
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Graphene encapsulated FexCoy nanocages derived from metal–organic frameworks as efficient activators for peroxymonosulfate†
Xuning
Li
ab,
Alexandre I.
Rykov
a,
Bo
Zhang
a,
Yanjie
Zhang
c and
Junhu
Wang
*a
aMössbauer Effect Data Center, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: wangjh@dicp.ac.cn; Tel: +86 411 84379159
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cResearch Institute of Photonics, Dalian Polytechnic University, Dalian 116034, China
First published on 18th August 2016
Abstract
A series of nitrogen-doped graphene encapsulated FexCoy bimetallic (FexCoy@C) nanocages were newly fabricated via a one-step thermal decomposition of Prussian blue analogue FeyCo1−y[Co(CN)6]0.67·nH2O nanospheres at different temperatures. The as-synthesized FexCoy@C nanocages were systematically characterized and applied as effective catalysts to activate peroxymonosulfate (PMS) for the removal of bisphenol A (BPA). The catalytic activity of Fe3Co7@C-650 obtained at 650 °C was found to be the highest and also much higher than that of metallic cobalt, cobalt oxide and cobalt–iron spinels. The nitrogen-doped graphene shells were demonstrated to be effective in preventing the release of metal ions, thus resulting in a better stability. Furthermore, the BPA degradation pathway was proposed based on GC-MS and LC-MS results. The involved radicals were identified through electron paramagnetic resonance and radical scavenger experiments. Based on the systematic characterization of the catalyst before and after the catalytic oxidation reaction, the overall mechanism of PMS activation over FexCoy@C nanocages was proposed. Results indicate that FexCoy@C nanocages are available to serve as promising materials for environmental remediation by activation of PMS.
1. Introduction
In recent years, Fenton and Fenton-like processes have been widely investigated and regarded as good alternatives for the treatment of recalcitrant organic pollutants in view of their high efficiency, simplicity and environmental friendliness.1,2 The hydroxyl radical (˙OH), generated from the Fenton or Fenton-like process, is highly efficient to degrade nearly all organic compounds. However, the narrow working pH range and low H2O2 utilization rate remarkably limit the practical application of ˙OH-based Fenton systems.3–7Compared to the ˙OH-based Fenton system, the sulfate radical (SO4˙−) based Fenton-like system by activation of peroxymonosulfate (PMS) was reported as one of the promising alternatives due to its high efficiency at a wide pH range and low catalyst dosage.8–10 Among various transition metal catalysts, Co was found to be very active for PMS activation to produce SO4˙−.11 However, Co is recognized as a toxic pollutant and the leached Co ions in water may induce adverse effects to the environment.12 The combination of Co with a suitable amount of Fe was reported to be an effective way to prevent the leaching of Co and to overcome the drawbacks of Fe.13–15
As an emerging new class of materials, bimetallic zero valent nanoparticles (BZVNs) have received considerable attention and been widely applied for the catalytic elimination of various pollutants.16,17 However, the long term reactivity due to the strong aggregation and surface passivation over time of BZVNs largely impeded their applications.18–20 Encapsulation of BZVNs into air-stable materials was suggested to be a promising way for the enhancement of transportation, suspension, and stability of BZVNs without significantly sacrificing the activity.21–23 Therefore, fabricating Fe–Co BZVNs encapsulated in carbon spheres may have great potential for efficient activation of PMS for pollutant degradation.
Prussian blue analogues (PBAs) have been regarded as promising precursors or sacrificial templates to construct various morphology-inherited nanomaterials.23–27 Previously, we have reported a facile strategy to synthesize porous FexCo3−xO4 nanocages by heating FeyCo1−y[Co(CN)6]0.67·nH2O (FeyCo1−y-Co PBAs) in air.15,25 In addition, thermal decomposition of PBAs in a N2 atmosphere has been reported as a general strategy to synthesize nanoparticles with nitrogen-doped carbon shells.23,24 Inspired by this, the thermal decomposition of FeyCo1−y-Co PBAs is expected as a facile strategy to synthesize nitrogen-doped graphene encapsulated FexCoy BZVNs (FexCoy@C). However, the heating temperature is usually considered as the main factor influencing both the crystallinity and porous structure of the catalysts, which largely affect the catalytic activity.28 Thus, heating at different temperatures is necessary to obtain FexCoy@C with an optimized nanostructure as well as the highest catalytic activity.
In the present work, a series of FexCoy@C nanocages were newly synthesized via a one-step thermal decomposition of FeyCo1−y-Co PBA nanospheres in a nitrogen atmosphere at different temperatures. The morphology, crystalline structure and textural properties of the prepared FexCoy@C nanocages were thoroughly characterized by various techniques. For the first time, a series of FexCoy@C nanocages were developed as Fenton-like catalysts to activate PMS for bisphenol A (BPA) removal. The concentrations of the leached metal ions during the reaction were detected and the catalytic stability was tested. SO4˙− was confirmed as the dominant radical involved and the BPA degradation pathway was further studied. Moreover, the overall mechanism of PMS activation over FexCoy@C nanocages was systematically investigated and proposed.
2. Experiments
2.1. Chemicals
Potassium hexacyanocobaltate(III) (98%) and cobaltosic oxide (98.5%) were purchased from Beijing J&K Co., Ltd, China. PMS (KHSO5·0.5KHSO4·0.5K2SO4) was purchased from Alfa Aesar. Cobalt (99.9%, 30 nm), iron powder (100 mesh), iron oxide(II,III) (99.5%, ∼20 nm), BPA, and 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) were purchased from Aladdin Co., China. Sodium sulfite, ferrous chloride, cobaltous chloride, poly(vinylpyrrolidone) (PVP), potassium iodide, sodium hydrogen carbonate, t-butanol (TBA) and methyl alcohol were purchased from Tianjin Kermel Chemical Reagent Co., Ltd, China. All chemical reagents were used without further purification.2.2. Sample preparation
FeyCo1−y-Co PBA nanospheres were prepared in a similar manner to our previous report.15 Typically, desired amounts of FeCl2·4H2O and CoCl2·6H2O (a total of 9 mM) were dissolved in 40 mL of deionized water with PVP (1.2 g) under vigorous stirring. Then 40 mL of K3[Co(CN)6] aqueous solution (5 mM) was added into the premixed solution slowly. The obtained colloidal solution was stirred for another 30 min and then aged for 20 h. The resulting precipitates were centrifuged and washed at least three times with a mixture of deionized water and ethanol, followed by drying in an oven at 333 K for 20 h. To obtain the Fe3Co7@C-T nanocages, Fe0.5Co0.5-Co PBA nanospheres were heated at different temperatures (500, 650 and 800 °C) and kept for 1 h in N2. Fe2Co8@C-500 nanocages were obtained by heating Fe0.3Co0.7-Co PBA nanospheres at 500 °C and keeping them for 1 h in N2.2.3. Characterization
The crystallographic structures of the samples were studied by powder X-ray diffraction (XRD) in a 2θ range from 10° to 90° on a PANalytical X'Pert-Pro X-ray diffractometer equipped with a Cu Kα (λ = 0.15406 nm) radiation source. The textural and morphological information was obtained by field-emission scanning electron microscopy (FESEM, JSM 7800F) and high-resolution transmission electron microscopy (HR-TEM, JEM-2100). The element compositions were measured by energy dispersive X-ray spectroscopy (EDS) measurements. The Fourier transform infrared spectra (FT-IR) were recorded on a Bruker spectrometer equipped with a MCT detector at a resolution of 4 cm−1. The specific surface areas were measured on a Micromeritics ASAP 2010 instrument by the Brunauer–Emmett–Teller (BET) N2 adsorption–desorption method. Thermo-gravimetric and differential scanning calorimetry (TG-DSC) analyses were conducted on a Setaram Setsys 16/18 thermo-analyzer in N2 or air flow and a heating rate of 10 K min−1. The Co K-edge X-ray absorption fine structure (XAFS) spectra were taken at Beijing Synchrotron Irradiation Facility (BSRF) with a stored electron energy of 2.2 GeV. The Co K-edge spectra were processed following the conventional procedure using the IFEFFIT package. The X-ray photoelectron spectra (XPS) fitted by XPSPEAK41 software with Shirley-type background were recorded on an ESCALAB 250 X-ray photoelectron spectroscope equipped with a monochromated Al Kα source. The room temperature 57Fe Mössbauer spectra were obtained using a Topologic 500A spectrometer and a proportional counter with 57Co (Rh) as a γ-ray radioactive source. The electron paramagnetic resonance (EPR) spectra were collected using a Bruker EPR I200 spectrometer with a center field at 3320 G and a sweep width of 100 G at room temperature. The detailed procedures of the Mössbauer and EPR measurements were similar to our previous work.292.4. Catalytic activity measurements
BPA degradation tests were carried out in a 200 mL reactor containing 20 mg L−1 BPA at pH = 6.0 in a dark box. The reaction temperature was maintained at 25 °C using a water bath. In a typical test, 5 mg of catalyst was added into a 50 mL BPA solution and stirred for 30 min to establish the adsorption–desorption equilibrium. The reaction was initiated by adding certain amounts of PMS aqueous solution. At predetermined time intervals, 1 mL samples were withdrawn and immediately quenched with 0.5 mL of methanol. To determine the reaction rate of the leached Fe/Co ions, the catalyst was separated immediately at different time intervals, and BPA was continuously degraded by the residual solution for another 15 min. For recycle tests of catalysts, the catalyst was recycled using a magnet and washed thoroughly with deionized water three times after each run.The concentration of BPA was analyzed using high performance liquid chromatography (HPLC, Agilent, 1260-Infinity) at a detection wavelength of 230 nm with a methanol/water mixture (70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30, v/v) as the mobile phase. The total organic carbon (TOC) values were analyzed by a total organic carbon analyzer (TOC-VCPH/CPN, Shimadzu, Japan). The details of the BPA degradation intermediates identified by GC-MS (Varian 450GC-320 MS) and LC-MS can be found in our previous work.15 The concentrations of leached cobalt and iron were detected by inductively coupled plasma atomic emission spectroscopy (ICP-OES). The PMS concentration was determined using an iodometric method.30–34 Briefly, 200 μL of sample was withdrawn and immediately mixed with 1.8 mL of KI stock solution (0.166 g of KI + 0.04 g of NaHCO3)/100 mL. After reaction for 5 min, the mixture was analyzed by a UV-vis spectrophotometer at λ = 352 nm. The calibration curve of PMS concentration is shown in Fig. S1.†
:30, v/v) as the mobile phase. The total organic carbon (TOC) values were analyzed by a total organic carbon analyzer (TOC-VCPH/CPN, Shimadzu, Japan). The details of the BPA degradation intermediates identified by GC-MS (Varian 450GC-320 MS) and LC-MS can be found in our previous work.15 The concentrations of leached cobalt and iron were detected by inductively coupled plasma atomic emission spectroscopy (ICP-OES). The PMS concentration was determined using an iodometric method.30–34 Briefly, 200 μL of sample was withdrawn and immediately mixed with 1.8 mL of KI stock solution (0.166 g of KI + 0.04 g of NaHCO3)/100 mL. After reaction for 5 min, the mixture was analyzed by a UV-vis spectrophotometer at λ = 352 nm. The calibration curve of PMS concentration is shown in Fig. S1.†
3. Results and discussion
3.1. Characterization of FexCoy@C nanocages
Fig. 1 shows the XRD patterns of Fe2Co8@C-500, Fe3Co7@C-500, Fe3Co7@C-650, and Fe3Co7@C-800. As can be seen, all of the four samples showed a main phase of Fe3Co7 (JCPDS No. 50-0795).35 In the patterns of the latter three samples, besides the peaks of Fe3Co7, two peaks at 44.2° and 51.5° were observed, which suggests the existence of Co with a cubic crystalline structure (JCPDS No. 89-7093).36 The other two weak peaks at 41.6° and 47.3° in the pattern of sample Fe2Co8@C-500 were possibly due to the existence of Co with a hexagonal crystalline structure (JCPDS No. 01-1278) with increasing content of Co. In addition, the increase of the peak intensities indicates the better crystallinity and larger crystal size of the four samples when the heating temperature increases.28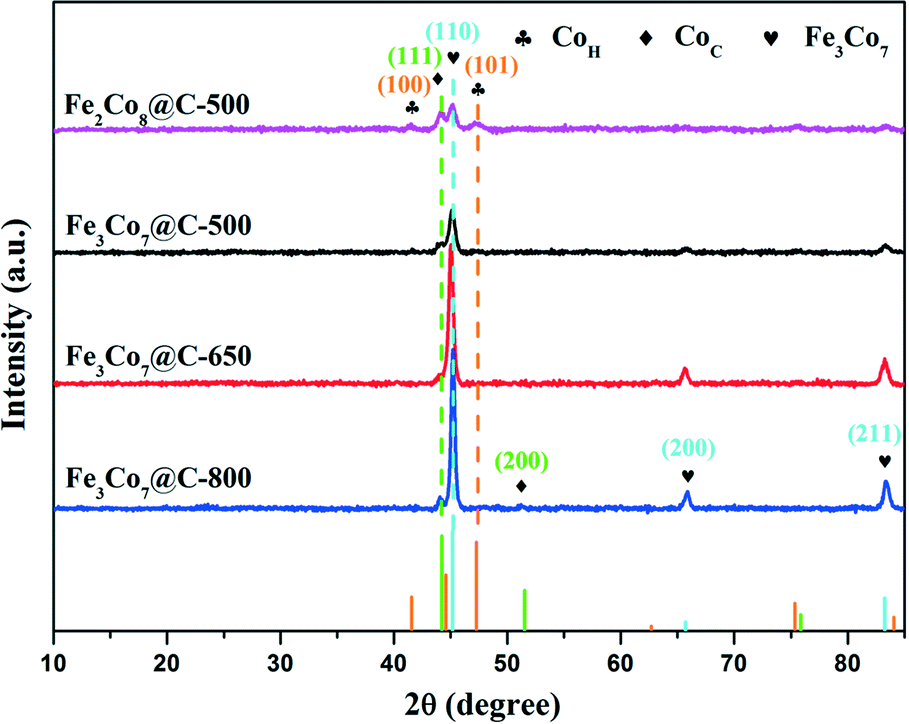 | ||
| Fig. 1 XRD patterns of FexCoy@C nanocages. | ||
Fig. 2 displays the SEM images of FexCoy@C, which reveal the typical morphology of the four samples to be nanocages. As can be seen, the FexCoy nanocrystals (white points) were all well dispersed on the nanocages and the crystal size became larger with increasing the heating temperature from 500 °C to 800 °C,37 which was consistent with the XRD analysis results calculated by the Debye–Scherrer equation (Table S1†). The average size of the Fe2Co8@C-500 nanocages, due to the smaller particle size of Fe0.3Co0.7-Co PBA nanospheres, appeared to be much smaller, which further suggests the significant influence of the iron doping amount on the final morphology. The TEM image of Fe3Co7@C-650 further confirmed the porous structure of the nanocages (Fig. 3A). The HR-TEM images of single nanocrystals (Fig. 3B–D) clearly show that the Fe3Co7 nanocrystals were completely coated with the graphene shells (5–20 layers) with an interlayer distance of 0.34 nm.23 The Fe3Co7 nanocrystal exhibits a d-spacing of 0.20 nm (Fig. 3B), corresponding to the (110) plane of the Fe3Co7 alloy. The chemical composition of FexCoy@C analyzed by EDS (Table S1†) indicates that all of the samples contain N, whose content was also changeable under the different heating temperatures. In addition, the EDS mapping (Fig. S2†) confirmed the homogeneous distribution of Fe and Co species in the FexCoy@C nanocages. The high resolution XPS spectrum of N 1s on the Fe3Co7@C-650 surface was deconvoluted into four individual peaks, which could be assigned to pyridinic N (398.5 eV), pyrrolic N (399.8, 400.6 eV), and quaternary N (401.5 eV), respectively (Fig. S3†).23 All these results suggest that the nitrogen-doped graphene encapsulated FexCoy BZVNs could be successfully synthesized through the one-step thermal decomposition of FeyCo1−y-Co PBA nanospheres.
 | ||
| Fig. 2 SEM images of FexCoy@C nanocages (A) Fe3Co7@C-500, (B) Fe3Co7@C-650, (C) Fe3Co7@C-800, and (D) Fe2Co8@C-500. | ||
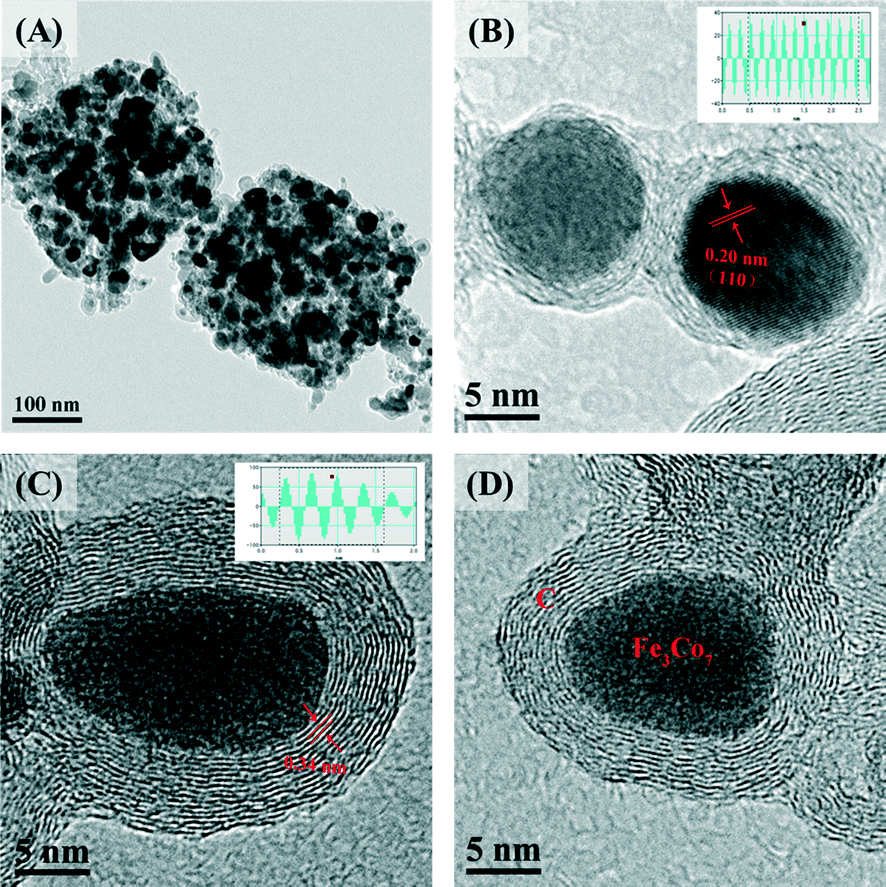 | ||
| Fig. 3 TEM image (A) and HR-TEM images (B, C, D) of Fe3Co7@C-650 nanocages. | ||
57Fe Mössbauer spectroscopy was applied to determine the coordination environment and oxidation state of Fe ions in the samples (Fig. 4). The spectra were fitted with two sextets, corresponding to the Fe3Co7 alloy in different chemical environments and/or with different crystallite sizes.38 The blue doublet in the spectra of Fe3Co7@C-500 and Fe2Co8@C-500 could be assigned to FeIII in Fe2O3. The corresponding Mössbauer parameters are shown in Table S2.† As can be seen, only Fe3Co7@C-650 and Fe3Co7@C-800 contain the pure phase of Fe metal. The other two samples obtained by heating FeyCo1−y-Co PBAs at 500 °C contain more than 20% Fe2O3, suggesting that the heating temperature largely affects the oxidation state of iron ions in the samples. Co K-edge XAFS was employed to confirm the coordination environment and chemical state of the Co ions in the FexCoy@C nanocages. In Fig. 5A, the similar pre-edge peak position suggests that the Co ions in FexCoy@C were present mainly in the form of Co0. The Fourier transforms of the Co K-edge EXAFS spectra are shown in Fig. 5B. The position of the peaks at around 0.21 and 0.48 nm could be correlated with the length of the Co–Fe/Co bond.39,40 The slight decrease of the Co–Co/Fe bond length at around 0.48 nm is most probably due to the increase of the Co–Fe bond content in the samples with increasing Fe content and heating temperature, which is consistent with the XRD results.
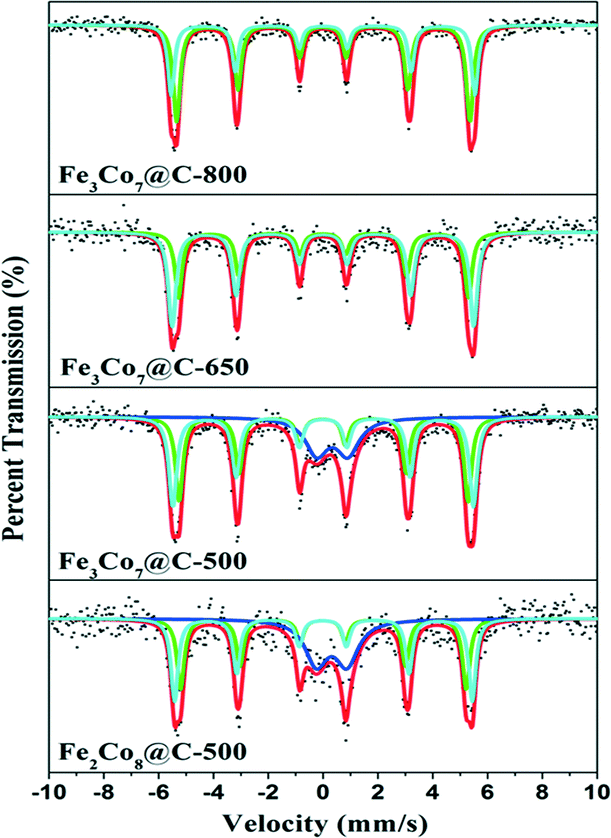 | ||
| Fig. 4 Room temperature 57Fe Mössbauer spectra of FexCoy@C nanocages. The spectra were fitted with two sextets, corresponding to two kinds of metallic irons in the Fe3Co7 alloy with different chemical environments and/or different crystallite sizes. The blue doublet in the spectra of Fe3Co7@C-500 and Fe2Co8@C-500 could be assigned to Fe3+ in Fe2O3. | ||
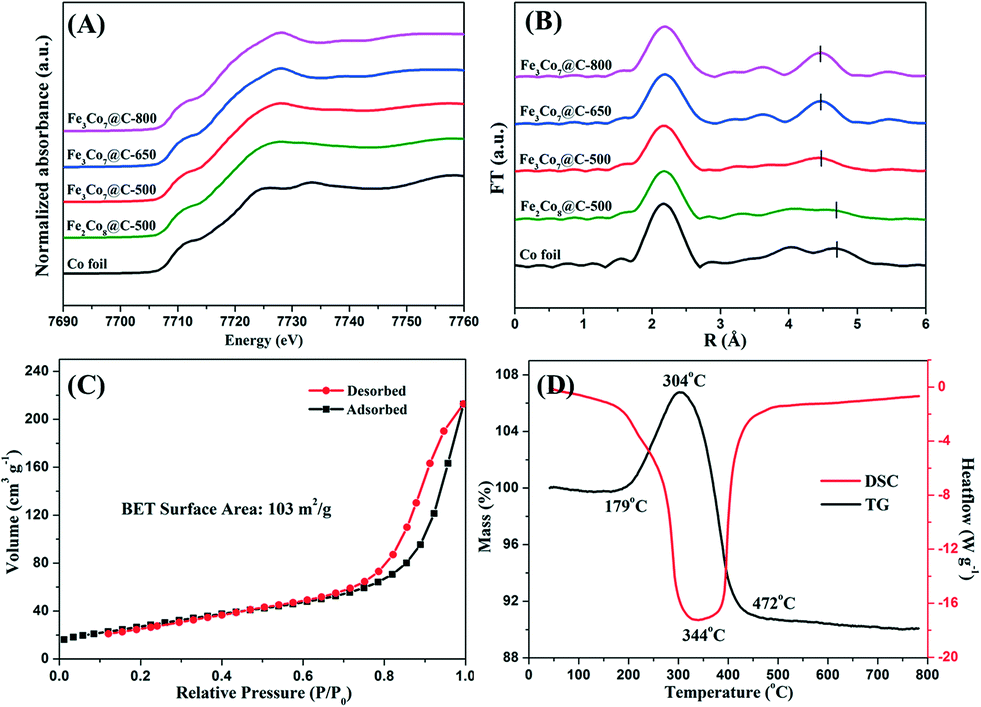 | ||
| Fig. 5 Normalized XANES of Co K-edge (A) and Fourier transforms of the Co K-edge EXAFS (B) of FexCoy@C nanocages; N2 adsorption–desorption isotherms (C) and TG-DSC curve (D) of Fe3Co7@C-650. | ||
Fig. 5C shows the N2 adsorption–desorption isotherms of the Fe3Co7@C-650 nanocages. The specific surface area of Fe3Co7@C-650 was calculated to be 103 m2 g−1, which was much larger than that of the recently reported Co@C catalysts.41 The TG-DSC profile of Fe3Co7@C-650 is shown in Fig. 5D. The increase of the weight at the first stage (179–304 °C) was due to the oxidation of Fe/Co to oxides. The following weight loss (over 304 °C) could be ascribed to the combustion of graphene layers. It was interesting to see that the weight remained unchanged until the temperature reached 179 °C, suggesting the good stability of the Fe3Co7@C-650 nanocages.
3.2. Catalytic performance and stability of FexCoy@C nanocages
The catalytic performance of the FexCoy@C nanocages was evaluated by activation of PMS for removal of BPA. As shown in Fig. 6A, only PMS could not induce significant BPA degradation. A control test using only Fe3Co7@C-650 nanocages shows that less than 3% of BPA was adsorbed in 30 min, suggesting its negligible adsorption capacity. The simultaneous presence of FexCoy@C nanocages and PMS led to a significant enhancement of the BPA removal efficiency, which was much higher than that of commercial metallic iron and Co3O4. At 30 min, the BPA removal efficiencies of Fe2Co8@C-500, Fe3Co7@C-500, Fe3Co7@C-650, and Fe3Co7@C-800 were 93%, 84%, 98%, and 87%, respectively. The comparison between the removal efficiencies of Fe2Co8@C-500 and Fe3Co7@C-500 suggests that a higher Co content would be beneficial for the catalytic performance, because Co is usually considered to be more active than Fe for PMS activation.13,15 The BPA removal efficiency of Fe3Co7@C-650 nanocages was found to be the highest one when compared with the others, indicating the less oxidation of the catalyst surface, which is consistent with the Mössbauer results. The lower catalytic performance of Fe3Co7@C-800 is most probably due to its larger crystal size, which could be also evidenced from the XRD characterization.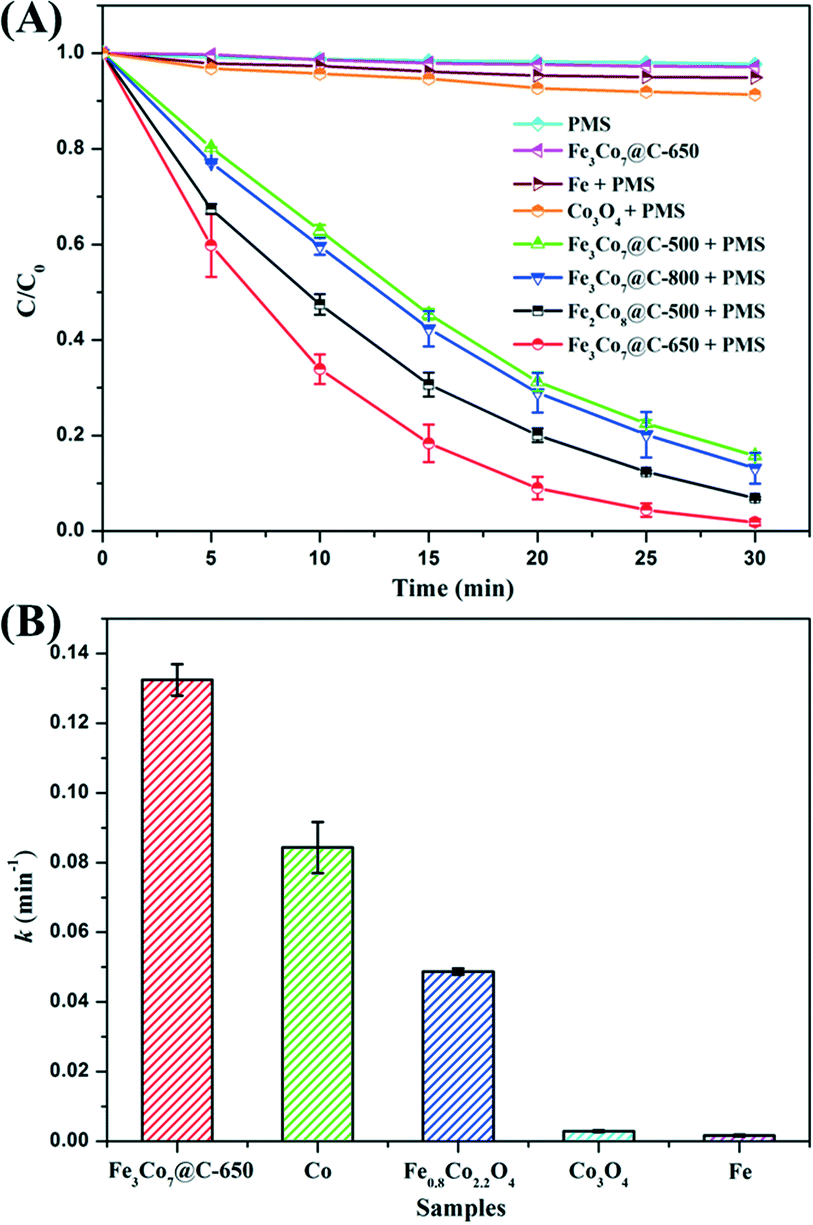 | ||
| Fig. 6 (A) Removal efficiency of BPA in different reaction systems within 30 min; (B) the rate constant of Fe3Co7@C-650 compared with that of metallic cobalt (the same content as Co in Fe3Co7@C-650), iron powder, Co2O3 and Fe0.8Co2.2O4. Reaction conditions: [BPA] = 20 mg L−1, [PMS] = 0.2 g L−1 (if needed), catalyst = 0.1 g L−1, T = 298 K, initial solution pH = 6.0. | ||
The BPA removal kinetics could be fitted well to first-order kinetics and the results are shown in Fig. 6B. The apparent rate constant (k) of Fe3Co7@C-650 (0.132 min−1) was much higher than that of the Fe0.8Co2.2O4 nanocages (0.049 min−1) reported in our previous work,15 suggesting the better PMS activation efficiency of the BZVNs than the bimetal oxides. The catalytic activity of Fe3Co7@C-650 was also higher than that of metallic cobalt (the same Co content as Fe3Co7@C-650), which is most probably because of the synergistic effect between Fe and Co.14 In addition, about 35% of BPA was mineralized in 30 min (Fig. 7A), suggesting that BPA was not only decomposed to small organic compounds, but even mineralized to inorganic carbon forms. The time courses of PMS remaining during the reaction are shown in Fig. S4.† As can be seen, the PMS remaining in the solution after reaction was less than 20%, further suggesting the excellent catalytic performance of Fe3Co7@C nanocages for PMS activation.
 | ||
| Fig. 7 (A) TOC removal efficiency of BPA at different reaction times; (B) the concentrations of the leached Fe/Co ions and the rate constants of the leached Fe/Co ions at different reaction times. (C) Effect of PMS concentration on BPA removal efficiency; (D) consecutive runs of the catalytic activities of Fe3Co7@C-650 and metallic cobalt. Reaction conditions: [BPA] = 20 mg L−1, [PMS] = 0.2 g L−1 (for A, B and D), catalyst = 0.1 g L−1, T = 298 K, initial solution pH = 6.0. | ||
To quantify the contribution of the homogeneous reaction to the BPA removal efficiency, the leached Fe/Co ions and the k of the homogeneous reaction catalyzed by the leached Fe/Co ions at different reaction times were measured (Fig. 7B). As can be seen, the concentration of the leached Fe was negligible when compared with that of the leached Co, which kept increasing to about 3.30 mg L−1 at 30 min. This result further suggests that Co is most probably the main active site. The k of the homogeneous reaction catalyzed by the leached Fe/Co ions was shown to increase along with their concentration. However, the k of the total reaction catalyzed by Fe3Co7@C-650 (0.132 min−1) was more than two times higher than that of the homogeneous reaction, suggesting the dominant role of the heterogeneous reaction.
Fig. 7C shows the effect of PMS concentration on the BPA removal efficiency. As can be seen, the BPA removal efficiency increased with the PMS concentration increasing from 0.1 to 0.2 g L−1, however, it decreased with the PMS concentration further increasing to 0.25 g L−1. The optimum PMS concentration could be explained by the competitive reactions which may adversely affect the generation of reactive species (SO4˙− + SO4˙− → S2O82−, HO˙ + SO4˙− → HSO5−).42–44
Finally, the operational stability of Fe3Co7@C-650 was explored and compared with commercial metallic cobalt. As shown in Fig. 7D, although the BPA removal efficiency keeps decreasing after each run due to the continuous loss of the active component, the operational stability of Fe3Co7@C-650 appears to be much better than that of the metallic cobalt as well as the recently reported Fe0/Fe3C@CS and Fe0@CS catalysts.21,45 In addition, the final leached Co ion concentration (3.30 mg L−1) of Fe3Co7@C-650 is much lower than that of the metallic cobalt (8.73 mg L−1). All these results suggest that the nitrogen-doped graphene shells were effective to prevent the release of the metal ions, thus resulting in a better stability.
3.3. Possible degradation pathway of BPA
The formed intermediates during BPA degradation were analyzed by GC-MS and LC-MS (Fig. S5–S7†). From the identified m/z determined by GC-MS and LC-MS, as well as compared with the results of previous studies,46–49 nine compounds (Products A–I) were identified as the main intermediates as listed in Table S3.† Among them, phenol (m/z = 94) and 4-isopropenylphenol (m/z = 134) were formed through the cleavage between one of the benzenes and the attack of radicals on the middle carbon atom (Route II in Fig. 8).4,46 However, unlike our previous work, more intermediates (Products C–I) through Route I were detected. As shown in Fig. 8, BPA-O-catechol (m/z = 244) was formed through the hydroxylation of the aromatic ring of BPA. Further oxidation of BPA-O-catechol could easily lead to the formation of Products D–F (m/z = 242, 290, and 276).47 The products at m/z = 232, 234, 250 and 222 were formed through the sequential decarboxylation of Product F (m/z = 276).48 The product at m/z = 460, with a high molecular weight, could result from secondary reactions between the products of m/z = 234 and m/z = 234.49 Based on all of these experimental results and the previous studies,46–49 the BPA degradation pathway by activation of PMS on Fe3Co7@C-650 nanocages was proposed as shown in Fig. 8. | ||
| Fig. 8 Proposed BPA degradation pathway by activation of PMS on Fe3Co7@C-650 nanocages. | ||
3.4. Reactive intermediates during PMS activation over FexCoy@C nanocages
To investigate the activation and oxidation mechanism, the involved radical intermediates produced in activation of PMS were probed by radical scavenger experiments. As shown in Fig. 9A, when TBA, a commonly used radical scavenger for ˙OH,50 was added to the reaction solution, only about 16% decrease of the reaction rate was observed. However, when methanol, an effective radical scavenger for both SO4˙− and ˙OH,42 was added to the reaction solution, more than 80% decrease of the reaction rate was observed. The production of ˙OH could be ascribed to the strong hydrolysis process of HSO5−.51 In addition, the more remarkable reaction rate decrease caused by methanol indicates that SO4˙− is most probably the dominant reactive intermediate during the activation of PMS by Fe3Co7@C-650 nanocages. | ||
| Fig. 9 (A) Comparison of the reaction rate with and without radical scavengers; (B) time-dependent EPR spectra for the activation of PMS by Fe3Co7@C-650 nanocages; reaction conditions: [BPA] = 20 mg L−1, [PMS] = 0.2 g L−1, catalyst = 0.1 g L−1, [scavenger] = 0.2 M (if needed), T = 298 K, initial solution pH = 6.0. | ||
EPR/DMPO experiments were carried out to further confirm the identity of the involved reactive intermediates. As shown in Fig. 9B, characteristic signals for both DMPO–˙OH adducts (with aN = aH = 14.9 G) and DMPO–SO4˙− adduct (with aN = 13.2 G and aH = 9.6 G) were observed at the different reaction times, revealing that both SO4˙− and ˙OH were generated during the activation of PMS by FexCoy@C nanocages.52,53 The stronger intensity of the signal for the DMPO–˙OH adduct was probably due to the instability of the DMPO–SO4˙− adduct, which was reported to be easily decomposed to DMPO–˙OH over time.31,54 Fig. S8† shows the time-dependent relationship between the BPA removal efficiency and the relative peak intensities of DMPO–˙OH/DMPO–SO4˙−. The DMPO–SO4˙− adduct reached the highest peak intensity at about 10 min, when saturated HSO5− was adsorbed on the catalyst surface. Then the gradually decreasing DMPO–SO4˙− intensity indicates the continuous consumption of the SO4˙− radicals for BPA removal. However, the evolution of the DMPO–˙OH intensity remained at a relatively higher value and exhibited almost no change during the first 15 min. Then the large decrease of the DMPO–˙OH intensity could also be ascribed to the continuous consumption of the SO4˙− radicals. All these results further confirm SO4˙− as the dominant radical involved during PMS activation over FexCoy@C nanocages.
3.5. Mechanism of PMS activation over FexCoy@C nanocages
To understand the mechanism of PMS activation over FexCoy@C nanocages, it is important to clarify the structural changes of the catalyst and the redox cycling of the Fe and Co species during the whole reaction process. To this end, a number of techniques were applied for the characterization of the catalyst after the reaction process.The XRD patterns of Fe3Co7@C-650 before and after a 4-cycle run are shown in Fig. S9.† As seen, besides the diffraction peaks of Fe3Co7, no other impurity peaks were detected. More important, the morphology of Fe3Co7@C-650 nanocages was found to remain unchanged even after the 4-cycle run (Fig. S10A and B†), and there were no new absorbance bands detected by FT-IR after reaction (Fig. S11†), further suggesting the good stability of the Fe3Co7@C-650 nanocages. The HR-TEM images of Fe3Co7@C-650 after the 4-cycle run are shown in Fig. S10C and D;† interestingly, the lattice spacing of 0.21 nm was found to correspond well to the (400) plane of γ-Fe2O3 (JCPDS No. 04-0755), suggesting that the oxidation of Fe occurred most probably during the PMS activation over the FexCoy@C nanocages.
The XPS spectra of Fe3Co7@C-650 nanocages before and after the 4-cycle run of the catalytic oxidation process were introduced to further explore the activation mechanism of PMS. Fig. 10A–C show the high resolution XPS spectra of Fe 2p3/2 on the Fe3Co7@C-650 surface before and after the reaction. The three peaks with binding energies located at 707.4, 710.4 and 712.5 eV, with relative contributions to the overall Fe intensity of 17%, 31% and 52%, could be assigned to Fe0, B-site, and A-site FeIII in γ-Fe2O3, respectively.55,56 However, after the catalytic oxidation reaction, the relative contributions of these three peaks change to 10%, 50%, and 40%, respectively. This result further confirms the oxidation of Fe0 to FeIII during the PMS activation over the FexCoy@C nanocages. Fig. 10B–D show the high resolution XPS spectra of Co 2p3/2 on the Fe3Co7@C-650 surface before and after the reaction. The two peaks with binding energies positioned at 778.6 and 780.4 eV, with relative contents of 41% and 59%, could be assigned to Co0 and CoIII, respectively.57 After the reaction, the relative contents of Co0 and CoIII change to 16% and 84%, respectively. The decrease of the Co0 content on the Fe3Co7@C-650 surface indicates the oxidation of Co0 to CoIII during the PMS activation process.
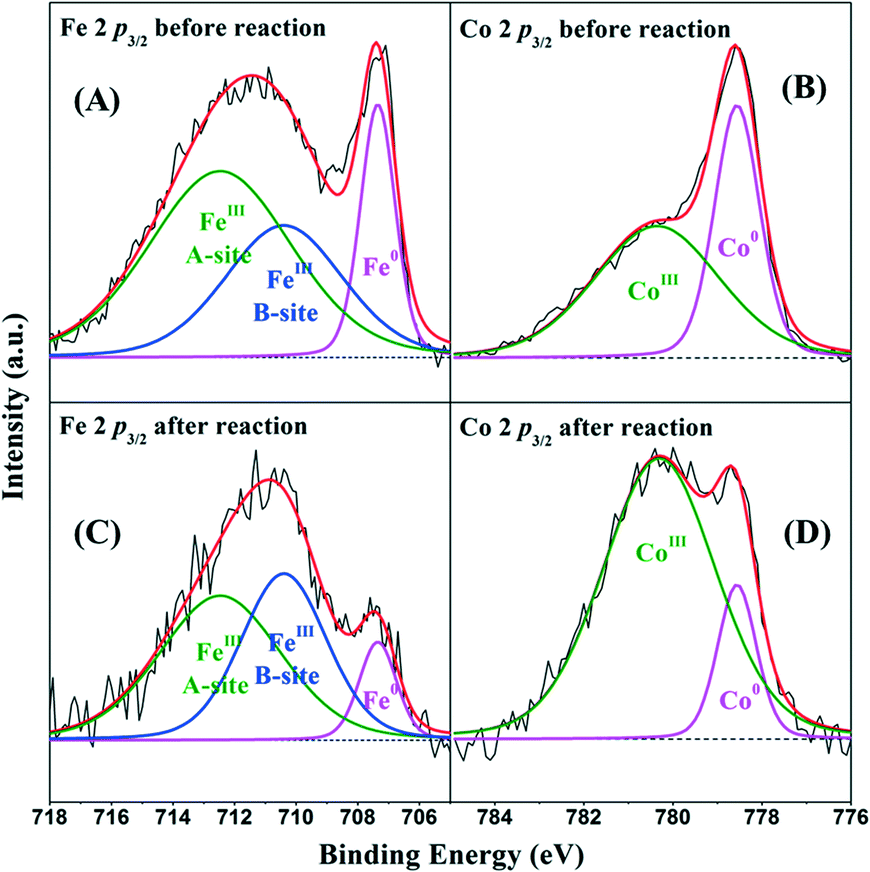 | ||
| Fig. 10 High resolution XPS spectra of Fe 2p3/2 (A–C) and Co 2p3/2 (B–D) of Fe3Co7@C-650 nanocages before and after reaction. | ||
Therefore, the overall mechanism of PMS activation over FexCoy@C nanocages was proposed as shown in Fig. 11. Firstly, FexCoy@C nanocages could release Co2+/Fe2+ ions at certain conditions and activate PMS to generate SO4˙− radicals through eqn (1)–(3).57,58 Meanwhile, Co0/Fe0 in FexCoy@C could activate PMS to produce SO4˙− radicals through eqn (4) with itself being oxidized to ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CoII/FeII.21,59 Secondly, the generated CoII/FeII could be quickly oxidized further by PMS witFurthermore, both Co0/Fe0 and PMS could reduce CoIII/FeIII to CoII/FeII and thus make the reaction proceed cyclically until PMS is completely consumed.44,57–59
CoII/FeII.21,59 Secondly, the generated CoII/FeII could be quickly oxidized further by PMS witFurthermore, both Co0/Fe0 and PMS could reduce CoIII/FeIII to CoII/FeII and thus make the reaction proceed cyclically until PMS is completely consumed.44,57–59
 | ||
| Fig. 11 The proposed mechanism for PMS activation using FexCoy@C nanocages as the catalysts. | ||
|
Co0/Fe0 → Co2+/Fe2+ + 2e−
| (1) |
| Co2+/Fe2+ + HSO5− → Co3+/Fe3+ + SO4˙− + OH− | (2) |
| Co3+/Fe3+ + HSO5− → Co2+/Fe2+ + SO5˙− + H+ | (3) |
|
Co0/Fe0 + 2HSO5− → CoII/FeII + 2SO4˙− + 2OH−
| (4) |
|
CoII/FeII + HSO5− → CoIII/FeIII + SO4˙− + OH−
| (5) |
|
CoIII/FeIII + HSO5− → CoII/FeII + SO5˙− + H+
| (6) |
|
Co0/Fe0 + 2 CoIII/FeIII → 3 CoII/FeII
| (7) |
| SO4˙− + BPA → CO2 + intermediates | (8) |
4. Conclusions
In summary, a series of FexCoy@C nanocages were successfully fabricated by simple thermal decomposition of FeyCo1−y-Co PBA nanospheres at 500, 650, and 800 °C. The heating temperature was found to largely affect the final crystallinity, porous structure, and even the oxidation state of FexCoy@C. Fe3Co7@C-650 obtained at 650 °C shows the highest catalytic activity for BPA removal by PMS activation, due to the less oxidation of the catalyst surface. The nitrogen-doped graphene shells were demonstrated to play a significant role in enhancing the stability of the Fe3Co7 catalyst, as well as in preventing the release of the metal ions which thus results in a better recycling performance. Through radical scavenger and EPR/DMPO experiments, SO4˙− radicals were proved to play a dominant role in the activation of PMS for BPA removal. The BPA degradation pathway, mainly through hydroxylation, oxidative skeletal rearrangement, and decarboxylation, was proposed. Moreover, based on the systematic characterization of Fe3Co7@C-650 before and after the reaction, the mechanism for PMS activation on the catalyst surface as well as in the bulk solution was proposed. These findings may pave the way towards the development of high-performance and long cycling BZVNs for catalytic elimination of various pollutants.Acknowledgements
This work was supported by the National Natural Science Foundation of China (11079036, 21476232) and the Chinese Academy of Sciences Visiting Professorships for Senior International Scientists (2011T1G15). X. N. Li also thanks Beijing Synchrotron Irradiation Facility for the XAFS measurement. Many thanks to the anonymous reviewers who have helped improve this paper.Notes and references
- X. J. Yang, X. M. Xu, J. Xu and Y. F. Han, J. Am. Chem. Soc., 2013, 135, 16058–16061 CrossRef CAS PubMed.
- X. Li, J. Liu, A. I. Rykov, H. Han, C. Jin, X. Liu and J. Wang, Appl. Catal., B, 2015, 179, 196–205 CrossRef CAS.
- L. Ma, H. He, R. Zhu, J. Zhu, I. D. R. Mackinnon and Y. Xi, Catal. Sci. Technol., 2016, 6, 6066–6075 CAS.
- X. Zhang, Y. Ding, H. Tang, X. Han, L. Zhu and N. Wang, Chem. Eng. J., 2014, 236, 251–262 CrossRef CAS.
- X. Li, A. I. Rykov and J. Wang, Catal. Commun., 2016, 77, 32–36 CrossRef CAS.
- L. Zhou, Y. Shao, J. Liu, Z. Ye, H. Zhang, J. Ma, Y. Jia, W. Gao and Y. Li, ACS Appl. Mater. Interfaces, 2014, 6, 7275–7285 CAS.
- J. C. Espinos, S. Navalon, M. Alvaro and H. Garcia, Catal. Sci. Technol., 2016 10.1039/c6cy00572a.
- Y. Yao, Y. Cai, G. Wu, F. Wei, X. Li, H. Chen and S. Wang, J. Hazard. Mater., 2015, 296, 128–137 CrossRef CAS PubMed.
- P. Hu and M. Long, Appl. Catal., B, 2016, 181, 103–117 CrossRef CAS.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2003, 37, 4790–4797 CrossRef CAS PubMed.
- G. P. Anipsitakis, D. D. Dionysiou and M. A. Gonzalez, Environ. Sci. Technol., 2006, 40, 1000–1007 CrossRef CAS PubMed.
- T. Zeng, X. Zhang, S. Wang, H. Niu and Y. Cai, Environ. Sci. Technol., 2015, 49, 2350–2357 CrossRef CAS PubMed.
- S. Su, W. Guo, Y. Leng, C. Yi and Z. Ma, J. Hazard. Mater., 2013, 244, 736–742 CrossRef PubMed.
- Q. Yang, H. Choi, S. R. Al-Abed and D. D. Dionysiou, Appl. Catal., B, 2009, 88, 462–469 CrossRef CAS.
- X. Li, Z. Wang, B. Zhang, A. I. Rykov, M. A. Ahmed and J. Wang, Appl. Catal., B, 2016, 181, 788–799 CrossRef CAS.
- W.-J. Liu, T.-T. Qian and H. Jiang, Chem. Eng. J., 2014, 236, 448–463 CrossRef CAS.
- F. Fu, Z. Cheng and J. Lu, RSC Adv., 2015, 5, 85395–85409 RSC.
- B. Gunawardana, N. Singhal and P. Swedlund, Environ. Eng. Res., 2011, 16, 187–203 CrossRef.
- C. Cai, L. G. Wang, H. Gao, L. W. Hou and H. Zhang, J. Environ. Sci., 2014, 26, 1267–1273 CrossRef CAS.
- M. A. Al-Shamsi, N. R. Thomson and S. P. Forsey, Chem. Eng. J., 2013, 232, 555–563 CrossRef CAS.
- H. Sun, G. Zhou, S. Liu, H. M. Ang, M. O. Tadé and S. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 6235–6241 CAS.
- C. Liu, J. Li, J. Qi, J. Wang, R. Luo, J. Shen, X. Sun, W. Han and L. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 13167–13173 CAS.
- Y. Yang, Z. Lun, G. Xia, F. Zheng, M. He and Q. Chen, Energy Environ. Sci., 2015, 8, 3563–3571 CAS.
- L. Hu, R. Zhang, L. Wei, F. Zhang and Q. Chen, Nanoscale, 2015, 7, 450–454 RSC.
- X. Li, L. Yuan, J. Wang, L. Jiang, A. I. Rykov, D. L. Nagy, C. Bogdan, M. A. Ahmed, K. Zhu, G. Sun and W. Yang, Nanoscale, 2016, 8, 2333–2342 RSC.
- L. Zhang, L. Shi, L. Huang, J. Zhang, R. Gao and D. Zhang, ACS Catal., 2014, 4, 1753–1763 CrossRef CAS.
- L. Zhang, H. B. Wu and X. W. Lou, J. Am. Chem. Soc., 2013, 135, 10664–10672 CrossRef CAS PubMed.
- N. Yan, L. Hu, Y. Li, Y. Wang, H. Zhong, X. Hu, X. Kong and Q. Chen, J. Phys. Chem. C, 2012, 116, 7227–7235 CAS.
- X. Li, J. Wang, A. I. Rykov, V. K. Sharma, H. Wei, C. Jin, X. Liu, M. Li, S. Yu, C. Sun and D. D. Dionysiou, Catal. Sci. Technol., 2015, 5, 504–514 CAS.
- H. Lee, H.-J. Lee, J. Jeong, J. Lee, N.-B. Park and C. Lee, Chem. Eng. J., 2015, 266, 28–33 CrossRef CAS.
- W.-D. Oh, Z. Dong and T.-T. Lim, Appl. Catal., B, 2016, 194, 169–201 CrossRef CAS.
- W.-D. Oh, Z. Dong, Z.-T. Hu and T.-T. Lim, J. Mater. Chem. A, 2015, 3, 22208–22217 CAS.
- W.-D. Oh, S.-K. Lua, Z. Dong and T.-T. Lim, J. Mater. Chem. A, 2014, 2, 15836–15845 CAS.
- W.-D. Oh, S.-K. Lua, Z. Dong and T.-T. Lim, J. Hazard. Mater., 2015, 284, 1–9 CrossRef CAS PubMed.
- R. Kumar Rana, I. Brukental, Y. Yeshurun and A. Gedanken, J. Mater. Chem., 2003, 13, 663–665 RSC.
- Y. Su, Y. Zhu, H. Jiang, J. Shen, X. Yang, W. Zou, J. Chen and C. Li, Nanoscale, 2014, 6, 15080–15089 RSC.
- L. Zhang, A. Wang, W. Wang, Y. Huang, X. Liu, S. Miao, J. Liu and T. Zhang, ACS Catal., 2015, 5, 6563–6572 CrossRef CAS.
- T. P. Braga, D. F. Dias, M. F. de Sousa, J. M. Soares and J. M. Sasaki, J. Alloys Compd., 2015, 622, 408–417 CrossRef CAS.
- Y. Masubuchi, H. Sato, T. Motohashi and S. Kikkawa, J. Ceram. Soc. Jpn., 2014, 122, 288–291 CrossRef.
- G. L. Wei, X. L. Liang, Z. S. He, Y. S. Liao, Z. Y. Xie, P. Liu, S. C. Ji, H. P. He, D. Q. Li and J. Zhang, J. Mol. Catal. A: Chem., 2015, 398, 86–94 CrossRef CAS.
- J. Gong, J. Liu, X. Chen, Z. Jiang, X. Wen, E. Mijowska and T. Tang, J. Mater. Chem. A, 2014, 2, 7461–7470 CAS.
- Y.-H. Guan, J. Ma, X.-C. Li, J.-Y. Fang and L.-W. Chen, Environ. Sci. Technol., 2011, 45, 9308–9314 CrossRef CAS PubMed.
- C. Qi, X. Liu, J. Ma, C. Lin, X. Li and H. Zhang, Chemosphere, 2016, 151, 280–288 CrossRef CAS PubMed.
- C. Cai, H. Zhang, X. Zhong and L. Hou, J. Hazard. Mater., 2015, 283, 70–79 CrossRef CAS PubMed.
- Y. Wang, H. Sun, X. Duan, H. M. Ang, M. O. Tadé and S. Wang, Appl. Catal., B, 2015, 172–173, 73–81 CrossRef CAS.
- J. Du, J. Bao, X. Fu, C. Lu and S. H. Kim, Appl. Catal., B, 2016, 184, 132–141 CrossRef CAS.
- W. Li, P.-X. Wu, Y. Zhu, Z.-J. Huang, Y.-H. Lu, Y.-W. Li, Z. Dang and N.-W. Zhu, Chem. Eng. J., 2015, 279, 93–102 CrossRef CAS.
- Y. Ding, L. Zhu, N. Wang and H. Tang, Appl. Catal., B, 2013, 129, 153–162 CrossRef CAS.
- M. Deborde, S. Rabouan, P. Mazellier, J.-P. Duguet and B. Legube, Water Res., 2008, 42, 4299–4308 CrossRef CAS PubMed.
- Y. Wang, Y. Xie, H. Sun, J. Xiao, H. Cao and S. Wang, Catal. Sci. Technol., 2016, 6, 2918–2929 CAS.
- Y. Wang, H. Sun, H. M. Ang, M. O. Tade and S. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 19914–19923 CAS.
- W. H. Ma, J. Li, X. Tao, J. He, Y. M. Xu, J. C. Yu and J. C. Zhao, Angew. Chem., Int. Ed., 2003, 42, 1029–1032 CrossRef CAS PubMed.
- G.-D. Fang, D. D. Dionysiou, S. R. Al-Abed and D.-M. Zhou, Appl. Catal., B, 2013, 129, 325–332 CrossRef CAS.
- P. L. Zamora and F. A. Villamena, J. Phys. Chem. A, 2012, 116, 7210–7218 CrossRef CAS PubMed.
- Z. Zhou, Y. Zhang, Z. Wang, W. Wei, W. Tang, J. Shi and R. Xiong, Appl. Surf. Sci., 2008, 254, 6972–6975 CrossRef CAS.
- S. Bae, S. Gim, H. Kim and K. Hanna, Appl. Catal., B, 2016, 182, 541–549 CrossRef CAS.
- Z. Xu, J. Lu, Q. Liu, D. Lian, X. Aihua, Q. Wang and Y. Li, RSC Adv., 2015, 5, 76862–76874 RSC.
- T. Zhou, X. Zou, J. Mao and X. Wu, Appl. Catal., B, 2016, 185, 31–41 CrossRef CAS.
- Y. Yao, C. Xu, J. Qin, F. Wei, M. Rao and S. Wang, Ind. Eng. Chem. Res., 2013, 52, 17341–17350 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cy01479h |
| This journal is © The Royal Society of Chemistry 2016 |