig. 6a–d), and changes in the PdPd bond length (R1PdPd) associated with this scattering interaction (Fig. 6e). As may be expected, considerable variations were observed, which are related to the nature of the starting phase present.
 | ||
| Fig. 6 (a–d) Changes in 1st shell fcc PdPd coordination number (N1PdPd) for each catalyst (as indicated) during heating under flowing 50% w/v EtOH/H2O to 353 K. The solid red lines denote the temperature (K). Figure (e) shows the corresponding evolution of the 1st shell fcc PdPd bond distance (R1PdPd): filled black circles = Pd/Al2O3 (IC); open black circles = Pd/Al2O3 (JM 325); solid red circles = Pd/C (JM490); open blue circles = Pd/TiO2. | ||
The two samples that initially comprised of PdO as the sole or majority phase [Pd/C (JM490) and Pd/Al2O3 (IC)] only appear to reduce and form Pd nanoparticles at around 350 K, as indicated by the appearance of a “metallic” PdPd scattering interaction and subsequent augmentation of the corresponding coordination number (N1PdPd), which is consistent with our previous study.17
By contrast, changes in the other two samples [Pd/Al2O3 (JM325) and Pd/TiO2] are observed to occur at much lower temperatures (ca. 310 K) such that the N1PdPd values associated with the fcc Pd phases present attain values of between 9 and 10 before the full temperature ramp is completed. For example, in the case of Pd/Al2O3 (JM325) the persistence of N1PdPd values of <6 is not observed above ca. 330 K. Thus, the solvent-driven agglomeration of highly dispersed and essentially metallic Pd is considerably more facile than the solvent-induced reduction of the PdO phases present in the other two samples. The EXAFS obtained at the end of the experiment over most of the catalyst beds is also now in accord with the Pd particle sizes indicated by TEM. In the case of Pd/C (JM 490), the maximal value of N1PdPd attained by the sample is significantly lower (8 versus ca. 10). This is also consistent with the microscopy image of widely distributed particle sizes (Fig. 1), which we have attributed to the porous structure of the carbon support.
As all of these systems evolve into reduced, fcc, Pd(0) phases the bond length R1PdPd associated with the PdPd scattering interaction also gets longer before settling down between values of ca. 2.75 and 2.77 Å by the end of the heating process (Fig. 6e).
The value of N1PdPd for fcc metal systems may be related to the number of atoms per average particle, calculated by using the method reported by Jentys.30 Assuming a spherical morphology, the implied average particle diameter was derived (denoted by the black line in Fig. 7a and allowing for a ±10% error in the value of N1PdPd). Results due to dry Pd/TiO2 and Pd/Al2O3 (JM325) catalysts are shown in light and dark blue respectively. The light and dark grey regions depict the state of each catalyst (as indicated) after reaction with aqueous ethanol at 353 K. Conversely, Fig. 7b shows the correlation of R1PdPd with N1PdPd derived from the experimental data obtained from each of the EXAFS spectra collected along the catalyst beds at the end of the experiment, compared to the average value of R1PdPd derived from a Pd foil measured in parallel with the samples (solid line).
 | ||
| Fig. 7 (a) Shows the relationship between N1PdPd and the average fcc Pd particle size of the initial (blue) and final (grey) states of the catalysts; Fig. 7 (b) the correlations obtained between R1PdPd and N1PdPd along with the average value of R1PdPd returned from analysis of the reference foil: black solid circles = Pd/Al2O3; black open circles = Pd/Al2O3 (JM325); red = Pd/C; blue = Pd/TiO2. | ||
Within the scheme of quantification advanced by Jentys,30 the agglomeration induced by the ethanol/water solvent mix in the very highly dispersed and essentially metallic samples [Pd/TiO2 and Pd/Al2O3 JM325], are shown to be considerable. The average number of atoms per Pd particle is implied to increase by up to a factor of 150.
As with our previous analyses,17 the values of R1PdPd returned show that in each of these samples the fcc Pd phase formed under the ethanol/water mixture is significantly contaminated with atomic hydrogen that resides in the bulk of the Pd nanoparticles. On the basis of the work of McCaulley,33 Davis34 and co-workers, we estimate an average particle composition of ca. PdH0.17, and a co-existence of α- and β-Pd hydrides. The presence of these species also implies that the final size of the Pd nanoparticles depicted in Fig. 7 are likely to be an under-estimate, as earlier work by McCaulley33 showed that hydride formation in similar catalysts reduced the apparent value of N1PdPd by about 20% compared to hydrogen free Pd particles. Taking this into account, the final state of all the catalysts is in good agreement with the TEM images derived from the fresh samples (Fig. 1).
Conclusions
Despite their impressive green credentials, ethanol and water cannot be regarded as inert as far as Pd catalysis is concerned. When exposed to aqueous ethanol, supported Pd can undergo reduction to Pd(0) and significant agglomeration into nanoparticles with an average size of 4 nm, irrespective of the support (γ-alumina, anatase titania, or carbon) or their initial structure (phase, size, oxidised or reduced).In this work, we have shown that the use of these solvents during the preparation of Pd catalysts for TEM can led to erroneous results. In light of this, we recommend that the use of alcoholic solvents such as ethanol and isopropanol be avoided for the preparation of Pd samples for analysis.
The results presented in this study also have profound implications for the development of ‘size-controlled’ and ‘shape-selective’ catalysis.35–37 We have shown that a mixture of ethanol and water can cause considerable sintering and loss of Pd dispersion in initially highly dispersed Pd particles, under relatively mild conditions. Hence, any foreseen benefits of preparing such samples for catalytic reactions will be lost by employing such alcoholic solvents in these cases.
Material and methods
Catalysts
5 wt% Pd/Al2O3 (IC) was synthesised using wet impregnation of Pd(NO3)2 solutions (15.05 wt% Pd, Johnson Matthey PLC) diluted as appropriate. This solution was added to γ-Al2O3 (Alfa Aesar; pellets pre-ground/sieved to afford a 75–90 μm mesh fraction) in a dropwise manner until incipiently wet. The resulting solid was then dried overnight in air at 373 K, washed in H2O, and then dried again overnight. The samples were calcined in flowing air (1 K min−1 ramp to 773 K and held at temperature for 4 hours before cooling at 1 K min−1). Once cooled the samples were sieved again to a 75–90 μm mesh fraction before use.The other 5 wt% Pd catalysts: Pd/C (JM490): Lot No. M09465, Pd/TiO2: Batch No. LR324, and Pd/Al2O3 (JM325): Lot No. LV0093, were provided by Johnson Matthey PLC and used as received.
Transmission electron microscopy
TEM measurements were carried out at Imperial College London. The catalysts were ground and dispersed in isopropanol prior to being applied to a TEM grid. Images were collected using a Jeol 2100F microscope. For each catalyst, between 150 and 200 particles were sampled to create the distribution graphs depicted in Fig. 1.Operando spatially resolved QEXAFS and overall experimental method
QEXAFS measurements were performed at the Swiss-Norwegian Beamline (SNBL) at the European Synchrotron radiation facility, using a Si (111) double crystal monochromator and ion chambers for detection of X-ray absorption, normalisation, and energy scale (Pd foil) calibration. The dimensions of the X-ray beam were 0.35 mm (vertical) × 3 mm (horizontal) sampling through the 4 mm i.d. catalyst bed. Bi-directional QEXAFS was employed with a time per (uni-directional) spectrum (24 to 25.5 keV) of between 6 to 20 seconds.QEXAFS spectra were collected along the catalyst bed at intervals of 0.5 mm, starting at the inlet and ending at the outlet (see Fig. 4). This process was then repeated throughout the remainder of the experiment. The continuous flow reactor and the overall experimental protocols followed in this study have been fully described elsewhere.1 The reactor was mounted vertically to aid smooth solvent flow and removal of air from the catalyst bed.
Data reduction was made using “Prestopronto”38 XAFS software or PAXAS39 prior to EXAFS analysis using EXCURV.28
Solvent components were individually degassed using nitrogen gas bubbled through the liquids followed by sonication after mixing. The sample was packed into a quartz tube – to yield beds of ca. 5 mm in length – and secured on either side using γ-Al2O3 of a smaller particle size and then quartz sand.
Once loaded into the reactor the sample was located and mapped in a dry state prior to the solvent being pumped through at a flow rate of 0.1 mL min−1 and the bed mapped again “wet”. The sample was then heated at 1 K min−1 under the flowing solvent to 353 K whereupon it was held at this temperature for the remainder of the experiment. Pd K-edge QEXAFS maps of the bed were continually collected with a single complete axial map of the bed being obtained in ca. 3–5 minutes.
Acknowledgements
This work was funded by EPSRC (UK), (EP/G070172/1), with support from the Pharmacat Consortium (AstraZeneca, GlaxoSmithKline and Pfizer). Data underlying this article can be accessed on figshare at 10.6084/m9.figshare.4233227, and used under the Creative Commons Attribution licence. EMB was supported by Xunta Galicia, and CJM by an EPSRC industrial Doctoral Training Grant (AstraZeneca). The Swiss-Norwegian Beamline at the ESRF are acknowledged for access to facilities. We thank Johnson Matthey plc for the provision of Pd salts and catalysts. Ekaterina Wade (Imperial College) is acknowledged for assistance with the TEM measurements. MAN is very grateful to the University of Warwick (Physics) for a visiting academic position.Notes and references
- C. Capello, U. Fischer and K. Hungerbuhler, Green Chem., 2007, 9, 927–934 RSC.
- K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31–36 RSC.
- R. K. Henderson, C. Jimenez-Gonzalez, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Green Chem., 2011, 13, 854–862 RSC.
- D. Prat, J. Hayler and A. Wells, Green Chem., 2014, 16, 4546–4551 RSC.
- P. J. Dunn, Chem. Soc. Rev., 2012, 41, 1452–1461 RSC.
- H. E. Eastman, C. Jamieson and A. J. B. Watson, Aldrichimica Acta, 2015, 48, 51–55 Search PubMed.
- P. D'Arrigo, L. Cerioli, A. Fiorati, S. Servi, F. Viani and D. Tessaro, Tetrahedron: Asymmetry, 2012, 23, 938–944 CrossRef.
- S. Gunzenhauser, E. Biala and P. Strazewski, Tetrahedron Lett., 1998, 39, 6277–6280 CrossRef CAS.
- M. R. Wiley, L. C. Weir, S. Briggs, N. A. Bryan, J. Buben, C. Campbell, N. Y. Chirgadze, R. C. Conrad, T. J. Craft, J. V. Ficorilli, J. B. Franciskovich, L. L. Froelich, D. S. Gifford-Moore, T. Goodson, D. K. Herron, V. J. Klimkowski, K. D. Kurz, J. A. Kyle, J. J. Masters, A. M. Ratz, G. Milot, R. T. Shuman, T. Smith, G. F. Smith, A. L. Tebbe, J. M. Tinsley, R. D. Towner, A. Wilson and Y. K. Yee, J. Med. Chem., 2000, 43, 883–899 CrossRef CAS PubMed.
- Y. Ito, H. Ohta, Y. M. A. Yamada, T. Enoki and Y. Uozumi, Tetrahedron, 2014, 70, 6146–6149 CrossRef CAS.
- G. An, H. Ahn, K. A. De Castro and H. Rhee, Synthesis, 2010, 477–485 CAS.
- Q. W. Du and Y. Q. Li, Beilstein J. Org. Chem., 2011, 7, 378–384 CrossRef CAS PubMed.
- F. Amoroso, S. Colussi, A. Del Zotto, J. Llorca and A. Trovarelli, Catal. Commun., 2011, 12, 563–567 CrossRef CAS.
- C. Duplais, A. J. Forman, B. A. Baker and B. H. Lipshutz, Chem. – Eur. J., 2010, 16, 3366–3371 CrossRef CAS PubMed.
- Y. Q. Yuan and S. R. Guo, Synth. Commun., 2012, 42, 1059–1069 CrossRef CAS.
- M. Hosseini-Sarvari, Z. Razmi and M. M. Doroodmand, Appl. Catal., A, 2014, 475, 477–486 CrossRef CAS.
- M. A. Newton, J. B. Brazier, E. M. Barreiro, S. Parry, H. Emmerich, L. A. Adrio, C. J. Mulligan, K. Hellgardt and K. K. Hii, Green Chem., 2016, 18, 406–411 RSC.
- S. M. Kozlov, H. A. Aleksandrov and K. M. Neyman, J. Phys. Chem. C, 2014, 118, 15242–15250 CAS.
- R. Dus, Surf. Sci., 1975, 50, 241–252 CrossRef CAS.
- D. Teschner, J. Borsodi, A. Wootsch, Z. Revay, M. Havecker, A. Knop-Gericke, S. D. Jackson and R. Schlogl, Science, 2008, 320, 86–89 CrossRef CAS PubMed.
- S. Nakamura and T. Yasui, J. Catal., 1970, 17, 366–374 CrossRef CAS.
- M. Zobel, R. B. Neder and S. A. J. Kimber, Science, 2015, 347, 292–294 CrossRef CAS PubMed.
- R. Mendil, Z. Ben Ayadi and K. Djessas, J. Alloys Compd., 2016, 678, 87–92 CrossRef CAS.
- V. R. Tapia, M. S. Tizapa, E. R. Mora, M. L. O. Martínez, A. Franco and E. B. Calva, Plasmonics, 2016, 1–10 Search PubMed.
- Q. Fu and T. Wagner, Surf. Sci. Rep., 2007, 62, 431–498 CrossRef CAS.
- S. Schauermann, N. Nilius, S. Shaikhutdinov and H. J. Freund, Acc. Chem. Res., 2013, 46, 1673–1681 CrossRef CAS PubMed.
- S. Penner and M. Armbruster, ChemCatChem, 2015, 7, 374–392 CrossRef CAS.
- N. Binsted, EXCURV98 CCLRC Daresbury Laboratory computer program, 1998 Search PubMed.
- R. E. Benfield, J. Chem. Soc., Faraday Trans., 1992, 88, 1107–1110 RSC.
- A. Jentys, Phys. Chem. Chem. Phys., 1999, 1, 4059–4063 RSC.
- R. A. Bennett, C. L. Pang, N. Perkins, R. D. Smith, P. Morrall, R. I. Kvon and M. Bowker, J. Phys. Chem. B, 2002, 106, 4688–4696 CrossRef CAS.
- J. B. Brazier, B. N. Nguyen, L. A. Adrio, E. M. Barreiro, W. P. Leong, M. A. Newton, S. J. A. Figueroa, K. Hellgardt and K. K. M. Hii, Catal. Today, 2014, 229, 95–103 CrossRef CAS.
- J. A. McCaulley, J. Phys. Chem., 1993, 97, 10372–10379 CrossRef CAS.
- R. J. Davis, S. M. Landry, J. A. Horsley and M. Boudart, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 39, 10580–10583 CrossRef CAS.
- P. Lignier, in Ionic Liquids, ed. J. Dupont and L. Kollar, 2015, vol. 51, pp. 55–78 Search PubMed.
- K. Na, Q. Zhang and G. A. Somorjai, J. Cluster Sci., 2014, 25, 83–114 CrossRef CAS.
- S. S. Cheong, J. D. Watt and R. D. Tilley, Nanoscale, 2010, 2, 2045–2053 RSC.
- S. J. A. Figueroa and C. Prestipino, J. Phys.: Conf. Ser., 2016, 712, 012012 CrossRef.
- N. Binsted, PAXAS: Programme for the analysis of X-ray absorption spectra, University of Southampton, 1988 Search PubMed.
| This journal is © The Royal Society of Chemistry 2016 |