
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Activation of [CrCl3{PPh2N(iPr)PPh2}] for the selective oligomerisation of ethene: a Cr K-edge XAFS study†‡
Stuart A.
Bartlett
ab,
Jerome
Moulin
c,
Moniek
Tromp
d,
Gillian
Reid
c,
Andy J.
Dent
e,
Giannantonio
Cibin
e,
David S.
McGuinness
f and
John
Evans
*ace
aResearch Complex at Harwell, Rutherford Appleton Laboratory, Didcot, OX11 0FA, UK. E-mail: stuart.bartlett@rc-harwell.ac.uk
bDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK
cChemistry, University of Southampton, Southampton, SO17 1BJ, UK. E-mail: je@soton.ac.uk
dVan't Hoff Institute for Molecular Sciences, University of Amsterdam, PO Box 94720, 1090 GS Amsterdam, Netherlands. E-mail: M.Tromp@uva.nl
eDiamond Light Source, Rutherford Appleton Laboratory, Didcot, OX11 0DE, UK. E-mail: john.evans@diamond.ac.uk
fSchool of Physical Sciences – Chemistry, University of Tasmania, Hobart 7001, Australia
First published on 18th May 2016
Abstract
The activation of the ethene tetramerisation catalyst system based upon [CrCl3(THF)3] and N(iPr)(PPh2)2 has been investigated in situ via the reaction of [CrCl3{PPh2N(R)PPh2}(THF)] 1a (R = iPr) with excess AlMe3 in toluene. The Cr K-edge XAFS spectrum of the solution freeze quenched after 1 min reaction time indicated monomethylation of the metal with the resultant product being [CrClMe(ClAlCl3){PPh2N(R)PPh2}(THF)] 4a (R = iPr). After 5 minutes reaction time the XAFS spectra indicate that ∼50% of 4a had been converted to a Cr(II) species, with the central core being high spin [CrCl2{PPh2N(R)PPh2}] 7a (R = iPr); a similar species, [CrClMe{PPh2N(R)PPh2}] 9a (R = iPr) was observed as its adduct with AlMe3 (10a) (R = iPr) when spectra were recorded on samples maintained at room temperature. Detailed analysis (EXAFS and XANES) indicated that 7a and 9a are stabilised by adduct formation of a Cr–Cl bond to the Lewis acids B(C6F5)3 and AlMe3, respectively. Modelling with DFT methods indicated that five-coordination was achieved, respectively by Cr–F (11a) and Cr–C (10a) interactions. In the presence of [Ph3C][Al{OC(tBuF)3}4], the Cr XAFS of the room temperature solution was inconsistent with the maintenance of a phosphine complex, but could be modelled with a site like [Cr2Me8]4− {Cr–Cr 2.01(2), Cr–C 2.14(4)}, thus demonstrating considerable variation in the effects of differing Lewis acids.
1. Introduction
Ethene oligomerisation is an attractive method to produce linear alpha-olefins (LAOs), which are further used in the production of synthetic polymers, detergents, plasticisers and lubricants.1 Employing metal catalysed oligomerisation of ethene can have a major drawback in the production LAOs, suffering from a statistical distribution of oligomer chain lengths.2 Selective oligomerisation is possible due to major breakthroughs using homogeneous chromium catalysts to produce 1-hexene and/or 1-octene.3,4 Using chromium with a bidentate diphenylphosphinoamine (PNP) in the presence of modified methylaluminoxane (MMAO) was seen to give 1-octene selectivity >70%.5Evidence suggests this occurs via a metallacycle ring after oxidative coupling of ethene to chromium, followed by further insertion of ethylene at the Cr–C bond and finally, reductive elimination of 1-octene.6,7 Despite this, the structure and oxidation state of the chromium intermediates are still not clear, with some studies favouring a Cr(II)/Cr(IV) synergy,8,9 and others suggesting a Cr(I)/Cr(III) cycle10–12 as the active pathway. One study has suggested a Cr-dimer, stabilised by PNP bridges.13
One previous study has used EPR and XAFS spectroscopy to help discriminate the active Cr species from Cr(acac)3 and PPh2N(iPr)PPh2 in the presence of MMAO, in which the authors proposed the active chromium species to be [CrII(Me)2(PPh2N(iPr)PPh2)].14 However, in this investigation the XAFS measurements were acquired at 30 minutes per spectrum, with 5–10 scans averaged to achieve a good signal-to-noise, thus limiting the time resolution to hours. Herein we report mixing of [CrCl3{PPh2N(R)PPh2}(THF)] 1a (R = iPr) with excess AlMe3 in toluene, swiftly followed by freeze-quenching and subsequent analysis by XAFS spectroscopy. The reaction was frozen at predetermined intervals down to 1 minute from mixing, using a technique used in previous experiments.15–18 The analysis of these reactions by XAFS spectroscopy also enabled a more thorough analysis of data previously collected from the same reaction during tens of minutes at room temperature, without freeze-quenching.19 The present analysis is aided by density functional theory (DFT) calculations, with a survey performed on [CrCl3{PPh2N(R)PPh2}(THF)] 1b (R = Me).
This analysis sheds new light on of the role of the aluminium and other co-catalysts during ethene tetramerisation, investigating the promotional effects previously reported.20 Here we present the first instance of conclusive structural data at much shorter time scales (1 and 5 minutes) for the species trapped in the solution phase, formed by the reaction of this highly valuable tetramerisation catalyst and its aluminum activator. This contrasts with previous studies that have allowed the reaction to proceed during analysis or have been crystallised out of solution over long time scales.
2. Results and discussion
2.1 Structure of complex 1
The Cr K-edge XANES and EXAFS data of complex 1a are presented in Fig. 1. The EXAFS analysis is consistent with the 6-fold coordination, with Cr–O, Cr–Cl and Cr–P shells at 2.05, 2.28(1) and 2.48(2) Å, respectively; the N atom in the chelate ring was also modelled in the fit presented (Fig. S1 and Tables 1 and S1†). Due to its low solubility in toluene, solution phase structural data for 1a were difficult to obtain and so the powder data have been used as a structural comparison.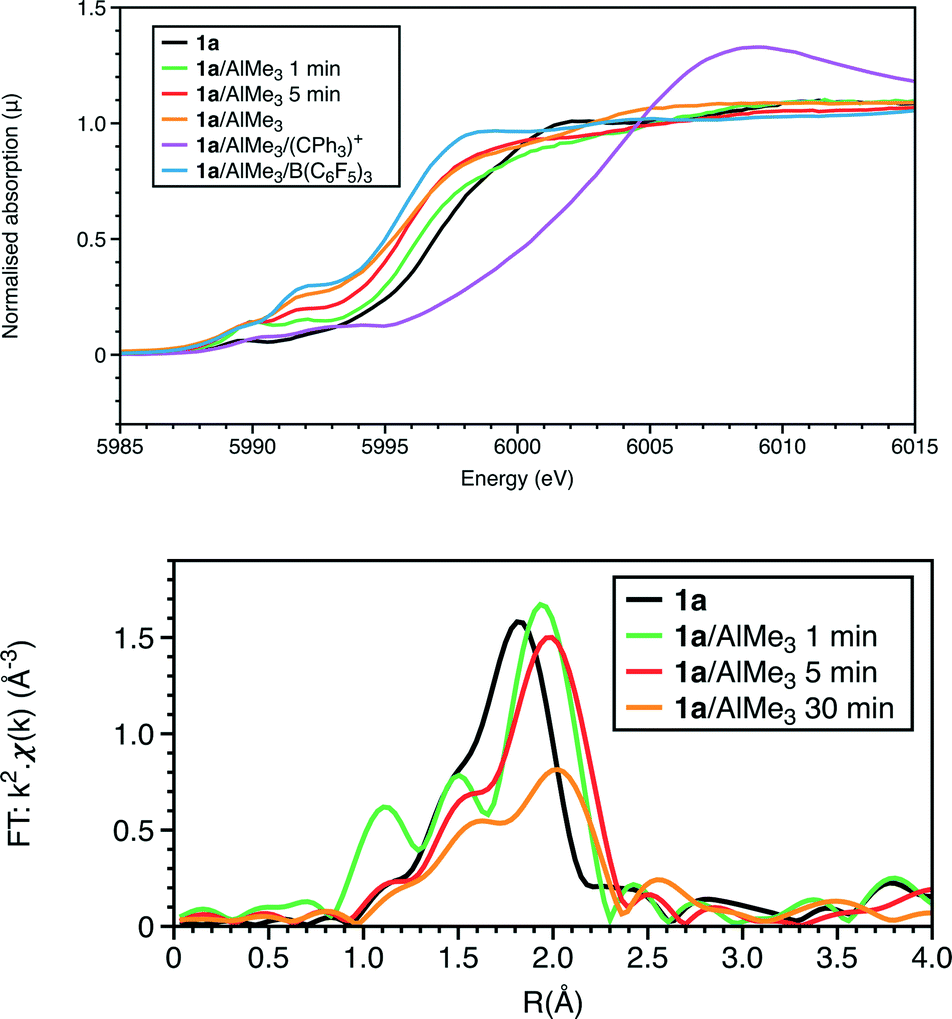 | ||
| Fig. 1 At the Cr K-edge: (top) XANES and (bottom) Fourier transforms of k2-weighted EXAFS data. Reactions performed at ambient temperature, some quenched by liquid nitrogen after a set reaction time. | ||
| Cr reaction | Coordination shell | d(Cr–X) (Å) |
|---|---|---|
| a Fixed parameters. | ||
| 1a in BN | 1Cr–O | 2.05a |
| 3Cr–Cl | 2.28(1) | |
| 2Cr–P | 2.48(2) | |
| 1Cr⋯N | 2.96(8) | |
| Quenched after 1 minute | 2Cr–C/O | 2.03(4) |
| 2Cr–Cl | 2.37(1) | |
| 2Cr–P | 2.54(6) | |
| Quenched after 5 minutes | 1Cr–C | 2.10(1) |
| 2Cr–Cl | 2.37(3) | |
| 2Cr–P | 2.49(2) | |
| Ambient temperature after 30 minutes | 1Cr–C | 2.14(2) |
| 2Cr–P | 2.49(1) | |
| Ambient temperature, with B(C6F5)3 | 1Cr–Cl | 2.73(3) |
| 2Cr–Cla | 2.36(2) | |
| 2Cr–Pa | 2.472(8) | |
| 1Cr–Ca | 3.04(1) | |
| Ambient temperature, [Ph3C][Al{OC(tBuF)3}4] | 1Cr–Cra | 2.01(2) |
| 3.3(7)Cr–C | 2.14(4) | |
A set of DFT models was compared in terms of predicting the Cr–E distances of complexes close to those in this study. Quartet states were adopted for Cr(III) species, and quintet states were established to be the most stable spin states for the Cr(II) complexes. Accordingly, [CrCl3{PPh2N(Cy)PPh2}(NCMe)] (Cy = cyclohexyl)21 was chosen as a base structure due to its similarity to complex 1a (R = iPr), albeit a fac rather than a mer isomer. The tendency for all methods was for an overestimate of the bond length compared to those determined in the crystal structure of this complex (Table S2†), but this difference was less for EDF2 and ωB97X-D functionals compared to the results using B3LYP. The EDF2/6-31G* method was employed in a wide survey of reaction possibilities starting from the reagents CrCl3(THF)3, PPh2N(Me)PPh2 and Al2Me6, and the more likely species probed by several DFT methods for R = Me and iPr. This showed that the mer isomers of both the tris THF complex and complex 1b were more stable than the fac ones (structures in Fig. 2 and ΔG° in Fig. 3).
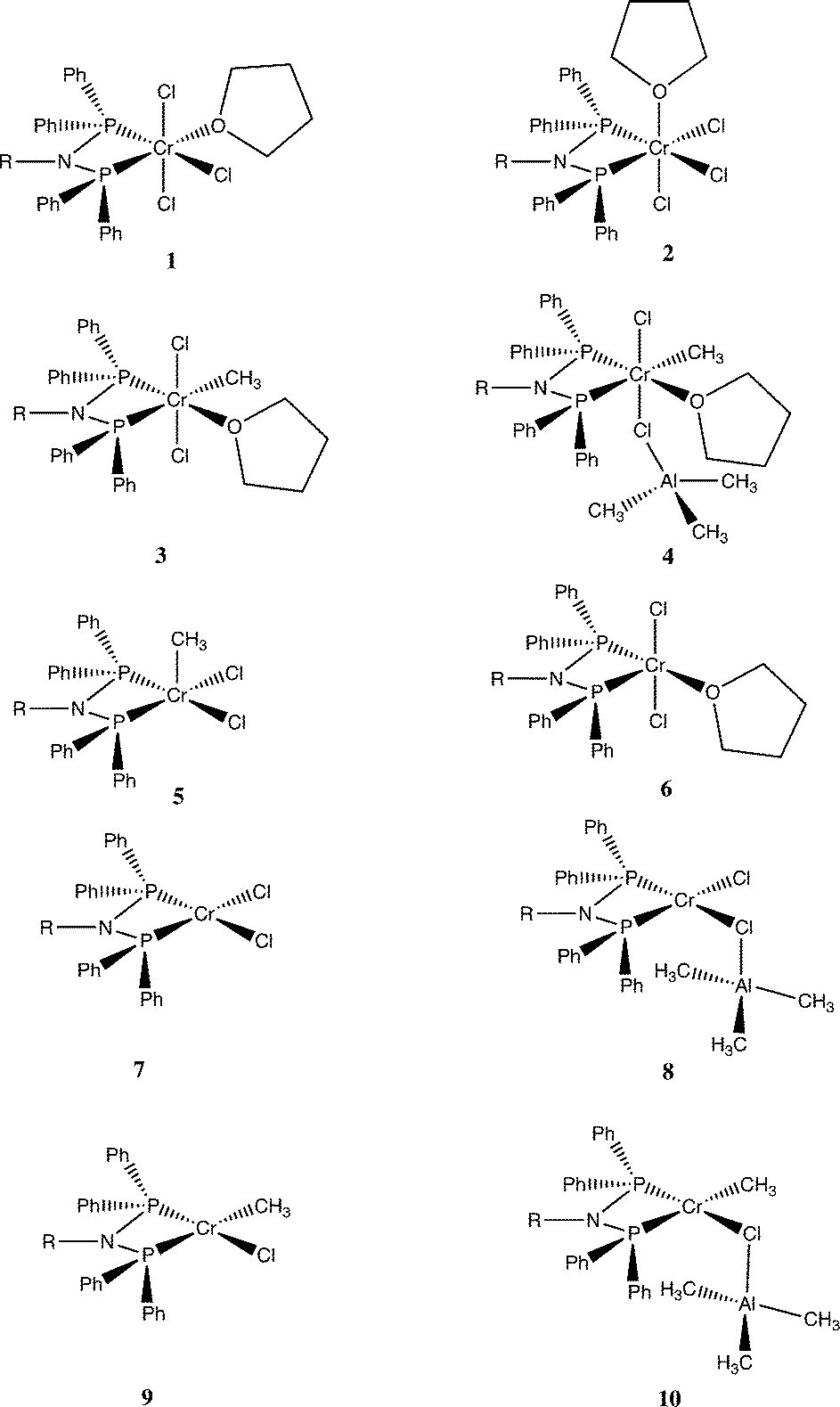 | ||
| Fig. 2 Structures considered for the reaction of [CrCl3{PPh2N(R)PPh2}(THF)] 1 (a R = iPr, b R = Me) with AlMe3. | ||
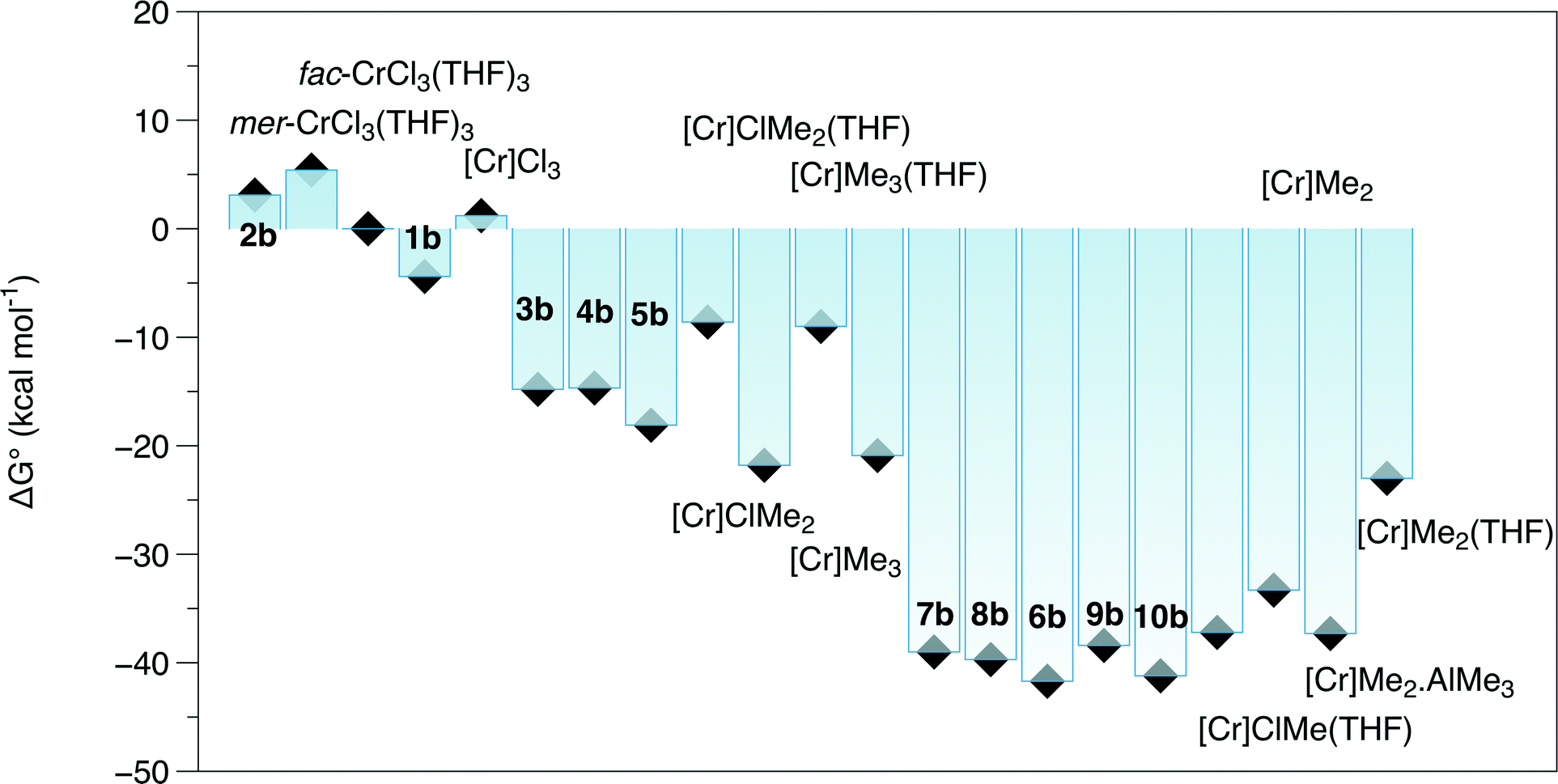 | ||
| Fig. 3 ΔG° calculated by EDF2/6-31G* for reactions from CrCl3(THF)3, (PPh2)2NMe and Al2Me6. Compound numbers as in Fig. 2, [Cr] = Cr(PPh2(NMe)PPh2). Cr(III) and Cr(II) complexes S = 3/2 and S = 2, respectively. | ||
(Yellow = Cr, green = Cl, red = O, brown = P, grey = C, purple = N and white = H).
A comparison of the EXAFS results for complex 1a and DFT models (Table 2) shows that the observed and calculated Cr–O and mean Cr–Cl distances are within 0.03 Å of each other. The fac isomer 2b displays a much longer Cr–O distance (2.18 Å) than observed experimentally, giving further support for the assignment of the mer isomer 1. The observed Cr–P bond length is similar to those previously reported fac-[CrCl3{PPh2N(Cy)PPh2}(NCMe)].21 The calculated structures display significant interdependent trans effects. There is a lengthening of the other Cr–P distance to 2.60 Å and a shortening of the Cr–Cl distance trans to it (2.27 Å); these distortions were not distinguishable from the EXAFS data.
| Distances (Å) | |||
|---|---|---|---|
| 1Cr–O | 3Cr–Cl | 2Cr–P | |
| EXAFS: 1a | 2.05 | 2.28(1) | 2.48(2) |
| 1b: EDF2/6-31G* | 2.064 | 2.274, 2.315, 2.331 | 2.461, 2.648 |
| EDF2/6-31G** | 2.065 | 2.277, 2.324, 2.326 | 2.469, 2.626 |
| ωB97X-D/6-31G* | 2.067 | 2.268, 2.303, 2.330 | 2.450, 2.634 |
| ωB97X-D/6-31G** | 2.066 | 2.268, 2.303, 2.333 | 2.454, 2.630 |
| 1a: EDF2/6-31G* | 2.072 | 2.277, 2.318, 2.316 | 2.464, 2.647 |
| EDF2/6-31G** | 2.059 | 2.280, 2.324, 2.328 | 2.455, 2.638 |
| ωB97X-D/6-31G* | 2.070 | 2.273, 2.309, 2.333 | 2.452, 2.620 |
| ωB97X-D/6-31G** | 2.070 | 2.271, 2.312, 2.330 | 2.453, 2.617 |
| 2b: EDF2/6-31G* | 2.178 | 2.283, 2.284, 2.286 | 2.550, 2.584 |
| EDF2/6-31G** | 2.174 | 2.283, 2.285, 2.286 | 2.551, 2.583 |
| ωB97X-D/6-31G* | 2.180 | 2.280, 2.282, 2.283 | 2.523, 2.567 |
| ωB97X-D/6-31G** | 2.178 | 2.281, 2.284, 2.284 | 2.524, 2.560 |
2.2 Reaction of complex 1 with excess AlMe3
The UV-visible spectrum of 1a displays two d–d transitions with maxima at 701 and 502 nm, which may be anticipated as arising from a 4A2 ground state to 4T2 and 4T1(F) excited states (Fig. S2†). TDDF calculations based upon the refined structure of 1a afforded clusters of peaks in the range of 620–720 and 450–550 nm (Fig. S3†). As we have reported for other ligand systems,17,22 the addition of AlMe3 to a solution of 1a in toluene at ambient temperature with spectra recorded within 2–3 minutes of the addition, results in a shift of the first absorption peak to shorter wavelength (660 nm) with a possible shoulder (∼450 nm) on the tail of an intense, charge-transfer band in the UV (Fig. S2†).2.3 Freeze-quench XAFS of the reaction of complex 1 with AlMe3 after 1 minute
Complex 1a is sparingly soluble in toluene, and decomposes to the dimer [CrCl3{PPh2N(iPr)PPh2}]2.5 This made the solution mixing by stopped flow methods from stock solutions, as employed previously in other chemical systems,15–18 an unsuitable technique. Instead, to investigate the early stages of the activation process using freeze-quenched solutions, a solution of AlMe3 in toluene was added to a suspension of 1a in toluene in a Schlenk tube, mixed rapidly and transferred quickly to a Kapton tube and the sample frozen to 77 K using liquid nitrogen, measured at 1.0 minute and 5.0 minutes elapsed time from point of mixing.The Cr K-edge XAFS spectra obtained from room temperature reaction of 1a in toluene with a 20 fold excess AlMe3 and frozen 1 minute after mixing {Fig. 1 (green)} show that a structural change is evident. EXAFS analysis (Fig. S5, Table S3†) is consistent with a substitution of 1 Cr–Cl ligand by a Cr–Me group. The combined Cr–O/C shell is refined to a similar distance to the Cr–C in 1a (2.03 Å), but there is a lengthening of both the Cr–Cl and Cr–P distances to 2.37(1) and 2.54(6) Å, respectively. This would suggest the product to be [CrCl2Me{PPh2N(R)PPh2}(THF)] (R = iPr) 3a.
There are 4 possible geometric isomers of the complex 3a; DFT calculations on the reaction of 1b with AlMe3 (eqn (1)) indicate that the most stable is that shown for 3b in Fig. 2, with trans chloride ligands, consistent with the substitution of a Cr–Cl in 1atrans to a Cr–P bond. Direct substitution of a mutually trans chloride in 1a provided an isomer calculated to be 6.2 kcal mol−1 less stable, and the two isomers requiring rearrangement of the THF ligand to becoming cis to both P atoms of the phosphine ligand were found to be less stable still (7.7 and 9.3 kcal mol−1).
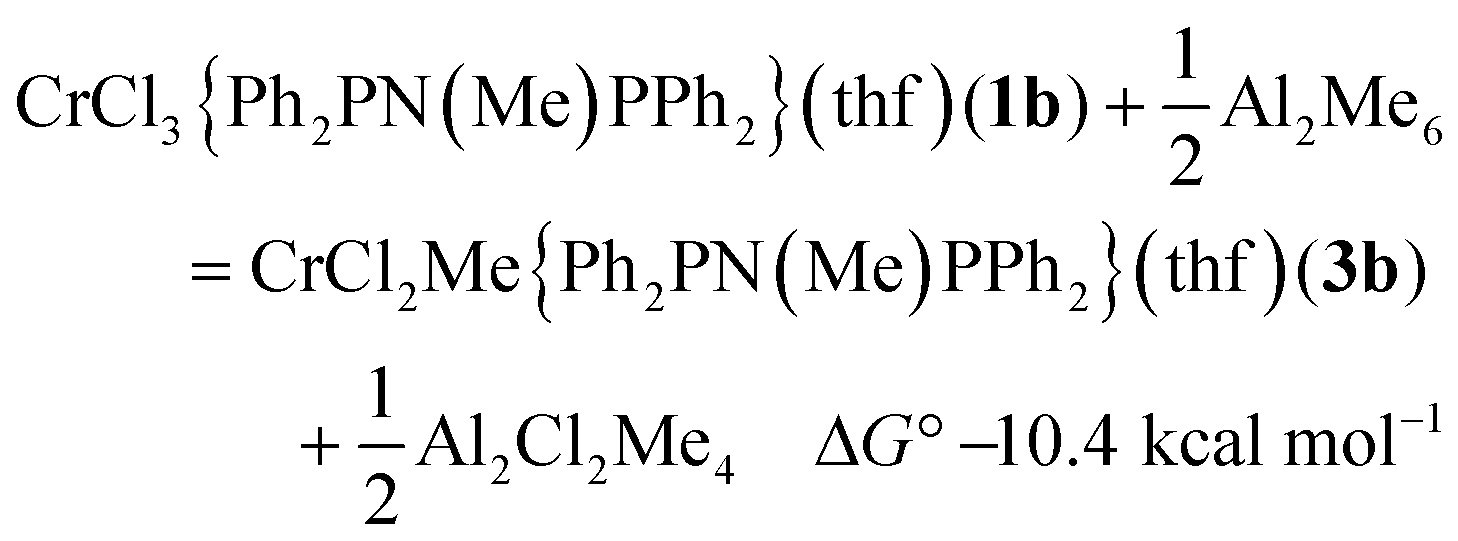 | (1) |
Calculated bond lengths from DFT methods are presented in Table 3, with the EXAFS-derived distances obtained from the freeze-quenched solution after 1 minute. The combined C/O shell agrees very closely with the calculated Cr–C distance from the most stable isomer of 3b (2.04 Å), and the Cr–O distance (2.08 Å) is within experimental error. Moving the methyl group to a position trans to Cr–Cl in the second most stable isomer increases the Cr–C distance to the region of 2.08 Å, towards the end of the extreme of the standard deviation of the EXAFS analysis. The mean of the calculated Cr–Cl distances (2.34 Å) is 0.04 Å less than the distance refined from the EXAFS by 2 standard deviations. Switching to the alternative isomer, with the Cr–Me trans to Cr–Cl, does increases that distance to Cl, as would be expected from the trans-influence of the alkyl group, but there is a compensating reduction in the other Cr–Cl bond length. An alternative way to increase the Cr–ligand bond lengths would be to form an adduct with AlMe3, which was in large excess. The binding of 1 AlMe3 to Cl, as in complex 4b is essentially neutral in terms of free energy (ΔG° = 0.1 kcal mol−1); the binding of a second AlMe3 to a Cr–Cl (ΔG° = 4.6 kcal mol−1), and the binding to the Cr–Me group (ΔG° = 8.0 kcal mol−1) are endoergic.
| Distances (Å) | |||
|---|---|---|---|
| Cr–C/O | Cr–Cl | Cr–P | |
| EXAFS: 4a | 2 at 2.04(5) | 2 at 2.38(2) | 2 at 2.54(6) |
| 3a Cl trans Cl | |||
| EDF2/6-31G* | 2.048 (C) | 2.333 | 2.503 |
| 2.088 (O) | 2.352 | 2.745 | |
| ωB97X-D/6-31G** | 2.043 (C) | 2.321 | 2.480 |
| 2.095 (O) | 2.361 | 2.703 | |
| 3b Cl trans Cl | |||
| EDF2/6-31G* | 2.047 (C) | 2.333 | 2.497 |
| 2.081 (O) | 2.337 | 2.816 | |
| ωB97X-D/6-31G** | 2.034 (C) | 2.317 | 2.484 |
| 2.079 (O) | 2.355 | 2.791 | |
| 3a Me trans Cl | |||
| EDF2/6-31G* | 2.076 (C) | 2.298 | 2.458 |
| 2.087 (O) | 2.411 | 2.669 | |
| ωB97X-D/6-31G** | 2.071 (C) | 2.296 | 2.441 |
| 2.106 (O) | 2.428 | 2.586 | |
| 3b Me trans Cl | |||
| EDF2/6-31G* | 2.088 (C) | 2.313 | 2.467 |
| 2.089 (O) | 2.414 | 2.552 | |
| ωB97X-D/6-31G** | 2.076 (C) | 2.308 | 2.453 |
| 2.095 (O) | 2.421 | 2.555 | |
| 3b Cl trans Cl; with 1Cr–Cl–AlMe3 (4b) | |||
| EDF2/6-31G* | 2.038 (C) | 2.314 | 2.509 |
| 2.076 (O) | 2.402 | 2.822 | |
| 3b Cl trans Cl; with 2Cr–Cl–AlMe3 | |||
| EDF2/6-31G* | 2.039 (C) | 2.365 | 2.518 |
| 2.057 (O) | 2.377 | 2.799 | |
| 3b Cl trans Cl; with 1Cr–C–AlMe3 | |||
| EDF2/6-31G* | 2.083 (C) | 2.325 | 2.514 |
| 2.073 (O) | 2.334 | 2.727 | |
In addition to lengthening the mean Cr–Cl distance by ∼0.02 Å, the effects of the AlMe3 binding to a Cr–Cl unit are to reduce the Cr–O and Cr–C bond lengths, affording a mean distance for that combined shell of 2.05 Å, within experimental error of the refined value. As compared to complex 1, the methyl group has a strong lengthening influence on the trans Cr–P bond, and thus making fitting the Cr–P shell potentially problematical due to the separation of the two distances by ∼0.3 Å. An alternative fitting model was attempted splitting this shell (Fig. S6, Table S4†) which afforded Cr–P distances of 2.45(6) and 2.60(2) Å. This had little effect on the R factor of the fit, but the Debye–Waller factors for the split Cr–P shells came into line with that of the Cr–Cl shell. The free energy calculations on these reactions places (Fig. 3) the structures for [CrCl2Me{PPh2N(R)PPh2}(THF)] (R = Me) 3b and its AlMe3 adduct 4b as both being 11 kcal mol−1 more favourable that the initial reagents. Given the excess of the aluminium reagent, assignment of structure 4a as the reaction product observed after 1 minute of reaction appears reasonable.
Given the challenges of establishing coordination numbers of minor shells, two alternatives with a lower coordination number for the Cr–O/C shell were considered: the Cr(III) complex 5a and the Cr(II) species 6a. The calculated Cr–Cl bond lengths (Table 4) of 5b were significantly shorter than the experimental ones (by 0.12 Å), rendering it less probable option. The XANES for the two 5-coordinate species 5b and 6b were calculated in addition to the 6-coordinate complex 3b (Fig. 4). It is the 6-coordinate centres 3b and 4b which more closely match the experimental features as both the 5 coordinate complexes are predicted to display a much larger shift to low energy as compared to the precursor, 1a. The model of 4b also replicates the two pre-edge features more closely than 3b, giving further support to this proposal.
| Distances (Å) | |||
|---|---|---|---|
| Cr–C/O | Cr–Cl | Cr–P | |
| EXAFS (after 5 minutes) | 1 at 2.10(1) | 2 at 2.37(3) | 2 at 2.49(2) |
| EXAFS (after 30 minutes) | 1 at 2.14(3) | 1 at 2.73(3) | 2 at 2.49(1) |
| EXAFS, with B(C6F5)3, ambient | 1 at 3.04(1) | 2 at 2.36(2) | 2 at 2.472(8) |
| 5b: EDF2/6-31G* | 2.036 (C) | 2.258, 2.267 | 2.513, 2.544 |
| ωB97X-D/6-31G** | 2.034 (C) | 2.268, 2.268 | 2.477, 2.498 |
| 6b: EDF2/6-31G* | 2.089 (O) | 2.339, 2.341 | 2.552, 2.768 |
| ωB97X-D/6-31G** | 2.089 (O) | 2.340, 2.356 | 2.555, 2.711 |
| 7a: EDF2/6-31G* | 2.281, 2.284 | 2.483, 2.486 | |
| ωB97X-D/6-31G** | 2.284, 2.293 | 2.470, 2.483 | |
| 7b: EDF2/6-31G* | 2.280, 2.282 | 2.488, 2.510 | |
| ωB97X-D/6-31G** | 2.286, 2.286 | 2.495, 2.500 | |
| 8b: EDF2/6-31G* | 2.850 (C) | 2.283, 2.378 | 2.492, 2.524 |
| ωB97X-D/6-31G** | 2.584 (C) | 2.286, 2.414 | 2.469, 2.533 |
| 9a: EDF2/6-31G* | 2.067 (C) | 2.289 | 2.445, 2.584 |
| ωB97X-D/6-31G** | 2.078 (C) | 2.299 | 2.457, 2.566 |
| 9b: EDF2/6-31G* | 2.064 (C) | 2.289 | 2.467, 2.606 |
| ωB97X-D/6-31G** | 2.072 (C) | 2.299 | 2.459, 2.596 |
| 10a: EDF2/6-31G* | 2.067 (C), 2.881 (C) | 2.396 | 2.456, 2.589 |
| ωB97X-D/6-31G** | 2.069 (C), 2.345 (C) | 2.596 | 2.435, 2.556 |
| 10b: EDF2/6-31G* | 2.067 (C), 2.862 (C) | 2.390 | 2.466, 2.641 |
| ωB97X-D/6-31G** | 2.070 (C), 2.592 (C) | 2.443 | 2.456, 2.589 |
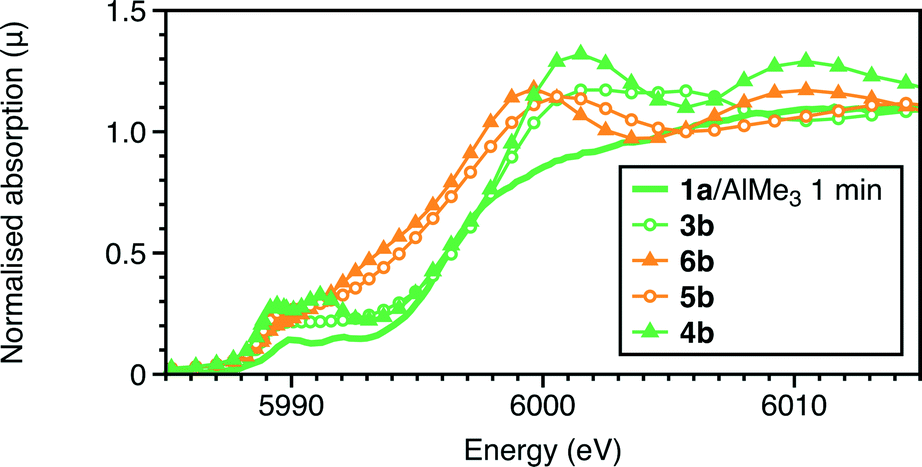 | ||
| Fig. 4 Comparison of the experimental Cr K-edge XANES of the reaction quenched after 1 minute with those calculated for complexes 3b, 4b, 5b and 6b. | ||
The calculated UV-visible spectra of 3b and 4b (Fig. S3†) both show an increase in intensity of the band near 400 nm, and a shift of the lowest energy band to lower energy, consistent with the experimental observations; the adduct formation with AlMe3 does reduce the magnitude of the shift. The calculated UV-visible spectrum of one of the most stable of the Cr(II) species, 8b, is radically different from that observed a few minutes after mixing. This also provides evidence for the first-observed species remaining Cr(III), in contrast to our observations using the RS(CH2CH2)2NH ligand system.17
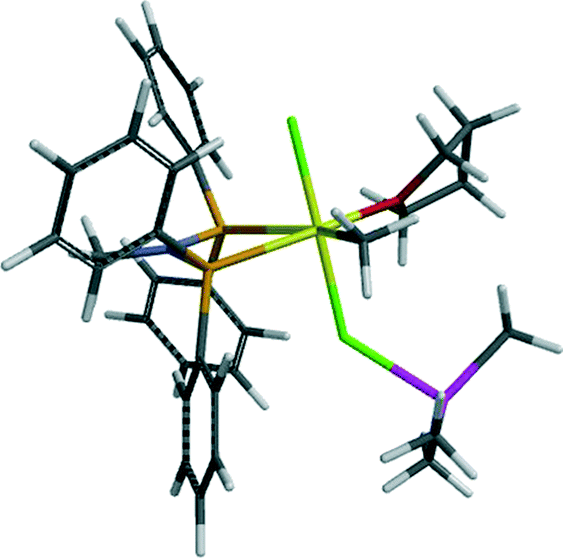
(Yellow = Cr, light green = Cl, red = O, brown=P, grey = C, purple = N, magenta = Al and white = H).
2.4 Freeze-quench XAFS of the reaction of complex 1 with AlMe3 after 5 minutes
The Cr K-edge XANES and EXAFS spectra of a freeze-quenched sample show a further change when the reaction time was increased to 5 minutes. When repeating the reaction and freezing at 1 minute in the presence of a Lewis acid, [Ph3C][Al{OC(tBuF)3}4], identical spectra to that of the reaction frozen at 5 minutes, without [Ph3C][Al{OC(tBuF)3}4], were observed. The main difference in the result of the EXAFS analysis (Fig. 1, S7 and S5, Tables 1, S5 and S3†) is the reduction of the coordination of the first shell from 2 to 1. This was modelled as 1Cr–C at 2.10(1) Å, but it may alternatively be due to an oxygen shell. Thus the possibilities for the coordination centre could include the 5 coordinate CrIII centre like [CrCl2Me{PPh2N(R)PPh2}] (R = iPr) 5a or a CrII site like [CrCl2{PPh2N(R)PPh2}(THF)] (R = iPr) 6a. Alternatively, this spectrum could represent a mixture of the first observed species, 4a, and the longer-lived product observable in room temperature samples. To test this, we performed linear combination fitting analysis (LCA) of the data from the reaction frozen after 5 minutes, starting by comparing 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixtures of the data collected at 1 minute reaction time with one of the ambient temperature reactions with and without B(C6F5)3, given as [CrCl2{PPh2N(R)PPh2}] or [CrClMe{PPh2N(R)PPh2}] complexes from the EXAFS analysis (Table 1), respectively. The LCA gave 3:2 mixture of product at 1 min and ambient temperature reaction without B(C6F5)3 (R = 0.06%). A better LCA fit is achieved when using the ambient temperature reaction with B(C6F5)3 as a standard, giving a 1:1 mixture with the product at 1 minute (R = 0.01%) (Fig. 5). The XANES features show a progressive shift of the main absorption edge to lower energy from that of 1a through the two freeze quench measurements to the room temperature scans.
:1 mixtures of the data collected at 1 minute reaction time with one of the ambient temperature reactions with and without B(C6F5)3, given as [CrCl2{PPh2N(R)PPh2}] or [CrClMe{PPh2N(R)PPh2}] complexes from the EXAFS analysis (Table 1), respectively. The LCA gave 3:2 mixture of product at 1 min and ambient temperature reaction without B(C6F5)3 (R = 0.06%). A better LCA fit is achieved when using the ambient temperature reaction with B(C6F5)3 as a standard, giving a 1:1 mixture with the product at 1 minute (R = 0.01%) (Fig. 5). The XANES features show a progressive shift of the main absorption edge to lower energy from that of 1a through the two freeze quench measurements to the room temperature scans.
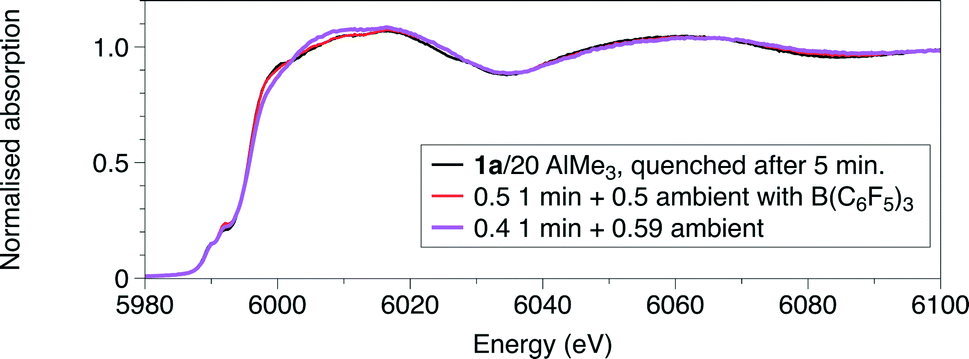 | ||
| Fig. 5 Comparison of the experimental Cr K-edge XANES of the reaction quenched after 5 minutes with spectra of a combination of a 2:3 combination of the spectrum from a sample quenched after 1 minute, and of the room temperature measurement and a 1:1 combination of the spectrum from a sample quenched after 1 minute, and of the room temperature measurement with solution containing B(C6F5)3. | ||
The free energy reaction survey (Fig. 3) indicated a very favourable reductive elimination reaction (2) to 7b (in a high spin state, S = 2). This is similar to the results obtained for the activation of [CrCl3{RS(CH2CH2)2NH}],17 with the ΔG° gain in forming and eliminating ethane providing the driving force for the reaction. We note that reductive elimination from Cr(III) alkyls has been previously reported,23 but have no further evidence to distinguish possible mechanisms. The additions of either THF to 7b to form 6b or AlMe3 (to 8b) were slightly exoergic.
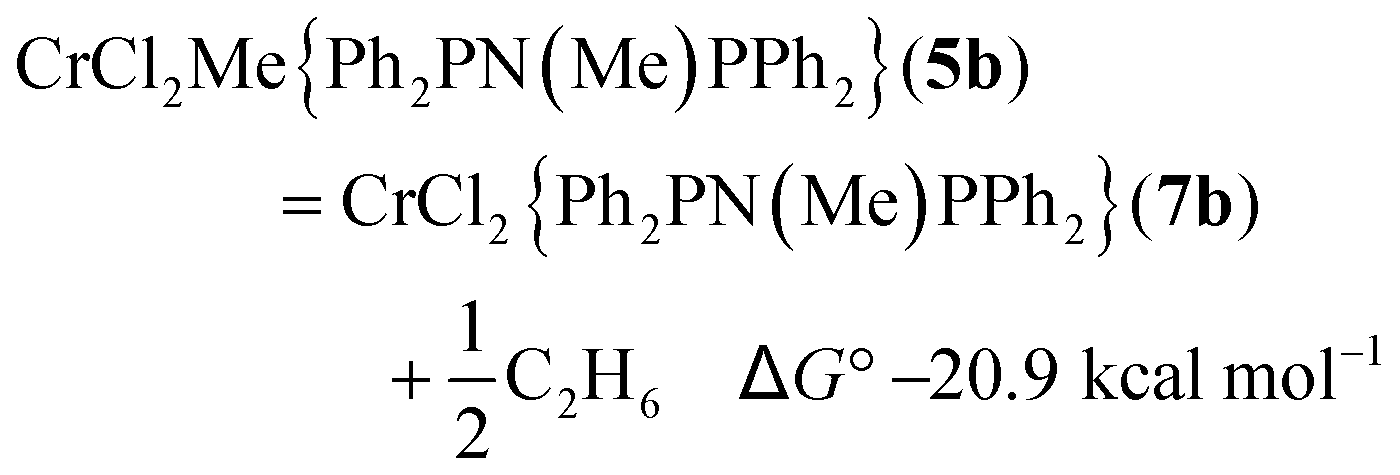 | (2) |
| 0.5 Al2Me6 + THF = AlMe3(THF) ΔG° −7.1 kcal mol−1 | (3) |
2.5 XAFS of complex 1 with excess AlMe3 at ambient temperature
The EXAFS also shows a marked change with the addition of the trityl-containing Lewis acid (Fig. 6, 7 and S9; Tables 1 and S7†). The results from this analysis suggest the initial chromium complex has undergone complete alkylation to lose the PNP ligand and form a short Cr–Cr multiple bond. The refined Cr–Cr bond length (2.01(2) Å) is close to that reported for the quadruple bond in the Cr(II) salt Li4[Cr2Me8)]·4THF,25 and the calculated shift in the edge energy for Li4[Cr2Me8)] also matches those of this reaction product. The EXAFS and XANES together do indicate that the effect of [Ph3C][Al{OC(tBuF)3}4] is the dissociation of the bidentate phosphine ligand and the formation of a highly alkylated Cr(II) metal–metal bonded dimer, but its complete formulation is uncertain. When freezing at 1 minute reaction time, we did not observe 4a in the presence of [Ph3C][Al{OC(tBuF)3}4] suggesting that it quickens the change from 1a to the phosphine-containing Cr(II) site prior to its dissociation.
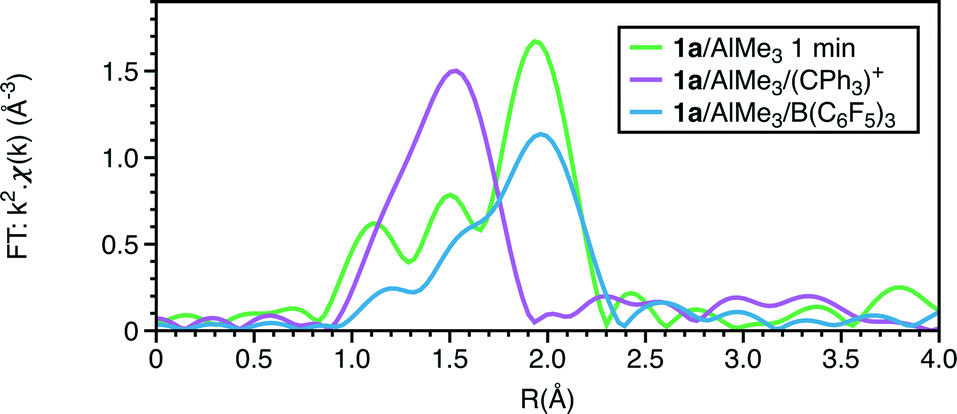 | ||
| Fig. 6 Cr K-edge Fourier transforms of k2-weighted EXAFS data of reactions of 1a with 20AlMe3 (1 min); 1a + 30AlMe3 + 1[Ph3C][Al{OC(tBuF)3}4]; 1a + 30AlMe3 + 1B(C6F5)3. | ||
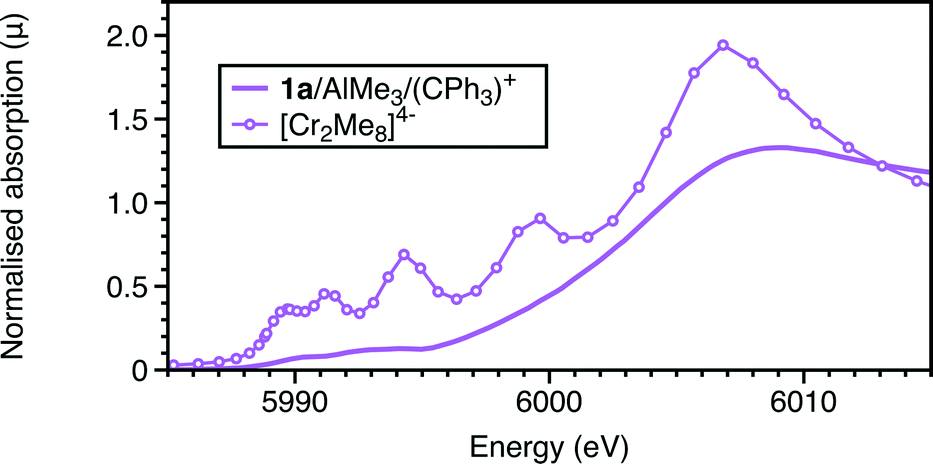 | ||
| Fig. 7 Cr K-edge XANES: experimental and calculated spectra of 1a with AlMe3 with (CPh3)[Al{OC(tBuF)3}4]. | ||
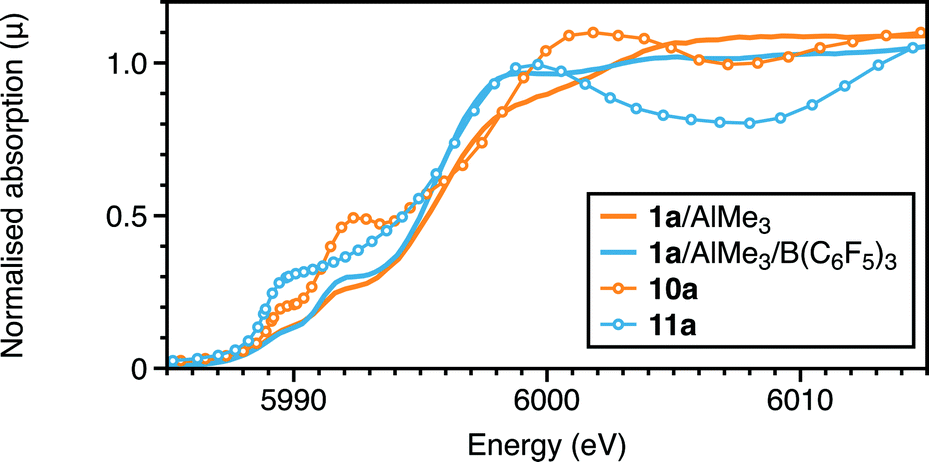 | ||
| Fig. 8 Cr K-edge XANES: experimental and calculated spectra of 1a with AlMe3 with and without B(C6F5)3. | ||
The calculated Cr–P distances for 7 are close to those provided by the EXAFS analysis, but the Cr–Cl shell is again shorter than the experimental value, by ∼0.08 Å. The EXAFS analysis has provided significant evidence, albeit inconclusive, for a light atom shell near 3.0 Å because when including this shell, the R-factor is greatly reduced to give a better overall fit. Accordingly, the interactions of BR3 (R = Me, Ph and C6F5) and AlMe3 with 7 at Cl and Cr were modelled using EDF2 and ωB97X-D functionals. Using EDF2/6-31G**, interaction of BMe3 to 7b was found to be extremely weak and involving a long Cr⋯HB distance (3.467 Å). This distance was reduced in the BPh3 adduct (3.135 Å), with an energy gain of 4.5 kcal mol−1; this would not be sufficient to offset the entropy loss by complexation at room temperature. The binding of AlMe3 to 7b, forming 8b, is stronger (ΔH° −8.6 kcal mol−1), and complex formation is slightly exoergic (Fig. 3). The structural effects are to increase the Cr–Cl distance for the bridging chloride to 2.378 Å, close to the experimental value. Also, one methyl group was calculated to bend back to take up an apical coordination site with a long Cr⋯C interaction (2.85 Å). This weaker interaction is strongly functional dependent. Using the ωB97X-D functional, which specifically includes long-range interactions26 and has proven effective in calculating reaction energies,27 the Cr–Cl(Al) is lengthened further (2.414 Å) and the Cr–C(Al) distance further reduced (2.58 Å). Inclusion of this interaction.
A different type of interaction to the metal is afforded by B(C6F5)3. Binding of this Lewis acid to 7a and 7b was two-fold in nature: boron to chloride (∼3.2 and 3.0 Å from EDF2 and ωB97X-D respectively), and a C–F group ortho to boron (Cr–F 2.55–2.65; Cr–C 3.19–3.35 Å) in complex 11 (a = iPr; b = Me). The effect of this coordination on the Cr–Cl and Cr–P distances is to increase the former by ∼0.1 Å and decrease the latter by ∼0.2 Å. Formation of 11b using EDF2/6-31G* was calculated to be exothermic (−13.2 kcal mol−1), but endoergic (4.8 kcal mol−1). However, using the alternative functional, which afforded the closer interactions of the C–F group to boron, rendered complexation more exothermic (−25 kcal mol−1), and thus the adduct 11a might be anticipated. Such interactions would be difficult to observe definitively by EXAFS from the Cr edge. There is though some evidence for a long light atom distance modelled as a Cr–C distance (3.04 Å). However, the calculated XANES for this aligns more closely to the experimental spectrum than that of complex 7a (Fig. 8 and S12†). What is also evident from the calculated structure of 11 is that the bidentate Lewis boron agent spatially protects the otherwise low-coordination chromium centre. This appears to retard the methylation of 7, which, in its absence appears to proceed to 9, stabilised as its AlMe3 adduct 10. The edge shift to higher energy in the absence of B(C6F5)3 (Fig. 8) is consistent with the substitution at chromium of Cl by CH3.
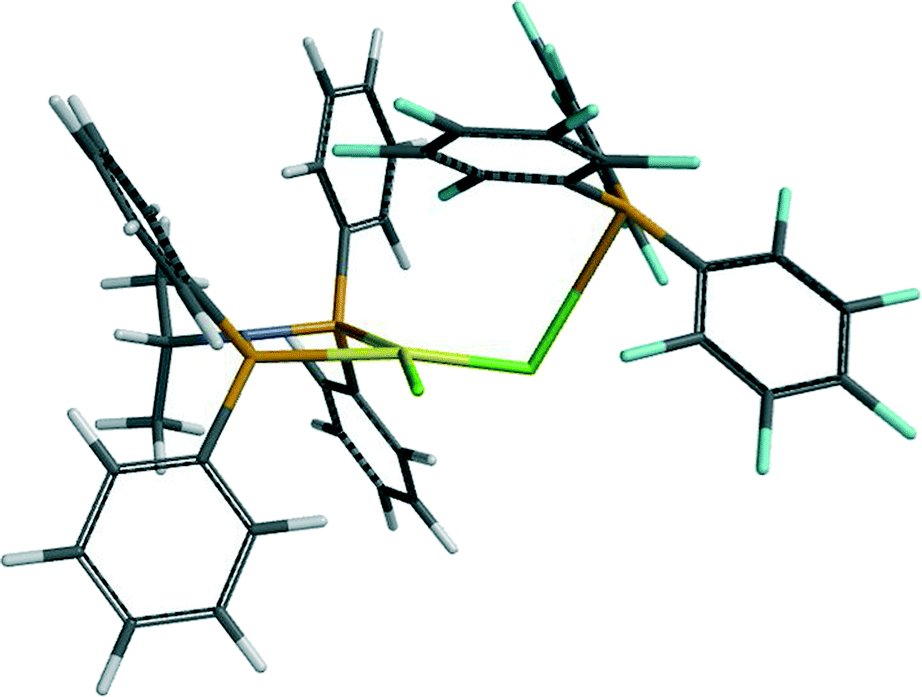
(Yellow = Cr, light green = Cl, dark brown = B, brown = P, grey = C, purple = N, blue = F and white = H).
3. Conclusions
These results provide strong evidence that, in the initial stages of the activation of the chromium tetramerisation catalyst, the (PPh2)2N(iPr) ligand allows rapid methylation of the chromium(III) centre. When comparing that to the homogeneous trimerisation chromium catalyst, [CrCl3{HN(CH2CH2S-decyl)2}],17 reaction with AlMe3 proceeds to a chromium(II) stabilised intermediate [CrCl{N(CH2CH2S-decyl)2}] in similar conditions after 1 second. A similar conversion to Cr(II), also driven by the formation of ethane by reductive elimination, takes several minutes with the aminophosphine ligand, and that complex probably exists as adducts with the Lewis acids AlMe3 and B(C6F5)3 (Scheme 1).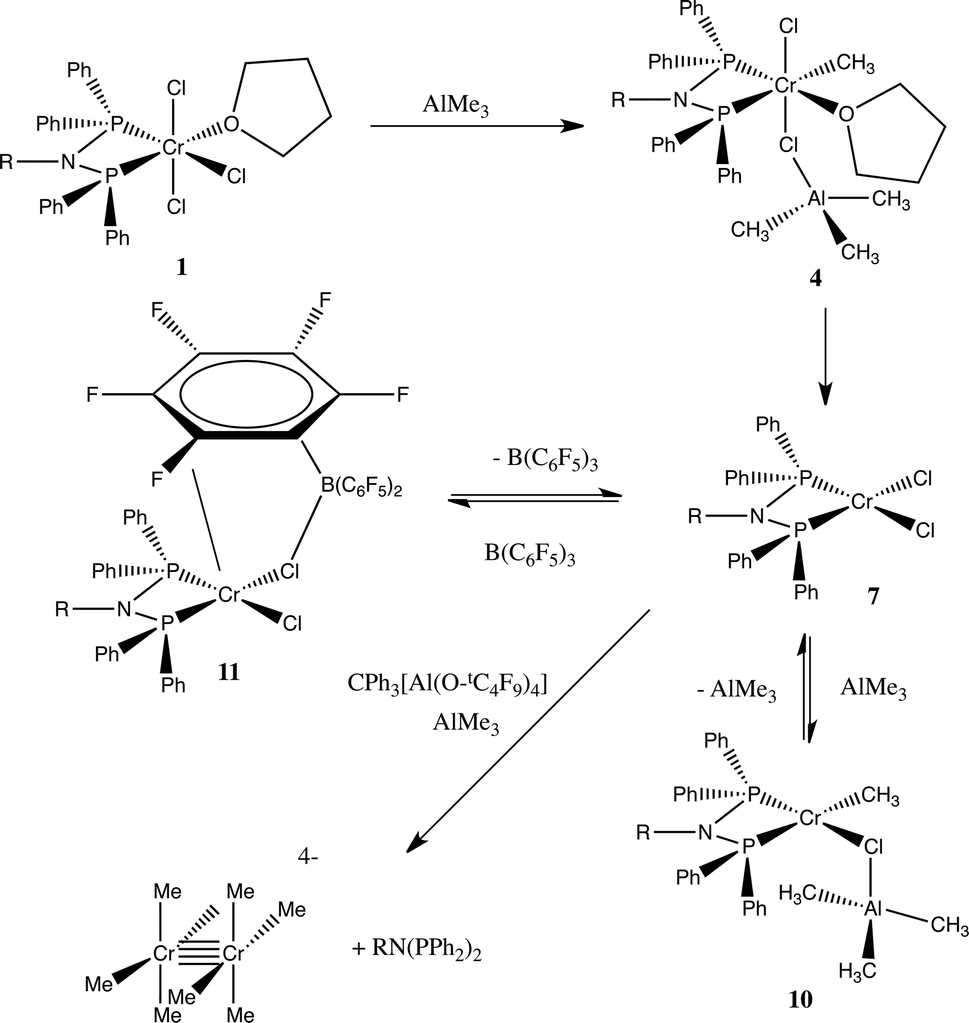 | ||
| Scheme 1 Proposed reaction scheme from the interaction of 1a with Al2Me6 and other Lewis acids. The formula [Cr2Me8]4− is only representative of the type of Cr site. | ||
A previous XAFS study on the reaction of Cr(acac)3, MMAO and (PPh2)2N(iPr) also proposed a Cr(II) product with a mean Cr–C coordination of 1.5 ± 0.6.14 Bond distances similar to our EXAFS analysis at (Cr–C 2.03 Å, Cr–P at 2.42 Å), were reported and also, there was no evidence in the EXAFS of Cr dimer formation. More closely related chemically is the study by Labinger and Bercaw using EPR and UV-visible spectroscopy to study the activation process by MAO with a bifunctional ligand bearing hemilabile donor groups e.g. [CrCl3-{P(C6H4OMe)2N(Me) P(C6H4OMe)2}].28 The initial product was reported as a trialkylated product, [CrIIIR3(PNP)] and was accompanied with a much larger shift of the lowest energy band o the visible region than observed in this work where the alkylation has not proceeded so far.
The Lewis acid, [Ph3C][Al{OC(tBuF)3}4], accelerates the reduction to Cr(II), but under ambient conditions it causes the loss of the (PPh2)N(iPr)PPh2 ligand, with the metal sequestered in a dimer related to [Cr2Me8]4− in terms of structure around Cr (Scheme 1). Indeed, the reaction of [Ph3C][Al{OC(tBuF)3}4] with (PPh2)N(iPr)PPh2 has been previously reported.20 The ligand is calculated to be relatively weakly bound to Cr(III), with complex formation of 1a for mer-CrCl3(THF)3 only slightly exoergic (4.4 kcal mol−1). Again this is in marked contrast to the coexistence of [CrCl{N(CH2CH2S-decyl)2}] and [Ph3C][Al{OC(tBuF)3}4].17 In contrast, the effect of B(C6F5)3 may be to retard the alkylation at chromium(II).
However, under ethene oligomerisation reaction conditions with an excess of aluminoxanes,5,29,30 or with this co-catalyst itself31 ligands like (PPh2)2N(iPr) do alter the catalytic selectivity from a phosphine free system. Detailed calculations of the oligomerisation mechanism have been carried out using a smaller ligand system [Cr{PMe2N(Me)PMe2}], based upon Cr(I)–Cr(III) catalytic cycle.32 As we observed for the [CrCl3{HN(CH2CH2S-decyl)2}] system,17 reduction to Cr(I) was not evident under our experimental conditions, although Cr(I) has been reported in a minor species using MAO as the aluminium reagent.28 In the [CrCl3{HN(CH2CH2S-decyl)2}] system,17 DFT calculations did show much stronger binding of ethene to Cr(I) rather than Cr(II). This suggests that ethene could extend the chromium chemistry beyond that in Scheme 1, in order to favour sites more suited to phosphine coordination and promote this further reduction.
4. Experimental
Synthesis of [CrCl3{PPh2N(R)PPh2}(THF)] 1a (R = iPr)
Ph2N(iPr)PPh2 (ref. 5) (0.20 g, 0.468 mmol) was added to a solution of [CrCl3(THF)3] (ref. 33) (0.175 g, 0.468 mmol) in dry/degassed CH2Cl2 (15 mL). The resulting mixture was stirred at room temperature under N2 for 1 hour, and the solvent removed in vacuo to afford 1a as a blue solid (0.194 g, 0.295 mmol, 63%). EA calc (found) %: C 56.49 (56.59), H 5.38 (5.36), N 2.09 (2.13). UV-vis (ε, M−1 cm−1): 14260 (300) and 19910 (233) cm−1. IR (cm−1) includes 1060, 1012, 860 (THF) and 372, 345, 315 (Cr–Cl).
Cr K-edge XAFS measurements
These were performed on freshly prepared samples of 1a were performed at the B18 beamline at Diamond Light Source in Didcot, England and BM26A (DUBBLE) of the ESRF. Si(111) double crystal monochromators were used in combination with a Ge 9 element detector for fluorescence acquisition on all samples. Cr K-edge scans were obtained in 20 min. All spectra were calibrated using a Cr foil. XAS data processing and EXAFS analysis were performed using IFEFFIT34 using the Hoare package.35 All reactions were carried out at ambient temperature. Freeze quench experiments were performed on B18 by addition of AlMe3 (with and without a Lewis acid) to [CrCl3{PPh2N(R)PPh2}(THF)] 1a (R = iPr) (5 mM) in toluene solutions and then by transfer under argon to a Kapton tube with a liquid nitrogen. The samples were maintained frozen as previously described.15Computational methods
The FEFF9 code was used to perform ab initio self-consistent field, real-space, full multiple scattering calculations.36 The calculations were performed using the Hedin–Lundquist exchange correlation potential. A (final state rule) core hole is included on the absorber atom to mimic the final state of the photon absorption process. To allow a direct comparison of calculated and experimental XANES spectra, all calculated XANES underwent the same energy shift (eV) which was established using the experimental [CrCl3(PNP)(THF)] data as a reference, shifting the calculated fac-[CrCl3(PNP)(THF)] spectrum to the same energy as the experimental data, using the first derivative of the absorption edge.Density functional theory calculations were performed using Spartan '14.37 B3LYP and EDF2 functionals appeared to overestimate the stiffness of the bidentate ligand, which have a small chelate angle in the crystal (P–Cr–P 66.0°).20 These methods also introduced a significant asymmetry to the Cr–P distances (0.08 Å), which was greatly reduced with the ωB97X-D method (0.05 Å). Some improvement was gained by adding the p-type polarisation functions on hydrogen atoms, and so candidate structures were investigated with EDF2 and also ωB97X-D functionals and 6-31G* and 6-31G** basis sets. The spin state for the Cr(III) was maintained as S = 3/2, consistent with the UV-visible spectra of 1a. For Cr(II) species, [CrXX′{Ph2N(Me)PPh2}] (X, X′ = Cl or Me), the S = 1 and S = 0 spin states were calculated to be ∼25 and ∼50 kcal mol−1 less stable than the high spin state (S = 2); hence all data presented here are for the S = 2 state.
Acknowledgements
We wish to thank EPSRC for funding this project (EP/F032463/1 and EP/I01974X/1), Diamond Light Source Ltd. and the ESRF for access to the synchrotron facilities, and the staff at B18 and BM26A for their assistance. We also thank Prof. R. P. Tooze and Dr. M. Hanton (Sasol Technology UK) for helpful discussions.Notes and references
- Nexant Process Evaluation Research Planning (PERP), Report-Alpha Olefins 06/07-5, 2008.
-
D. Vogt, B. Cornils and W. A. Herrmann, Oligomerization of Ethylene to Higher Linear α-Olefins: In Applied Homogeneous Catalysis with Organometallic Compounds, VCH, New York, 1996, vol. 1, pp. 245–258 Search PubMed
.
- D. S. McGuinness, Chem. Rev., 2011, 111, 2321–2341 CrossRef CAS PubMed
- T. Agapie, Coord. Chem. Rev., 2011, 255, 861–880 CrossRef CAS
- A. Bollmann, K. Blann, J. T. Dixon, F. M. Hess, E. Killian, H. Maumela, D. S. McGuinness, D. H. Morgan, A. Neveling, S. Otto, M. Overett, A. M. Z. Slawin, P. Wasserscheid and S. Kuhlmann, J. Am. Chem. Soc., 2004, 126, 14712–14713 CrossRef CAS PubMed
- T. Agapie, S. J. Schofer, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2004, 126, 1304–1305 CrossRef CAS PubMed
- A. K. Tomov, J. J. Chirinos, D. J. Jones, R. J. Long and V. C. Gibson, J. Am. Chem. Soc., 2005, 127, 10166–10167 CrossRef CAS PubMed
- A. Jabri, P. Crewdson, S. Gambarotta, I. Korobkov and R. Duchateau, Organometallics, 2006, 25, 715–718 CrossRef CAS
- D. S. McGuinness, D. B. Brown, R. P. Tooze, F. M. Hess, J. T. Dixon and A. M. Z. Slawin, Organometallics, 2006, 25, 3605–3610 CrossRef CAS
- A. J. Rucklidge, D. S. McGuinness, R. P. Tooze, A. M. Z. Slawin, J. D. A. Pelletier, M. J. Hanton and P. B. Webb, Organometallics, 2007, 26, 2782–2787 CrossRef CAS
- I. Y. Skobelev, V. N. Panchenko, O. Y. Lyakin, K. P. Bryliakov, V. A. Zakharov and E. P. Talsi, Organometallics, 2010, 29, 2943–2950 CrossRef CAS
- L. McDyre, E. Carter, K. J. Cavell, D. M. Murphy, J. A. Platts, K. Sampford, B. D. Ward, W. F. Gabrielli, M. J. Hanton and D. M. Smith, Organometallics, 2011, 30, 4505–4508 CrossRef CAS
- S. Peitz, B. R. Aluri, N. Peulecke, B. H. Müller, A. Wöhl, W. Müller, M. H. Al-Hazmi, F. M. Mosa and U. Rosenthal, Chem. – Eur. J., 2010, 16, 7670–7676 CrossRef CAS PubMed
- J. Rabeah, M. Bauer, W. Baumann, A. E. C. McConnell, W. F. Gabrielli, P. B. Webb, D. Selent and A. Brückner, ACS Catal., 2013, 3, 95–102 CrossRef CAS
- S. A. Bartlett, P. P. Wells, M. Nachtegaal, A. J. Dent, G. Cibin, G. Reid, J. Evans and M. Tromp, J. Catal., 2011, 284, 247–258 CrossRef CAS
- S. A. Bartlett, G. Cibin, A. J. Dent, J. Evans, M. J. Hanton, G. Reid, R. P. Tooze and M. Tromp, Dalton Trans., 2013, 42, 2213–2223 RSC
- S. A. Bartlett, J. Moulin, M. Tromp, G. Reid, A. J. Dent, G. Cibin, D. S. McGuinness and J. Evans, ACS Catal., 2014, 4, 4201–4204 CrossRef CAS
-
S. A. Bartlett, PhD Thesis, University of Southampton, 2012 Search PubMed
-
J. Moulin, PhD Thesis, University of Southampton, 2006 Search PubMed
- D. S. McGuinness, M. Overett, R. P. Tooze, K. Blann, J. T. Dixon and A. M. Z. Slawin, Organometallics, 2007, 26, 1108–1111 CrossRef CAS
- S. Teo, Z. Weng and T. S. A. Hor, Organometallics, 2008, 27, 4188–4192 CrossRef CAS
- J. O. Moulin, J. Evans, D. S. McGuinness, G. Reid, A. J. Rucklidge, R. P. Tooze and M. Tromp, Dalton Trans., 2008, 1177–1185 RSC
- R. A. Heintz, S. Leelasubcharoen, L. M. Liable-Sands, A. L. Reingold and K. H. Theopold, Organometallics, 1998, 17, 5477–5485 CrossRef CAS
- M. Tromp, J. Moulin, G. Reid and J. Evans, AIP Conf. Proc., 2007, 882, 699–701 CrossRef CAS
- J. Krausse, G. Marx and G. Schödl, J. Organomet. Chem., 1970, 21, 159–168 CrossRef CAS
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC
- K. Yang, J. Zheng, Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2010, 132, 164117 CrossRef PubMed
- L. H. Do, J. A. Labinger and J. E. Bercaw, ACS Catal., 2013, 3, 2582–2585 CrossRef CAS PubMed
- M. J. Overett, K. Blann, A. Bollmann, J. T. Dixon, D. Haasbroek, E. Killian, H. Maumela, D. S. McGuinness and D. H. Morgan, J. Am. Chem. Soc., 2005, 127, 10723–10730 CrossRef CAS PubMed
- Y. Shaikh, K. Albahily, M. Sutcliffe, V. Fomitcheva and S. Gambarotta, Angew. Chem., Int. Ed., 2012, 51, 1366–1369 CrossRef CAS PubMed
- D. S. McGuinness, A. J. Rucklidge, R. P. Tooze and A. M. Z. Slawin, Organometallics, 2007, 26, 2561–2569 CrossRef CAS
- D. S. McGuinness, B. Chan, G. J. P. Britovsek and B. F. Yates, Aust. J. Chem., 2014, 67, 1481–1490 CrossRef CAS
- R. J. Kern, J. Inorg. Nucl. Chem., 1962, 24, 1105–1109 CrossRef CAS
- M. J. Newville, J. Synchrotron Radiat., 2001, 8, 322–324 CrossRef CAS PubMed
- B. Ravel and M. J. Newville, J. Synchrotron. Radiat., 2005, 12, 537–541 CrossRef CAS PubMed
- J. J. Rehr, J. J. Kas, M. P. Prange, A. P. Sorini, Y. Takimoto and F. Vila, C. R. Phys., 2009, 10, 548–559 CrossRef CAS
-
Spartan '14, Wavefunction Inc., Irvine, CA10 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cy00902f |
| ‡ Additional data available: XAFS spectra, XANES simulations and DFT calculations for all complexes are available to download from http://www.eprints.soton.ac.ukvia doi: 10.5258/SOTON/395279. |
| This journal is © The Royal Society of Chemistry 2016 |