
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric sulfoxidation by engineering the heme pocket of a dye-decolorizing peroxidase†
Dolores
Linde‡
a,
Marina
Cañellas‡
bc,
Cristina
Coscolín§
a,
Irene
Davó-Siguero
a,
Antonio
Romero
a,
Fátima
Lucas
bc,
Francisco J.
Ruiz-Dueñas
a,
Victor
Guallar
*bd and
Angel T.
Martínez
*a
aCentro de Investigaciones Biológicas, CSIC, Ramiro de Maeztu 9, E-28040 Madrid, Spain. E-mail: ATMartinez@cib.csic.es
bJoint BSC-CRG-IRB Research Program in Computational Biology, Barcelona Supercomputing Center, Jordi Girona 29, E-08034 Barcelona, Spain. E-mail: victor.guallar@bsc.es
cAnaxomics Biotech, Balmes 89, E-08008 Barcelona, Spain
dICREA, Passeig Lluís Companys 23, E-08010 Barcelona, Spain
First published on 23rd May 2016
Abstract
The so-called dye-decolorizing peroxidases (DyPs) constitute a new family of proteins exhibiting remarkable stability. With the aim of providing them new catalytic activities of biotechnological interest, the heme pocket of one of the few DyPs fully characterized to date (from the fungus Auricularia auricula-judae) was redesigned based on the crystal structure available, and its potential for asymmetric sulfoxidation was evaluated. Chiral sulfoxides are important targets in organic synthesis and enzyme catalysis, due to a variety of applications. Interestingly, one of the DyP variants, F359G, is highly stereoselective in sulfoxidizing methyl-phenyl sulfide and methyl-p-tolyl sulfide (95–99% conversion, with up to 99% excess of the S enantiomer in short reaction times), while the parent DyP has no sulfoxidation activity, and the L357G variant produces both R and S enantiomers. The two variants were crystallized, and their crystal structures were used in molecular simulations to provide a rational explanation for the new catalytic activities. Protein energy landscape exploration (PELE) showed more favorable protein–substrate catalytic complexes for the above variants, with a considerable number of structures near the oxygen atom of the activated heme, which is incorporated into the substrates as shown in 18O-labeling experiments, and improved affinity with respect to the parent enzyme, explaining their sulfoxidation activity. Additional quantum mechanics/molecular mechanics (QM/MM) calculations were performed to elucidate the high stereoselectivity observed for the F359G variant, which correlated with higher reactivity on the substrate molecules adopting pro-S poses at the active site. Similar computational analyses can help introduce/improve (stereoselective) sulfoxidation activity in related hemeproteins.
Introduction
There is increasing interest in the synthesis and use of molecules containing stereogenic centers. The sulfur atom of sulfoxides bearing two different substituents is a chiral center, since it adopts tetrahedral sp3 hybridization, with a lone electron pair occupying the fourth quadrant. Chiral sulfoxides have a wide range of applications, from chiral auxiliaries to pharmaceuticals.1 Among enzymes of interest in chiral synthesis,2 flavoenzymes (including Baeyer–Villiger monooxygenases) and hemeperoxidases are used for sulfoxidation reactions.3–5 Cytochrome P450 enzymes also catalyze sulfoxidations, but the requirement of an auxiliary flavin-containing enzyme/domain (and a source of reducing power, as in the case of NAD[P]H-dependent flavoenzymes) limits their biotechnological applicability.6 The enzymatic production of active S omeprazole, a multibillion dollar drug, by a modified cyclohexanone monooxygenase is a good example of these biotransformations.7In peroxidases, the oxidation reaction is mediated by a peroxygenase mechanism rather than a peroxidase mechanism, and sulfoxidation of thioanisole (methyl-phenyl sulfide, MPS) and methyl-p-tolyl sulfide (MTS) yielding the corresponding sulfoxides has been used as a probe of oxygen transfer to organic sulfides. In this way, sulfoxidation has been reported for the well-known fungal chloroperoxidase (CPO)8–10 and horseradish peroxidase (HRP),10–12 as well as for other haloperoxidases (including vanadium peroxidases),13 several animal peroxidases,14 and also for cytochrome c peroxidase (CcP),15Coprinopsis cinerea peroxidase (CiP),16 and lignin peroxidase (LiP),17 with different conversion rates and stereoselectivities. Recently, a robust peroxidase/peroxygenase of the HTP protein superfamily has been described from the basidiomycete Agrocybe aegerita with predominant monooxygenase activity (unspecific peroxygenase, UPO),18 which is able to selectively convert MPS into the R enantiomer of methyl-phenyl sulfoxide.19,20 Another UPO, from the related fungus Coprinellus varians, is especially efficient in sulfoxidizing dibenzothiophene.21
Although the natural substrate/s of the so-called dye-decolorizing peroxidases (DyPs), forming part of the new CDE protein superfamily,22 are still to be identified, these enzymes present considerable catalytic versatility and exceptional stability under extreme pH, temperature, and even pressure conditions.23,24 DyPs, as some fungal ligninolytic peroxidases of the peroxidase–catalase superfamily (LiP and versatile peroxidase), have the ability to oxidize substrates at two catalytic sites, one located at an exposed protein radical and a second one at the heme pocket, which has a more axial access in DyPs than in ligninolytic and related peroxidases.25
In this work, we engineer the heme pocket of the Auricularia auricula-judae DyP, one of the few members of this new family fully characterized to date,26–28 with the aim of providing new catalytic properties to the enzyme. The native recombinant DyP is unable to perform sulfoxidation reaction, but interestingly, two individual mutations at the heme pocket provide sulfoxidation activity on MPS and MTS. More interestingly, one of them was stereoselectively forming the S enantiomer, while the second variant yielded similar percentages of both isomers. With the use of molecular modeling techniques, we aim to provide a rational explanation at the atomic level of the differences in yield and selectivity observed during sulfoxidation of MPS and MTS by the native DyP and its two heme pocket variants, whose crystal structures were solved.
Results and discussion
Experimental enzyme engineering and sulfoxidation reactions
The heme pockets of A. auricula-judae DyP and other peroxidases were compared, and variants with enlarged access to the enzyme cofactor were obtained and evaluated for sulfoxidation of two organic sulfides, as described in the next three sections.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O is located in compound I after H2O2 activation) in peroxidases from three different superfamilies is shown in Fig. 1. In HRP (Fig. 1A), the so-called distal histidine occupies an axial position (above the heme iron) contributing to the reaction with H2O2 together with a neighbor arginine.29 In UPO (Fig. 1B), a glutamate/arginine couple plays a similar function,30 while the couple is aspartate/arginine in DyP (Fig. 1C).27
O is located in compound I after H2O2 activation) in peroxidases from three different superfamilies is shown in Fig. 1. In HRP (Fig. 1A), the so-called distal histidine occupies an axial position (above the heme iron) contributing to the reaction with H2O2 together with a neighbor arginine.29 In UPO (Fig. 1B), a glutamate/arginine couple plays a similar function,30 while the couple is aspartate/arginine in DyP (Fig. 1C).27
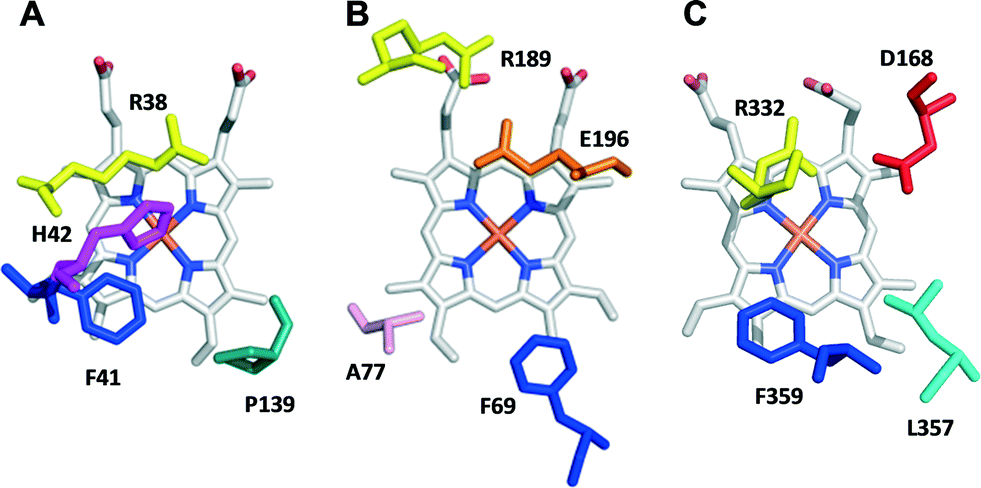 | ||
| Fig. 1 Upper side of the heme pocket in the crystal structures of (A) HRP (PDB 1ATJ), (B) A. agerita UPO (PDB 2YP1), and (C) A. auricula-judae DyP (PDB 4W7J) (this heme side is also known as the distal side due to the presence of the distal histidine of peroxidase–catalases, His42 in A). | ||
Due to their crucial role in the formation of reactive compound I, site-directed mutagenesis of the above-mentioned residues in A. auricula-judae DyP drastically decreased its catalytic efficiency reducing H2O2 (measured with 2,2′-azino-bis[3-ethylbenzothiazoline-6-sulfonic acid], ABTS, as the enzyme reducing substrate), which passed from 2050 s−1 mM−1 in native DyP to only 4 and 1 s−1 mM−1 in the D168N and R332L variants, respectively. Therefore, other residues contributing to a confined heme pocket in DyP, such as Phe359 (homologous to Phe41 in HRP) and Leu357 (Fig. 1C), were modified, and the enlarged pocket variants were expressed in E. coli, in vitro activated, purified to homogeneity (with specific activities of 400–500 U mg−1 measured with ABTS), and crystallized, and their sulfoxidation activity was evaluated, as described below.
Although the L357G variant showed slightly decreased thermal and pH stabilities (T50 = 59 °C and 50% activity at pH 9), the F359G variant did not show significantly modified stability in the range of pH 4–9 (being always higher than 85%) and had only one degree lower T50 (61 °C) than the native recombinant DyP (62 °C).
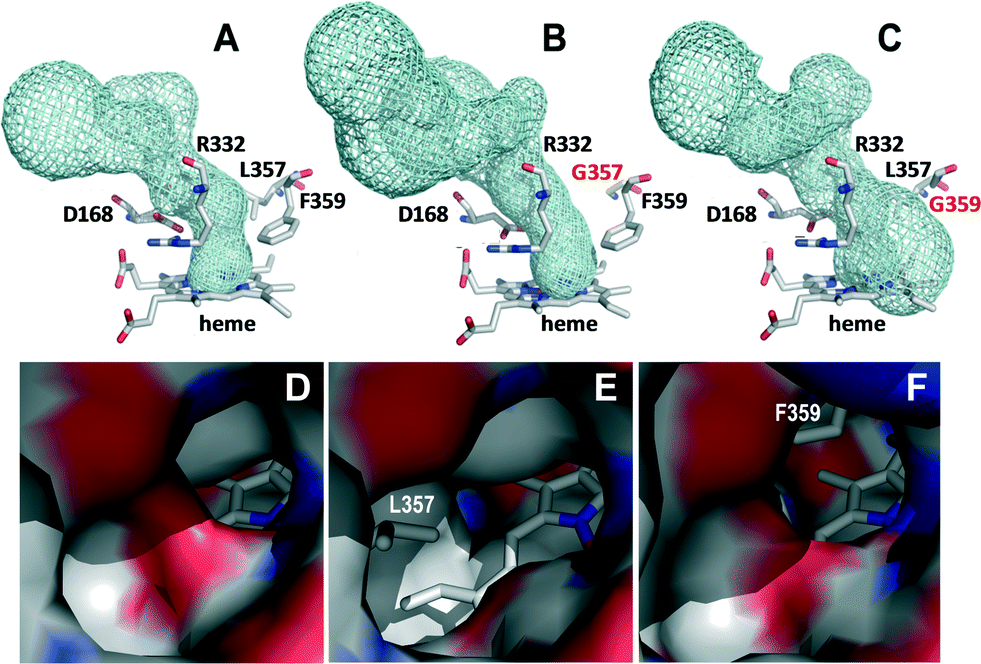 | ||
| Fig. 2 Details of the access channel to the heme pocket (top) and its opening at the protein surface (bottom) in the crystal structures of the native DyP (A and D), L357G variant (B and E) and F359G variant (C and F). In A–C, the heme access channel is shown as cyan meshes and the heme and neighbor residues as CPK-colored sticks (the two mutated residues in B and C with red labels), while in D–F an electrostatic potential surface is shown, with the heme as CPK sticks and the removed side chains in the two variants as gray sticks. | ||
A close-up view of the heme pocket shows an asymmetric distribution of the cavity according to the volume and position of the mutated residue. Thus, in the F359G variant, the heme pocket is larger allowing the substrate to be properly positioned, with a potential effect on the stereoselectivity of sulfoxidation.
 | ||
| Fig. 3 Results from chiral HPLC analysis of MPS (A–C) and MTS (D–F) reactions with native DyP and L357G and F359G variants and controls (C) without the enzyme (after 0, 30, 60 and 360 min incubation) showing the remaining substrate (A and D) and the resulting R (B and E) and S (C and F) sulfoxides. | ||
These long-term incubation experiments were complemented by estimating kinetic constants for MPS and MTS oxidation under steady-state conditions. F359G showed higher catalytic efficiency (kcat/Km) than L357G due to 2-fold higher turnover number (kcat) and lower Km (Table 1). This agrees with the higher catalytic efficiency of the F359G variant oxidizing the standard substrate ABTS at the heme channel (1040 ± 80 s−1 mM−1) compared with L357G (175 ± 19 s−1 mM−1). The sulfoxidation catalytic efficiency (and other kinetic constants) of F359G DyP is 2–3 orders of magnitude higher than that of wild-type HRP,11,12 in the same order of those reported for the best HRP variants,31–34 and lower than that reported for wild-type CPO (the classic sulfoxidation biocatalysts),35 and especially for the recently discovered UPO.20
| MPS | MTS | |||||
|---|---|---|---|---|---|---|
| k cat | K m | k cat/Km | k cat | K m | k cat/Km | |
| DyP | 0 | — | — | 0 | — | — |
| L357G | 8.0 ± 0.5 | 0.60 ± 0.08 | 13.3 ± 1.0 | 0 | — | — |
| F359G | 17.2 ± 0.9 | 0.37 ± 0.01 | 45.9 ± 7.4 | 3.6 ± 0.4 | 0.13 ± 0.03 | 26.5 ± 3.8 |
Interestingly, in the MPS (Fig. 3B and C) and MTS (Fig. 3E and F) reactions, the enantiomeric production by the two variants is not the same. L357G produces nearly racemic mixtures of the methyl-phenyl and methyl-p-tolyl sulfoxides, while F359G is stereoselectively producing the S sulfoxide. From these values, enantiomeric excesses (ee) of 92% and 99% were calculated for MPS and MTS oxidation by the F359G variant, respectively, while the L357G was scarcely stereoselective, as shown in Table 2. On the other hand, F359G showed a higher total conversion rate than L357G, in agreement with its higher catalytic efficiency shown in Table 1.
Sulfoxidation had been reported for a bacterial (Thermobifida fusca) DyP, but the conversion was poor (no rate provided) and the reaction was only slightly selective (61/49% ee after 36 h reaction with MPS/MTS).36 On the other hand, improvements in sulfoxidation ability have been reported by engineering the heme pocket of HRP,31–34 and CcP.15 These included the W51A and F41L variants, in which the bulky tryptophan/phenylalanine side chains in the active sites of CcP and HRP, respectively, were replaced by smaller groups. This agrees with the present results showing that enlarging the heme pocket of DyP, as found in the F359G variant, resulted in efficient (and stereoselective) sulfoxidation. Protein engineering has also been reported to improve other sulfoxidation reactions, such as in the Codexis patent for the already mentioned production of S omeprazole by a bacterial monooxygenase.37
Computational analyses
For a deeper inquiry on the atomic mechanistic details responsible for the observed differences in sulfoxidation (rate and selectivity) by the native DyP and its two variants, we turned to molecular modeling using the crystal structures solved in this work, as described in the next two sections. | ||
| Fig. 4 Interaction energies (in kcal mol−1) vs. ligand distances (in Å) from PELE simulations for MPS (left) and MTS (right) substrates in the (A) native DyP, (B) L357G variant, and (C) F359G variant. The distances are between the reactive O atom in the heme compound I and the sulfur (S) atom of the substrates; each color corresponds to a different trajectory. The structures used for the selectivity analysis are delimited with a red box. | ||
Moreover, we see a higher number of trajectories at catalytic distances for those conditions where we observe over 90% conversion in 30 min: F359G reaction with MTS and MPS and L357G reaction with MPS. Notice that although in native-DyP MPS interaction energies are quite favorable, the ligand is positioned too far from the heme. These differences in the energy profiles, which indicate an unfavorable ligand–protein interaction and ligand positioning on native DyP, can explain its undetectable (on MTS) or very low (on MPS) activity, which was only observed in long-term incubation experiments.
 | ||
| Fig. 5 R/S enantiomer classification criterion. The Φ dihedral is computed from the compound I catalytic oxygen (O) atom and substrate S and carbon C1 and C2 atoms. When Φ is between 40° and 140°, the substrate is classified as pro-R, and when Φ is between −40° and −140°, it is classified as pro-S. Otherwise, the substrate is considered to be able to form both R and S enantiomers equally. | ||
First, it should be mentioned that there was a higher number of structures in a good position towards the heme and with an optimal energy to react in the F359G variant than in L357G, in agreement with the higher catalytic efficiency of the phenylalanine variant. The L357G and, especially, the F359G mutations cause a binding site enlargement, which improves MPS and MTS positioning in comparison with native DyP, as shown by PELE.
Fig. 6 shows the different pro-R (cyan) and pro-S (green) positions for the native DyP and F359G, the best variant. In DyP, the presence of large (and bulky) residues (Arg332, Leu357 and Phe359) in the heme pocket hinders the proper positioning of both MPS and MTS substrates. Otherwise, for the two mutant proteins, we find an easier positioning of the substrate ring, in correlation with the binding energy analysis shown above. However, contrary to experimental results, substrates on both DyP variants show a preference to adopt pro-R positions, even though pro-S and pro-R/S positions are also possible.
 | ||
| Fig. 6 Substrate positioning at the heme pocket: A) pro-R and pro-S MPS (left) and MTS (right) positioning on native DyP. B) Pro-R and pro-S MPS (left) and MTS (right) positioning on the F359G variant. Pro-S and pro-R substrate positions are shown as CPK sticks with the C atoms in green and cyan color, respectively. The spin density population on the substrate, from QM/MM calculations, is indicated in each image. | ||
To clarify this apparent contradiction with the experimental results, substrate spin densities were computed with quantum mechanics/molecular mechanics (QM/MM) calculations on the best (substrate) positioned structures. The spin density originates from substrate oxidation, which directly correlates with sulfoxidation.38,39 In addition to having better protein–ligand interaction energies, as described above, the DyP variants showed a significantly larger spin density than the native DyP, again confirming the improvement in catalytic activity upon mutation. Moreover, in the F359G variant, we observe a substantial difference between the spin density values for the two enantiomers provided in Fig. 6, which might discriminate against the R enantiomer oxidation. Thus, even though a higher percentage of structures are placed favoring the formation of an R enantiomer, pro-S structures are more easily oxidized, which would explain the S stereoselectivity experimentally observed for this variant. In agreement with the racemic mixture seen experimentally for the L357G variant, we see a higher relative frequency of the pro-R/S position for L357G compared to F359G (Fig. S1A†).
Interestingly, changes in spin density, associated with the different enantiomers, correlate with the position adopted by the substrate with respect to Arg332. Mutations induce a change in the positioning of the ligand that leads to a closer interaction of the pro-R structures with this arginine residue (Fig. 7). As seen in previous publications,40 the electrostatic environment changes caused by this positive charge could lead to an altered substrate oxidation. This effect is larger in the F359G variant, where differences in spin density are more pronounced, while L357G results do not show a significant population of the pro-S conformer at large distances (Fig. S1B†). The correlation between the substrate spin density and its charge distribution was further verified by QM/MM calculations on 10 selected structures with different charge distribution (Fig. S2†).
 | ||
| Fig. 7 Frequency–distance (in Å) distribution of MPS and MTS S atom to Arg332 CZ atom on the F359G variant for the selected reactive structures: last 10 kcal mol−1 interaction energies and distances to the heme below 5 Å. Histograms for pro-R structures are shown in cyan, for pro-S structures in green, and for pro-R/S structures in grey. | ||
In connection with the above computational results, improvement in the HRP sulfoxidation ability has been reported by removing the distal arginine residue (Fig. 1A).33 A similar change is not possible in DyP engineering, since Arg332 (Fig. 1C) is necessary for DyP activation by H2O2, the catalytic efficiency being 2000-fold lower in the R332L variant, as shown above.
Conclusions
Stereoselective sulfoxidation ability could be introduced in a model (robust) peroxidase (A. auricula-judae DyP) by directed mutagenesis of residues at the distal side of the heme cofactor. The best variant (F359G) converted 95–99% MPS and MTS into the corresponding sulfoxides in 30 min reactions yielding the S enantiomer with 90–99 ee. The crystal structure of the F359G variant revealed an enlarged heme pocket enabling better accommodation of the sulfide substrates near the Fe4+O of the activated heme cofactor, whose oxygen atom is transferred to the sulfide substrate as shown by 18O-labeling (in H218O2 reactions).
Computational modeling, based on the crystal structures of the parent enzyme and several variants, showed that the sulfoxidation ability of the F359G variant (no reaction was produced by the parent enzyme) is due to better substrate access (closer distance) to the reactive cofactor with better interaction energies, as shown by the PELE software. Moreover, the sulfoxidation stereoselectivity could be rationalized by QM/MM calculations predicting a higher reactivity of the sulfide molecules adopting pro-S poses at the active site (which was affected by a heme pocket arginine residue).
The study also demonstrates how molecular simulations can help obtain the required oxidation yields and stereoselectivities in sulfoxidation engineering.
Materials and methods
Chemicals
Hexane (HPLC quality), 4′-methoxyacetophenone, methyl-phenyl sulfoxide, R-(+)-methyl-p-tolyl sulfoxide, R/S methyl-p-tolyl sulfoxide, MPS and MTS were from Sigma-Aldrich. Isopropyl alcohol (LC-MS quality) was from Fluka. Dichloromethane (HPLC quality) was from Merck.Directed mutagenesis
Simple DyP variants were produced by PCR using the pET23a-DyPI vector harboring the mature protein-coding sequence of A. auricula-judae DyP as a template.24 For each mutation, a direct primer and a reverse primer were designed complementary to opposite strands of the DNA region containing the desired mutation. The sequences of the direct primers (with the mutated codons in italics) used for mutagenic PCR were the following: D168N mutation, 5′-G TTC GGC TTC CTT AAC GGA ATT GCT CAG CC-3′; R332L mutation, 5′-GCT AAC TCT ATC ATG CTC AGC GGC ATC CC-3′; L357G mutation, 5′-CT CAG GAG CGC GGC GGA GCG TTT GTG GCA TAC-3′; F359G, F359H or F359W mutations, 5′-GGC CTT GCG (GGA, CAC or TGG, respectively) GTG GCA TAC-3′.PCR reactions were carried out in an Eppendorf (Hamburg, Germany) Mastercycler Pro using 10 ng of template DNA, 250 μM of each dNTP, 125 ng of direct and reverse primers, 2.5 units of polymerase (Expand Long Template PCR System), and the manufacturer's reaction buffer. Reaction conditions were as follows: i) a “hot start” of 95 °C for 1 min; ii) 18 cycles at 95 °C for 50 s, 55 °C for 50 s, and 68 °C for 10 min; and iii) a final cycle at 68 °C for 10 min. The mutated sequences were confirmed by DNA sequencing using an ABI 3730 DNA analyzer (Applied Biosystem). pET23a-DyPI plasmids containing the mutations described above were digested with endonuclease DpnI and transformed into E. coli DH5α for propagation.
Enzyme production
E. coli BL21(DE3)pLysS cells with the pET23a-DyPI vector containing the A. auricula-judae mature DyP sequence (and those of DyP variants) were grown overnight at 37 °C and 170 rpm in Luria Bertani broth (with 100 μg mL−1 ampicillin and 34 μg mL−1 chloramphenicol) and used to inoculate 2 L flasks containing 1 L of Terrific Broth (TB) (with 100 μg mL−1 ampicillin and 34 μg mL−1 chloramphenicol) that were grown for 3 h at 37 °C and 200 rpm, induced with 1 mM isopropyl-β-D-thiogalactopyranoside, grown for further 4 h, and harvested by centrifugation. The apoenzyme, accumulated in inclusion bodies, was solubilized in 50 mM Tris-HCl (pH 8.0) containing 8 M urea, 1 mM EDTA, and 1 mM dithiothreitol for 1 h at 4 °C. In vitro activation of the native DyP and its D168N, R332L and L357G variants was carried out as previously described,24 and specific activities were determined using 7.5 mM ABTS as a substrate and 2.5 mM H2O2.For the three Phe359 variants, in vitro activation was performed at 4 °C using 0.15 M urea, 10 μM hemin, 0.02 mM dithiothreitol, 0.4 mM GSSG, 20% glycerol, 0.1 mM EDTA and 0.1 mg mL−1 protein in 50 mM phosphate (pH 6.5). After 144 h, the folding mixture was concentrated using a Millipore Pellicon ultrafiltration system (10 kDa cutoff) and centrifuged for 18 h at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm. Once concentrated, samples were dialyzed against 20 mM sodium acetate (pH 4.3), and the insoluble material was eliminated by centrifugation (13000 rpm, 30 min).
000 rpm. Once concentrated, samples were dialyzed against 20 mM sodium acetate (pH 4.3), and the insoluble material was eliminated by centrifugation (13000 rpm, 30 min).
Active DyPs (native enzyme and four directed variants) were purified using a Resource Q column (GE Healthcare) coupled to an ÄKTA liquid chromatography system, using a gradient from 0 to 0.3 M NaCl in 10 mM Tris-HCl, pH 7 (15 min, 2 mL min−1). DyPs were analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis to confirm the purity of the proteins. The absorption spectra were recorded in 10 mM sodium tartrate (pH 5) at 25 °C using a Thermo Spectronic diode-array spectrophotometer. The DyP molar absorption coefficient (ε405 117000 M−1 cm−1) was used for protein concentration determination.
Enzyme kinetics
Steady-state kinetic studies were performed by measuring the initial rates for MPS and MTS oxidation at 25 °C in 100 mM acetate, pH 5 (in triplicate). A 500 mM MPS and MTS stock in ethanol was serially diluted in ethanol to obtain final substrate concentrations from 250 mM to 2 mM. 10 μl of substrate were used to measure activity in 1 mL of reaction, in 1 cm light path cuvettes (when the initial absorbance was greater than 1, 0.33 mL reactions in 0.1 cm light path cuvettes were performed). 200 nM and 100 nM concentrations of native DyP and its mutated variants were used, respectively, and the reactions were started by adding 2 mM H2O2. The initial rates (absorbance decrease) were calculated using the differences in the molar absorption coefficients (Δε) of MPS and methyl-phenyl sulfoxide (Δε254 7870 M−1 cm−1) and MTS and methyl-p-tolyl sulfoxide (Δε253 8040 M−1 cm−1).41 Kinetic constants for H2O2 reduction and ABTS oxidation were estimated by following the formation of the ABTS cation radical (ε436 29 300 M−1 cm−1). In the former case, a saturation concentration of ABTS (7.5 mM for native DyP and the L357G variant and 2.5 mM for the D168N, R332L, F359G, F359H and F359W variants), 10 nM enzyme, and different concentrations of H2O2 (from 20 μM to 25 mM) in 100 mM tartrate, pH 3, were used, while in the second case oxidation of different ABTS concentrations, in the presence 2.5 mM H2O2, was measured. Plotting and analysis of kinetic curves were carried out using SigmaPlot 11.0. Apparent affinity, turnover number and catalytic efficiency were estimated by non-linear least-squares fitting to the Michaelis–Menten model.Chromatographic analyses
Reactions (at 25 °C) were performed using 2 mL of 50 mM sodium acetate (pH 5) containing substrate (1 mM MPS or 0.5 mM MTS), 4 μM enzyme, and 1 mM H2O2. At 0, 30, 60 and 360 min of reaction, 0.5 mL aliquots were taken, and after addition of 4′-methoxyacetophenone as internal standard, the solution was extracted with hexane and analyzed by chiral HPLC.The reaction products were analyzed using an Agilent HPLC equipment fitted with a Chiralpack IB column (dimensions of 4.6 mm × 250 mm and particle size of 5 μm) and as a mobile phase isocratic hexane:isopropyl alcohol (95:5, v/v) for 40 min for analysis of reactions with MPS and 98:2 (v/v) for 70 min for reactions with MTS, at a flow rate of 1 mL min−1, at room temperature. Elution was monitored at 207, 216, 237, 248 and 263 nm. Calibration curves were obtained for identification and quantification of substrates and their sulfoxidation products. The retention times for the R (62 min) and S (64 min) methyl-p-tolyl sulfoxides were obtained from the corresponding standards, while those for the R (31 min) and S (33 min) methyl-phenyl sulfoxides were obtained from enzymatic oxidation of MPS assuming the published elution order,42 which coincided with that observed for the two methyl-p-tolyl sulfoxides.
Enzymatic reactions with 18O-labeled hydrogen peroxide (H218O2, 90% isotopic content, 2% w/v solution) and water (H218O, 97% isotopic content) from Sigma-Aldrich were performed under the same conditions, extracted with dichloromethane, and analyzed by GC-MS using a gas chromatograph equipped with an HP-5MS column (Agilent, Santa Clara, CA, USA; 30 m × 0.25 mm internal diameter; 0.25 μm film thickness) coupled to a quadrupole mass detector. Helium was used as the carrier gas, and the oven program started at 110 °C for 2 min and increased at 20 °C min−1 until 240 °C. 18O-labeling was calculated as the ratio between the abundances of the 18O- to 16O sulfoxide peaks, in their molecular ion (m/z 142 and 140, respectively, in the MPS reactions) chromatograms.
pH and temperature stability
The pH stability of the L357G and F359G variants and the native recombinant DyP was determined by incubating the enzymes (1 μM) for 24 h in 50 mM Britton–Robinson buffer (pH 2–12) at 25 °C. To evaluate their temperature stability, the enzymes in 10 mM sodium tartrate (pH 5) were incubated in the range from 25 to 80 °C for 10 min, followed by 2 min at 4 °C. The remaining activities were measured (in triplicate reactions) using 2.5 mM ABTS in 100 mM sodium tartrate (pH 3), as described above. The activity immediately after adding the enzyme to the buffer was taken as 100%. The T50 values, defined as the temperature at which 50% of activity is lost in 10 min incubation, were calculated.Crystallization, data collection and refinement
Crystallization of the L357G and F359G variants was performed in 96-well sitting drop plates (Swissci MRC, England) at 22 °C using a Cartesian Honeybee robot (Digital, USA) and commercially available kits: JBScreen Classic (Jenna Bioscience, Germany), Wizard Classic Screen (Emerald Biostructures, USA) and ProPlex HT-96 (Molecular Dimensions, UK). Each droplet was 0.4 μL in size, containing 0.2 μL of protein (3 μg) solution and 0.2 μL of precipitant and was equilibrated over 50 μL of reservoir solution. Crystals of the L357G mutant were obtained in 5% MPD (v/v), 100 mM MES (pH 6.5) and 15% PEG 6000 (w/v). For the F359G variant, crystals were obtained in 2 M magnesium formate and 20% PEG 3350 (w/v). Crystals were cryoprotected using Paratone-N (Hampton Research).X-ray diffraction images were collected at the ESRF (Grenoble, France) and ALBA (Barcelona, Spain) synchrotrons. Diffraction data were processed using XDS43 and scaled using AIMLESS.44,45 The structures were solved by molecular replacement using PHASER,46 with the A. auricula-judae native DyP (PDB 4W7J) as the search model. The initial model was first refined with REFMAC5 (ref. 47) and alternating manual building with COOT.48 The final model was obtained by repetitive cycles of refinement using PHENIX.46 Subsequent refinement, introduction of solvent molecules and structure validation were as described for the native DyP26 (data collection, refinement and final statistics of the two DyP variants are summarized in Table S1†).
The L357G structure did not show electron density for the first residue at the N-terminus, but the whole sequence could be solved for the F359G variant. In contrast, the C-terminal region showed good electron density for both structures. The coordinates and structure factors have been deposited in the Protein Data Bank.
System preparation for molecular modeling
The starting structures for the computational simulations were the native DyP crystal at a resolution of 1.79 Å (PDB 4W7J)26 and the crystal structures of the L357G and F359G variants (PDB 5IKG and 5IKD, respectively). Protein structures were prepared accordingly under pH 5 conditions, optimal for MPS and MTS sulfoxidation, using Schrödinger's Protein Preparation Wizard,49 and the H++ web server.50 Under these mild acidic conditions, histidines were double-protonated, with the exception of His-115 (ε-protonated) and His-304 (δ-protonated), and all other acidic residues were deprotonated. The heme site was modeled as thiolate-ligated compound I after being fully optimized in the protein environment with quantum mechanics/molecular mechanics (QM/MM) using QSite (see below for the level of theory).51 Finally, MPS and MTS molecules were optimized with Jaguar,52 at the density functional theory (DFT) M06 level with the 6-31G** basis set and Poisson–Boltzmann Finite element (PBF) implicit solvent in order to obtain their electrostatic potential atomic charges.PELE computational analysis
Once the protein structures had been prepared and ligands optimized, heme binding site exploration was performed with PELE, a Monte Carlo-based algorithm capable of effectively sampling the protein–ligand conformational space.53,54 The substrates were placed manually in identical positions at the entrance of the heme-access channel of each protein. PELE simulations were carried out in two stages: first, ligands were requested to move from the solvent to the heme site, and once the center of mass of the ligand was within 5 Å of the heme catalytic oxygen, it was free to explore the active site pocket with a 15 Å restrain. The results presented here are based on 240 trajectories × 48 h for each ligand.QM/MM simulations
Hybrid QM/MM calculations were carried out in order to investigate the mutation effect on substrate sulfoxidation. PELE minima snapshots of MPS and MTS substrates on native DyP, L357G and F359F variant binding sites were selected for quantum calculations. For each case, at least two structures were investigated: a structure with the ligand in an optimal orientation toward the heme reactive oxygen to produce an R-(−)-sulfoxide product and a structure with the ligand placed correctly to give an S-(+)-sulfoxide product. QM/MM calculations were performed by including the heme (modeled as compound I), its axial ligand and the substrate in the quantum region and computing the spin density. In order to prepare the system for QM/MM, 0.5 ns molecular dynamics were performed with Desmond,55 using SPC (simple point charge) solvent and an ionic force of 0.15 M. Calculations were performed at the DFT M06-L(lacvp*)/OPLS level with QSite.52Acknowledgements
This work was supported by the INDOX (KBBE-2013-7-613549) EU project and by the BIO2014-56388-R (NOESIS), BFU2014-55448-P and CTQ2013-48287-R projects of the Spanish Ministry of Economy and Competitiveness (MINECO). Pedro Merino (University of Zaragoza, Spain) is acknowledged for his suggestions on chiral HPLC analyses, and Alicia Prieto and Leonor Rodríguez (CIB, Madrid, Spain) for their help in GC-MS analyses. We cordially thank the staff at ID23-1 beamline (ESRF) and the BL13-XALOC beamline (ALBA). F. J. R.-D. acknowledges a MINECO Ramón & Cajal contract.Notes and references
- I. Fernández and N. Khiar, Chem. Rev., 2003, 103, 3651–3706 CrossRef PubMed.
- H. Gröger, in Catalytic Asymmetric Synthesis, ed. I. Ojima, John Wiley & Sons, Inc., 2010, pp. 269–341 Search PubMed.
- W. J. H. van Berkel, N. M. Kamerbeek and M. W. Fraaije, J. Biotechnol., 2006, 124, 670–689 CrossRef PubMed.
- S. Colonna, N. Gaggero, C. Richelmi and P. Pasta, Trends Biotechnol., 1999, 17, 163–168 CrossRef PubMed.
- A. Matura and K. H. van Pee, in Enzyme catalysis in organic synthesis: A comprehensive handbook, ed. K. Drautz, H. Gröger and O. May, Wiley, Weinheim, 2012, pp. 1553–1567 Search PubMed.
- E. G. Hrycay and S. M. Bandiera, Arch. Biochem. Biophys., 2012, 522, 71–89 CrossRef PubMed.
- T. Matsui, Y. Dekishima and M. Ueda, Appl. Microbiol. Biotechnol., 2014, 98, 7699–7706 CrossRef PubMed.
- S. Colonna, N. Gaggero, L. Casella, G. Carrea and P. Pasta, Tetrahedron: Asymmetry, 1992, 3, 95–106 CrossRef.
- M. P. J. van Deurzen, F. van Rantwijk and R. A. Sheldon, Tetrahedron, 1997, 53, 13183–13220 CrossRef.
- S. Kobayashi, M. Nakano, T. Goto, T. Kimura and A. P. Schaap, Biochem. Biophys. Res. Commun., 1986, 135, 166–171 CrossRef PubMed.
- S. Colonna, N. Gaggero, G. Carrea and P. Pasta, J. Chem. Soc., Chem. Commun., 1992, 357–358 RSC.
- R. Z. Harris, S. L. Newmyer and P. R. Ortiz de Montellano, J. Biolumin. Chemilumin., 1993, 268, 1637–1645 Search PubMed.
- V. M. Dembitsky, Tetrahedron, 2003, 59, 4701–4720 CrossRef.
- M. Sono, M. P. Roach, E. D. Coulter and J. H. Dawson, Chem. Rev., 1996, 96, 2841–2888 CrossRef PubMed.
- V. P. Miller, G. D. DePillis, J. C. Ferrer, A. G. Mauk and P. R. Ortiz de Montellano, J. Biolumin. Chemilumin., 1992, 267, 8936–8942 Search PubMed.
- W. Adam, C. Mock-Knoblauch and C. R. Saha-Möller, J. Organomet. Chem., 1999, 64, 4834–4839 Search PubMed.
- E. Baciocchi, M. F. Gerini, P. J. Harvey, O. Lanzalunga and S. Mancinelli, Eur. J. Biochem., 2000, 267, 2705–2710 CrossRef PubMed.
- M. Hofrichter, H. Kellner, M. J. Pecyna and R. Ullrich, Adv. Exp. Med. Biol., 2015, 851, 341–368 CrossRef PubMed.
- A. Horn, R. Ullrich, M. Hofrichter and K. Scheibner, Abstracts BioTrans-2007, Oviedo, 2007, p. 63 Search PubMed.
- A. Horn, Der Einsatz einer neuartigen Peroxidase aus dem Basidiomyceten Agrocybe aegerita am Beispiel der enantioselektiven Sulfoxidation, PhD thesis, Universität Rostock, Rostock, 2009 Search PubMed.
- E. Aranda, M. Kinne, M. Kluge, R. Ullrich and M. Hofrichter, Appl. Microbiol. Biotechnol., 2009, 82, 1057–1066 CrossRef PubMed.
- B. Goblirsch, R. C. Kurker, B. R. Streit, C. M. Wilmot and J. L. Dubois, J. Mol. Biol., 2011, 408, 379–398 CrossRef PubMed.
- M. Puhse, R. T. Szweda, Y. Y. Ma, C. Jeworrek, R. Winter and H. Zorn, Biochim. Biophys. Acta, Proteins Proteomics, 2009, 1794, 1091–1098 CrossRef PubMed.
- D. Linde, C. Coscolín, C. Liers, M. Hofrichter, A. T. Martínez and F. J. Ruiz-Dueñas, Protein Expression Purif., 2014, 103, 28–37 CrossRef PubMed.
- D. Linde, F. J. Ruiz-Dueñas, E. Fernández-Fueyo, V. Guallar, K. E. Hammel, R. Pogni and A. T. Martínez, Arch. Biochem. Biophys., 2015, 574, 66–74 CrossRef PubMed.
- D. Linde, R. Pogni, M. Cañellas, F. Lucas, V. Guallar, M. C. Baratto, A. Sinicropi, V. Sáez-Jiménez, C. Coscolín, A. Romero, F. J. Medrano, F. J. Ruiz-Dueñas and A. T. Martínez, Biochem. J., 2015, 466, 253–262 CrossRef PubMed.
- E. Strittmatter, C. Liers, R. Ullrich, S. Wachter, M. Hofrichter, D. A. Plattner and K. Piontek, J. Biolumin. Chemilumin., 2013, 288, 4095–4102 Search PubMed.
- C. Liers, C. Bobeth, M. Pecyna, R. Ullrich and M. Hofrichter, Appl. Microbiol. Biotechnol., 2010, 85, 1869–1879 CrossRef PubMed.
- J. N. Rodríguez-López, A. T. Smith and R. N. F. Thorneley, J. Biolumin. Chemilumin., 1996, 271, 4023–4030 Search PubMed.
- K. Piontek, E. Strittmatter, R. Ullrich, G. Grobe, M. J. Pecyna, M. Kluge, K. Scheibner, M. Hofrichter and D. A. Plattner, J. Biolumin. Chemilumin., 2013, 288, 34767–34776 Search PubMed.
- S. L. Newmyer and P. R. Ortiz de Montellano, J. Biolumin. Chemilumin., 1995, 270, 19430–19438 Search PubMed.
- S. Ozaki and P. R. Ortiz de Montellano, J. Am. Chem. Soc., 1995, 117, 7056–7064 CrossRef.
- M. I. Savenkova, J. M. Kuo and P. R. Ortiz de Montellano, Biochemistry, 1998, 37, 10828–10836 CrossRef PubMed.
- M. I. Savenkova and P. R. Ortiz de Montellano, Arch. Biochem. Biophys., 1998, 351, 286–293 CrossRef PubMed.
- F. van Rantwijk and R. A. Sheldon, Curr. Opin. Biotechnol., 2000, 11, 554–564 CrossRef PubMed.
- E. van Bloois, D. E. T. Pazmino, R. T. Winter and M. W. Fraaije, Appl. Microbiol. Biotechnol., 2010, 86, 1419–1430 CrossRef PubMed.
- Y. K. Bong, M. D. Clay, S. J. Collier, B. Mijts, M. Vogel, X. Zhang, Y. Zhu, J. Nazor, D. Smith and S. Song, Patent (International), WO071982, 2011 Search PubMed.
- S. Shaik, Y. Wang, H. Chen, J. Song and R. Meir, Faraday Discuss., 2010, 145, 49–70 RSC.
- Y. Goto, T. Matsui, S. I. Ozaki, Y. Watanabe and S. Fukuzumi, J. Am. Chem. Soc., 1999, 121, 9497–9502 CrossRef.
- P. Molina-Espeja, M. Cañellas, F. Plou, M. Hofrichter, F. Lucas, V. Guallar and M. Alcalde, ChemBioChem, 2016, 17, 341–349 CrossRef PubMed.
- C. Capeillere-Blandin, C. Martin, N. Gaggero, P. Pasta, G. Carrea and S. Colonna, Biochem. J., 1998, 335(Pt 1), 27–33 CrossRef PubMed.
- Y. Wang, H. Li, W. Qi, Y. Yang, Y. Yan, B. Li and L. Wu, J. Mater. Chem., 2012, 22, 9181–9188 RSC.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef PubMed.
- P. R. Evans, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 282–292 CrossRef PubMed.
- P. R. Evans and G. N. Murshudov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 1204–1214 CrossRef PubMed.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, J. Appl. Crystallogr., 2007, 40, 658–674 CrossRef PubMed.
- G. N. Murshudov, A. A. Vagin and D. J. Dodson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1997, 53, 255 CrossRef PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef PubMed.
- G. M. Sastry, M. Adzhigirey, T. Day, R. Annabhimoju and W. Sherman, J. Comput.-Aided Mol. Des., 2013, 27, 221–234 CrossRef PubMed.
- R. Anandakrishnan, B. Aguilar and A. V. Onufriev, Nucleic Acids Res., 2012, 40, W537–W541 CrossRef PubMed.
- Schrödinger, QSite 5.7, LCC, New York, 2011 Search PubMed.
- Schrödinger, Jaguar 8.1, LCC, New York, 2013 Search PubMed.
- K. W. Borrelli, A. Vitalis, R. Alcantara and V. Guallar, J. Chem. Theory Comput., 2005, 1, 1304–1311 CrossRef PubMed.
- K. Borrelli, B. Cossins and V. Guallar, J. Comput. Chem., 2010, 31, 1224–1235 Search PubMed.
- Desmond, Maestro-Desmond Interoperability Tools, version 2. 4, Schrödinger, New York, NY, 2010 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Table S1 shows the X-ray data collection and refinement statistics, Fig. S1 shows the simulation position and distance analysis for the L357G variant, and Fig. S2 shows correlation between substrate spin density and charge distribution. See DOI: 10.1039/c6cy00539j |
| ‡ These two authors contributed equally to this work. |
| § Current address: Instituto de Catálisis y Petroleoquímica, CSIC, Marie Curie 2, 28049 Madrid, Spain. |
| This journal is © The Royal Society of Chemistry 2016 |