
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Water oxidation catalysis with ligand substituted Ru–bpp type complexes†
Stephan
Roeser
a,
Fernando
Bozoglian
a,
Craig J.
Richmond
a,
Aaron B.
League
b,
Mehmed Z.
Ertem
b,
Laia
Francàs
a,
Pere
Miró
b,
Jordi
Benet-Buchholz
a,
Christopher J.
Cramer
*b and
Antoni
Llobet
*ac
aInstitute of Chemical Research of Catalonia (ICIQ), Barcelona Institute of Science and Technology, Av. Països Catalans 16, E-43007 Tarragona, Spain. E-mail: allobet@iciq.cat
bDepartment of Chemistry, Chemical Theory Center, and Supercomputing Institute, University of Minnesota, 207 Pleasant St. SE, Minneapolis, MN 55455, USA. E-mail: cramer@umn.edu; Web: http://www.twitter.com/ChemProfCramer
cDepartament de Química, Universitat Autònoma de Barcelona (UAB), Cerdanyola del Vallès, E-08193 Barcelona, Spain
First published on 11th March 2016
Abstract
A series of symmetric and non-symmetric dinuclear Ru complexes of general formula {[Ru(R2-trpy)(H2O)][Ru(R3-trpy)(H2O)](μ-R1-bpp)}3+ where trpy is 2,2′:6′,2′′-terpyridine, bpp− is 3,5-bis(2-pyridyl)-pyrazolate and R1, R2 and R3 are electron donating (ED) and electron withdrawing (EW) groups such as Me, MeO, NH2 and NO2 have been prepared using microwave assisted techniques. These complexes have been thoroughly characterized by means of analytical (elemental analysis), spectroscopic (UV-vis, NMR) and electrochemical (CV, SQWV, CPE) techniques. The single crystal X-ray structures for one acetate- and one chloro-bridged precursor have also been solved. Kinetic analysis monitored by UV-vis spectroscopy reveals the electronic effects exerted by the ED and EW groups on the substitution kinetics and stoichiometric water oxidation reaction. The catalytic water oxidation activity is evaluated by means of chemically (CeIV), electrochemically, and photochemically induced processes. It is found that, in general, ED groups do not strongly affect the catalytic rates whereas EW groups drastically reduce catalytic rates. Finally, DFT calculations provide a general and experimentally consistent view of the different water oxidation pathways that can operate in the water oxidation reactions catalyzed by these complexes.
Introduction
Water splitting by sunlight is considered a contributing approach to solving today's increasing energy-demand challenges as it could eventually lead to an economy based on clean, sustainable energy sources such as hydrogen and/or methanol.1 Overall water splitting with light was first accomplished in an artificial manner in 1972 by Fujishima and Honda, however this required high energy UV irradiation due to the high bandgap of the TiO2 semiconductor used.2 In addition to shifting light absorbance from the UV to the visible region, catalytic water oxidation is also one of the key reactions that needs to be optimized in order to produce viable technological devices.3 Two primary factors make water oxidation a challenging task for a catalyst: i) the large endoergonicity of the reaction, 2H2O → O2 + 4H+ + 4e−E° = 1.23 V (vs. NHE at pH = 0); and ii) the molecular complexity from a mechanistic point of view, since two protons and two electrons have to be removed from each of the two water molecules and an oxygen–oxygen bond has to be formed between them. The first well-characterized molecular catalyst to achieve this complex reaction was presented in 1982 by Meyer and coworkers, involving the archetypal complex {[(bpy)2(OH2)Ru]2(μ-O)}4+, (bpy is 2,2′-bipyridine), the so called “Blue-dimer”.4 This dinuclear ruthenium based complex has been thoroughly studied mechanistically, although its activity is relatively low in terms of turnover number (TON) and turnover frequency (TOF). Substantial progress was made when in 2004 Llobet et al. presented a dinuclear ruthenium based complex, bridged by a pyrazolato based dinucleating tetraaza ligand, 3,5-bis(2-pyridyl)pyrazolato (bpp−) and containing the tridentate meridional trpy (trpy is 2,2′:6′:2′′-terpyridine) of formula {[Ru(trpy)(H2O)]2(μ-bpp)}3+ and labeled as 153+ in Scheme 1.5 This more rugged framework presented a record efficiency of up to 70% at that time under catalytic conditions and high TONs of up to 510 were achieved.6 A number of dinuclear and trinuclear Ru complexes with different levels of performance have subsequently been reported.7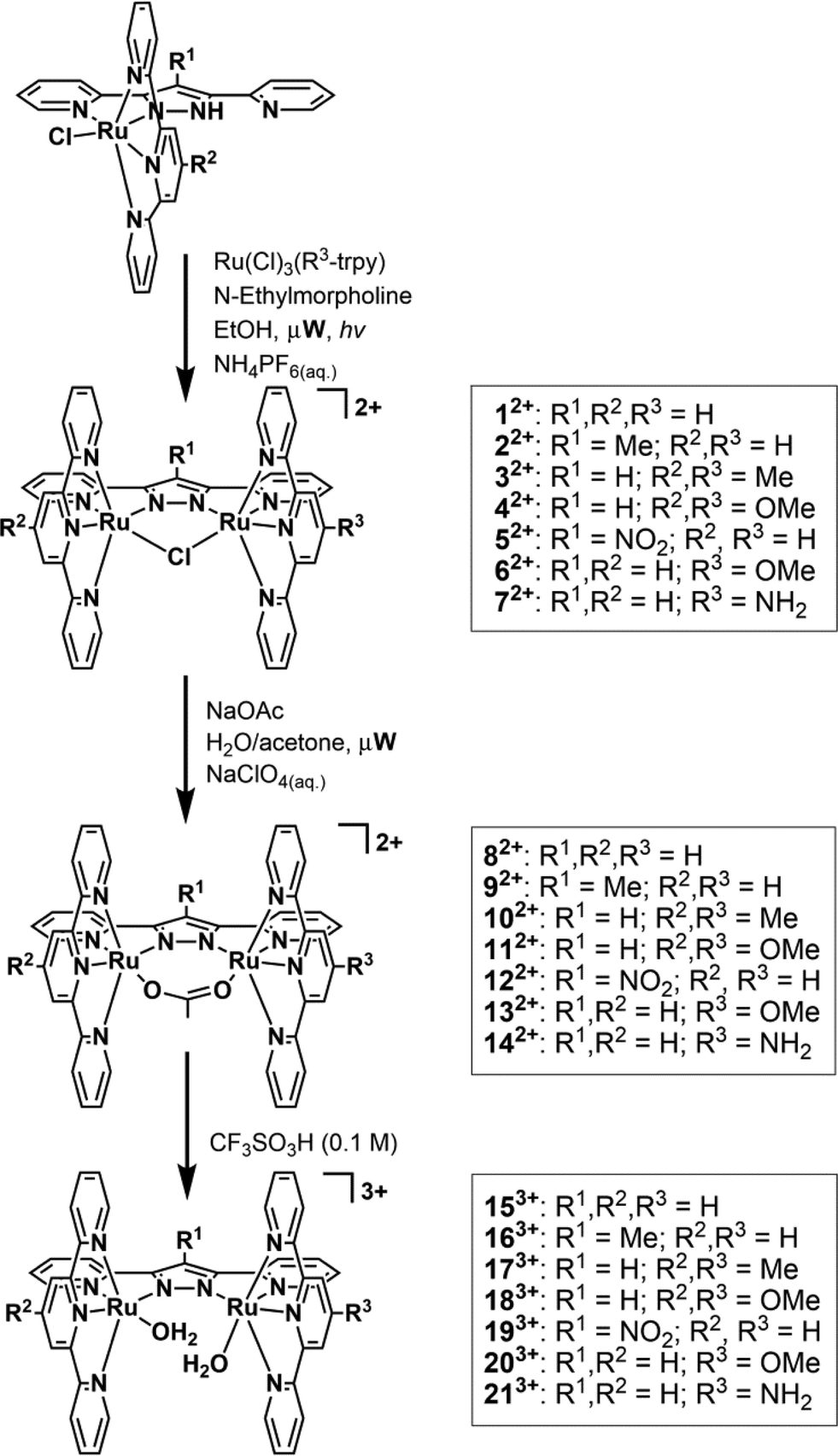 | ||
| Scheme 1 General synthetic strategy for the synthesis of complexes 12+–213+. | ||
In this paper, we explore the effect of electronic perturbations exerted by the ligand framework on the Ru–bpp type of water oxidation catalyst. For this purpose we have prepared new complexes of general formula {[Ru(R2-trpy)(H2O)][Ru(R3-trpy)(H2O)](μ-R1-bpp)}3+ (163+–213+) whose structures are presented in Scheme 1 together with a labeling scheme for these complexes and their μ-Cl and μ-OAc precursors (12+–142+). In these new complexes the bpp− and trpy ligands have been substituted with electron-donating and electron-withdrawing groups at the central pyrazolate unit and/or at the 4′ position of the trpy ligands. In this way we have prepared symmetric (153+–193+) and non-symmetric (203+–213+) Ru–aqua dinuclear complexes with different types of electronic perturbations exerted by the auxiliary ligand framework.
Results and discussion
Synthesis and structure
The synthesis of complexes 12+–52+ and 82+–132+ are described elsewhere.8 The procedure used for the synthesis of complexes 62+ and 72+ is based on the previously described5 synthetic route for complex 12+ adapted for microwave irradiation and is outlined in Scheme 1. The process consists of the reaction between [RuCl3(4′-R3-trpy)] (R3 = OMe, NH2) and complex out-[Ru(Cl)(Hbpp)(trpy)]2+ in the presence of N-ethylmorpholine (serving as both reducing agent and base) in ethanol, generating a mixture of isomeric dinuclear compounds with the general composition of {[Ru2(Cl)2(R2-trpy)(R3-trpy)](μ-R1-bpp)}2+ in addition to the chloro-bridged dinuclear complexes 62+ (R1, R2 = H, R3 = OMe) or 72+ (R1, R2 = H, R3 = NH2). The isomeric mixture of dinuclear complexes was completely converted into the desired chloro-bridged compounds through light irradiation overnight.Despite no effort having been made to optimize the duration of the light driven isomerization process, the microwave synthesis described here still presents significant advantages over the conventional synthesis, in terms of both reaction time and work-up steps. The reaction time of the original published procedure is cut in half, as the first step is completed within a few hours instead of refluxing overnight. The work-up is also much quicker, involving only filtration and washing of the residue with DCM, instead of column chromatography, to obtain the desired complexes with a high degree of purity. The bridging chloro-ligand is easily replaced with an acetato ligand by refluxing complexes 62+ or 72+ in a mixture of acetone/water (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with excess sodium acetate to give complexes 132+ and 142+ respectively. Finally, acid hydrolysis of the μ-OAc Ru dinuclear complexes leads to the in situ formation of complexes 153+–213+.
:1) with excess sodium acetate to give complexes 132+ and 142+ respectively. Finally, acid hydrolysis of the μ-OAc Ru dinuclear complexes leads to the in situ formation of complexes 153+–213+.
The bridged compounds reported here for the first time – namely 62+, 72+, 132+ and 142+ − were thoroughly characterized using a variety of spectroscopic (NMR, MS, UV-vis) and electrochemical techniques. Additionally, suitable crystals for single crystal X-ray diffraction were obtained for complexes 62+ and 142+. Fig. 1 shows the Ortep plot for the cationic moiety of these two complexes, whose metric parameters are similar to previously reported Ru(II) complexes with related ligands.5,9 It is worth mentioning that complex 62+ presents a pseudo-σ symmetry plane containing the bpp− ligand and the Ru metal centers, that bisects the perpendicular trpy ligands. The Me group of the MeO substituent is located outside of this plane, although at room temperature it rotates very fast. From an NMR perspective this fast rotation simplifies 1H- and 13C-NMR assignments. On the other hand, in complex 142+ the replacement of the Cl-bridge by an OAc-bridge produces a distortion in the planarity that eliminates the mentioned pseudo-σ symmetry plane, due to the large binding angle of the acetato bridge acting in a κ-O2 fashion. All 1D and 2D NMR spectra are reported in the ESI† with their corresponding assignments.
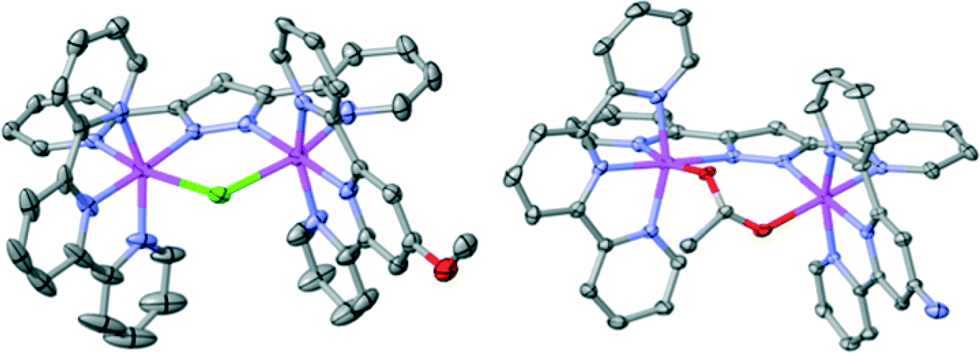 | ||
| Fig. 1 Ortep plot (ellipsoids at 30%) of complex 62+ (left) and 142+ (right). Hydrogen atoms are not shown. Color code: Ru, magenta; N, blue; O, red; Cl, green; C, grey. | ||
UV-vis spectroscopy
Optical properties of the complexes described in this work were analyzed by UV-vis spectroscopy. Table 1 summarizes the main features of the Ru–aqua complexes 153+–213+ whereas Fig. 2 shows their UV-vis spectra in a 4:1 volumetric ratio mixture of 0.1 M triflic acid and acetone (data also in Table 1). Acetone was used as a cosolvent to overcome the very low solubility of these complexes in plain aqueous solutions.
:CF3SO3H (0.1 M) (1:4)
| Complex | Assignment | λ max (nm) (ε (M−1 cm−1)) |
|---|---|---|
| 15 3+ | π → π* | 261 (55000), 304 (59300), |
| dπ → π* | 355 (18000), 460 (12500), 485 (10500) |
|
| 16 3+ | π → π* | 263 (57800), 305 (57900) |
| dπ → π* | 354 (18800), 454 (11800), 486 (9200) |
|
| 17 3+ | π → π* | 225 (52500), 263 (55600), 303 (54300) |
| dπ → π* | 353 (20600), 465 (12400), 489 (11200) |
|
| 18 3+ | π → π* | 241 (48000), 272 (55300), 308 (47100) |
| dπ → π* | 357 (20200), 478 (12500), 502 (11500) |
|
| 19 3+ | dπ → π* | 360 (13000), 452 (11900), 491 (13600), 545 (8300) |
| 20 3+ | π → π* | 232 (47000), 272 (58200), 310 (52900) |
| dπ → π* | 358 (20900), 471 (12000), 500 (10800) |
|
| 21 3+ | π → π* | 232 (51300), 273 (61100), 312 (51700) |
| dπ → π* | 366 (18200), 468 (11800) |
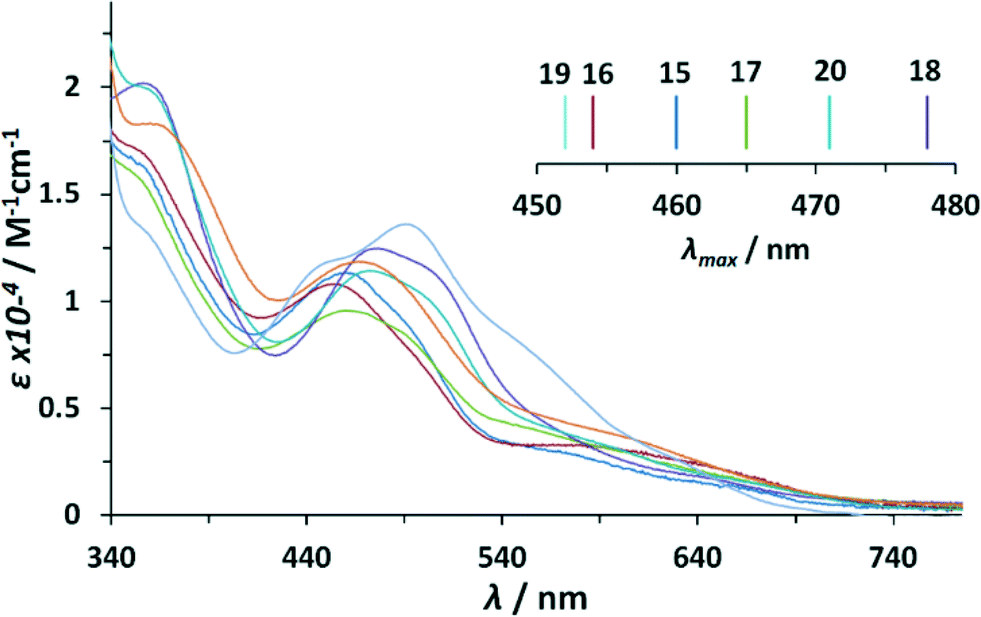 | ||
| Fig. 2 UV-vis absorption spectra recorded in acetone:CF3SO3H (0.1 M) (1:4) of complexes 153+–213+. Color code: blue, 153+; red, 163+; green, 173+; violet, 183+; light blue, 193+; turquoise 203+; orange, 213+. Inset, λmax of the first MLCT transition as a function of substituent. | ||
In the UV-vis absorption spectra of the dinuclear Ru–bpp compounds, three main regions can be distinguished: one between 200 and 350 nm, in which very intense bands are observed due to intraligand π–π* transitions; another between 350 and 550 nm, in which there are mainly broad unsymmetrical Ru(dπ)-bpp/trpy (π*) metal-to-ligand charge transfer (MLCT) bands; and finally, the region above 550 nm, in which d–d transitions occur. The transition bands in this region of the μ-Cl-, μ-OAc-complex are clearly red shifted (≈10 nm and ≈20 nm respectively) with regard to the analogous Ru–aqua complex (see ESI†). This is in accordance with the capacity of the anionic chlorido/acetato ligand to destabilize dπ-orbitals, with the effect being slightly higher for the acetato ligand. The MLCT-transitions of the μ-Cl dinuclear compounds 12+–72+ are of very high similarity to each other. For the μ-OAc dinuclear compounds 82+–142+, only minor bathochromic shifts can be observed by adding electron donating (ED) substituents to the ligand backbone. This can be explained by the relatively similar energies of the trpy and bpp− π* orbitals, that might dilute the destabilizing effect associated with substitution on only a single ligand, or by the potential hybridization (i.e. covalent mixing) of the dπ(Ru) and π(trpy and/or bpp−) orbitals.8
Noteworthy differences in the MLCT region are observed for the Ru-dinuclear aqua complexes. A general trend is identified concerning the addition of electron donating substituents onto the trpy ligand as shown in Fig. 2 and its inset. Each MeO-group added (1× complex 203+, 2× complex 183+), causes a red shift of about 10 nm with respect to unsubstituted complex 153+. The addition of two methyl groups in complex 173+ generates only a shift of about 5 nm, reflecting their less electron-donating character. In sharp contrast, the addition of a methyl group onto the 4′-position of the pyrazole ring bridging ligand in complex 163+, induces a blue shift of about 8 nm, indicating that the π*-orbital is strongly affected (destabilized) by ED-substituents on the bpp− moiety. An even larger blue shift combined with a slightly different absorption pattern can be observed for complex 193+ containing the electron withdrawing NO2 group. The blue shift can be explained by the large stabilizing effect of the electron withdrawing (EW) nitro-substituent on dπ-orbitals.8
The UV-vis spectroscopic properties of the higher oxidation states derived from the Ru–aqua-complex 203+ were investigated via spectrophotometric redox titration using CeIV as oxidant and their UV-vis spectra are presented in Fig. 3. As observed previously,5 the absorption intensity in the visible region decreases with increasing oxidation state until a broad plain band is observed in the highest accessible stable redox state III,IV. Upon further oxidation to the IV,IV redox state a series of kinetic events is triggered accompanied with the release of molecular oxygen and thus its spectrum was calculated using singular value decomposition (SVD) analysis.
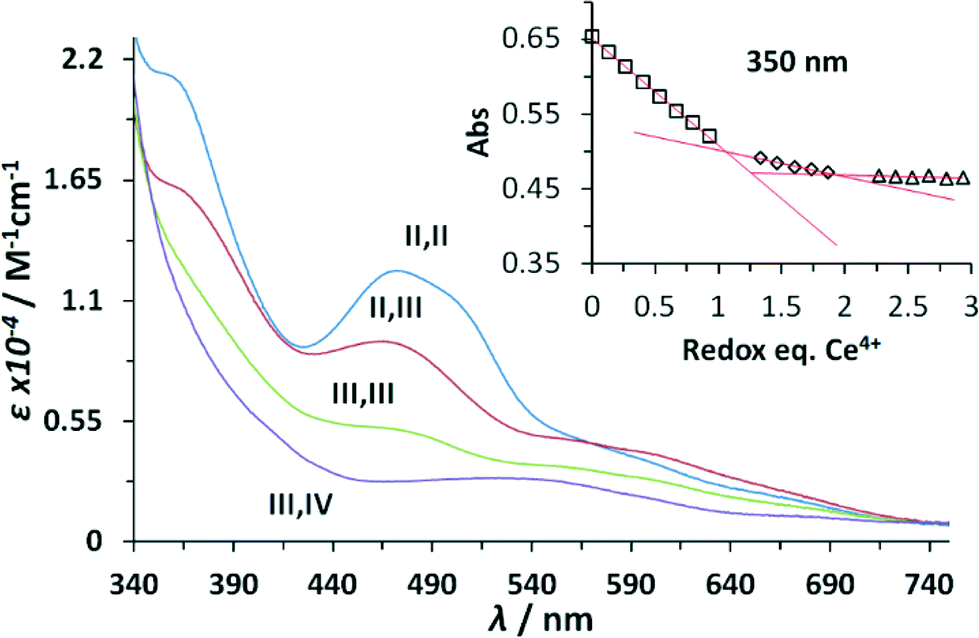 | ||
| Fig. 3 Absorption spectra (MLCT region) of complex 203+ and its higher oxidation state species in CF3SO3H (0.1 M). Oxidation was achieved by consecutive addition of Ce(IV). Inset shows absorption change at 350 nm during each oxidation step (black squares, Ru2II,II → Ru2II,III, diamonds Ru2II,III → Ru2III,III, triangles Ru2III,III → Ru2III,IV), plus the corresponding linear fitting (red lines). | ||
Redox properties
The redox properties of the complexes described in this work were investigated by means of cyclic voltammetry (CV) and square wave voltammetry (SQWV) using a sodium saturated calomel electrode (SSCE) as a reference electrode, Pt wire as an auxiliary electrode and a glassy carbon disk as a working electrode. All the potentials reported here are vs. SSCE. Table 2 displays a summary of the redox potentials obtained for the first four oxidative processes associated with the transition from Ru2II,II to Ru2IV,IV for Ru–aqua complexes (153+–213+). Their voltammograms are shown in Fig. 4 for 203+ and in the ESI† for the others. From Table 2 it can be observed that, in general, electron donating groups decrease redox potentials and electron withdrawing groups increase them as expected, although the overall influence is smaller than expected. The cyclic voltammogram shown in Fig. 4 shows a set of four waves up to 1.3 V that we associate with 4 consecutive one-electron processes. A Pourbaix diagram for 203+ was also built and is displayed in Fig. 5 within the pH range 0–2. At higher pH the low solubility of the complexes prevented further electrochemical analysis. Nevertheless it allowed the full assignment of the degree of oxidation and protonation of the species derived from 203+ from its initial II,II oxidation state. In addition, in the CV for 203+ in Fig. 4, above 1.3 V a large electrocatalytic wave appears associated with the oxidation of water to molecular dioxygen upon reaching the [O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) RuVRuIVO] oxidation state. The high reactivity of such a [RuVO] moiety has already been described for mononuclear ruthenium compounds and it has also been proposed recently for dinuclear species.7f,10 These new findings suggest that an additional redox process should be included in the mechanism previously proposed,6 which is discussed in the following section. Bulk electrolysis experiments were carried out by dissolving complex 203+ (5.4 × 10−5 M) in CF3SO3H (0.1 M). The catalyst produced a total of 23 μmol of O2 at Eapp = 1.45 V and 13 μmol at Eapp = 1.1 V after 10 hours, corresponding to a final TON of 47 and 27 respectively (see ESI†). The results obtained here clearly show that complex 203+ can act as a catalyst both at oxidation state IV,IV and IV,V with the latter being more active.
:CF3SO3H (0.1 M) (1:4) vs. SSCE
RuVRuIVO] oxidation state. The high reactivity of such a [RuVO] moiety has already been described for mononuclear ruthenium compounds and it has also been proposed recently for dinuclear species.7f,10 These new findings suggest that an additional redox process should be included in the mechanism previously proposed,6 which is discussed in the following section. Bulk electrolysis experiments were carried out by dissolving complex 203+ (5.4 × 10−5 M) in CF3SO3H (0.1 M). The catalyst produced a total of 23 μmol of O2 at Eapp = 1.45 V and 13 μmol at Eapp = 1.1 V after 10 hours, corresponding to a final TON of 47 and 27 respectively (see ESI†). The results obtained here clearly show that complex 203+ can act as a catalyst both at oxidation state IV,IV and IV,V with the latter being more active.
:CF3SO3H (0.1 M) (1:4) vs. SSCE
| Complex | E 1/2(1) ΔE | E 1/2(2) ΔE | E 1/2(3) ΔE | E 1/2(4) ΔE |
|---|---|---|---|---|
| 15 3+ | 0.56 (80) | 0.625 (70) | 0.855 (70) | 1.07 (60) |
| 16 3+ | 0.50 (60) | 0.635 (60) | 0.85 (60) | 0.96 (60) |
| 17 3+ | 0.51 (60) | 0.61 (60) | 0.83 (60) | 1.06 (60) |
| 18 3+ | 0.50 (60) | 0.59 (60) | 0.84 (60) | 1.02 (60) |
| 19 3+ | 0.74 (60) | 0.74 (60) | 1.00 (60) | 1.10 (60) |
| 20 3+ | 0.50 (60) | 0.59 (80) | 0.88 (100) | 0.98 (80) |
| 21 3+ | 0.48 (60) | 0.64 (60) | 0.80 (60) | 0.97 (80) |
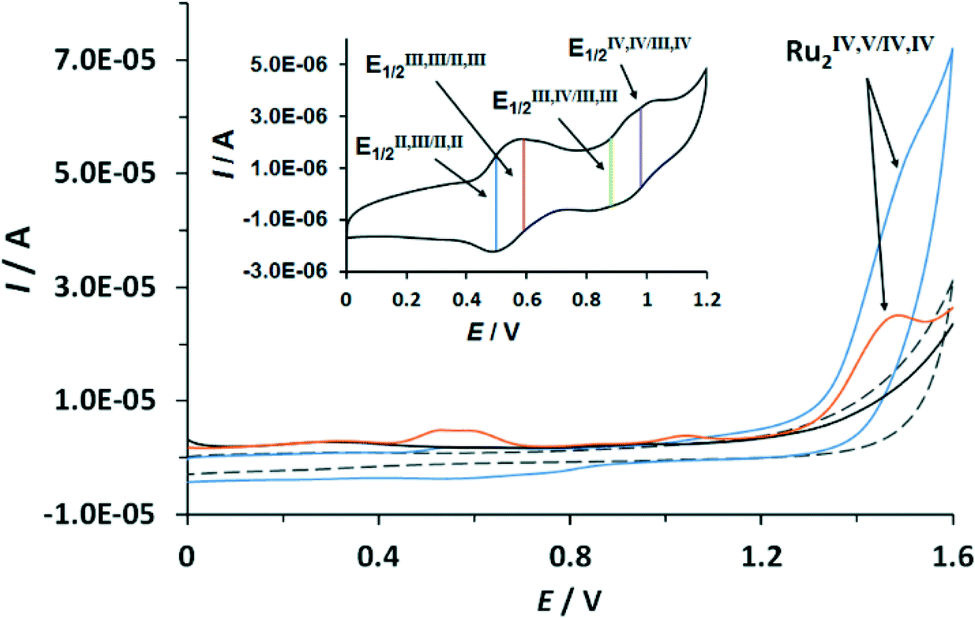 | ||
| Fig. 4 CV (blue line) and SWV (orange line) of complex 203+vs. SSCE in CF3SO3H (0.1 M). Blank CV (dashed grey line) and blank SWV (black line) of pure CF3SO3H (0.1 M) is additionally presented. Inset, CV of complex 203+ acetone:CF3SO3H (0.1 M) (1:4) at a scan rate of 50 mV s−1 with the reverse potential at 1.2 V. | ||
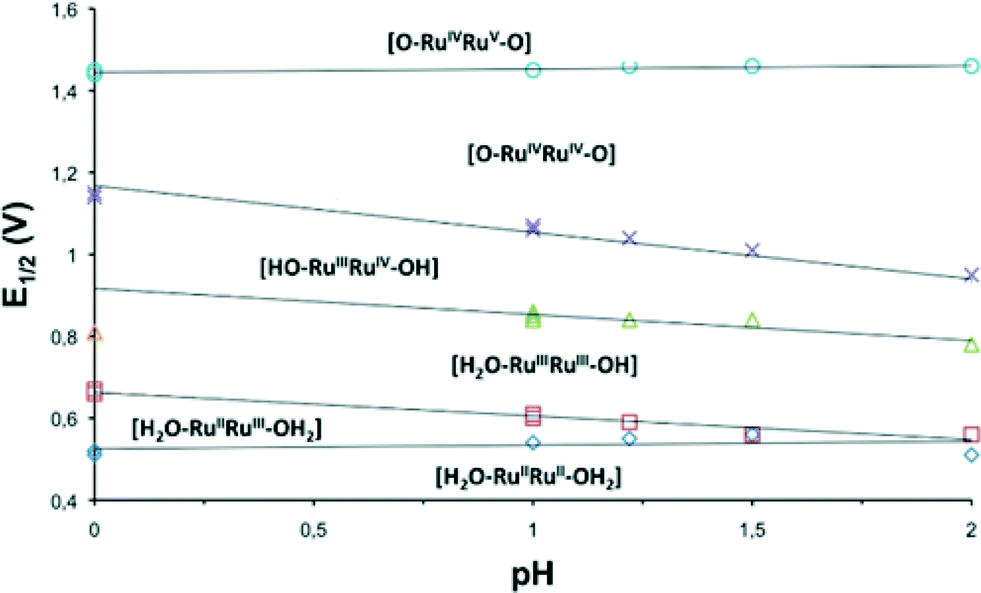 | ||
| Fig. 5 Pourbaix diagram in the pH range 0–2 for 203+ obtained from square wave voltammetry experiments. The dominant species at the different pH-E zones are indicated. | ||
Substitution kinetics
Whereas it is known that in high oxidation states the kinetics of substitution reactions slow down,6 there is no information on how different substituents on the ligand backbone influence the reaction rates (ks) in dinuclear systems such as the ones in the present work. Therefore, we measured the substitution kinetics of the aqua ligands by MeCN in CF3SO3H (0.1 M) solution, according to eqn (1) and (2), for complexes 193+ and 203+ and compared the rate constants with those of the parental complex 153+.| H2O−RuIIRuII−H2O + MeCN → H2O−RuIIRuII−NCMe + H2O | (1) |
| H2O−RuIIRuII−NCMe + MeCN → MeCN−RuIIRuII−NCMe + H2O | (2) |
Substitution reactions described in eqn (1) and (2) generate complexes {[Ru(trpy)(H2O)](μ-bpp)[Ru(trpy)(MeCN)]}3+ and {[Ru(trpy)(MeCN)]2(μ-bpp)}3+ as final products, respectively. Substitution reactions were carried out under pseudo-first-order reaction conditions ([MeCN]/[dinuc] ≥ 10) and were monitored by UV-vis spectroscopy. The change in absorption over time for complex 203+ is shown in the ESI.† The resulting rate constants for the first and second substitution reactions are given in Table 3. As can be observed from the data in Table 3, the first substitution takes place relatively quickly for oxidation state RuII–RuII, with a corresponding half-life of 440 s for complex 193+ and 21 s for complex 203+. While the rate of substitution for complex 203+ is almost identical to the one obtained for complex 153+, the one obtained for complex 193+ is more than one order of magnitude slower. Thus suggesting that electron donating substituents practically do not affect the first substitution process at oxidation state II,II whereas the electron withdrawing group sharply slows it down. Concerning the second substitution process, the rate constant for the non-symmetric complex 203+ increases by 24 times with regard to the symmetric 153+.
| Complex | First MeCN substitution | Second MeCN substitution | ||
|---|---|---|---|---|
| k S1 (s−1) | t 1/2 (s) | k S2 (s−1) | t 1/2 (s) | |
| 15 3+ | 4.0 × 10−2 | 1.7 × 101 | 5.8 × 10−5 | 1.2 × 104 |
| 19 3+ | 1.5 × 10−3 | 4.4 × 102 | — | — |
| 20 3+ | 3.3 × 10−2 | 2.1 × 101 | 1.4 × 10−3 | 4.8 × 103 |
Water oxidation under stoichiometric conditions
Kinetic studies on the stepwise oxidation of [H2O–RuIIRuII–OH2] to [ORuIVRuIVO] (see eqn (3)) for complex 203+ were performed in order to determine the influence of the ED groups over the rate constants and were monitored using UV-vis spectroscopy. The same methodology was applied as to the one reported previously for complex 153+.6 After addition of four equivalents of CeIV to the catalyst, a series of kinetic events leads to the formation of molecular oxygen as shown in the upper part of Scheme 2. Since the system is catalytic, addition of more than four equivalents of CeIV repeats the catalytic cycle. In order to isolate these kinetic events, the experiments at this point were performed up to a 1:4 ratio of complex 203+:CeIV. Fig. 6 shows the change in absorption spectra together with the assigned species distribution and calculated absorption spectra (see also ESI†). A summary of all the obtained ET- and reaction rate constants, together with the ones for complex 153+ are presented in Table 4.
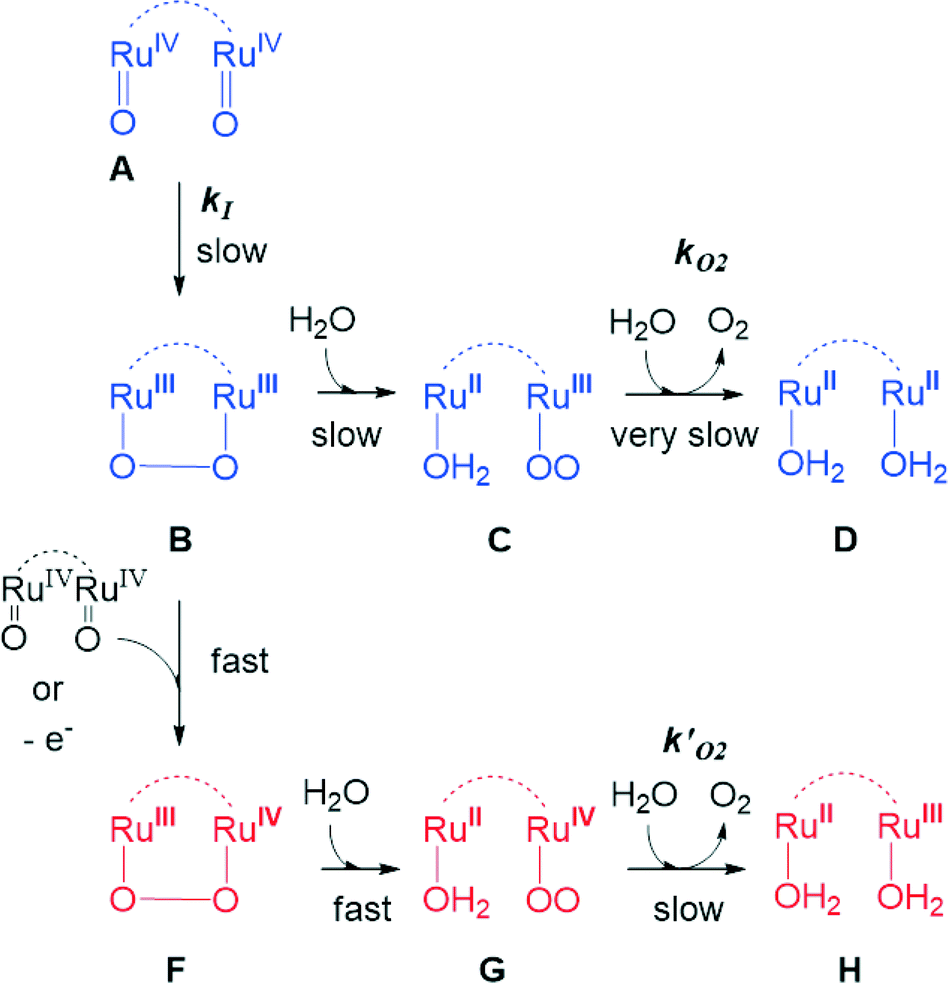 | ||
| Scheme 2 Upper part (blue), simplified reaction scheme for oxygen evolution under stoichiometric conditions starting from species [ORuIVRuIVO], A, through B, C and D. Lower part (red), additional pathways operating under excess CeIV starting from B and involving species F, G and H. | ||
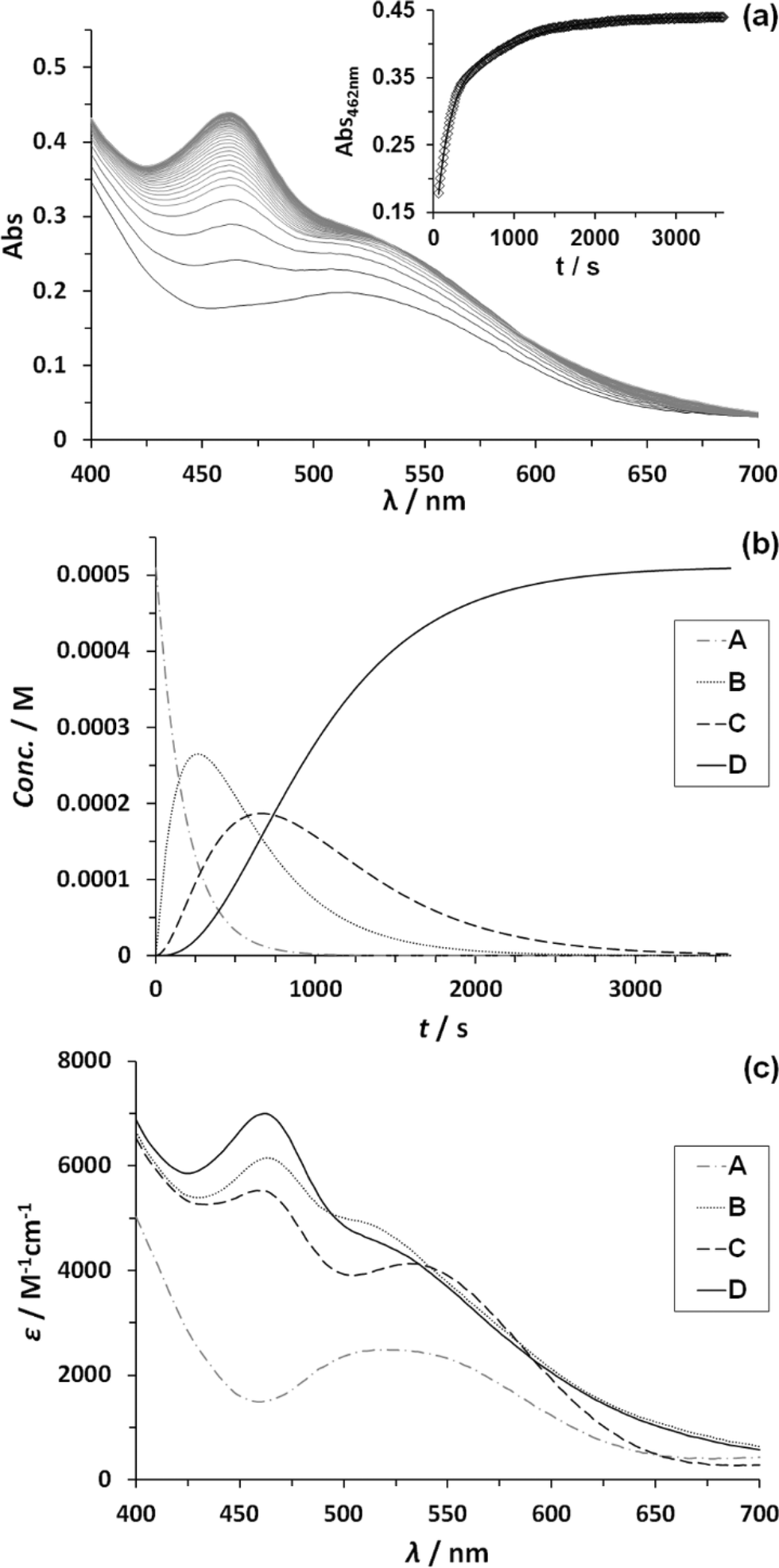 | ||
| Fig. 6 (a) Change of absorption spectra with time after addition of 4 eq. CeIV to a 5.12 × 10−5 M solution of complex 203+ in CF3SO3H (0.1 M) at 25 ± 0.1 °C. Inset shows absorbance change over time at 462 nm (black diamonds) plus its mathematical fit for a reaction model of three consecutive first order reactions (black line). (b) Corresponding species distribution diagram as denoted in Scheme 2.6 (c) Calculated spectra of participating species. Line code: dashed grey, species A; solid grey, species B; dashed black, species C; solid black, species D, as denoted in Scheme 2.6 | ||
| k | k 20 3+ | ΔH‡ | ΔS‡ | k 15 3+, | ΔH‡ | ΔS‡ | ||
|---|---|---|---|---|---|---|---|---|
| a Values obtained from ref. 6a. | ||||||||
| k ETn | k ET1 | Eqn (3a) | (9.5 ± 0.2) × 104 | 36 ± 4 | −30 ± 8 | 8.6 × 104 | 22 ± 1 | −84 ± 3 |
| (M−1 s−1) | k ET2 | Eqn (3b) | (6.5 ± 0.3) × 104 | 25 ± 3 | −70 ± 8 | 6.8 × 104 | 34 ± 2 | −45 ± 7 |
| k ET3 | Eqn (3c) | (3.9 ± 0.4) × 104 | 20 ± 1 | −90 ± 4 | 5.8 × 104 | 29 ± 2 | −65 ± 5 | |
| k ET4 | Eqn (3d) | (1.5 ± 0.1) × 103 | 21 ± 2 | −150 ± 8 | 9.8 × 102 | 23 ± 3 | −122 ± 10 | |
| k (s−1) | k I | Scheme 2 | (6.7 ± 0.1) × 10−3 | 38 ± 5 | −157 ± 16 | 1.7 × 10−3 | 53 ± 3 | −137 ± 9 |
| k O2 | Scheme 2 | (1.5 ± 0.1) × 10−3 | 78 ± 6 | −37 ± 20 | 5.5 × 10−4 | 85 ± 8 | −39 ± 27 | |
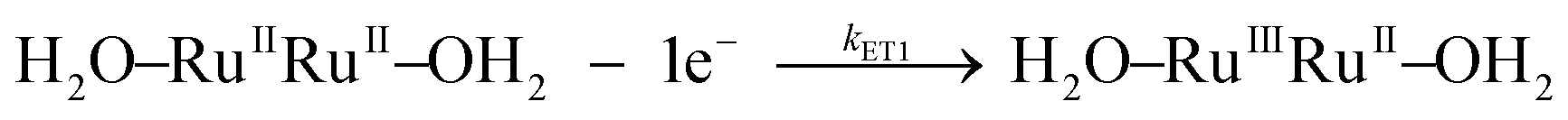
| (3a) |
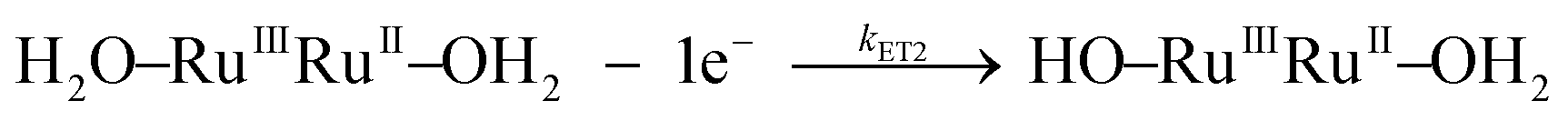
| (3b) |
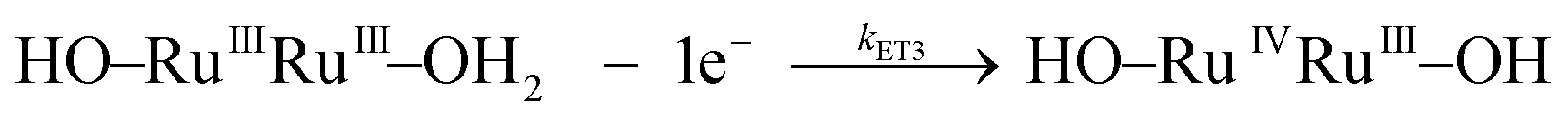
| (3c) |
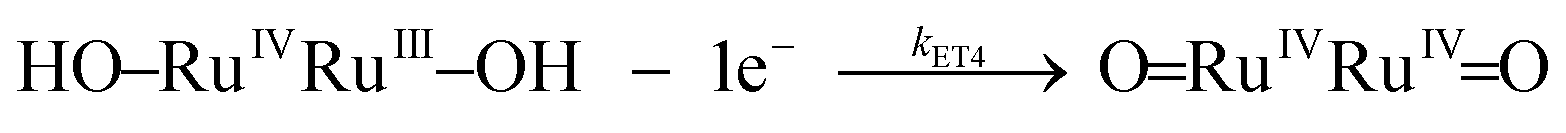
| (3d) |
From Table 4, it can be observed that all ET and reaction rates are larger for complex 203+ compared to 153+ which is attributed to the increase in thermodynamic driving force, since 203+ has lower redox potentials than 153+, and the same oxidant (CeIV) is used. The following steps involve the O–O bond formation (A → B, in Scheme 2), the oxidation of the metal peroxide to superoxide (B → C) and finally the dioxygen ejection (C → D). For complex 203+ the rate determining step (kO2) is more than one order of magnitude faster than for complex 153+. This step involves the formal reduction of the complex from III,II to II,II and the substitution of the oxygen species by water. The fact complex 203+ with lower redox potentials has higher kO2 values, points out that the main factor influencing this constant is the substitution process. This is in perfect agreement with the substitution trends described in the previous section, which highlights the influence of both electronic and symmetry effects (see also theory section later).
Interpretation of activation parameters is a complex issue when highly polar solvents and multiple oxidation states and electron transfer processes are involved in the TS or reaction intermediates. However some conclusions can be drawn from the data obtained for complexes 203+ and 153+. The high negative value for the activation entropy calculated for kI is consistent with electrostriction dominating this step and agrees with the crucial role of water and hydrogen bonding in stabilizing intermediates or transition-state structures. Activation enthalpies for the kO2 step are significantly larger compared to those of kI. This can be seen as a consequence of breaking/forming more and/or stronger bonds to reach the final release of O2 than to reach the intermediate step. It is also in agreement with the fact that the inductive effect exerted by the methoxy group in 203+ is responsible for the slightly lower activation enthalpy compared to catalyst 153+.
Chemically and photochemically induced catalytic water oxidation
In order to determine the influence of ED- or EW-substituents on the reactivity towards water oxidation, complexes 153+–213+ were reacted with an excess of CeIV in CF3SO3H (0.1 M) in a ratio 1:100 (1 mM Cat:100 mM CeIV) (see Fig. 7 and the ESI†). Oxygen evolution was observed and quantified by manometric measurements and on-line mass-spectrometry. Molecular oxygen was identified as the only product for all dinuclear systems. Modest enhancement of oxygen evolution was observed for complex 203+ compared to complex 153+, but a much slower rate was observed for complex 193+ bearing the EW nitro-substituent on the bpp− ligand. The results obtained for 153+, 193+ and 203+ are in agreement with the results found in the stoichiometric conditions. In addition, under excess of CeIV additional pathways can exist involving the oxidation of the peroxo intermediate B to F, as shown in the lower part of Scheme 2. This will generate the III,II species (H) upon oxygen evolution, which would end up being the initial reactant species of the next catalytic cycle. As in the stoichiometric case the substitution factor at the rds seems to dominate the kinetic barriers, as will be discussed later on in the theory section.
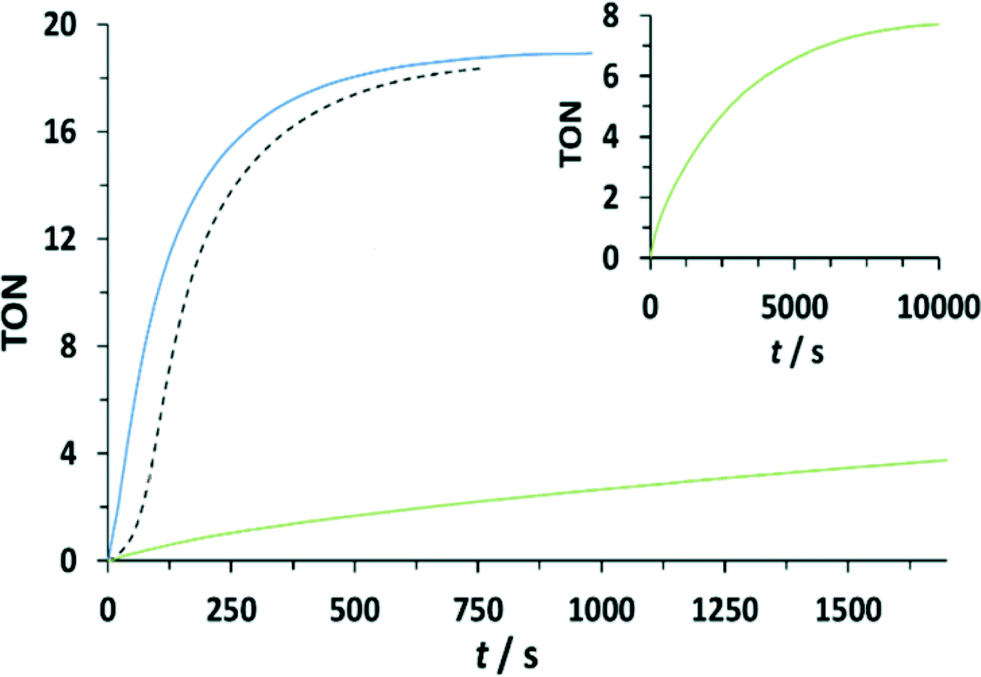 | ||
| Fig. 7 Catalytic activity of complexes 153+, 193+ and 203+, followed by manometry. Reaction conditions: dinuc:CeIV (1 mM:100 mM) in CF3SO3H (0.1 M). Line code: dashed black, 153+; green, 193+; blue 203+. Inset shows full time span for complex 193+. | ||
An additional pathway for the water oxidation reaction involves the formation of the [ORuVRuIVO] species, as has been shown by CV for complex 203+ which occurs at 1.40–1.45 V (Fig. 4) with the onset of the wave at 1.30 V. In the case of CeIV the redox potentials are not sufficiently high to enable this pathway (E1/2 = 1.20 V at pH = 1; vs. SSCE)11 but oxidants with higher redox potentials such as [Ru(debpy)2(bpy)]2+ (debpy is diethyl-[2,2′-bipyridine]-4,4′-dicarboxylate) (E1/2 = 1.38 V vs. SSCE))12 could achieve it. For this reason light induced water oxidation was carried out using [Ru(bpy)3]2+ (E1/2 = 1.03) and [Ru(debpy)2(bpy)]2+ (E1/2 = 1.38 V) complexes as photosensitizers (PS) in combination with Co(III) as sacrificial electron acceptor (SEA) at pH = 7.0 using a phosphate buffer.7a,13 Water oxidation catalysts (WOCs) 203+ and 153+ were tested under these conditions using a Clark electrode to monitor the generation of oxygen over time and the results obtained are depicted in Fig. 8.
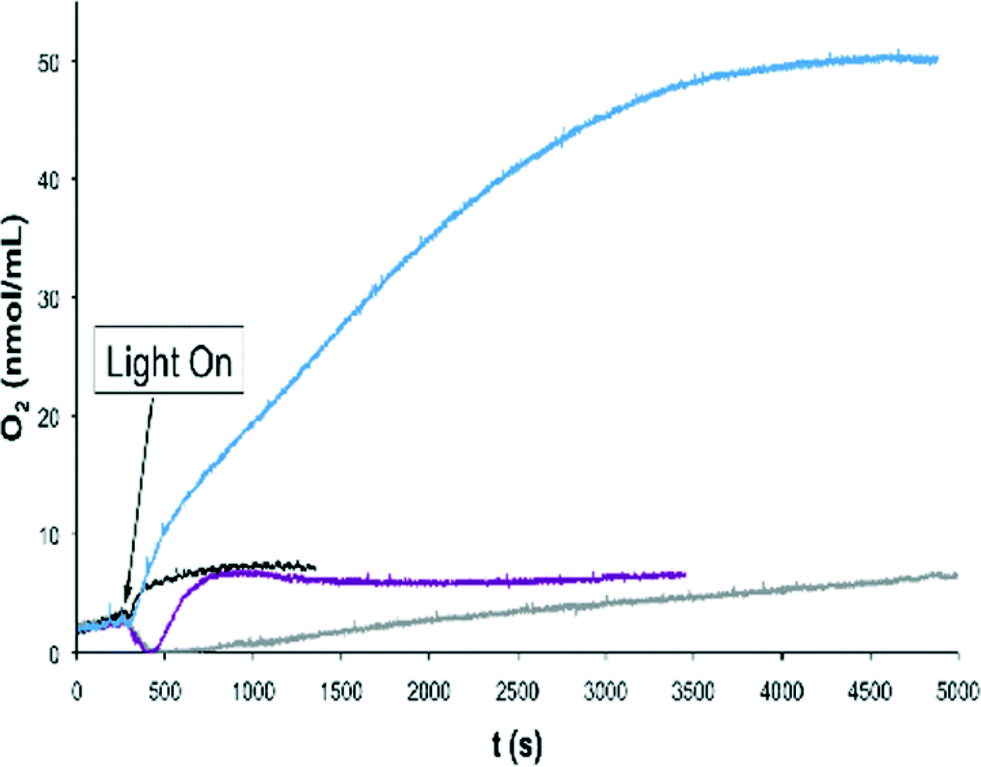 | ||
| Fig. 8 Oxygen evolution profiles of different catalysts at pH 7.0 in a phosphate buffer solution at 25.0 °C, upon irradiation with a 150 W xenon lamp with a 400 nm cut-off filter. Color code: 203+ and [Ru(debpy)2(bpy)]2+, blue; 203+ and [Ru(bpy)3]2+, violet; 153+ and [Ru(debpy)2(bpy)]2+; black; [Ru(debpy)2(bpy)]2+ alone gray. [Co(OTf)(NH3)5](OTf)2 was used as a sacrificial electron acceptor in all cases. | ||
Several points can be deduced from the plot: a) the combination of [Ru(bpy)3]2+ and 203+ generates a very small amount of oxygen, close to the amount generated by a blank experiment with no catalyst; b) the combination of 203+ and the [Ru(debpy)2(bpy)]2+ generates a much larger amount of oxygen. This indicates that [Ru(debpy)2(bpy)]2+ is a sufficiently powerful oxidant to drive the oxygen evolution reaction while regular [Ru(bpy)3]2+ is not. However, the amount of O2 generated is relatively low, possibly due to the need for using an even stronger oxidant to perform this job and c) the 153+ catalyst that has slightly higher redox potentials than 203+ does not generate any oxygen at all with [Ru(bpy)3]2+ but a small amount (again close to base line) was produced with the [Ru(debpy)2(bpy)]2+. This highlights again the importance of properly matching the redox potentials of the PS and the catalyst in order to build successful systems for light induced water oxidation catalysis. Also note that the observed oxygen consumption at the initial stages of the reaction using [Ru(bpy)3]2+ as PS and in the blank with [Ru(debpy)2(bpy)]2+ is likely due to residual oxygen quenching by the [Ru(bpy)3]2* excited state as has been reported earlier.14 Faster oxygen evolution processes such as the ones using [Ru(debpy)2(bpy)]2+ and 153+ or 203+ preclude the observation of this phenomenon.
Water oxidation mechanisms
Density functional theory (DFT) calculations at the M06-L level of theory (see computational methods for details) were carried out in order to examine the water oxidation mechanisms and compare the energetics of the possible catalytic pathways of the parent and substituted Ru–Hbpp complexes. For a substituted complex containing an electron donating (ED) group, 203+ with a methoxy group at the central position of the trpy ligands was chosen while for the case of a complex with an electron withdrawing (EW) group, the analogue of 203+ containing a nitro group, {[Ru(NO2-trpy)(H2O)][Ru(trpy)(H2O)](μ-bpp)}3+, 223+ was used. 223+ has not been prepared in this work but it is useful for direct comparative purposes to assess the effects of varying substitution at a specific position.As a first step, we explored both the water nucleophilic attack mechanism and intramolecular O–O bond formation pathways for 153+ starting from the common reactant [ORuIVRuIVO] in Fig. 9. The free energy of activation (ΔG‡) associated with the WNA is computed to be 39.0 kcal mol−1 in aqueous solution, which is much too high to be consistent with experimental kinetics. The WNA pathway is also not consistent with the available 18O isotope labelling experiments, which demonstrate that the evolved O2 originates from oxygen atoms of Ru–O units. The intramolecular direct coupling O–O bond formation between the two terminal oxo moieties features a ΔG‡ of just 15.5 kcal mol−1 in aqueous solution (23.5 kcal mol−1 lower than in the WNA pathway). The optimized transition-state (TS) structure has an O–O distance of 1.71 Å and results in the formation of a μ-peroxo intermediate with O–O distance of 1.39 Å. Next, we considered the steps leading to O2 evolution from the η2-peroxo intermediate. The displacement of peroxo by water molecules to generate a η1-superoxo intermediate and the consequent release of dioxygen are found to require high free energies of activation on the (IV,IV) potential energy surface (ΔG‡ = 28.6 and 40.6 kcal mol−1 respectively) (Fig. 9). In earlier studies, the role of explicit water molecules in the first solvation shell in stabilizing intermediates or TS structures was investigated. For instance, introduction of an additional water molecule in the transition state structure for generation of the η1-superoxo intermediate (TS2) enables optimization of a hydrogen-bonding pattern compatible with the proton transfer to the departing peroxide oxygen. Inclusion of explicit water molecules in the models permits especially favorable microsolvation that is not accounted for in the continuum solvation model (at the cost of additional computational overhead associated with searching for, and averaging over, all possibly relevant microsolvated structures).
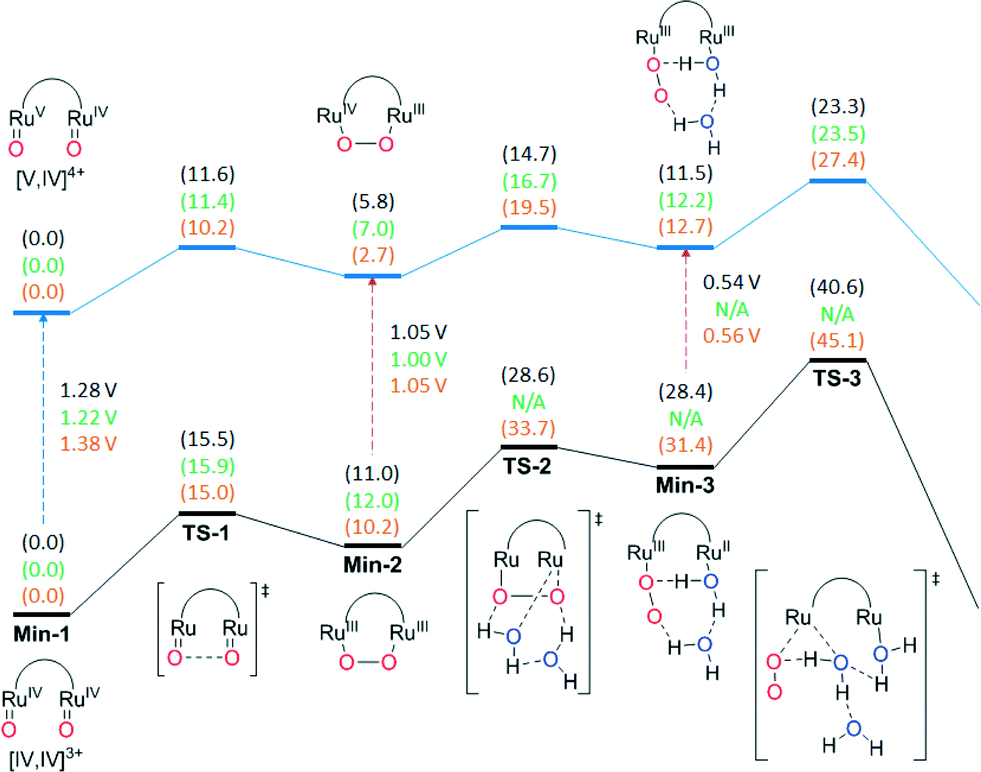 | ||
| Fig. 9 Previously reported free energies for stationary points relevant to water oxidation in complex 153+ (black text, above) both in the [IV,IV]+3 and the [V,IV]+4 oxidation state, compared against those of complex 203+ (green text, middle) and complex 223+ (orange text, below). Redox potentials are reported vs. NHE. | ||
Next, we investigated the remaining steps in water oxidation pathways for all complexes starting from the [ORuIVRuVO] intermediate obtained by one electron oxidization of [ORuIVRuIVO], for which the computed redox potential is 1.28 V vs. NHE (Fig. 9). Similar to the [ORuIVRuIVO] case, intramolecular O–O bond formation is favored significantly over a WNA pathway for 153+ (ΔG‡ = 11.6 and 27.4 kcal mol−1 respectively). Continuing from the mixed valence η2-peroxo intermediate, the displacement of peroxo by a water molecule to yield the superoxo intermediate and subsequent O2 evolution step was found to proceed with ΔG‡ values of 14.7 and 23.3 kcal mol−1 respectively (Fig. 9). It should be noted that in addition to one-electron oxidation of [ORuIVRuIVO], the one electron oxidized pathway can also be accessed by oxidation of the peroxide intermediate [RuIII(O–O)RuIII] to generate [RuIV(O–O)RuIII], or oxidation of the superoxide intermediate [(OO–RuIII–RuII(H2O)] to generate [(OO–RuIII–RuIII(H2O)] (Fig. 9). In general for the complexes reported here, the potential at the foot of the catalytic wave for the V,IV/IV,IV redox couple is just at the edge of what may be reached by CeIV, however once the [RuIII(O–O)RuIII] intermediate is generated then it can more readily access the one-electron-further oxidized pathway, and indeed even under conditions of limited stoichiometry of oxidant, disproportionation reactions may permit access to the most highly oxidized potential energy surface with its lower rate-determining free energy of activation. Direct oxidation of the [ORuIVRuIVO] intermediate can, of course, be accomplished either electrochemically or by using more powerful chemical oxidants. Thus, for the parent system 153+, intramolecular O–O bond formation is favored over a WNA mechanism and the rate-determining step is predicted to be the release of oxygen from the mixed valent peroxo intermediate with a free energy of activation of 23.3 kcal mol−1 on the potential energy surface for the most highly oxidized species.
Fig. 9 also presents the energetics for the corresponding catalytic pathways involving the trpy-substituted Ru complexes 203+ (methoxy) and 223+ (nitro). For 203+ the free energy of activation of O2 release is predicted to be very similar (within 0.2 kcal mol−1) to that of unsubstituted 153+, in agreement with the very similar experimental rates obtained under catalytic conditions using CeIV as oxidant. On the other hand, the use of electron withdrawing substituents as in the case of 223+ increases ΔG‡ for this step by 4.1 kcal mol−1, which is in agreement with the experimental observation that nitro-substituted 193+ has a much lower rate of reaction than 153+ or 203+ (see Fig. 7), assuming inductive effects through the bpp ligand to be qualitatively similar to those through the trpy ligand. The fact that for 203+ the rds involves both substitution and reduction clearly indicates that in this case the substitution effect is the dominant factor.
Experimental
Instrumentation and measurements
UV-vis spectroscopy was performed on a Cary 50-Bio (Varian) spectrophotometer in 1 cm quartz cuvettes in either CF3SO3H (0.1 M), CH2Cl2 (DCM) or CH3CN (MeCN). For acid–base spectrophotometric titration, a solution containing 203+ in CF3SO3H (0.1 M) was added to phosphate buffer (pH = 6.1) to yield a 5 × 10−5 M solution. The pH was adjusted by adding small volumes of 4 M NaOH in order to produce negligible overall volume change. pH measurements were taken with a Micro-pH-2000 from Crison. Redox spectrophotometric titrations were performed by sequential addition of small volumes (2 μL) of 3.68 mM aqueous solution of (NH4)2Ce(NO3)6 in CF3SO3H (0.1 M) to a 5.78 × 10−5 M solution of the complex in the same solvent. Kinetic studies on the stepwise oxidation of RuIIRuII to RuIIIRuIV were performed at various concentrations of dinuc and CeIV, but always with [CeIV] lower or equal to [dinuc] (second order conditions) using a fast mixing stopped flow module Bio-Logic SFM300 and a J&M TIDAS UV-vis spectrophotometer with diode array detector. Temperature was adjusted by a Huber CCE-905 VPCw cryostat. In a typical experiment, a solution of Ru complex (1 × 10−4–5 × 10−5 M) in 0.1 M CF3SO3H was mixed with stoichiometric or less than stoichiometric amounts of a solution of CeIV in the same media and the changes in absorbance vs. time were monitored. The same experiment was performed starting from Ru oxidation states II,III and III,III. Oxidation of RuIIIRuIV to RuIVRuIV were measured under pseudo-first order conditions with concentration of CeIV at least 10 times that of dinuc. For slow reactions a Cary 50-Bio (Varian) was used. In all cases, the temperature was maintained at ±0.1 °C. In a typical experiment, to a 2.5 mL solution of complex 20 (2 × 10−4–5 × 10−5 M) 3.9 eq. of CeIV were added. First and second order rate constants were calculated by global fitting or single wavelength fitting method using Specfit.12 SVD analysis of the spectral data was used to obtain the significant components. A global fit to the obtained model was used to extract the spectrum of the intermediate species. In all cases, rate constants were measured between 10.0 °C and 40.0 °C. Errors shown correspond to one-sigma of the models respect to experimental points.Online manometric measurements were carried out on a Testo 521 differential pressure manometer with an operating range of 1–100 mbar and accuracy within 0.5% of the measurement. The manometer was coupled to thermostated reaction vessels for dynamic monitoring of the headspace pressure. The manometer's secondary ports were connected to thermostated reaction vessel containing the same solvent and headspace volumes as the sample vials. The composition of the gas phase was monitored on-line by an OmnistarTM GSD 301 C (Pfeiffer) quadrupol mass spectrometer. In this case, all solutions were degassed with argon (99.999%) prior to use. For photochemical oxygen evolution, irradiation was carried out with a 150 W xenon arc lamp equipped with a 400 nm cutoff filter to remove UV and IR radiation. The intensity of the radiation was approximately 0.3 W cm−2. The temperature of the cell was maintained constant at 20 °C thanks to water circulation through a jacketed compartment attached to the main cell.
Oxygen evolution was analyzed with a liquid-phase Clark-type oxygen electrode (Hansatech DW2/2 electrode system). The photochemical experiments involved a three-component system; catalyst, sensitizer and sacrificial electron acceptor dissolved in 2 mL of a 50 mM phosphate buffer solution (pH 7.04). A typical experiment involved degassing, the above mentioned solution and dissolving the catalysts (complex 153+, or 203+), the PS ([Ru(bpy)3]2+ or [Ru(debpy)2(bpy)]2+; bpy = 2,2′-bipyridine, debpy = diethyl [2,2′-bipyridine]-4,4′-dicarboxylate and the SEA ([Co(OTf)(NH3)5](OTf)2) with concentration of 1.0 × 10−5 M, 1.0 × 10−4 M, and 2.9 × 10−2 M, respectively.
Electrochemical experiments were performed on an IJ-Cambria HI-660 potentiostat using a three-electrode cell. A glassy carbon (2 mm diameter) or platinum disk (1 mm) electrode were used as working electrode, platinum disk (1 mm diameter) as auxiliary electrode and SSCE as reference electrode. Glassy carbon working electrodes were thoroughly polished with 0.05 micron alumina paste and washed consecutively with distilled water and acetone followed by blow-drying, before each measurement. The complexes were either dissolved in acetone-CF3SO3H (0.1 M) (1:4) or CH2Cl2, CH3CN, containing the necessary amount of (n-Bu4N)(PF6) (TBAH) as supporting electrolyte to yield a 0.1 M ionic strength solution. All E1/2 values reported in this work were estimated from cyclic voltammetry (CV) experiments (scan rate = 50 mV s−1), as the average of the oxidative and reductive peak potentials (Ep,a + Ep,c)/2 and verified by square wave voltammetry (SWV) experiments (incr E(V) = 0.002, amplitude (V) = 0.025, frequency (Hz) = 15). Bulk electrolysis experiments were performed on a Biologic SP-150 potentiostat connected to an external high current booster (5 A) using Pt-meshes as working and counter electrode and Ag/AgCl (3 M KCl) as reference electrode. A homemade two chamber electrochemical reactor, separated by Vycor frit. Additional inlets were closed by Suba-seal® septa and further tagged with Blue-Tack® to ensure hermitic sealing. Oxygen evolution was analyzed with a gas-phase Clark-type oxygen electrode (Unisense Ox-N needle microsensor). The electrode was calibrated using nitrogen saturated and air saturated water.
NMR spectroscopy experiments were performed on a Bruker Avance 400 spectrometer. Samples were run in [d6]-acetone with internal references (residual protons). Elemental analyses were performed using a Fisons CHNS-O EA-1108 elemental analyzer. ESI-MS experiments were recorded on a Water LCT Premier spectrometer; MALDI-TOF experiments were performed on a Bruker Autoflex spectrometer.
Materials
Trifluoromethanesulfonic acid (99%) was purchased from STREM/CYMIT. 4′-Amino-2,2′:6′,2′′-terpyridine (4′NH2-trpy) was purchased from HetCat. All other reagents used in this work were obtained from Aldrich Chemical Co. and were used without further purification. Ammonium cerium(IV) nitrate was purchased as ≥99.99% trace metal basis quality from Sigma-Aldirch® and stored in the glove box to avoid decomposition. Acetonitrile Chromasolve HPLC grade was obtained from Sigma-Aldrich®. Acetone RS and methanol, both HPLC grade and absolute ethanol were purchased from PANREAC. Methanol was freshly distilled (Mg/I2) before use. Highly purified water was achieved by passing it through an UltraClear water purifier from SG Wasseraufbereitung und Regenerierstation GmbH.Preparations
Compounds 4′-methoxy-2,2′:6′,2′′-terpyridine (4′MeO-trpy),15 out-[Ru(Cl)(Hbpp)(trpy)]2+,16 [RuCl3(4′MeO-trpy)]17 and [RuCl3(4′NH2-trpy)]17 were prepared according to previous published procedures. [{Ru(trpy)}2(μ-Cl)(μ-bpp)]2+ (12+),5 [{Ru(trpy)}2(μ-Cl)(μ-(Mebpp))]2+ (22+),8 [{Ru(4′Me-trpy)}2(μ-Cl)(μ-bpp)]2+ (32+),8 [{Ru(4′MeO-trpy)}2(μ-Cl)(μ-bpp)]2+ (42+),8 [{Ru(trpy)}2(μ-Cl)(μ-(NO2bpp))]2+ (52+),8 [{Ru(trpy)}(μ-OAc)(μ-bpp){Ru(trpy)}]2+ (82+),5 [{Ru(trpy)}2(μ-OAc)(μ-(Mebpp))]2+ (92+),8 [{Ru(4′Me-trpy)}2(μ-OAc)(μ-bpp)]2+ (102+),8 [{Ru(4′MeO-trpy)}2(μ-OAc)(μ-bpp)]2+ (112+),8 [{Ru(trpy)}2(μ-OAc)(μ-(NO2bpp))]2+ (122+).8600), 314 (46800), 386 (22000), 480 (14300), 505 (13500), 655 (1800); Echem (DCM-TBAH (0.1 M), vs. SSCE): E1/2II,III/II,II = 0.685 V, ΔE = 60 mV; E1/2III,III/II,III = 1.110 V, ΔE = 60 mV; ΔE1/2 = 425 mV; ESI-MS (MeOH): m/z 1100.9 (M-PF6)2+, 478.0 (M-2PF6)3+; anal. calcd. for C44H33ClF12N10OP2Ru2·3.5H2O: C, 40.39; H, 3.08; N, 10.71. Found: C, 40.72; H, 2.78; N, 10.41.
200), 316 (44800), 387 (21700), 480 (15300), 505 (14700), 651 (1800); Echem (DCM-TBAH (0.1 M), vs. SSCE): E1/2II,III/II,II = 0.610 V, ΔE = 60 mV; E1/2III,III/II,III = 1.075 V, ΔE = 90 mV; ΔE1/2 = 465 mV; ESI-MS (MeOH): m/z 1085.9 (M-PF6)2+, 470.5 (M-H-2PF6)3+; anal. calcd. for C43H32ClF12N11P2Ru2·1.5H2O: C, 41.98; H, 2.81; N, 12.25. Found: C, 40.93; H, 2.64; N, 11.93.
:1) solvent mixture are added and the resulting brown suspension is put for 1 h at 90 °C under microwave irradiation. Consecutively, 2 mL of NaClO4 (aq., sat.) and 5 mL of H2O are added and the resulting violet precipitate is collected by filtration. The solid is washed with minimum volume of H2O followed by Et2O and is dried in vacuum. Yield = 27.3 mg (23.2 μmol (93%)); 1H-NMR ([d6]acetone): δ (ppm) 8.75 (d, 3J = 8.0 Hz, 2H, H35, H37); 8.64 (m, 4H, H4, H12, H32, H40); 8.52 (s, 1H, H22); 8.45 (d, 3J = 5.6 Hz, 2H, H1, H15); 8.43 (d, 3J = 5.6 Hz, 2H, H29, H43); 8.41 (s, 2H, H7, H9); 8.20 (m, 3H, H19, H25, H36); 8.02 (t, 3J = 7.8 Hz, 2H, H31, H41); 8.00 (t, 3J = 7.8 Hz, 2H, H3, H13); 7.74 (t, 3J = 7.8 Hz, 2H, H18, H26); 7.50 (m, 5H, H2, H14, H16 or H28, H30, H42); 7.38 (d, 3J = 5.6 Hz, 1H, H16 or H28); 6.87 (t, 3J = 6.6 Hz, 1H, H17 or H27); 6.83 (t, 3J = 6.6 Hz, 1H, H17 or H27); 4.26 (s, 3H, CH3 (4′MeO-trpy)); 0.48 (s, 3H, CH3 (OAc)). Labeling is keyed in Chart S1;†13C-NMR ([d6]acetone): δ (ppm) 191.4, 166.1, 160.2, 160.0, 159.9, 159.8, 154.2, 153.8, 153.0, 152.8, 137.3, 137.1, 135.8, 135.5, 133.8, 127.4, 127.3, 123.8, 123.6, 122.6, 122.0, 119.5, 119.4, 109.7, 103.9, 56.0; UV-vis (DCM): λmax, nm (ε, M−1 cm−1) 277 (69000), 317 (47300), 379 (23800), 502 (14600), 526 (14000); Echem (DCM-TBAH (0.1 M)), vs. SSCE): E1/2II,III/II,II = 0.685 V, ΔE = 70 mV; E1/2III,III/II,III = 1.030 V, ΔE = 60 mV; ΔE1/2 = 345 mV; ESI-MS (MeOH): m/z 1078.9 (M-ClO4)2+, 490.0 (M-2ClO4)3+; anal. calcd. for C46H36Cl2N10O11Ru2·NaClO4: C, 42.49; H, 2.79; N, 10.77. Found: C, 42.74; H, 2.99; N, 10.41.
:1) solvent mixture are added and the resulting brown suspension is put for 1 h at 90 °C under microwave irradiation. Consecutively, 2 mL of NaClO4 (aq., sat.) and 5 mL of H2O are added and the resulting violet precipitate is collected by filtration. The solid is washed with minimum volume of H2O followed by Et2O and is dried in vacuum. Yield = 42.9 mg (36.9 μmol (91%)); 1H-NMR ([d6]acetone): δ (ppm) 8.74 (d, 3J = 8.0 Hz, 2H, H35, H37); 8.62 (d, 3J = 8.0 Hz, 2H, H32, H40); 8.48 (s, 1H, H22); 8.40 (d, 3J = 5.6 Hz, 4H, H1, H15, H29, H43); 8.31 (d, 3J = 8.0 Hz, 2H, H4, H12); 8.19 (t, 3J = 8.0 Hz, 1H, H36); 8.18 (d, 3J = 7.6 Hz, 2H, H19, H25); 8.02 (t, 3J = 8.0 Hz, 2H, H31, H41); 8.00 (s, 2H, H7, H9); 7.94 (t, 3J = 8.0 Hz, 2H, H3, H13); 7.72 (t, 3J = 7.6 Hz, 2H, H18, H26); 7.61 (d, 3J = 5.6 Hz, 1H, H16 or H28); 7.52 (t, 3J = 6.6 Hz, 2H, H30, H42); 7.41 (t, 3J = 6.6 Hz, 2H, H2, H14); 7.36 (d, 3J = 5.6 Hz, 1H, H16 or H28); 6.90 (t, 3J = 7.6 Hz, 1H, H17 or H27); 6.81 (t, 3J = 7.6 Hz, 1H, H17 or H27); 6.46 (s, 2H, NH2); 0.51 (s, 3H, CH3 (OAc)). Labeling is keyed in Chart S1;†13C-NMR ([d6]acetone): δ (ppm) 190.2, 160.4, 160.0, 159.8, 158.2, 157.3, 156.6, 155.0, 154.4, 153.7, 153.1, 152.8, 152.6, 137.2, 136.8, 135.8, 135.0, 133.7, 127.3, 126.6, 123.5, 122.9, 122.6, 122.0, 119.4, 119.3, 108.0, 103.8; UV-vis (DCM): λmax, nm (ε, M−1 cm−1) 277 (69000), 317 (47300), 379 (23800), 502 (14600); Echem (DCM-TBAH (0.1 M)), vs. SSCE): E1/2II,III/II,II = 0.59 V, ΔE = 60 mV; E1/2III,III/II,III = 1.00 V, ΔE = 60 mV; ΔE1/2 = 410 mV; MALDI-MS (dctb, DCM, acetone): m/z 1064.0 (M-ClO4)2+, 963.1 (M-H-2ClO4)2+; anal. calcd. for C45H35Cl2N11O10Ru2·0.5NaClO4·H2O: C, 43.51; H, 3.00; N, 12.40. Found: C, 43.69; H, 2.91; N, 12.26.
Complexes 153+–213+ are prepared in situ by hydrolysis of compounds 8–14 in acidic aqueous media. Only complex 203+ was isolated in the solid state.
000), 304 (59300), 355 (18000), 460 (12500), 485 (10500); Echem (acetone:CF3SO3H (0.1 M) (1:4), vs. SSCE): E1/2II,III/II,II = 0.54 V, ΔE = 60 mV; E1/2III,III/II,III = 0.63 V, ΔE = 60 mV; E1/2III,IV/III,III = 0.86 V, ΔE = 60 mV; E1/2IV,IV/III,IV = 1.08 V, ΔE = 60 mV.
800), 305 (57900), 354 (18800), 454 (11800), 486 (9200); Echem (acetone:CF3SO3H (0.1 M) (1:4), vs. SSCE): E1/2II,III/II,II = 0.50 V, ΔE = 60 mV; E1/2III,III/II,III = 0.635 V, ΔE = 70 mV; E1/2III,IV/III,III = 0.85 V, ΔE = 60 mV; E1/2IV,IV/III,IV = 0.96 V, ΔE = 60 mV.
500), 263 (55600), 303 (54300), 353 (20600), 465 (12400), 489 (11200); Echem (acetone:CF3SO3H 0.1 M (1:4), vs. SSCE): E1/2II,III/II,II = 0.51 V, ΔE = 60 mV; E1/2III,III/II,III = 0.61 V, ΔE = 60 mV; E1/2III,IV/III,III = 0.83 V, ΔE = 60 mV; E1/2IV,IV/III,IV = 1.06 V, ΔE = 60 mV.
000), 272 (55300), 308 (47100), 357 (20200), 478 (12500), 502 (11500); Echem (acetone:CF3SO3H 0.1 M (1:4), vs. SSCE): E1/2II,III/II,II = 0.50 V, ΔE = 60 mV; E1/2III,III/II,III = 0.59 V, ΔE = 60 mV; E1/2III,IV/III,III = 0.84 V, ΔE = 60 mV; E1/2IV,IV/III,IV = 1.02 V, ΔE = 60 mV.
000), 452 (11900), 491 (13600), 545 (8300); Echem (acetone:CF3SO3H (0.1 M) (1:4), vs. SSCE): E1/2III,III/II,II = 0.74 V, ΔE = 60 mV; E1/2III,IV/III,III = 1.00 V, ΔE = 60 mV; E1/2IV,IV/III,IV = 1.10 V, ΔE = 60 mV.
000), 272 (58200), 310 (52900), 358 (20900), 471 (12000), 500 (10800); Echem (CF3SO3H (0.1 M) (1:4), vs. SSCE): E1/2II,III/II,II = 0.50 V, ΔE = 80 mV; E1/2III,III/II,III = 0.56 V, ΔE = 80 mV; E1/2III,IV/III,III = 0.84 V, ΔE = 100 mV; E1/2IV,IV/III,IV = 1.03 V, ΔE = 80 mV; MALDI-MS (pyrene, CH2Cl2): m/z 1101.0 (M-2PF6)2+, 956.0 (M-3PF6)2+, 940.1 (M-3PF6-OH)2+, 921.0 (M-3PF6-2OH)2+; anal. calcd. for C44H37F18N10O3P3Ru2·2,5H2O: C, 36.80; H, 2.95; N, 9.75. Found: C, 36.63; H, 2.72; N, 9.66.
300), 273 (61100), 312 (51700), 366 (18200), 468 (11800); Echem (acetone:CF3SO3H (0.1 M) (1:4), vs. SSCE): E1/2II,III/II,II = 0.48 V, ΔE = 60 mV; E1/2III,III/II,III = 0.64 V, ΔE = 60 mV; E1/2III,IV/III,III = 0.80 V, ΔE = 60 mV; E1/2IV,IV/III,IV = 0.97 V, ΔE = 80 mV.
Computational methods
Standard reduction potentials were calculated for various possible redox couples to assess the energetic accessibility of different intermediates at various oxidation states. For a redox reaction of the form
| O(aq) + ne−(g) → R(aq) |
where ΔG0O|R is the free energy change associated with the equation below (using Boltzmann statistics for the electron), ΔG0NHE is the free energy change associated with,
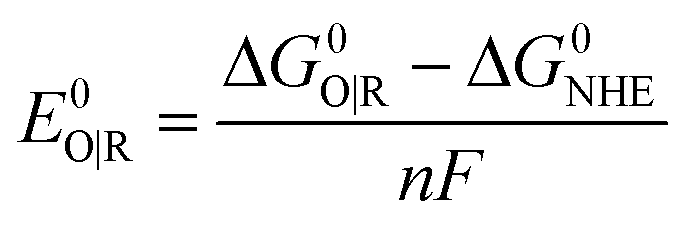
which is −4.28 eV with Boltzmann statistics for the electron,24c,25 and F is the Faraday constant.
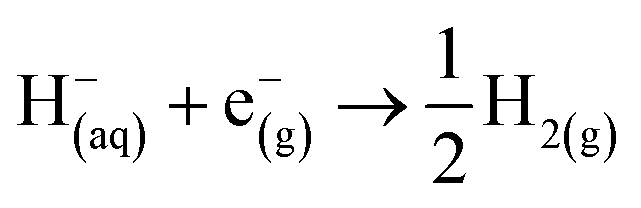
where HS refers to the single-determinantal high-spin coupled state that is related to the low-spin state by spin flip(s) and <S2> is the expectation value of the total spin operator applied to the appropriate determinant. This broken-symmetry DFT approach has routinely proven effective for the prediction of state-energy splittings in metal coordination compounds.26b,28
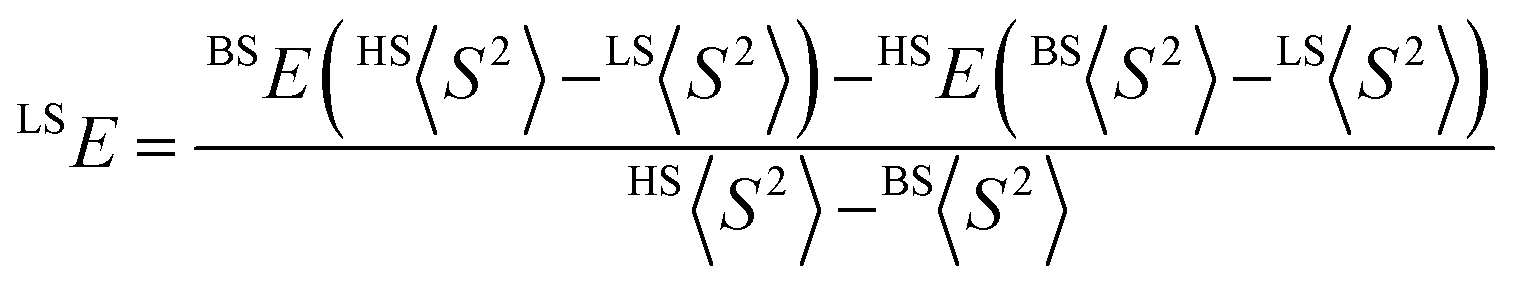
Conclusions
We present a facile microwave based synthetic procedure to obtain symmetric and non-symmetric dinuclear ruthenium complexes, structurally based on the so called “Ru–bpp”-catalyst. The procedure presents an increase in overall yield in combination to a decrease of reaction time and workup steps. The electronic perturbation effects of different substituents on the ligand backbone on electronic transitions are examined and explained. Their effect on metal based redox potentials follows the expected trend of intrinsic electron donating and withdrawing properties. The position of the substituents on the ligand backbone has a major influence on the magnitude of these potentials. We have also analyzed the influence of the substitution kinetics as a function of ED and EW groups and found that while the former enhances substitution kinetics the latter drastically reduces them. This in turn seems to be the major effect for the rds of the water oxidation reactions under stoichiometric conditions analyzed for the case of 203+. We have also shown that while complex 203+ is an active catalyst at oxidation state IV,IV as previously reported, it is a much better catalyst at the oxidation state IV,V, as shown by its large electrocatalytic wave. Further supporting this point, whilst the IV,IV oxidation state can be accessed with CeIV oxidant, the IV,V can only be accessed electrochemically or with stronger oxidants such as [Ru(debpy)2(bpy)]2+. Indeed efficient light induced water oxidation is obtained with the PS complex containing the debpy ligand while with regular [Ru(bpy)3]2+ is practically inactive. Finally, DFT calculations nicely complement the experimental results giving a consistent global description of the different reactions pathways that can operate in the water oxidation reactions catalyzed by the complexes described in this work.Acknowledgements
A. L. thanks MINECO (CTQ-2013-49075-R, SEV-2013-0319; CTQ-2014-52974-REDC; ENE2014-52280-REDT) and “La Caixa” foundation for financial support. COST actions, CM1202 and CM1205 and Feder funds from the EU are also gratefully acknowledged. C. J. R. is grateful for a Marie Curie IEF grant. C. J. C. and co-workers thank the NSF (CHE-1361595) and acknowledge the Minnesota Supercomputing Institute (MSI) at the University of Minnesota for providing resources that contributed to the research results reported within this paper.Notes and references
- (a) N. Armaroli and V. Balzani, ChemSusChem, 2011, 4, 21 CrossRef CAS PubMed; (b) S. Berardi, S. Drouet, L. Francas, C. Gimbert-Surinach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501 RSC; (c) D. Kim, K. K. Sakimoto, D. Hong and P. Yang, Angew. Chem., Int. Ed., 2015, 54, 2 CrossRef; (d) J. M. Thomas, Energy Environ. Sci., 2014, 7, 19 RSC.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS PubMed.
- (a) K. Sanderson, Nature, 2008, 452, 400 CrossRef CAS PubMed; (b) M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072 CrossRef CAS PubMed; (c) T. Arai, S. Sato, T. Kajino and T. Morikawa, Energy Environ. Sci., 2013, 6, 1274 RSC.
- S. W. Gersten, G. J. Samuels and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4029 CrossRef CAS.
- C. Sens, I. Romero, M. Rodríguez, A. Llobet, T. Parella and J. Benet-Buchholz, J. Am. Chem. Soc., 2004, 126, 7798 CrossRef CAS PubMed.
- (a) F. Bozoglian, S. Romain, M. Z. Ertem, T. K. Todorova, C. Sens, J. Mola, M. Rodriguez, I. Romero, J. Benet-Buchholz, X. Fontrodona, C. J. Cramer, L. Gagliardi and A. Llobet, J. Am. Chem. Soc., 2009, 131, 15176 CrossRef CAS PubMed; (b) A. Poater, Catal. Commun., 2014, 44, 2 CrossRef CAS.
- (a) J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974 CrossRef CAS PubMed; (b) Y. Xu, A. Fischer, L. Duan, L. Tong, E. Gabrielsson, B. Åkermark and L. Sun, Angew. Chem., Int. Ed., 2010, 49, 8934 CrossRef CAS PubMed; (c) T. M. Laine, M. D. Kärkäs, R.-Z. Liao, P. E. M. Siegbahn and B. Åkermark, Chem. – Eur. J., 2015, 21, 10039 CrossRef CAS PubMed; (d) L.-Z. Zeng, C.-J. Wang, T.-T. Li, X. Gan, C. Li and W.-F. Fu, Catal. Commun., 2015, 68, 84 CrossRef CAS; (e) A. C. Sander, A. Schober, S. Dechert and F. Meyer, Eur. J. Inorg. Chem., 2015, 4348 CrossRef CAS; (f) S. Neudeck, S. Maji, I. López, S. Meyer, F. Meyer and A. Llobet, J. Am. Chem. Soc., 2014, 136, 24 CrossRef CAS PubMed; (g) T. Wada, K. Tsuge and K. Tanaka, Inorg. Chem., 2001, 40, 329 CrossRef CAS PubMed; (h) J. Mola, C. Dinoi, X. Sala, M. Rodriguez, I. Romero, T. Parella, X. Fontrodona and A. Llobet, Dalton Trans., 2011, 40, 3640 RSC; (i) S. Maji, L. Vigara, F. Cottone, F. Bozoglian, J. Benet-Buchholz and A. Llobet, Angew. Chem., Int. Ed., 2012, 51, 5967 CrossRef CAS PubMed; (j) Z. Deng, H.-W. Tseng, R. Zong, D. Wang and R. Thummel, Inorg. Chem., 2008, 47, 1835–1848 CrossRef CAS PubMed; (k) Y. Jiang, F. Li, B. Zhang, X. Li, X. Wang, F. Huang and L. Sun, Angew. Chem., Int. Ed., 2013, 52, 3398 CrossRef CAS PubMed.
- S. Roeser, M. Z. Ertem, C. Cady, R. Lomoth, J. Benet-Buchholz, L. Hammarström, B. Sarkar, W. Kaim, C. J. Cramer and A. Llobet, Inorg. Chem., 2012, 51, 320 CrossRef CAS PubMed.
- (a) J. Mola, E. Mas-Marza, X. Sala, I. Romero, M. Rodríguez, C. Viñas, T. Parella and A. Llobet, Angew. Chem., Int. Ed., 2008, 47, 5830 CrossRef CAS PubMed; (b) L. Francàs, X. Sala, J. Benet-Buchholz, L. Escriche and A. Llobet, ChemSusChem, 2009, 2, 321 CrossRef PubMed; (c) J. Aguilo, L. Francas, H. J. Liu, R. Bofill, J. Garcia-Anton, J. Benet-Buchholz, A. Llobet, L. Escriche and X. Sala, Catal. Sci. Technol., 2014, 4, 190 RSC; (d) L. Francàs, X. Sala, E. Escudero-Adán, J. Benet-Buchholz, L. Escriche and A. Llobet, Inorg. Chem., 2011, 50, 2771 CrossRef PubMed.
- (a) D. J. Wasylenko, C. Ganesamoorthy, M. A. Henderson, B. D. Koivisto, H. D. Osthoff and C. P. Berlinguette, J. Am. Chem. Soc., 2010, 132, 16094 CrossRef CAS PubMed; (b) J. J. Concepcion, J. W. Jurss, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2008, 130, 16462 CrossRef CAS PubMed.
- A. G. Stromberg, Zh. Obshch. Khim., 1955, 25, 639 Search PubMed.
- S. Berardi, L. Francàs, S. Neudeck, S. Maji, J. Benet-Buchholz, F. Meyer and A. Llobet, ChemSusChem, 2015, 8, 3688–3696 CrossRef CAS PubMed.
- (a) S. Roeser, P. Farràs, F. Bozoglian, M. Martínez-Belmonte, J. Benet-Buchholz and A. Llobet, ChemSusChem, 2011, 4, 197 CrossRef CAS PubMed; (b) H. D. Gafney and A. W. Adamson, J. Am. Chem. Soc., 1972, 94, 8238 CrossRef CAS.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351 CrossRef CAS.
- J. Chambers, B. Eaves, D. Parker, R. Claxton, P. S. Ray and S. J. Slattery, Inorg. Chim. Acta, 2006, 359, 2400 CrossRef CAS.
- C. Sens, M. Rodriguez, I. Romero, A. Llobet, T. Parella and J. Benet-Buchholz, Inorg. Chem., 2003, 42, 8385 CrossRef CAS PubMed.
- S. J. Stoessel, C. M. Elliott and J. K. Stille, Chem. Mater., 1989, 1, 259 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- D. Andrae, U. Häussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123 CrossRef CAS.
- W. J. Hehre, L. Radom, P. V. R. Schleyer and J. A. Pople, Ab Initio Molecular Orbital Theory, Wiley, New York, 1986 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- C. J. Cramer, Essentials of Computational Chemistry: Theories and Models, John Wiley & Sons, Chichester, 2nd edn, 2004 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
- (a) M. D. Tissandier, K. A. Cowen, W. Y. Feng, E. Gundlach, M. H. Cohen, A. D. Earhart, J. V. Coe and T. R. Tuttle, J. Phys. Chem. A, 1998, 102, 7787 CrossRef CAS; (b) D. M. Camaioni and C. A. Schwerdtfeger, J. Phys. Chem. A, 2005, 109, 10795 CrossRef CAS PubMed; (c) C. P. Kelly, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2006, 110, 16066 CrossRef CAS PubMed; (d) V. S. Bryantsev, M. S. Diallo and W. A. Goddard, J. Phys. Chem. B, 2008, 112, 9709 CrossRef CAS PubMed.
- (a) A. Lewis, J. A. Bumpus, D. G. Truhlar and C. J. Cramer, J. Chem. Educ., 2004, 81, 596 CrossRef CAS; (b) P. Winget, C. J. Cramer and D. G. Truhlar, Theor. Chem. Acc., 2004, 112, 217 CrossRef CAS.
- (a) T. Ziegler, A. Rauk and E. J. Baerends, Theor. Chim. Acta, 1977, 43, 261 CrossRef CAS; (b) L. Noodleman, J. Chem. Phys., 1981, 74, 5737 CrossRef CAS; (c) C. J. Cramer and D. G. Truhlar, Phys. Chem. Chem. Phys., 2009, 11, 10757 RSC.
- (a) K. Yamaguchi, F. Jensen, A. Dorigo and K. N. Houk, Chem. Phys. Lett., 1988, 149, 537 CrossRef CAS; (b) T. Soda, Y. Kitagawa, T. Onishi, Y. Takano, Y. Shigeta, H. Nagao, Y. Yoshioka and K. Yamaguchi, Chem. Phys. Lett., 2000, 319, 223 CrossRef CAS.
- (a) L. Noodleman, C. Y. Peng, D. A. Case and J.-M. Mouesca, Coord. Chem. Rev., 1995, 144, 199 CrossRef CAS; (b) I. Ciofini and C. A. Daul, Coord. Chem. Rev., 2003, 238, 187 CrossRef; (c) J. N. Harvey, Struct. Bonding, 2004, 112, 151 CrossRef CAS; (d) F. Neese, Coord. Chem. Rev., 2009, 253, 526 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC numbers for 62+ and 142+ are 1432987 and 1432988 respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cy00197a |
| This journal is © The Royal Society of Chemistry 2016 |