
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Substrate and cofactor binding to nitrile reductase: a mass spectrometry based study†
Lorina
Gjonaj
a,
Martijn
Pinkse
b,
Elena
Fernández-Fueyo
a,
Frank
Hollmann
a and
Ulf
Hanefeld
*a
aGebouw voor Scheikunde, Biokatalyse, Afdeling Biotechnologie, Technische Universiteit Delft, Julianalaan 136, 2628BL Delft, The Netherlands. E-mail: U.Hanefeld@tudelft.nl
bAnalytische Biotechnologie, Afdeling Biotechnologie, Technische Universiteit Delft, Julianalaan 67, 2628BC Delft, The Netherlands
First published on 23rd June 2016
Abstract
Nitrile reductases catalyse a two-step reduction of nitriles to amines. This requires the binding of two NADPH molecules during one catalytic cycle. For the nitrile reductase from E. coli (EcoNR) mass spectrometry studies of the catalytic mechanism were performed. EcoNR is dimeric and has no Rossman fold. It was demonstrated that during catalysis each active site binds one substrate molecule. NADPH binds independent of the substrate. The PreQ0 binding pocket of the active site is not involved in the binding of NADPH; this is in conflict with an earlier hypothesis.
Introduction
Nitrile reductases (NR) have been discovered as participants in the biosynthetic pathway of queuosine, displaying an unusual activity. They are the first enzymes able to catalyse the reduction of a nitrile group to a primary amine.1 NR has emerged as a potential tool to overcome and replace traditional chemical approaches and harsh conditions needed to accomplish nitrile hydrogenation.2 The nitrile reductase is a NADPH-dependent enzyme which catalyses the reduction of the nitrile group of 7-cyano-7-deazaguanine (PreQ0) to an amino group, 7-aminomethyl-7-deazaguanine (PreQ1) (Scheme 1). This chemoselective enzyme has been characterised in several organisms confirming this particular activity.3 The crystal structures of NR from Bacillus subtilis (BsNR)4 and Vibro cholerae (VcNR)5 have been resolved, providing more information about the mechanism of the catalytic reaction. While the active site in all known NR's is highly conserved, the overall structure of the enzymes can be different. To date two types of structure have been elucidated. In the case of BsNR the enzyme forms a homodecamer (dimer of pentamers) and for VcNR a homotetramer (dimer of dimers) was observed (Fig. 1). In the former the active site is established at the interfaces of the five subunits, in the latter the active sites are within one protein and two monomers face each other in the dimer. Nonetheless, the active sites are virtually identical in both types of structures (Fig. 1). The covalently bound substrate is clearly visible in the active sites. The reaction mechanism for all NRs is proposed to proceed in two reduction steps.3–6 In order to reduce one molecule of PreQ0, two consecutive molecules of NADPH will act as hydride donor within the active site. The first overall step involves the formation of the covalent adduct of the PreQ0 within the active site. By introducing mutations in the active site of the NR from Escherichia coli (EcoNR) in the positions previously elucidated for VcNR and BsNR, the essential amino acids for the catalytic cycle were confirmed.3c They are shown to be Cys190, Asp197 and His229 in the schematic representation of the catalytic cycle (Scheme 2).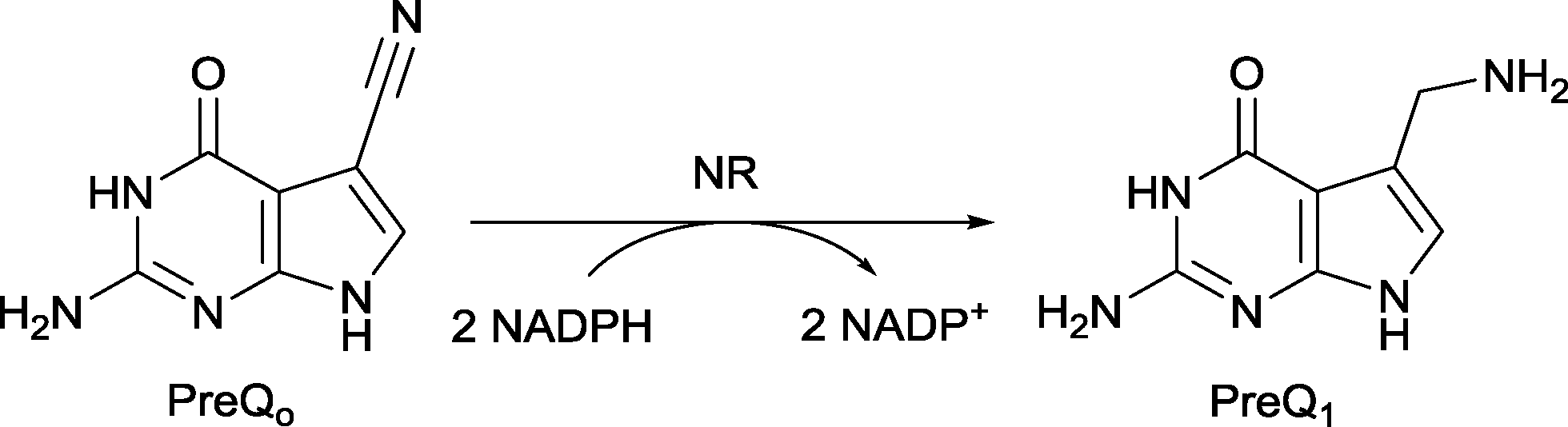 | ||
| Scheme 1 Catalytic reduction of PreQ0 with NADPH catalysed by NR. | ||
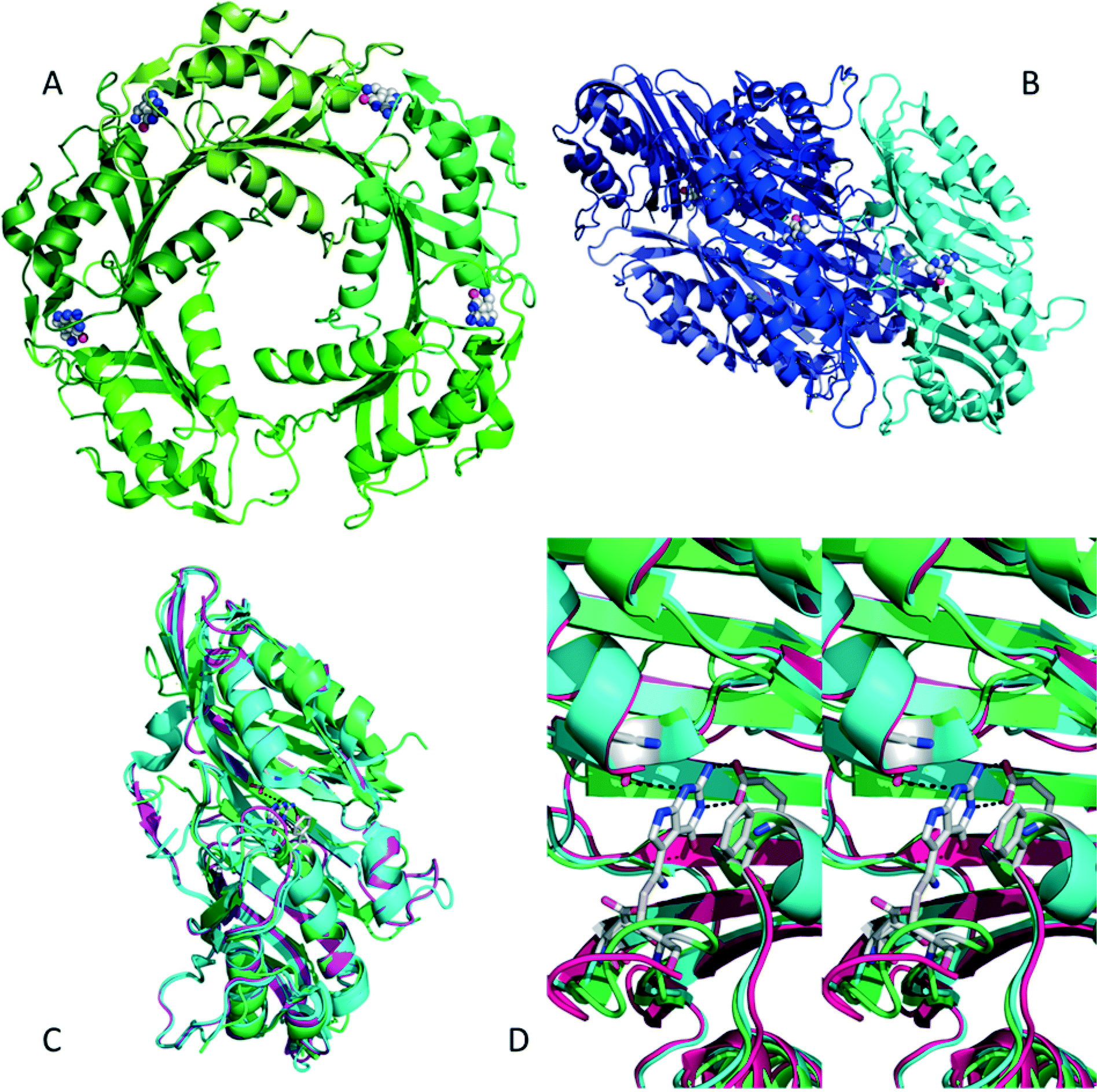 | ||
| Fig. 1 A) BsNR is pentameric, with the active sites located at the interfaces of the subunits, the substrate is visible in four active sites (pdb 4F8B). B) VcNR is dimeric, with two dimers assembling to a tetramer, the substrate is visible in the active sites which are within the subunits (pdb 3BP1). C) Overlay of VcNR (blue, pdb 3BP1), BsNR (green, pdb 4F8B) and EcoNR (magenta), indicating the very high degree of similarity, even though the subunits might be different. D) Stereoview of the three different active sites, with PreQ0 covalently bound; colours as in C. All pictures were generated using the PyMOL molecular graphics system. | ||
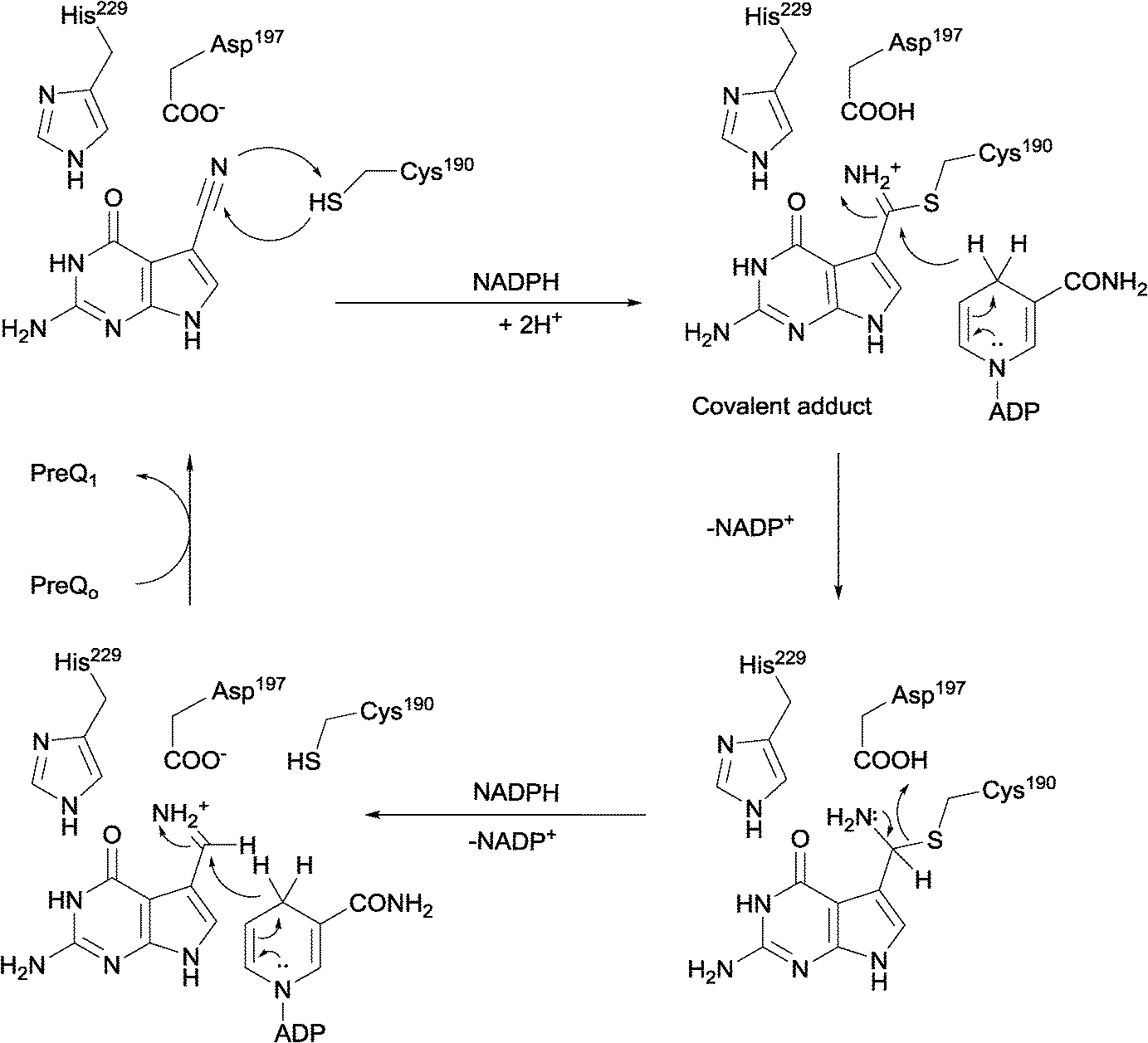 | ||
| Scheme 2 Cys190 is the nucleophile that attacks the nitrile, Asp197 and His229 have been suggested to take part in the catalytic mechanism of NR, His229 in particular in the positioning of the substrate. | ||
Remarkably, no determined structure of an NR contains a Rossman fold, characteristic for NAD(P)H dependent enzymes. Moreover, no structures with a complete NADPH bound to the enzyme are available (see also pdb 3UXJ and 3UXV). This raises the question how the two NADPH cofactor molecules per catalytic cycle bind to the NR. Molecular dynamic simulation performed on VcNR suggest that the cofactor will be positioned in one of the active sites of the dimeric enzyme.5 The adenine moiety of the NADPH is believed to bind within the PreQ0 pocket of one monomer of the dimer. The docking studies suggest that this occurs due to great similarity between the adenine moiety and PreQ0. This is supposed to occur in combination with PreQ0 docking in the second active site of the dimer.5 This implies that per dimer of VcNR only one substrate molecule can bind in an active site as the other active site acts as anchor for the NADPH.
To probe this, direct detection of the binding of PreQ0 and NADPH needs to be monitored. With mass-spectrometry the proposed intermediates can ideally be identified directly.7,8 This would allow insight into the mechanism of the reduction reaction and the binding mode of the cofactor. In our previous research a production system for EcoNR was successfully established3dEcoNR was purified via His-tag affinity column. We found for PreQ0 a Km value of 2.84 μM and kcat = 5.24 min−1 and for the cofactor NADPH Km = 6.42 μM and kcat = 5.2 min−1 for the enzyme monomer, in line with previous results (ESI† Fig. S1 and S2).1,3
Based on the sequence analysis, EcoNR shows a 63.6% homology with VcNR (Fig. 1). As for VcNR, dimer and tetramer are the main conformations of EcoNR (Fig. S3†). This allows for electrospray ionisation mass-spectrometry (ESI-MS) and detection of the covalent adduct (Scheme 2), particularly as the active site is inside one monomer. The pentameric and decameric enzymes such as that from BsNR are less suitable for this purpose. Indeed, attempts with ESI-MS to characterise the covalent PreQ0 adduct with BsNR had earlier been unsuccessful.3a
Results and discussion
Feasibility studies
As expected the dimeric structure of EcoNR is essentially not affected by ESI and the MS shows the enzyme to occur predominantly as a dimer (see Fig. 2A). In ESI-MS experiments multiple charged ions of the proteins are obtained. Therefore the dimeric EcoNR that has a large mass (>60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da) can still be measured straightforwardly. The signals represent different mass to charge ratios (m/z). All peaks in the spectra represent an intact protein or dimer in our case. From the m/z value the molecular weight of the protein can be determined. As shown in Fig. 2A, the dominate peak m/z = 4015 (z = 16+) is correlated to the mass of dimeric EcoNR, in which the His-tag is lost from both of the monomers during ionisation. Also the m/z peak (m/z = 4079, z = 17+) of the dimeric EcoNR with one tag still attached to the N-terminus of one of the monomers is clearly visible. The reason for the loss of the His-tag is unknown. The other peaks relate to the same dimers of EcoNR yet with a higher or lower number of charges (for the complete spectrum up to m/z = 9000 see Fig. S4†). Unlike the pentameric BsNR, EcoNR is thus a suitable system to study the binding of the substrates. This being PreQ0 bound to the Cys190 as an imidothioate and NADPH of which the mode of binding still needs to be unravelled. In the currently proposed mechanism PreQ0 and NADPH would each bind to one active site of the dimer.
000 Da) can still be measured straightforwardly. The signals represent different mass to charge ratios (m/z). All peaks in the spectra represent an intact protein or dimer in our case. From the m/z value the molecular weight of the protein can be determined. As shown in Fig. 2A, the dominate peak m/z = 4015 (z = 16+) is correlated to the mass of dimeric EcoNR, in which the His-tag is lost from both of the monomers during ionisation. Also the m/z peak (m/z = 4079, z = 17+) of the dimeric EcoNR with one tag still attached to the N-terminus of one of the monomers is clearly visible. The reason for the loss of the His-tag is unknown. The other peaks relate to the same dimers of EcoNR yet with a higher or lower number of charges (for the complete spectrum up to m/z = 9000 see Fig. S4†). Unlike the pentameric BsNR, EcoNR is thus a suitable system to study the binding of the substrates. This being PreQ0 bound to the Cys190 as an imidothioate and NADPH of which the mode of binding still needs to be unravelled. In the currently proposed mechanism PreQ0 and NADPH would each bind to one active site of the dimer.
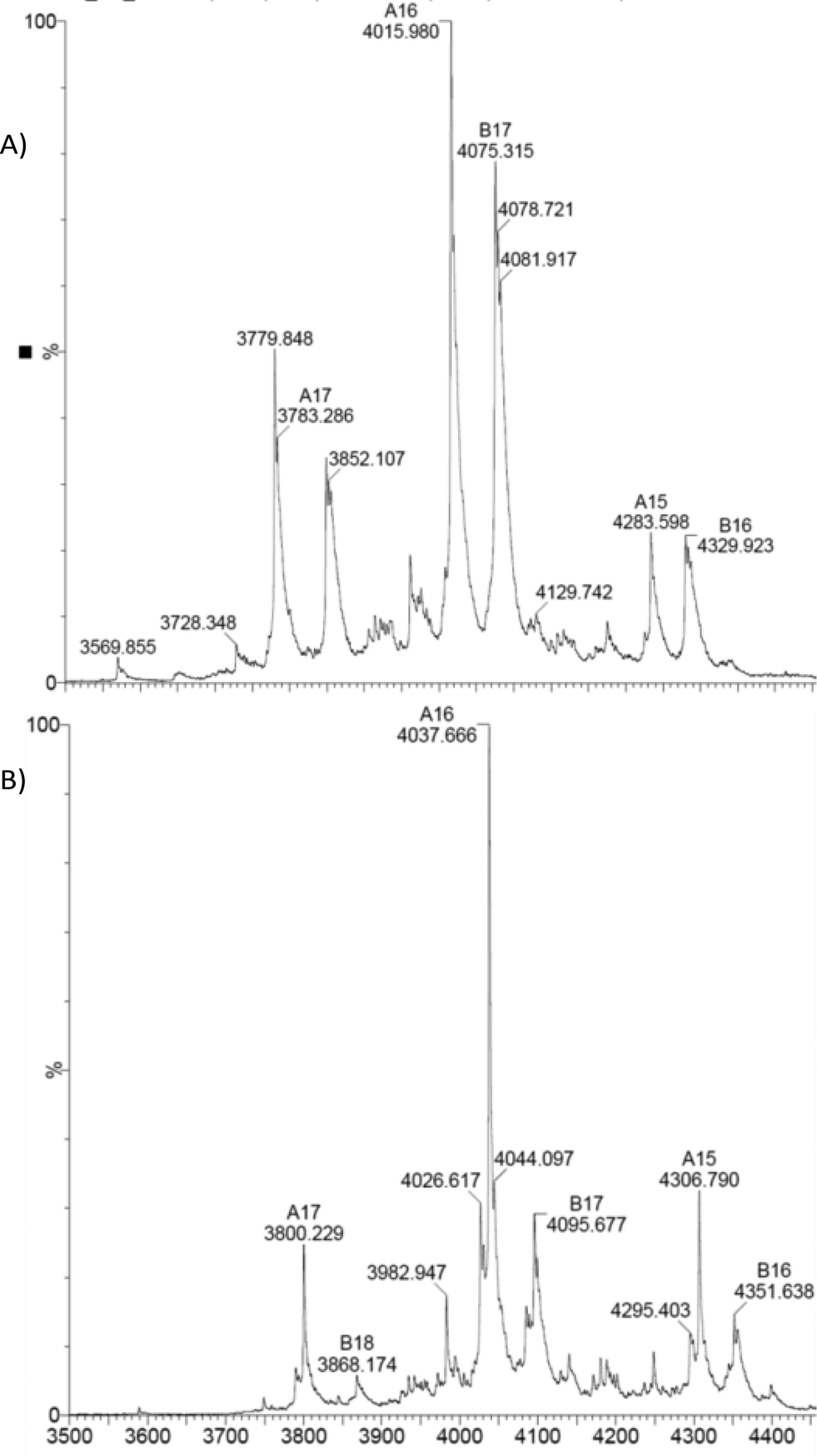 | ||
| Fig. 2 A) Native EcoNR and B) EcoNR treated with one equivalent of substrate PreQ0 per monomer (m/z = 4037). | ||
For BsNR, adduct formation with PreQ0 and its reduction were followed by UV-vis spectroscopy.3a,4b While inconclusive for the NADPH binding, this allows ESI-MS independent proof of adduct formation. In line with those results the characteristic absorbance of the new band appearing at 376 nm was observed for EcoNR. Using 2–3 fold excess of PreQ0 per active site of EcoNR (concentration 40–60 μM), gentle shaking for 3 h, followed by dialysis to remove the excess of the substrate the adduct formation was confirmed by UV-vis spectrometry. Based on these results the PreQ0EcoNR should be detectable by ESI-MS (Fig. 3).
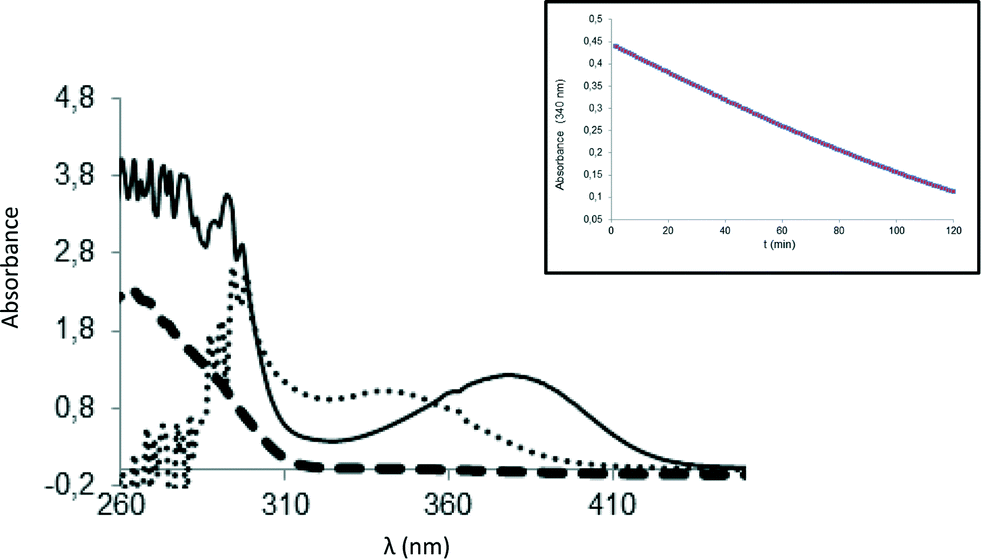 | ||
| Fig. 3 UV-vis characterisation of the covalent PreQ0 adduct. Solid line: covalent adduct; (·) covalent adduct after addition of an excess of NADPH; (-) EcoNR. Inset: Consumption of the substrate over time in presence of NADPH. | ||
ESI-MS characterisation of the intermediates of the catalytic cycle
Initial experiments to detect the PreQ0EcoNR adduct by ESI-MS were inconclusive. Conditions of the UV-vis experiments were not suitable for ESI-MS measurements, possibly due to hydrolysis of the delicate imidothionate bond of the covalent adduct. To circumvent this, the nitrile reductase was treated with one equivalent PreQ0 per active site and analysed immediately. No mass peak of free EcoNR was found. Instead a new peak m/z = 4037 is observed, the multiply charged ion (z = 16+) of the dimeric EcoNR with one molecule of PreQ0 covalently bound to each of the monomers. Complete saturation of the enzyme with the substrate (Fig. 2B) is thus achieved by treatment with one equivalent of substrate per active site. This confirms the earlier crystallographic results for VcNR and BsNR (Fig. 1).Based on the similar Km values for NADPH and PreQ0 one equivalent of NADPH per active site was added to EcoNR. Molecular modelling and crystal structures had suggested that only one molecule of cofactor is bound to the dimeric enzyme, as the minimum catalytic unit.5 Analysis of the mass-spectra revealed the appearance of a new signal (m/z = 4062, z = 16+), with a deconvoluted mass 752 amu higher than the native EcoNR (Fig. 4). The new peak corresponds to the dimeric enzyme with one molecule of NADPH bound to it. This would be in line with the proposed binding of one molecule of NADPH in one active site per dimer but contradict our results from the PreQ0 binding studies.5 Alternatively, the NADPH could bind to the dimeric EcoNR, however, in a different binding pocket than that of PreQ0.
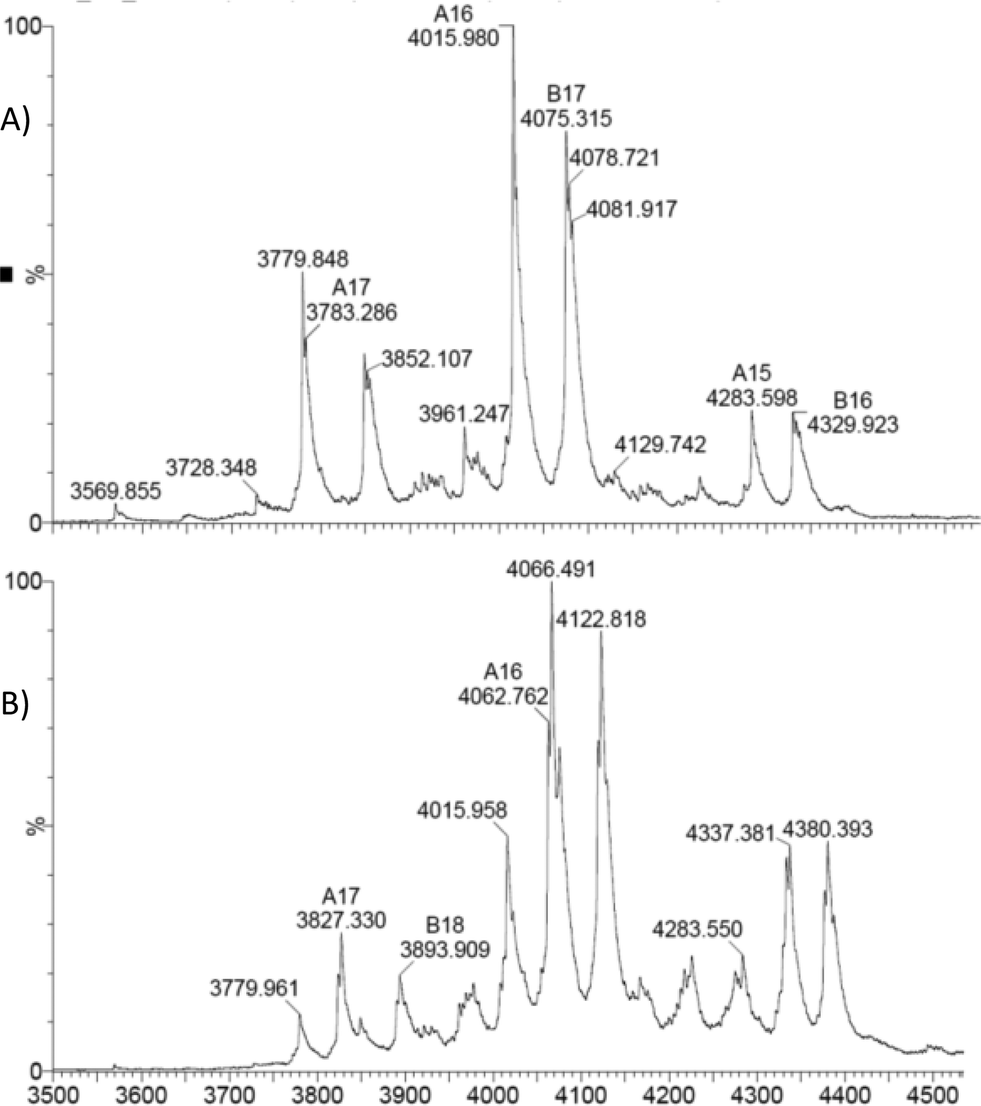 | ||
| Fig. 4 A) native EcoNR and B) EcoNR treated with one equivalent of NADPH per monomer (m/z = 4062). | ||
To probe the mode of binding of NADPH and PreQ0 to EcoNR both were added. First, EcoNR was treated with one equivalent of PreQ0 per monomer followed by the addition of one equivalent of NADPH per active site/monomer. In a second experiment, NADPH and PreQ0 were added in the reverse order to EcoNR. Both experiments led to the same mass spectra (Fig. 5 and Fig. S5 and S6, ESI†). When interpreting ESI mass-spectra it needs to be taken into account that spectra are recorded over time. From the moment the enzyme is treated with PreQ0 and cofactor, the catalytic reaction will proceed. This includes the time needed for sample preparation and the measurements, which represent several scans over the entire measured mass range. Therefore, signals for various species can be expected to be present in the spectra. In more detail (Fig. 5A), the difference between the molecular weight of the new peaks appearing in the spectra compared to the mass of the free EcoNR, led to the identification of the following multiple charge ions: a mass peak correlating to two molecules of PreQ0 and one molecule of NADPH bound to the dimeric enzyme (m/z = 4085, z = 16+). This multiple-charged ion m/z = 4085 proves our earlier hypothesis that the cofactor and the two substrate molecules bind, independent of each other to the enzyme. NADPH cannot bind to the PreQ0 binding pocket, as suggested earlier.5 Therefore, they must bind to two different sites and not both to the PreQ0 binding pocket. The second peak correlates to one substrate and one cofactor molecule bound to the dimer (m/z = 4074, z = 16+). Additionally, a small peak correlating to only a NADPH molecule bound to the enzyme (m/z = 4063, z = 16+) is visible. It may be assumed that each of the m/z peaks identified represents a step of the catalytic cycle. However, given the time delay in the measurements it is not clear whether both active sites act in concert or not. Initially a substrate molecule adds per active site. A possible intermediate with also two NADPH molecules per dimer is not visible. Although unlikely, since only one NADPH molecule bound per dimer was detected when only NADPH was added (see above, Fig. 4), it cannot be ruled out as it might have already reacted before measurements. The signal corresponding to two PreQ0 molecules and one molecule of NADPH in the dimer might represent a mixture of two substrate molecules bound and one equivalent of NADPH or already represent an intermediate in which one preQ0 or both are partially reduced. This is followed by the complete reduction of one of the substrates resulting in the peak at m/z = 4074. Subsequently complete reduction of the other preQ0 leaves only one NADPH molecule bound to the dimeric enzyme (m/z = 4063, z = 16+).
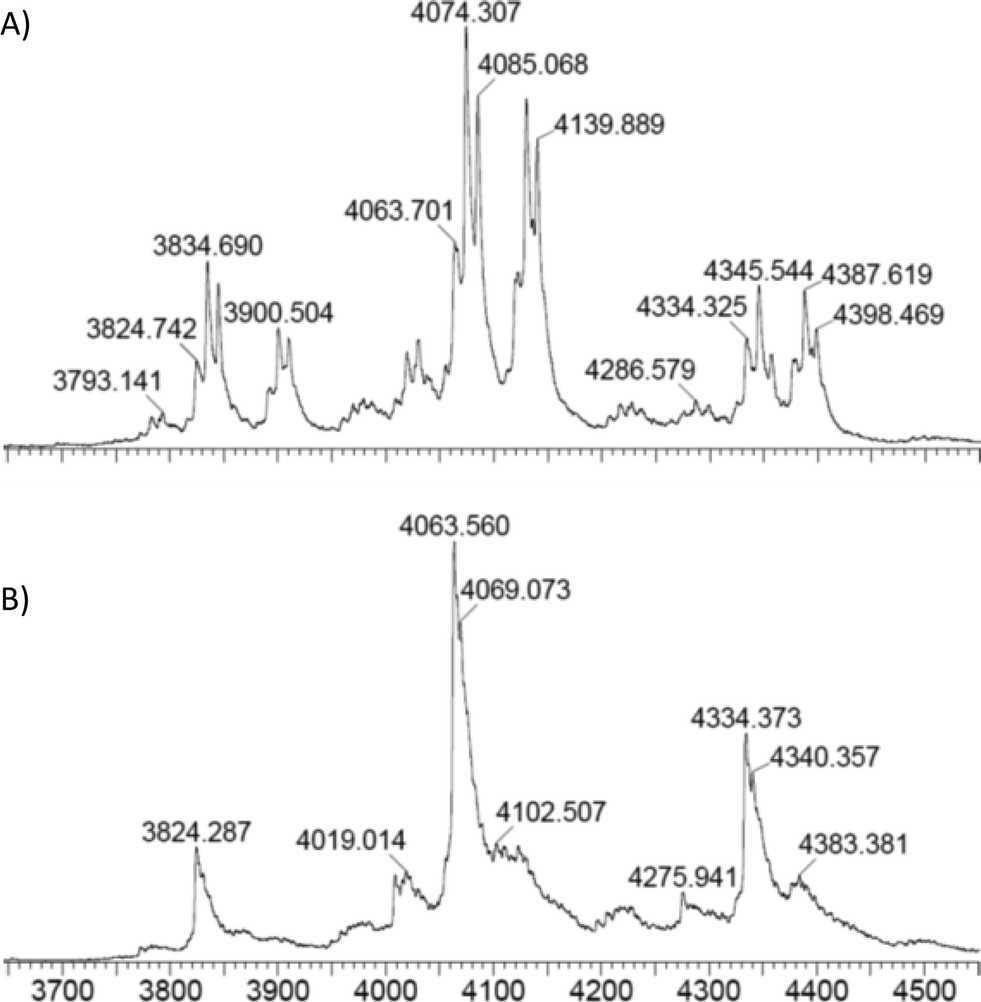 | ||
| Fig. 5 A) EcoNR treated with one equivalent of PreQ0 and NADPH each per monomer; three main peaks are present: m/z = 4085 (dimeric enzyme + 2 PreQ0 + NADPH); m/z = 4074 (dimeric enzyme + PreQ0 + NADPH). B) The mass peak m/z = 4063 is correlated to the mass of the dimeric EcoNR + NADPH when an excess of cofactor was used. | ||
In the following experiments, an excess of NADPH was added to an aliquot of the reaction mixture used to acquire the mass-spectrometry data reported in Fig. 5A. Complete consumption of PreQ0 (Fig. 5B) verified that the enzyme remains active under the reaction conditions. Remarkably, even with the excess of NADPH remaining at the end of this experiment only one NADPH molecule is bound to the dimeric enzyme.
The absence of the Rossmann fold in the NRs left the mode of NADPH binding unanswered, in particular since the suggested binding to one active site can now be ruled out. Therefore, even the importance of the adenosine moiety of NADPH is in question.
In order to elucidate whether the cofactor binds via the nicotinamide and/or the adenosine portion, EcoNR was treated with the AMP and/or a nicotinamide analogue (Fig. 6), followed by ESI-MS. No adduct was observed when the enzyme was treated with AMP using equimolar or higher concentrations per monomer of EcoNR. Subsequently, NADPH and substrate were added to the mixture of enzyme and AMP (Fig. S7†). After the addition, the same mass-spectra were obtained as in the absence of AMP (Fig. 4A). This demonstrated that the presence of AMP has no influence on the NADPH binding to the enzyme. Thus the binding of the cofactor seems to be independent of the adenosine moiety. Binding to the EcoNR via the nicotinamide moiety of NAPDH was probed via nicotinamide cofactor analogues. NR was treated with a nicotinamide mimic (Fig. 6).9 Again no change in the mass of EcoNR was observed, nor did any reaction occur. Also, no difference was seen when a mixture of AMP and nicotinamide analogue was employed, ruling out a co-operation of both of these moieties in binding.
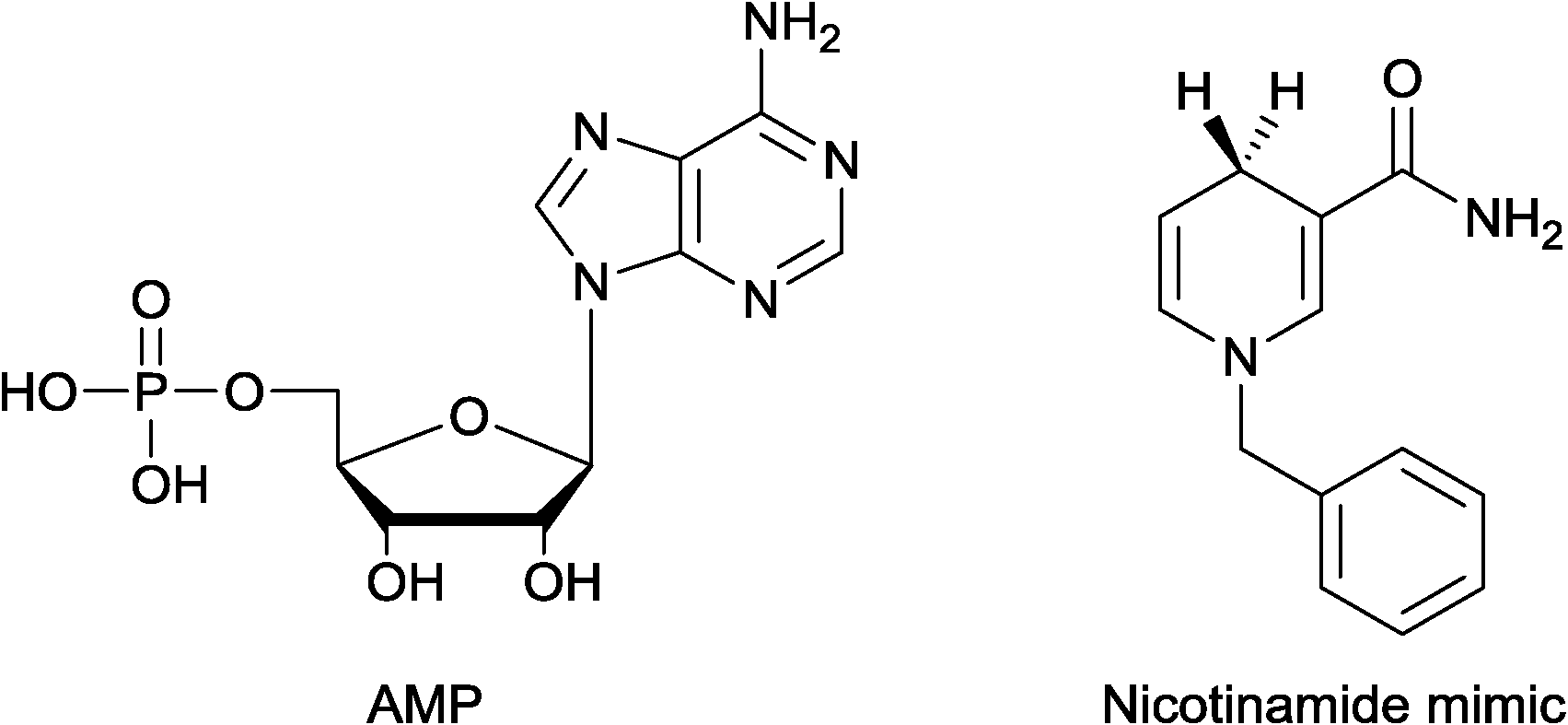 | ||
| Fig. 6 Cofactor mimics. | ||
Conclusion
Here, we provide new evidence for the mechanism of action of nitrile reductases. Mass-spectrometry proved to be a successful technique to gain insight in the binding mode of the cofactor and the natural substrate. Additionally, it allowed surveying the steps of the catalytic cycle. Indeed, the first step of the catalytic cycle is the covalent binding of the substrate, one to each monomer of the dimeric enzyme. This dimeric enzyme was the only catalytic unit we detected. ESI-MS analysis suggested that only one molecule of NADPH binds to the dimeric EcoNR. This is independent of the other two molecules of substrate bound to the two active sites. This would contradict the previously proposed docking of one molecule of substrate and one molecule of NADPH per dimeric enzyme. Furthermore, the binding of NADPH is independent of the adenosine moiety.Experimental section
Materials and methods
UV-vis measurements were performed on a UV/vis spectrometer UV-2401 PC from Shimadzu with a Julano type F12 thermostat. Liquid chromatography purification was done on a UFLC equipment from Shimadzu. Ni-NTA spin nickel column were provided from Qiagen. Bicinchoninic acid assay (BCA) kit was purchased from ThermoFisher Scientific. All equipment was used according to the provided instructions. The chemicals were purchased from Sigma-Aldrich, if not mentioned otherwise. PreQ0, was synthesised as described earlier.3b,c:buffer, 7:3), which was further diluted in Tris buffer mentioned above, to obtain the desired final concentration. Fresh stock solution of NADPH [100 μM in buffer] and PreQ0 (final concentration of 500 μM containing 30% DMSO in buffer) were prepared prior the experiments. In order to determine the Km for PreQ0, the concentration of the cofactor was kept constant at 100 μM, while changing the substrate concentration from 1–75 μM. In order to determine the Km for NADPH, the concentration of PreQ0 was kept constant at 50 μM, while changing the cofactor concentration from 1–400 μM.
Protein mass spectrometry
Native protein mass spectra were recorded on a Waters quadrupole time-of-flight mass spectrometer (Q-TOF premier, Waters, Manchester, UK) equipped with an 8 kDa quadrupole mass filter. Mass calibration in the range of m/z 1000 to 10000 was done by electrospraying a solution of cesium iodide (2 mg ml−1 in 50% 2-propanol and 50% water). Prior to analysis, proteins were buffer-exchanged to 50 mM ammonium acetate in MilliQ water (pH = 6.8) using ultra centrifugation filters (Amicon® Ultra) with a molecular weight cut-off of 10 kDa. Protein ions were formed in the positive ion mode from a ∼2–20 μM solution mode via nanoelectrospray using borosilicate spray capillaries (ES383 from ThermoFisher Scientific, Breda, The Netherlands). The applied voltage was optimised for each capillary (range 0.9 to 1.5 kV). The pressure in the first stage of the source region of the MS system was set to 6.0 mbar (standard 1.8 mbar) by reducing the pumping efficiency of the scroll pump (Edwards XDS 35i) for optimised transmission of ions with high m/z values. The sample cone voltage was set to 150 V, the extraction cone was set to 5 V and the quadrupole was set as a broad-band mass filter, transmitting ions from m/z 2000 to 10000. The scan rate was set to 3 s with a 0.1 s interscan time. Protein mass spectra were analyzed using the MassLynx 4.1 software package (Waters).
:ammonium acetate 50 mM pH = 7.2, 7:3) of PreQ0 and the other compounds tested were made. They were further diluted in ammonium acetate buffer (50 mM) until the final concentration of 10–40 μM was reached. A stock solution of NADPH [1.0 mM] was freshly made prior to each set of experiments and further diluted until the desired final concentration was reached (10–40 μM). The final concentration of DMSO was kept the same in all the samples. The enzyme was treated with equimolar amount of PreQ0 or any of the other compounds, gently mixing for 15 min. Prior to analysis, the samples were buffer-exchanged to 50 mM ammonium acetate in MilliQ water (pH = 6.8) using ultra centrifugation filters (Amicon® Ultra) with a molecular weight cut-off of 10 kDa.
Acknowledgements
The authors gratefully acknowledge financial support by STW (grant 11142). The authors wish to thank K. Moeller and Dr. G. S. Nguyen for help with initial experiments and literature synthesis. They thank C. E. Paul for kindly providing the nicotinamide cofactor.References
- S. G. Van Lanen, J. S. Reader, M. A. Swairjo, V. de Crécy-Lagard, B. S. Lee and D. Iwata-Reuyl, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4264 CrossRef CAS PubMed.
- (a) S. Gomez, J. A. Peters and T. Maschmeyer, Adv. Synth. Catal., 2002, 344, 1037 CrossRef CAS; (b) L. Veum, S. R. M. Pereira, J. C. van der Waal and U. Hanefeld, Eur. J. Org. Chem., 2006, 1664 CrossRef CAS.
- (a) B. W. K. Lee, S. G. Van Lanen and D. Iwata-Reuyl, Biochemistry, 2007, 46, 5833 Search PubMed; (b) B. Wilding, M. Winkler, B. Petschacher, R. Kratzer, A. Glieder and N. Klempier, Adv. Synth. Catal., 2012, 354, 2191 CrossRef CAS; (c) B. Wilding, M. Winkler, B. Petschacher, R. Kratzer, S. Egger, G. Steinkellner, A. Lyskowksi, B. Nidetzky, K. Gruber and N. Klempier, Chem. – Eur. J., 2013, 19, 7007 CrossRef CAS PubMed; (d) K. Moeller, G. S. Nguyen, F. Hollmann and U. Hanefeld, Enzyme Microb. Technol., 2013, 52, 129 CrossRef CAS PubMed; (e) L. Yang, S. L. Koh, P. W. Sutton and Z.-X. Liang, Catal. Sci. Technol., 2014, 4, 2871 RSC.
- (a) M. A. Swairjo, R. R. Reddy, B. Lee, S. G. Van Lanen, S. Brown, V. de Crécy-Lagard, D. Iwata-Reuyl and P. Schimmel, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2005, 61, 945 CrossRef CAS PubMed; (b) V. M. Chikwana, B. Stec, B. W. K. Lee, V. de Crécy-Lagard, D. Iwata-Reuyl and M. A. Swairjo, J. Biol. Chem., 2012, 287, 30560 CrossRef CAS PubMed.
- Y. Kim, M. Zhou, S. Moy, J. Morales, M. A. Cunningham and A. Joachimiak, J. Mol. Biol., 2010, 404, 127 CrossRef CAS PubMed.
- A. J. M. Ribeiro, L. Yang, M. J. Ramos, P. A. Fernandes, Z.-X. Liang and H. Hirao, ACS Catal., 2015, 5, 3740 CrossRef CAS.
- (a) A. J. R. Heck and R. H. H. van den Heuvel, Mass Spectrom. Rev., 2004, 23, 368 CrossRef CAS PubMed; (b) U. Hanefeld, A. J. J. Straathof and J. J. Heijnen, Biochim. Biophys. Acta, 1999, 1432, 185 CrossRef CAS PubMed; (c) Y. M. Efimova, A. A. van Well, U. Hanefeld, B. Wierczinski and W. G. Bouwman, Phys. B, 2004, 350, e877 CrossRef CAS.
- C. O'Reilly and P. D. Turner, J. Appl. Microbiol., 2003, 95, 1161 CrossRef.
- C. E. Paul, S. Gargiulo, D. J. Opperman, I. Lavandera, V. Gotor-Fernández, V. Gotor, A. Taglieber, I. W. C. E. Arends and F. Hollmann, Org. Lett., 2013, 15, 180 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cy01140c |
| This journal is © The Royal Society of Chemistry 2016 |