
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supported bimetallic nano-alloys as highly active catalysts for the one-pot tandem synthesis of imines and secondary amines from nitrobenzene and alcohols†
Meenakshisundaram
Sankar
*ab,
Qian
He
c,
Simon
Dawson
b,
Ewa
Nowicka
b,
Li
Lu
c,
Pieter C. A.
Bruijnincx
a,
Andrew M.
Beale
de,
Christopher J.
Kiely
c and
Bert M.
Weckhuysen
*a
aInorganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands. E-mail: B.M.Weckhuysen@uu.nl
bCardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff CF10 3AT, UK. E-mail: Sankar@cardiff.ac.uk; Tel: +44 (0)29 2087 5748
cDepartment of Material Science and Engineering, Lehigh University, 5 East Packer Avenue, Bethlehem, PA 18015-3195, USA
dResearch Complex at Harwell, Rutherford Appleton Laboratory, Oxfordshire, OX11 0FA, UK
eDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK
First published on 23rd March 2016
Abstract
The synthesis and functionalization of imines and amines are key steps in the preparation of many fine chemicals and for pharmaceuticals in particular. Traditionally, metal complexes are used as homogeneous catalysts for these organic transformations. Here we report gold–palladium and ruthenium–palladium nano-alloys supported on TiO2 acting as highly efficient heterogeneous catalysts for the one-pot synthesis of the imine N-benzylideneaniline and the secondary amine N-benzylaniline directly from the easily available and stable nitrobenzene and benzyl alcohol precursors using a hydrogen auto-transfer strategy. These reactions were carried out without any added external hydrogen, sacrificial hydrogen donor or a homogeneous base. The bimetallic catalysts were prepared by the recently developed modified impregnation strategy, giving efficient control of size and nano-alloy composition. Both bimetallic catalysts were found to be far more active than their monometallic analogues due to a synergistic effect. Based on the turnover numbers the catalytic activities follow the order Ru < Pd < Au ≪ Au–Pd < Ru–Pd. Aberration corrected scanning transmission electron microscopy (AC-STEM) and X-ray absorption spectroscopy (XAFS) studies of these catalysts revealed that the reason for the observed synergistic effect is the electronic modification of the metal sites in the case of the Au–Pd system and a size stabilisation effect in the case of the Ru–Pd catalyst.
Introduction
Supported bimetallic nano-alloys exhibit substantially different, often enhanced, properties compared to their monometallic analogues, which makes them very attractive for various technical applications, especially as catalysts.1 Precise control of their structural parameters, such as (a) size-dependent compositional variation, (b) morphology and (c) mono-dispersity of particle size, is crucial to fully harness the catalytic power of these bimetallic nano-alloys.2,3 Conventionally, supported bimetallic nano-alloys are prepared by a wet-impregnation (CIm) method using aqueous solutions containing both metal precursors and the support. Catalysts prepared by this route normally contain metal particles ranging from sub-nm metal clusters to a few micron-sized particles.3 To overcome this problem of the broad size distribution, size-controlled nano-alloys have been prepared in aqueous solutions in the presence of stabilizer ligands, such as polyvinyl alcohol (PVA) or poly(vinylpyrrolidone) (PVP).4 These particles are then immobilized onto a solid oxide support (sol-immobilization (SIm) method) to form catalysts with almost all the metal particles existing within the 2–10 nm range.5 However, supported nano-alloys prepared by the SIm method still tend to exhibit a size-dependent composition variation.6,7Recently, Sankar et al. reported an excess anion modification of the CIm method (the modified impregnation (MIm) method) for the synthesis of supported gold–palladium nano-alloys, which affords a more precise control over size, composition and morphology.8 By adding excess hydrochloric acid to the Au and Pd precursors in the wet-impregnation stage, and subsequent direct gas-phase reduction, Au–Pd alloy particle size can be controlled to be within the 1–6 nm range with no size significant dependent composition variation of Au and Pd. Occasionally, some micron-scale Au-rich particles were found in some of the MIm derived Au–Pd catalysts. Such micron-sized particles could be completely eliminated, though, by increasing the hydrochloric acid concentration during the preparation stage. However, the latter catalyst, i.e., without any micron-sized gold-rich particles, was found to be unstable for catalytic applications, whereas the former catalyst with occasional micron-scale particles was found to be stable for catalytic re-use.8 The stable bimetallic catalysts showed significant improvements in the catalytic activities for the direct synthesis of hydrogen peroxide and the aerobic benzyl alcohol oxidation reactions.10 Very recently, we also reported the synthesis of supported Ru–Pd catalysts, using the same MIm route; no micron-scale particles were detected for these catalysts, suggesting all of the metal precursors are converted into alloyed nanoparticles with controlled size and composition. These supported bimetallic Ru–Pd nano-alloy catalysts were exceptionally active, selective and stable in the hydrogenation of biomass derived levulinic acid to γ-valerolactone.9
Dehydrogenation reactions are often carried out in the presence of stoichiometric amounts or a large excess of oxygen, peroxides, iodates, metal oxides or sacrificial hydrogen acceptors to circumvent the thermodynamic restrictions associated with such dehydrogenations.10 In all these processes, the liberated hydrogen is “wasted” and ends up in hydrogenated by-products (e.g., H2O). An example of this kind of reaction is the oxidative dehydrogenation of benzyl alcohol to benzaldehyde, where oxygen is used as the sacrificial hydrogen acceptor to form water.11 From an atom-economic perspective, the expensive hydrogen produced by the dehydrogenation reaction should be more effectively used (in situ), for instance in a coupled hydrogenation reaction. Here, we demonstrate such an efficient use of the hydrogen liberated in the dehydrogenation of benzyl alcohol (1), by using nitrobenzene (3) as the hydrogen-acceptor rather than O2 thus reducing nitrobenzene effectively to aniline (4). The products of these reactions, i.e., benzaldehyde (2) and aniline, readily couple to form N-benzylideneaniline (5). When 1 is used in excess, 5 is further hydrogenated to N-benzylaniline (6) (Fig. 1). Furthermore, the rate of the overall reaction increases with the increase in the amount of alcohol. This sequence of reactions has been previously reported and are known as acceptor-less dehydrogenation reactions, hydrogen auto-transfer reactions or as a borrowing hydrogen strategy.10,12,13 Such reactions have proven extremely valuable for the synthesis of an assortment of useful compounds, including amines and amides, without stoichiometric reagents and harmful by-products. However in order to compete with the economically more advantageous conventional strategies using molecular H2, the catalysts for these hydrogen-auto transfer processes have to be exceptionally active and selective. Various heterogeneous catalysts have been reported for the N-alkylation of aniline using benzyl alcohol using this strategy.14,15 However, only a few catalytic systems have been reported for the direct synthesis of imines and N-alkylamines from nitroarenes and alcohols using this hydrogen auto transfer strategy. Most of these use homogeneous Ru, Ir or Pd-based metal complexes.15–18 To the best of our knowledge, only a few research groups have used heterogeneous catalysts for this transformation process, and most of these systems require a homogeneous base for this direct transformation of nitrobenzene and benzyl alcohol to the corresponding benzyl imines and amines.19,20 Two groups have reported the use of supported monometallic gold nanoparticles for this one-pot synthesis.21,22
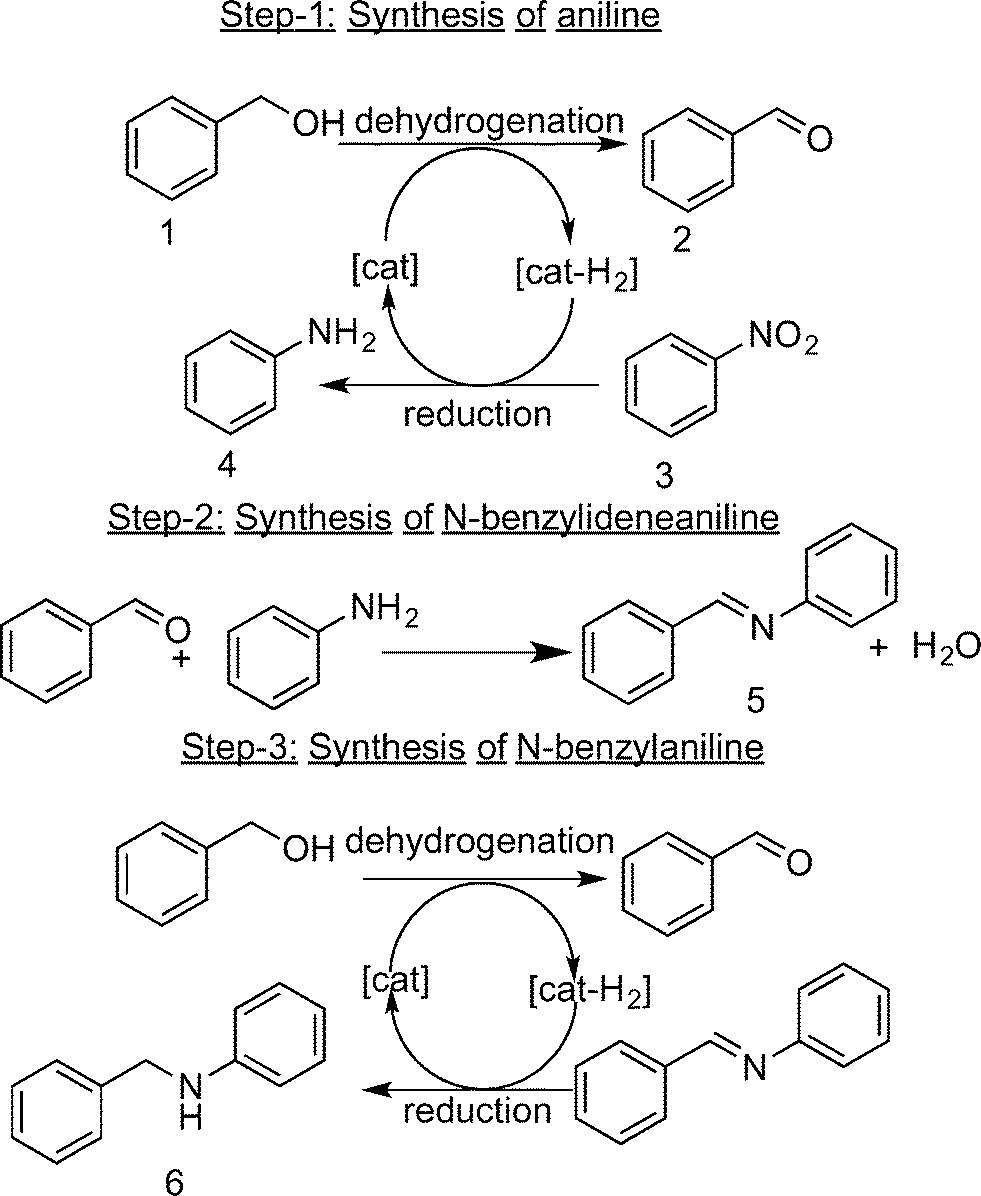 | ||
| Fig. 1 Sequence of reactions involved in the one-pot tandem synthesis of N-benzylideneaniline (5) and N-benzylaniline (6) from nitrobenzene (3) and benzyl alcohol (1). | ||
Here, we show that supported gold–palladium and ruthenium–palladium nano-alloys are efficient catalysts for this hydrogen auto-transfer reaction, greatly surpassing the catalytic activities of their supported monometallic counterparts when prepared and tested under identical reaction conditions. Using abberation corrected scanning transmission electron microscopy (AC-STEM) and X-ray absorption spectroscopy (XAFS) characterisation data, we rationalise the observed catalytic behaviour of these supported gold–palladium and ruthenium–palladium nano-alloy catalysts.
Experimental
Synthesis of catalysts
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 80% hydrolysed) and an aqueous solution of 0.1 M NaBH4 were also prepared. To an aqueous solution of the precursors, the requisite amount of the PVA solution was added (PVA/Au + Pd (wt/wt) = 1.3) and stirred for 30 min. Following this, the requisite amount of the freshly prepared aqueous solution of NaBH4 (NaBH4/Au + Pd (mol/mol) = 5) was added all at once to the stirring solution to instigate bimetallic sol formation. The solution was then stirred for a further 30 min to complete the sol formation. Subsequently, the required amount of support (TiO2, Evonik-Degussa P25) was added. After several minutes of stirring, a drop of concentrated H2SO4 was added under vigorous stirring. The total target metal loading was calculated to be 1 wt% with equimolar metal amounts for the bimetallic catalysts. After 2 h of stirring, the slurry was filtered and the solid catalyst was washed thoroughly with 2 L of distilled water until the mother liquor became neutral. The filtered solid was then dried at 393 K overnight under static air and was used as a catalyst without any further modification. All the catalysts prepared by this method are labelled (SIm).
000, 80% hydrolysed) and an aqueous solution of 0.1 M NaBH4 were also prepared. To an aqueous solution of the precursors, the requisite amount of the PVA solution was added (PVA/Au + Pd (wt/wt) = 1.3) and stirred for 30 min. Following this, the requisite amount of the freshly prepared aqueous solution of NaBH4 (NaBH4/Au + Pd (mol/mol) = 5) was added all at once to the stirring solution to instigate bimetallic sol formation. The solution was then stirred for a further 30 min to complete the sol formation. Subsequently, the required amount of support (TiO2, Evonik-Degussa P25) was added. After several minutes of stirring, a drop of concentrated H2SO4 was added under vigorous stirring. The total target metal loading was calculated to be 1 wt% with equimolar metal amounts for the bimetallic catalysts. After 2 h of stirring, the slurry was filtered and the solid catalyst was washed thoroughly with 2 L of distilled water until the mother liquor became neutral. The filtered solid was then dried at 393 K overnight under static air and was used as a catalyst without any further modification. All the catalysts prepared by this method are labelled (SIm).
| Total TON = [mol(3) converted/moltotal metal in the catalyst]. |
The amine turnover number (Amine TON) was calculated using the formula
| Amine TON = [mol(6) formed/moltotal metal in the catalyst]. |
Product selectivity is defined as the fraction of the identified products. The products from benzyl alcohol dehydrogenation and disproportionation (benzaldehyde and toluene) have not been quantified.
Results and discussion
All the monometallic and bimetallic catalysts (1% Au/TiO2 (MIm), 1% Ru/TiO2 (MIm), 1% Pd/TiO2(MIm), 1% Au–Pd/TiO2 (MIm) and 1% Ru–Pd/TiO2 (MIm)), prepared by the MIm method, were tested for the one-pot tandem synthesis of 5 and 6 from 3 and excess of 1 at 433 K under an inert atmosphere. From the results (Fig. 2) it is evident that the bimetallic catalysts greatly out perform their monometallic counterparts. The total turnover number (TONtot), based on molar conversion of 3, and the amine turnover number (TONamine), based on the yield of 6, were used as a measure of catalytic activity. After 3 h, the TONtot values of these monometallic catalysts follow the order 1% Au/TiO2 (340) > 1% Pd/TiO2 (102) > 1% Ru/TiO2 (53) and this catalytic activity trend matches that previously reported for this reaction.21,22 The TONamine values for these monometallic 1% Au/TiO2, 1% Pd/TiO2, and 1% Ru/TiO2 catalysts are 2, 1 and 1, respectively. Notably, the bimetallic catalysts showed a dramatic increase in the TONtot values with 475 and 787 for the 1% RuPd/TiO2 and 1% AuPd/TiO2 catalysts respectively. Based on the TONamine values the reactivity order follows 1% Ru–Pd/TiO2 (174) > 1% Au–Pd/TiO2 (93) ≫ monometallic catalysts. A time-on-line study for the most active supported ruthenium palladium bimetallic catalyst (1% Ru–Pd/TiO2) at 433 K, clearly indicates the reduction of 3 and the formation of 5 to be kinetically more favourable than the reduction of 5 to 6 (Fig. 3). Notably, the selectivity to 5 is always maintained high (>90%) until the conversion level reaches 99%. Throughout this reaction, the selectivity for aniline (4) remained less than 1%, which indicates that the coupling of 4 and 2 is very fast. The corresponding time-on-line evolution of the TONs is presented in the ESI† (Fig. S1). A few examples of bimetallic catalysts have been reported that are superior for the oxidative dehydrogenation of benzyl alcohol compared to their monometallic analogues.1,6,23 Based on this fact, and the sequence of reactions presented in Fig. 1, we hypothesize that the primary effect of the bimetallic catalyst is to enhance the rate of the initial dehydrogenation step, which has knock-on benefits for steps 2 and 3 (i.e. the production of N-benzylideneaniline and the secondary amine N-benzylaniline respectively). | ||
| Fig. 2 Comparison of TONs of monometallic and bimetallic catalysts for the tandem synthesis of 5 and 6. (a): Ru and Pd for total TON; (b): Au and Pd for total TON; (c): Ru and Pd for amine TON and (d): Au and Pd for amine TON; key; C1: 1% Ru/TiO2; C2: 1% Pd/TiO2; C3: 1% Au/TiO2; C4: 1% Au–Pd/TiO2; C5: 1% Ru–Pd/TiO2. Reaction conditions: catalyst: 0.1 g; nitrobenzene: 4.5 mmol; benzyl alcohol: 45 mmol; mesitylene (solvent): 5 mL; Ar: 20 bar; T: 433 K; t: 3 h. | ||
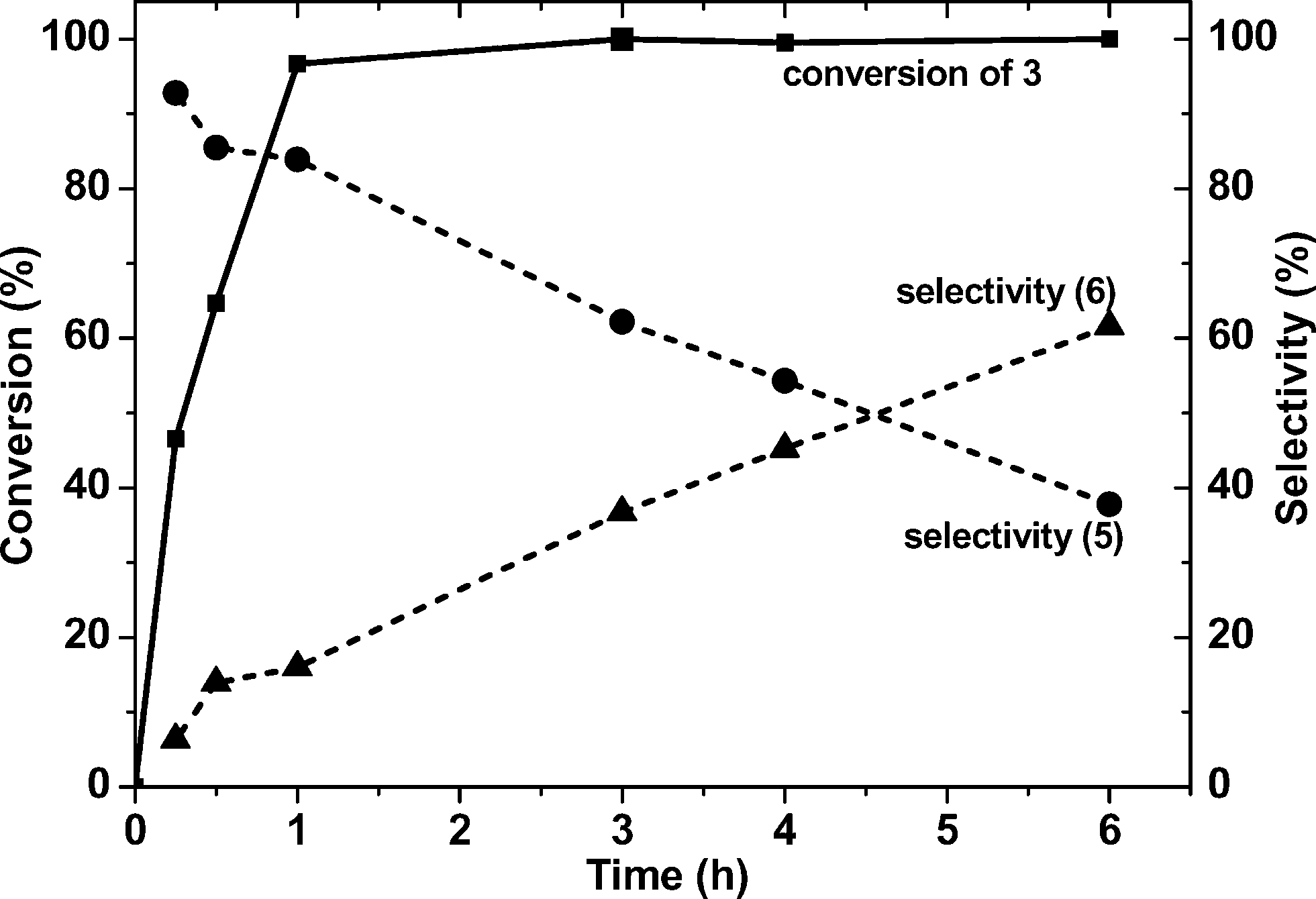 | ||
| Fig. 3 Time-on-line profile of the nitrobenzene conversion and product selectivity for the tandem synthesis of 5 and 6 using the 1% RuPd/TiO2 (MIm) catalyst. Reaction conditions: catalyst: 0.1 g; nitrobenzene: 4.5 mmol; benzyl alcohol: 45 mmol; mesitylene (solvent): 5 mL; Ar: 20 bar; T: 433 K. | ||
Earlier, Sankar et al. have reported that, for the oxidative dehydrogenation of 1, TiO2-supported Au–Pd nano-alloys also catalyse an unwanted disproportionation reaction, resulting in the production of toluene and benzaldehyde, besides the desired dehydrogenation reaction to form benzaldehyde.23 It was further reported that MgO-supported gold–palladium catalysts exclusively favour the dehydrogenation reaction by switching-off the disproportionation reaction.23 A 1% AuPd/MgO (MIm) catalyst was therefore also tested for the current hydrogen auto-transfer reaction, but was found to have a much lower activity (23% conversion) compared to the TiO2-supported catalyst (99% conversion). An activated carbon-supported Ru–Pd catalyst also proved to be less active (9% conversion) (Table 1). In addition to the choice of support, the preparation method is also expected to strongly influence the activity, selectivity and stability of these supported bimetallic catalysts.3,8 Prati et al. demonstrated the sol-immobilization (SIm) technique as an effective way of preparing supported Au–Pd catalysts.4 Hence, 1% Au–Pd/TiO2 (SIm) and 1% AuPd/MgO (SIm) catalysts were also therefore tested in the hydrogen auto-transfer reaction for comparative purposes, but neither of these catalyst preparations proved to be very effective (Table 1) displaying only 4% and 3% conversion levels respectively. The catalytic results thus clearly show that the TiO2-supported bimetallic catalysts prepared by the MIm route perform best in the direct synthesis of 5 and 6 from 1 and 3. To further understand the role that the amount of 1 has on the resultant activity, we performed reactions with different 1 to 3 molar ratios (i.e. 5 & 3) using the best catalyst 1% RuPd/TiO2 (MIm) and the results are presented in Table 1. The results clearly indicate that the activity depends on the amount of benzyl alcohol present, or in other words, the extent of the dehydrogenation reaction. For the reaction with a 1 to 3 molar ratio of 5, the molar conversion of 3 is 60% with a selectivity of 93% to product 5. For the corresponding reaction performed with a molar ratio of 3, the conversion is found to be 32% with 97% selectivity to product 5. It is important to note that for all the other standard reactions reported in this article a molar ratio of 10 was used. Another approach to synthesize substituted amines is the reductive alkylation of nitrobenzene using benzyl alcohol (equimolar) as the alkylating agent under H2 atmosphere. To test our bimetallic 1% RuPd/TiO2 (MIm) catalyst for this reductive alkylation reaction, we used an equimolar mixture of 1 & 3 under 20 bar of H2. After 3 h of reaction, a 78% molar conversion of 3 was achieved with an 84% selectivity to product 6. This result clearly indicates that this bimetallic catalyst is effective for the reductive alkylation reaction as well. Further optimization studies are currently in progress for this reaction.
| Catalyst | Preparation method | Nitrobenzene conversion (%) | Product selectivity (%) | |
|---|---|---|---|---|
| 5 | 6 | |||
| a Reaction conditions: catalysts: 0.1 g; nitrobenzene: 4.5 mmol; benzyl alcohol: 45 mmol; mesitylene (solvent): 5 mL; Ar: 20 bar; T: 433 K; t: 3 h. b Nitrobenzene: 4.5 mmol; benzyl alcohol: 22.5 mmol (benzyl alcohol vs. nitrobenzene = 5). c Nitrobenzene: 4.5 mmol; benzyl alcohol: 13.5 mmol (benzyl alcohol vs. nitrobenzene = 3). d Nitrobenzene: 4.5 mmol; benzyl alcohol: 4.5 mmol (benzyl alcohol vs. nitrobenzene = 1); reaction performed at 20 bar H2. | ||||
| 1% AuPd/TiO2 | SIm | 4 | >99 | — |
| 1% Au–Pd/MgO | SIm | 3 | >99 | — |
| 1% Au–Pd/TiO2 | MIm | 99 | 88 | 12 |
| 1% Au–Pd/MgO | MIm | 23 | >99 | — |
| 1% Ru–Pd/TiO2 | MIm | 99 | 54 | 45 |
| 1% Ru–Pd/MgO | MIm | 26 | >99 | — |
| 1% RuPd/C | MIm | 9 | >99 | — |
| 1% RuPd/TiO2b | MIm | 60 | 93 | 7 |
| 1% RuPd/TiO2c | MIm | 32 | 97 | 3 |
| 1% RuPd/TiO2d | MIm | 78 | 16 | 84 |
The heterogeneous nature of the most active catalyst (1% Ru–Pd/TiO2 (MIm)) was demonstrated by three methods; namely (a) hot filtration, (b) ICP-MS analysis of the reaction mixture for metal content and (c) reusability of the recovered catalyst. These catalytic tests were performed in a 50 mL Radley's glass reactor held at 413 K and a pressure of 1 bar of He. In the hot filtration method, the catalyst was filtered off after 30 min (8% conversion) and the reaction mixture was allowed to react for a further 150 min under standard reaction conditions. No increase in conversion (9%) and selectivity was seen after filtration (Fig. 4a). The reaction without catalyst removal had a conversion of 23% over the same time-period. ICP-MS measurements showed the filtered reaction mixture to contain negligible amounts of Ru and Pd (1.5% for Ru and 0.6% for Pd of the original amounts used in the reaction). Furthermore, the catalyst could be re-used three times without any loss in activity (8–9%). Interestingly however, the selectivity to 6 increased progressively from 27% for the fresh catalyst to 40% for the three-times used catalyst at the expense of 5 for the spent catalysts (Fig. 4b).
 | ||
| Fig. 4 (a) Confirming the heterogeneous nature of 1% Ru–Pd/TiO2 (MIm) catalyst by hot-filtration. For all the points shown the selectivity was found to be around 73% (imine) and 27% (amine). (b) Re-usability study of 1% Ru–Pd/TiO2. Reaction conditions: nitrobenzene: 0.9 mmol; benzylalcohol: 8.6 mmol; mesitylene (solvent): 1 mL; 1% Ru–Pd/TiO2: 0.020 g; temperature: 413 K; stirring speed: 500 rpm; time: 0.5 h; this reaction-set was performed in a moderate pressure glass reactor (Radleys®) fitted with a magnetic stirrer bar. | ||
To characterize the composition and nanostructure of these highly active bimetallic MIm catalysts and to arrive at a structure/activity correlation, aberration corrected scanning transmission electron microscopy (AC-STEM) studies were carried out; the SIm catalysts have previously been extensively characterized and are not reported here.6,7,23 Analysis of the 1% AuPd/TiO2 MIm sample by high-angle annular dark-field (HAADF) – STEM imaging indicated that the mean size of the supported metal particles was ∼1.5 nm (Fig. 5(a) and (b)). Even smaller sub-nm clusters were also apparent in this sample (Fig. S2(a)†) as were occasional very large μm-scale Au particles. X-ray energy dispersive spectroscopic (XEDS) analysis of individual nm-scale particles confirmed them to be Au–Pd alloys (Fig. 5(c) and (d)). A comparative HAADF-STEM study of the corresponding 1% RuPd/TiO2 MIm sample shows that the mean size of the supported metal particles was slightly smaller at ∼1.2 nm (Fig. 5(e) and (f)). Once again, many sub-nm clusters were apparent (Fig. S2(b)†), but in this case SEM analysis showed that the sample was devoid of any μm-scale metal particles. XEDS analysis confirmed that the nm-scale particles were indeed Ru–Pd alloys (Fig. 5(g) and (h)). To better understand how representative these structures are of the entirety of the catalyst sample, and to complement the STEM data, these catalysts were also characterised by X-ray absorption spectroscopy (XAS).26
 | ||
| Fig. 5 Representative STEM analyses of the 1% Au–Pd/TiO2 (a, b, c & d) and 1% Ru–Pd/TiO2 (e, f, g & h) samples prepared by the MIm route. (a) HAADF image of 1% Au–Pd/TiO2 showing typical particle sizes; (b) corresponding particle size distribution derived from measurement of over 500 particles; (c) HAADF image of an individual Au–Pd metal nanoparticle and (d) its corresponding XEDS spectrum confirming it is an Au–Pd alloy; (e) HAADF image showing typical particle sizes of 1% RuPd/TiO2; (f) corresponding particle size distribution derived from measurement of over 500 particles; (g) HAADF image of an individual metal nanoparticle and (h) its corresponding XEDS spectrum confirming it is a Ru–Pd alloy. The scale bar in each image represents 2 nm. | ||
The STEM observations of the presence of bimetallic species are confirmed by the XAFS data recorded at the Au L3, Pd K and Ru K edges, respectively.9 The Au and Pd XANES data for selected catalysts are given in Fig. 6, whilst the results from the analysis of the EXAFS data are given in Table 2 along with some example EXAFS spectra (including the results from the least squares fitting of the data) given in the ESI† (Fig. S3). The EXAFS data for the 1% Au–Pd/TiO2 (MIm) and 1% Ru–Pd/TiO2 catalysts are reported in our recent article.9 The Au L3-edge data shown arise from a dipole-allowed transition of an initial 2p3/2 electron to a 5d state and are therefore sensitive to changes in the electronic density of states (both initial and final).27–29 When comparing the reference 1% Au/TiO2 (MIm) sample with that of the bimetallic 1% Au–Pd/TiO2 samples (MIm and SIm) we observe an enhancement of the feature at ∼11935 eV in the latter two samples which appears consistent with the formation of bimetallic species. This signal is proposed to arise as a result of charge transfer/electron donation from Au to the more electropositive Pd. Such charge transfer between Au and Pd leading to a slight positive charge build-up on Pd has previously been confirmed by XPS analysis of these materials.9 This is accompanied by a ‘blue shift’ in the EXAFS oscillations, as well as a reduced oscillation frequency consistent with the formation of shorter Au–Pd bonds (as compared to the longer Au–Au bonds ∼2.85 Å). That the particles are bimetallic can be seen immediately from the Fourier transform (FT) data. Two intense peaks in the FTs are observed as a consequence of a ‘π phase flip’ in the backscattering amplitude from 6 Å−1 for Au (or indeed for all elements where Z > 78) resulting in a splitting of the major contribution in the FT into a high and low R (distance) component. This occurs when two elements are present in equivalent amounts; often the splitting and intensity of the low r-space contribution becomes more intense with an increasing number of bimetallic bonds.30–32 A similar effect is seen in the Pd K-edge FTs of the EXAFS data although a mismatch in the total coordination number from an analysis of the two edges has previously been shown to be due to the presence of large (∼μm sized) Au particles in addition to bimetallic species and for the 1% Au–Pd/TiO2 (MIm) sample, atomically dispersed Pd species.9 The Pd K-edge XANES data are less revealing regarding the extent of alloy formation, but are however, sensitive to the degree of Pd–O interactions. A greater rising absorption edge (lower density of states) is observed in the 1% Ru–Pd/TiO2 (MIm) sample which EXAFS data suggest contains the highest number of Pd–O neighbours (they are therefore the smallest particles and possess fewest number of metal–metal (M–M) bonds). It should be noted however that the similarity in X-ray scattering contrast between Pd and Ru precludes distinguishing between the two components of the bimetal in the EXAFS data although a first shell analysis assuming one M–M scatterer type could be performed on the basis that XEDS confirms the bimetallic nature of the sample.
 | ||
| Fig. 6 (a) Normalised Au L3-edge XANES and (b) Pd K-edge XANES recorded for select monometallic and bimetallic catalysts. The arrow in (a) illustrates a greater transition intensity at ca. 11935 eV for the bimetallic Au–Pd/TiO2 samples due to electron transfer from Au to Pd. | ||
| Sample | Au–Au R (Å) | (N) | 2σ2 (Å2) | Au–Pd (Å) | (N) | 2σ2 (Å2) | Pd–Pd R (Å) | (N) | 2σ2 (Å2) | Pd–Au R (Å) | N | 2σ2 (Å2) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
E
f ∼ ±15 eV; R values for all data range from 28–38%.
a A Pd–O contribution at 2.02 Å, N = 0.3, 2σ2 = 0.013 Å2 is also present.
b The EXAFS data and the associated Fourier transform data for this catalyst is presented in the ESI (Fig. S3).
c A Pd–O contribution at 2.02 Å, N = 1, 2σ2 = 0.0013 Å2 is also present.
d A Pd–O contribution at 1.99 Å, N = 1.3, 2σ2 = 0.013 Å2 is also present.
e A Pd–O contribution at 1.99 Å, N = 2, 2σ2 = 0.007 Å2 is also present.
Afac values, 0.94 (Pd/Ru) and 0.98 Au. Debye–Waller factors were initially determined from the AuPd/TiO2 SIm sample and not refined for the remaining AuPd samples. |
|||||||||||||
| 1% Au–Pd/TiO2 (SIm)a | 2.8 | 7.8 | 0.017 | 2.78 | 4.1 | 0.014 | 2.75 | 4 | 0.014 | 2.78 | 3.9 | 0.016 | Current Workb |
| 1% Au–Pd/TiO2 (MIm)c | 2.8 | 4.8 | 0.017 | 2.77 | 2.5 | 0.014 | 2.75 | 1 | 0.014 | 2.78 | 2.0 | 0.016 | 9 |
| 1% Au–Pd/MgO (MIm)d | 2.79 | 8.0 | 0.017 | 2.73 | 3.3 | 0.014 | 2.72 | 3.5 | 0.014 | 2.75 | 2.9 | 0.016 | Current Work |
| Ru–O | Ru–Ru(Pd) | Pd–O | Pd–Pd(Ru) | ||||||||||
| 1% Ru–Pd/TiO2 (MIm)e | 2.0 | 3.0 | 0.011 | 2.69 | 2 | 0.023 | 2.0 | 2.7 | 0.009 | 2.72 | 1.8 | 0.019 | 9 |
The closeness in the coordination numbers of both species from an analysis of both edges supports the notion that the Pd and Ru exist in a similar environment (i.e. within intimately mixed bimetallic particles). Furthermore, the low coordination numbers obtained are consistent with the STEM analysis in that the particles are on average smaller than those seen for 1% Au–Pd/TiO2 (MIm). These observations from EXAFS and electron microscopy clearly indicate that they are complimentary techniques for characterizing these supported bimetallic nano-alloys. From the above characterization data, it is evident that the bimetallic catalysts have a homogeneous random alloy structure.
Previously, we applied the most active bimetallic catalysts (1% AuPd/TiO2 (MIm), 1% RuPd/TiO2) in a different reaction, namely the selective hydrogenation of levulinic acid, and reported in detail on their characterization.9 Methods used included X-ray photoelectron spectroscopy (XPS), FF-IR of CO adsorption in addition to XAS and AC-STEM data of this material. FT-IR CO adsorption data clearly revealed that the electronic structure of both Au and Pd are modified by their close proximity to each other. This modification of the electronic structures of Au and Pd, where the Au atoms have a slight negative charge and the Pd atoms have slight positive charge, could be a contributing factor for the observed enhanced catalytic activity for the bimetallic Au–Pd catalyst. In the case of the Ru–Pd catalyst, it was very difficult to differentiate between Ru and Pd, however indirect evidence from all the characterisation techniques employed suggest that this material has a slightly smaller particle size compared to the Au–Pd catalyst. This stabilization effect, where the second metal stabilizes the smaller size of the first metal, could be the underlying reason for the enhanced catalytic activity of this latter material.
Conclusions
Supported gold–palladium and ruthenium–palladium bimetallic catalysts were prepared by a modified impregnation (MIm) method. These catalysts were found to be exceptionally active for the direct synthesis of imines and secondary amines from nitroarenes and aromatic alcohols without using any hydrogen, sacrificial hydrogen donor and/or base using a hydrogen auto transfer strategy. This hydrogen auto transfer strategy is an alternative to those using H2. Supported gold–palladium and ruthenium–palladium bimetallic catalysts were found to be several times more active (in terms of turnover number (TON)) than their monometallic counterparts. Bimetallic catalysts prepared by this MIm method are also found to be more effective than their comparable catalyst formulations prepared by the more elaborate yet time-consuming, stabilizer-ligand assisted sol-immobilization (SIm) method. TiO2 was identified to be the best support for these bimetallic catalysts after comparing the catalytic activities of these bimetallics on a variety of support materials. AC-STEM and EXAFS characterization of the most active bimetallic catalysts prepared by MIm method have revealed that these catalysts comprise of very small alloy particles with a narrow particle size distribution. They further suggest that the bimetallic particles have a homogeneous random alloy structure. In the case of Au–Pd system the charge transfer between Au and Pd, and in the case of Ru–Pd size stabilization effect have been correlated with the observed synergistic effect for the bimetallic catalysts. From a broader perspective, here we have demonstrated that the deliberate alloying of two metals in a highly controlled fashion can significantly enhance catalytic activity for a new class of hydrogen auto-transfer reactions as well as the more widely reported oxidation, reduction and hydrochlorination type reactions. This work implies that alloying using an appropriate combination of second metal choice and synthesis strategy could potentially increase the catalytic activity of many supported metal catalyst systems.Acknowledgements
MS thanks the Research Executive Agency (REA) of the European Commission for granting a Marie Curie Intra-European Fellowship for Career Development (FP7-PEOPLE-IEF-2010, Grant Agreement No. 275755). MS further thanks Cardiff University for awarding a University Research Fellowship. The authors also thank NWO and ESRF (Grenoble, France) for the beam-time at DUBBLE and BM23 (Proposal numbers: 26-01/949 BM26A & CH-3639). The authors also thank Dr. Wenhao Luo and Dr. Ines Lezcano-Gonzales, both from Utrecht University, for their help during the XAS measurements.References
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS PubMed.
- M. Sankar, N. Dimitratos, P. J. Miedziak, P. P. Wells, C. J. Kiely and G. J. Hutchings, Chem. Soc. Rev., 2012, 41, 8099–8139 RSC.
- P. Paalanen, B. M. Weckhuysen and M. Sankar, Catal. Sci. Technol., 2013, 2, 2869–2880 Search PubMed.
- A. Villa, D. Wang, D. S. Su and L. Prati, Catal. Sci. Technol., 2015, 5, 55–68 CAS.
- L. Prati and A. Villa, Acc. Chem. Res., 2014, 47, 855–863 CrossRef CAS PubMed.
- J. Pritchard, L. Kesavan, M. Piccinini, Q. He, R. Tiruvalam, N. Dimitratos, J. A. Lopez-Sanchez, A. F. Carley, J. K. Edwards, C. J. Kiely and G. J. Hutchings, Langmuir, 2010, 26, 16568–16577 CrossRef CAS PubMed.
- R. C. Tiruvalam, J. C. Pritchard, N. Dimitratos, J. A. Lopez-Sanchez, J. K. Edwards, A. F. Carley, G. J. Hutchings and C. J. Kiely, Faraday Discuss., 2011, 152, 63–86 RSC.
- M. Sankar, Q. He, M. Morad, J. Pritchard, S. J. Freakley, J. K. Edwards, S. H. Taylor, D. J. Morgan, A. F. Carley, D. W. Knight, C. J. Kiely and G. J. Hutchings, ACS Nano, 2012, 6, 6600–6613 CrossRef CAS PubMed.
- W. Luo, M. Sankar, A. M. Beale, Q. He, C. J. Kiely, P. C. A. Bruijnincx and B. M. Weckhuysen, Nat. Commun., 2015, 6, 6540 CrossRef PubMed.
- C. Gunanathan and D. Milstein, Science, 2013, 341, 249 CrossRef CAS PubMed.
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037–3058 CrossRef CAS PubMed.
- G. Guillena, D. J. Ramón and M. Yus, Chem. Rev., 2009, 110, 1611–1641 CrossRef PubMed.
- A. J. A. Watson and J. M. J. Williams, Science, 2010, 329, 635–636 CrossRef CAS PubMed.
- K.-I. Shimizu, Catal. Sci. Technol., 2015, 5, 1412–1427 CAS.
- B. Chen, L. Wang and S. Gao, ACS Catal., 2015, 5, 5851–5876 CrossRef CAS.
- A. Zanardi, J. A. Mata and E. Peris, Chem. – Eur. J., 2010, 16, 10502–10506 CrossRef CAS PubMed.
- X. Cui, Y. Zhang, F. Shi and Y. Deng, Chem. – Eur. J., 2011, 17, 2587–2591 CrossRef CAS PubMed.
- C. Feng, Y. Liu, S. Peng, Q. Shuai, G. Deng and C.-J. Li, Org. Lett., 2010, 12, 4888–4891 CrossRef CAS PubMed.
- R. Cano, D. J. Ramón and M. Yus, J. Org. Chem., 2011, 76, 5547–5557 CrossRef CAS PubMed.
- L. Tang, H. Sun, Y. Li, Z. Zha and Z. Wang, Green Chem., 2012, 14, 3423–3428 RSC.
- Q. Peng, Y. Zhang, F. Shi and Y. Deng, Chem. Commun., 2011, 47, 6476–6478 RSC.
- C.-H. Tang, L. He, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, Chem. – Eur. J., 2011, 17, 7172–7177 CrossRef CAS PubMed.
- M. Sankar, E. Nowicka, R. Tiruvalam, Q. He, S. H. Taylor, C. J. Kiely, D. Bethell, D. W. Knight and G. J. Hutchings, Chem. – Eur. J., 2011, 17, 6524–6532 CrossRef CAS PubMed.
- S. Nikitenko, A. M. Beale, A. M. J. van der Eerden, S. D. M. Jacques, O. Leynaud, M. G. O'Brien, D. Detollenaere, R. Kaptein, B. M. Weckhuysen and W. Bras, J. Synchrotron Radiat., 2008, 15, 632–640 CrossRef CAS PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- A. I. Frenkel, Chem. Soc. Rev., 2012, 41, 8163–8178 RSC.
- R. J. Davis and M. Boudart, J. Phys. Chem., 1994, 98, 5471–5477 CrossRef CAS.
- S. N. Reifsnyder and H. H. Lamb, J. Phys. Chem. B, 1999, 103, 321–329 CrossRef CAS.
- T. Balcha, J. R. Strobl, C. Fowler, P. Dash and R. W. J. Scott, ACS Catal., 2011, 1, 425–436 CrossRef CAS.
- A. M. Beale and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2010, 12, 5562–5574 RSC.
- A. G. McKale, B. W. Veal, A. P. Paulikas, S. K. Chan and G. S. Knapp, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 38, 10919–10921 CrossRef CAS.
- J. M. Cowley, Phys. Rev., 1965, 138, A1384–A1389 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cy00425c |
| This journal is © The Royal Society of Chemistry 2016 |