
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect C–H bond activation of ethers and successive C–C bond formation with benzene by a bifunctional palladium–titania photocatalyst†
Akanksha
Tyagi
a,
Tomoya
Matsumoto
a,
Tatsuhisa
Kato
ab and
Hisao
Yoshida
*ac
aGraduate School of Human and Environmental Studies, Kyoto University, Yoshida Nihonmatsu-cho, Sakyo-ku, Kyoto 606-8501, Japan. E-mail: yoshida.hisao.2a@kyoto-u.ac.jp
bInstitute for Liberal Arts and Sciences, Kyoto University, Yoshida Nihonmatsu-cho, Sakyo-ku, Kyoto 606-8501, Japan
cElements Strategy Initiative for Catalysts and Batteries (ESICB), Kyoto University, Kyotodaigaku-Katsura, Nishikyo-ku, Kyoto 615-8520, Japan
First published on 11th February 2016
Abstract
Palladium-loaded titanium oxide was found to work as a bifunctional photocatalyst for functionalization of benzene with ether upon photoirradiation, without using any special reagents. The metal-loaded TiO2 photocatalyst activated a C–H bond of ethers and the heterogeneous Pd metal nanoparticle catalyst promoted the successive C–C bond formation between benzene and the radical species. In this reaction, benzene reacted very selectively with the α-carbon of various ethers at least in the initial stage of the reaction. Kinetic and ESR studies revealed a detailed mechanism for the reaction.
1. Introduction
Activation of sp3 C–H bonds is one of the most desired reactions in current chemistry because it provides an opportunity to utilize and convert abundant and inexpensive alkanes to valuable products.1 Especially in the view of green and sustainable chemistry, the direct activation of these bonds promises efficient synthetic practices because the desired compounds can be prepared in a single step instead of the currently used multistep processes consuming extra reagents. However, this is not an easy reaction since these bonds have very low reactivity due to their high bond strength and attainment of electronic saturation.2Some of the earliest methods for sp3 C–H bond activation have included the usage of strong acids or bases, but the harsh reaction conditions would limit the substrates available for the reaction. Since then, among notable advancements in this area, employing metal catalysts has become the most popular method, where the key idea is the formation of a new bond between the metal and the sp3 carbon.2 Even though these methods show higher efficiency and selectivity over the earlier methods, they all have inherent limitations. One is the sensitive nature of the involved reagents, i.e., the preparation, storage and usage of these metal complexes should require air- or moisture-free conditions, which limits the applicability of these methods. The other is that some kinds of harmful and reactive reagents are often required. Hence, an innovative methodology for general and mild activation of sp3 C–H bonds has been desired to be established.
In this regard, heterogeneous photocatalysis is a promising alternative.3 In addition to the obvious advantages like simple product separation and reusability of heterogeneous catalysts, the use of photoenergy as the driving force would enable us to realize unique synthetic routes. Since the photoexcited electrons and holes themselves are very reactive, they can simultaneously but independently induce the reductive and oxidative reactions, respectively, thereby eliminating the use of other harmful and reactive chemicals. Thus, the reactions can be performed under mild conditions, making the process green and sustainable.
In our previous studies, we found that metal-loaded TiO2 photocatalysts (M/TiO2, M = Pd or Pt) can activate various types of chemicals to radical species under UV light. Based on this concept, we have reported the photocatalytic hydroxylation4 and amination5 of aromatic rings as well as the hydration of alkenes,6 where we realized the activation of an O–H bond in water and an N–H bond in ammonia for the formation of rather difficult O–C and N–C bonds, respectively. It was considered that this strategy should be applicable to the activation of sp3 C–H bonds in a similar way. To facilitate the reaction, we focused on molecules consisting of a functional group adjacent to the target sp3 carbon, which would result in a decrease of the bond dissociation energy. For example, the bond dissociation enthalpy of a C–H bond in methane is ca. 426.8 kJ mol−1 while it becomes 401.6 kJ mol−1 in acetonitrile due to the adjacent cyano group.7 This methodology has been firstly demonstrated in our previous study for the direct cyanomethylation of the benzene ring over Pd/TiO2 photocatalysts,8 where the adjacent cyano group would assist the C–H bond dissociation and stabilize the generated cyanomethyl radical to enhance the reaction efficiency for the successive C–C bond formation with benzene.
To expand this concept, ethers were examined in the present study, which were expected to become α-oxyalkyl radicals to realize the successive C–C bond formation for functionalization of benzene. We found for the first time that a bifunctional Pd/TiO2 photocatalyst could successfully promote the direct sp3 C–H bond activation in different ethers and the successive sp3 C–sp2 C bond formation with benzene to give ether-substituted benzenes (α-arylated ethers), an important class of compounds,9 with almost complete selectivity and high yields. The yields obtained here are notable since these kinds of photocatalytic organic transformations have been crippled by low yields unless auxiliary chemicals or long reaction times are used.4–6,8,10
2. Experimental
2.1. Catalyst preparation
Various TiO2 powder samples employed for the experiments were donated by the Catalysis Society of Japan as JRC-TiO-8 (anatase phase, 338 m2 g−1), JRC-TiO-6 (rutile phase, 100 m2 g−1) and JRC-TiO-4 (mixture of rutile and anatase phases, 50 m2 g−1). All metal-loaded TiO2 catalysts were prepared by the photodeposition method using the desired TiO2 powder and an appropriate metal precursor solution of PdCl2 (Kishida, 99%), H2PtCl6·6H2O (Wako, 99.9%), RhCl3·3H2O (Kishida, 99%) or HAuCl4·4H2O (Kishida, 99.9%). The TiO2 powder (4 g) was dispersed in ion-exchanged water (300 ml) and was irradiated with a ceramic xenon lamp (PE300 BUV) for 30 min. Then, methanol (100 ml) and the desired amount of the metal precursor solution were added to the suspension and the contents were stirred for 15 min without irradiation, followed by 1 h of stirring under light. It was then filtered off with suction, washed with ion-exchanged water and dried at 323 K for 12 h so as to obtain the metal-loaded TiO2 photocatalysts. The catalysts were referred to as M(x)/TiO2, where M indicates Pd, Pt, Rh or Au, and x indicates the loading amount of the metal species in weight%.2.2. Catalyst characterization
Pd K-edge XAFS (X-ray absorption fine structure) analysis was carried out to determine the state of metal nanoparticles loaded over TiO2 after the reaction. The spectra were recorded at NW10A of Photon Factory at the Institute of Materials Structure Science, High Energy Accelerator Research Organization (KEK-PF, Tsukuba, Japan) with a Si(311) double-crystal monochromator at room temperature. For the prepared catalyst samples, the spectra were recorded in the fluorescence mode by using a Lytle detector filled with a krypton (100%) flow equipped with a Ru filter (μt = 6) for the fluorescence and an ion chamber filled with an argon (100%) flow for the incident X-ray. The spectra of the reference samples were measured in the transmission mode. The spectra were analysed with REX 2000 software (Rigaku).2.3. Reaction test
The durability and reusability of the M(x)/TiO2 sample was determined using a flow reactor, the details of which are explained in the ESI.†
Further information about the mechanism was obtained from the results of reaction tests using isotopic compounds such as deuterated diethyl ether, (C2D5)2O, and deuterated benzene, C6D6, and temperature-controlled reaction tests.
3. Results and discussion
3.1. Catalytic reaction between benzene and diethyl ether over M(x)/TiO2 samples
The reaction between benzene and diethyl ether (DEE, 1a) with a Pd(0.2)/TiO2 sample gave 1-ethoxyethylbenzene (1-EEB, 2a) as the major product through the sp2 C–sp3 C bond formation between benzene and the α-position of DEE (Table 1, entry 1), while a trace amount of 2-ethoxyethylbenzene (2-EEB), a product of the reaction with the β-position of DEE, was also obtained (not shown). The benzene conversion to the main product, 1-EEB, reached 23% for the initial 3 h. Caronna et al.11 reported the C–C bond formation between a reactive heteroarene and ether over a TiO2 photocatalyst with the aid of H2O2 as an additional radical generator. In contrast, our system employing the Pd/TiO2 sample was free from such an auxiliary reagent and could work under much milder conditions to give the desired products with complete selectivity and high yields.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ether | Product | Reaction time (h) | Amount of producta (μmol) | Yb (%) | Sc (%) |
| a GC-MS yield. b Y (%): yield of the listed product = 100 × (the amount of the product)/(the initial amount of benzene). c S (%): selectivity for the listed product among the benzene-containing products = 100 × (amount of the product)/(total amount of benzene-containing products). d 0.1 g of the Pd(0.2)/TiO2 sample was used; the wavelength of incident light was ≥350 nm; the light intensity was 56 mW cm−2 when measured at 365 ± 20 nm wavelength using a UV radiometer (Topcon, UVR-2, UD-36); 5 μl (56 μmol) of benzene, 2 ml (19.2 mmol) of DEE and 0.4 μl of water were used; 1-EEB: 1-ethoxyethylbenzene; the amount of 1-EEB was determined from the calibration curve of 1-EEB. e 5 μl (56 μmol) of benzene, 2 ml (20 mmol) of THP and 0.4 μl of water were used; other conditions were the same as those mentioned above; 2-PTHP: 2-phenyltetrahydropyran; the amount of 2-PTHP was tentatively determined from the calibration curve of 1-methoxybutylbenzene (1-MBB). f 2 μl (22 μmol) of benzene, 1 ml (12 mmol) of THF and 0.4 μl of water were used; the remaining conditions were the same as those mentioned above; 2-PTHF: 2-phenyltetrahydrofuran; the amount of 2-PTHF was tentatively determined from the calibration curve of 1-EEB. g 100 μl (1.12 mmol) of benzene, 0.35 ml (1.19 mmol) of BME and 4 μl of water were used; the remaining conditions were the same as those before; BMB: butoxymethylbenzene, 1-MBB: 1-methoxybutylbenzene; the amounts of 1-MBB and BMB were determined from the calibration curve of 1-MBB. | ||||||
| 1d | 1a | 2a | 3 | 12.9 | 23 | 99 |
| 2e | 1b | 2b | 3 | 16.8 | 30 | 99 |
| 3f | 1c | 2c | 3 | 4.1 | 18 | 99 |
| 4g | 1d | 2d1 | 1 | 4.1 | 1.1 | 99 |
| 2d2 | 7.9 | |||||
The high selectivity for α-substitution can be attributed to the lower bond dissociation enthalpy of the α-C–H bonds (397.4 kJ mol−1 for DEE) compared to their β-counterparts (426.8 kJ mol−1 for DEE).12 Although some homocoupling products of DEE such as 2,3-diethoxybutane and its derivatives were also formed, the quantity was small. This means that the generated α-oxyalkyl radical would preferentially react with benzene before reacting with DEE or another α-oxyalkyl radical under these conditions. It is important to note that biphenyl, the dimer of the phenyl radical, was not detected at all under the examined reaction conditions. Among the gaseous products, hydrogen was the major product while very small amounts of methane and ethane were also detected.
Among the various M(x)/TiO2 samples tested, the ones loaded with Pd, Pt or Rh were active while the Au(0.1)/TiO2 sample was inactive for the C–C bond formation (Table 2, entries 1–4). Also, among the various TiO2 tested for this reaction, with Pd as the co-catalyst, anatase phase TiO2 (JRC-TiO-8) having a large specific surface area exhibited maximum activity (Table S1, ESI†). The selectivity for the α-arylated ether (1-EEB) was always high over these metal-loaded TiO2 samples (more than 88% in Table 2 and Table S1†). The desired product was not formed in the absence of light (Table 2, entry 8) or the M(x)/TiO2 sample (Table 2, entry 9), indicating that the reaction needed both light and the M(x)/TiO2 sample. Moreover, when the reaction was performed with a pristine TiO2 sample, only a very small amount of products was obtained (Table 2, entry 10), highlighting the importance of metal loading. It is notable that the Pd(x)/TiO2 samples exhibited the highest activities for this reaction and the highest selectivity, more than 98%, for the α-arylated ether among the M(x)/TiO2 samples (Table 2, entries 1, 5–7). For the Pd(0.1)/TiO2 sample, the amount of 1-EEB was as high as 18.8 μmol (Table 1, entry 1) which corresponds to a TON (turnover number) of ca. 20 for 1 h per Pd atom in the system and this value further increased with time. These results confirmed that this reaction should be a photoinduced catalytic reaction.
| Entry | Sample | Productsb (μmol) | S1-EEBc (%) | |
|---|---|---|---|---|
| 1-EEB | 2-EEB | |||
| a Reaction conditions: 3.3 ml (31.7 mmol) of DEE, 0.7 ml (7.8 mmol) of benzene and 0.1 g of the M(x)/TiO2 sample were used, reaction time was 1 h, λ ≥ 350 nm, and I = 40 mW cm−2 (at 365 ± 20 nm). b 1-EEB: see Table 1, 2-EEB: 2-ethoxyethylbenzene; the amount of 2-EEB was tentatively determined from the calibration curve of 1-EEB. c S1-EEB (%): selectivity for 1-EEB = 100 × (amount of 1-EEB)/(total amount of benzene-containing products). d No light was supplied during the reaction. e No photocatalyst was used and only light was supplied. f 0.1 g of pristine TiO2 (anatase) was used for the reaction. | ||||
| 1 | Pd(0.1)/TiO2 | 18.8 | 0.3 | 98 |
| 2 | Pt(0.1)/TiO2 | 1.8 | 0.2 | 90 |
| 3 | Rh(0.1)/TiO2 | 1.6 | 0.2 | 88 |
| 4 | Au(0.1)/TiO2 | 0 | 0 | 0 |
| 5 | Pd(0.05)/TiO2 | 10.8 | 0.2 | 98 |
| 6 | Pd(0.2)/TiO2 | 19.2 | 0.4 | 98 |
| 7 | Pd(0.5)/TiO2 | 12.8 | 0.1 | 99 |
| 8d | Pd(0.1)/TiO2 | 0 | 0 | 0 |
| 9e | — | 0 | 0 | 0 |
| 10f | TiO2 | 0.6 | 0.1 | 86 |
In fact, the amount of products did not increase drastically when the reaction time increased from those indicated in Table 1 to the longer one such as 5 h in these conditions. This might be due to the fact that the present reactor is a closed batch reactor which has some limitations. In a closed reactor, the reactants or the products including an undetectable amount of the by-products could be adsorbed on the catalyst surface which might cover the active sites and thus decrease the activity. Thus, the reaction seemed to stop after a certain period and no significant increase in product amount was observed. To support this proposal, the same reaction was also performed in a fixed-bed flow reactor as shown in the ESI.† The results evidenced that the photocatalyst had a long life as it maintained a constant activity for almost 10 h (Fig. S1†). This can be explained by considering the fact that, in a flow reactor, the reactants are being constantly removed from the catalyst bed which does not allows the attainment of adsorption–desorption equilibrium. Thus, the catalyst surface would be relatively uncovered in a flow reactor than a closed batch reactor. In fact, the catalyst was active enough to be reused for the next cycle without any pre-treatment (Fig. S2†).
3.2. Catalytic reaction between benzene and other ethers over the Pd(0.2)/TiO2 sample
This reaction proceeded well with other ethers too (Table 1, entries 2–4). The reaction between benzene and THP (1b) gave 2-phenyltetrahydropyran (2-PTHP, 2b) selectively, where the benzene conversion to the product reached 30% for the initial 3 h. THF (1c) also reacted with benzene selectively at the α-position to give 2-phenyltetrahydrofuran (2-PTHF, 2c) with high benzene conversion (18%) for 3 h. BME (1d), with two α-carbons, gave two kinds of α-substituted products, butoxymethylbenzene (BMB, 2d1) and 1-methoxybutylbenzene (1-MBB, 2d2). In this case, the yield of the α-substituted products was not high and many by-products were observed, which could be because the α-oxymethylene radical formed from BME, which would produce BMB, would be less stable and preferentially converted to other compounds before reacting with benzene.3.3. Mechanistic studies
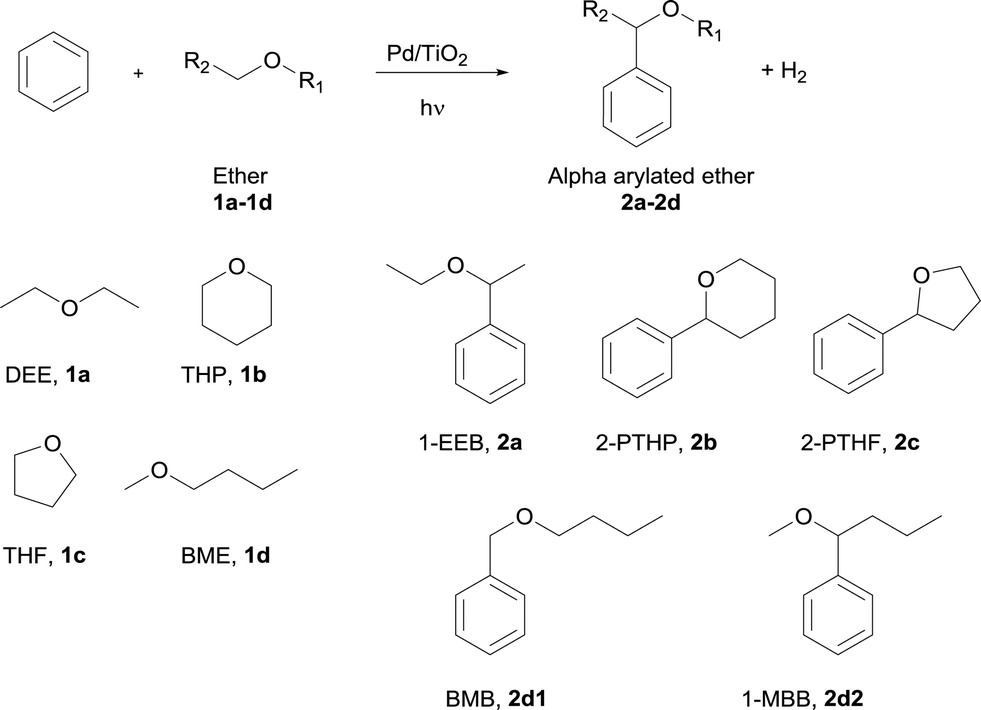
 | ||
| Fig. 1 ESR spectrum of the DEE radical with PBN as a spin trap. | ||
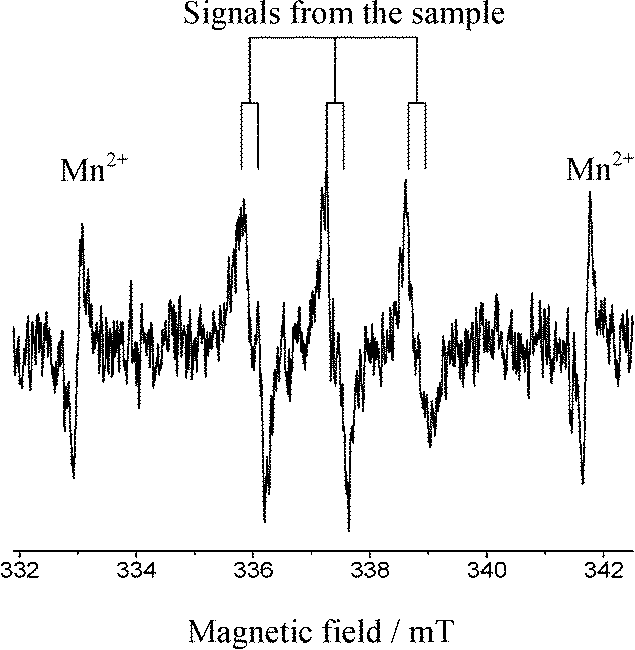
These signals were different from those observed when the photodissociation of PBN took place under UV light (ESI†). Moreover, no signals were observed in the absence of DEE, the Pd/TiO2 photocatalyst and light. Thus, it was confirmed that the reaction involves the oxidation of ether molecules to the radical species in the presence of the Pd(0.1)/TiO2 photocatalyst under UV light irradiation (eqn (1)).
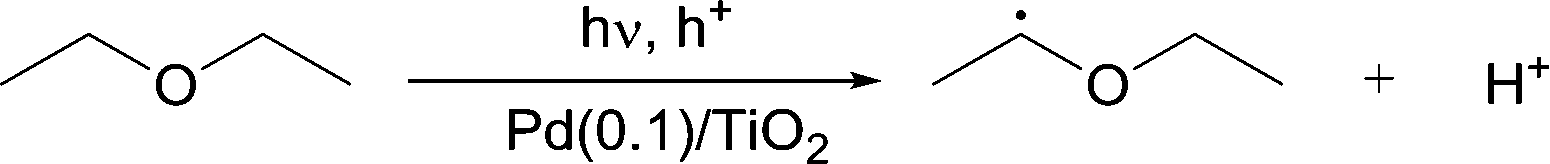 | (1) |
| Entry | Reactants | Product, 1-EEB (μmol) | k H/kDb | |
|---|---|---|---|---|
| a Reaction conditions: 0.1 ml (1.1 mmol) of benzene, 0.4 ml (3.8 mmol) of DEE and 0.0125 g of the Pd(0.2)/TiO2 catalyst were used, reaction time was 1 h, λ ≥ 350 nm, and I = 40 mW cm−2 (at 365 ± 20 nm). b k H/kD was determined from the amounts of 1-EEB produced in entries 1 and 2 for entry 2 and those in entries 1 and 3 for entry 3. | ||||
| 1 | Benzene | DEE | 7.0 | — |
| 2 | Benzene-d6 | DEE | 7.6 | 0.92 |
| 3 | Benzene | DEE-d10 | 7.5 | 0.93 |
However, it is important to consider the small inverse kinetic isotope effect (KIE) observed for each reaction. In order to give the α-arylated product, the reaction must involve the attack of the α-oxyalkyl radical on the benzene ring. This should involve a transition state where the hybridization of the benzene carbon would change from sp2 to sp3. This change mainly effects the out-of-plane bending vibration of the C–H bond which is observed at 1350 cm−1 for the sp3 C atom and 800 cm−1 for the sp2 C atom.14 Thus, when deuterated benzene was used, the change of the zero-point energies from sp2 C to sp3 C should be considered. Quantitatively, the difference in the zero-point energies for the C–H bonds in the reactants and the transition state is 550 cm−1 (800 cm−1 and 1350 cm−1, respectively) while that for the C–D bonds is 403.2 cm−1 (586.8 cm−1 and 990 cm−1, respectively). Thus, because the difference in the zero-point energies at the change from sp2 to sp3 for the C–D bonds is smaller than that for the C–H bonds, an inverse kinetic isotope effect should be observed. This could exactly explain what was observed for the reaction test between deuterated benzene and DEE (Table 3, entry 2). On the other hand, as for the inverse KIE observed for the reaction between benzene and deuterated DEE (Table 3, entry 3), other reasons should be considered. As discussed above, the attack of the DEE radical on benzene would generate a sp3 like transition state. Since D atom is heavier than H atom, the C–D bond is stronger than the C–H bond. This causes the C–D bond to be slightly shorter than C–H bond. Thus, the transition state would be less sterically-crowded in the case of D than H, because of which the reaction of benzene with deuterated DEE will give an inverse KIE (Table 3, entry 3). Thus, from the above results, it can be concluded that the α-oxyalkyl radical would attack the carbon in the benzene molecule to form the reaction intermediate having the sp3-like carbon centre in the aromatic ring (eqn (2)) and this step is most likely to be the rate-determining step for this reaction.
 | (2) |
This neutral reaction intermediate would release a hydrogen radical to give the product (eqn (3)).
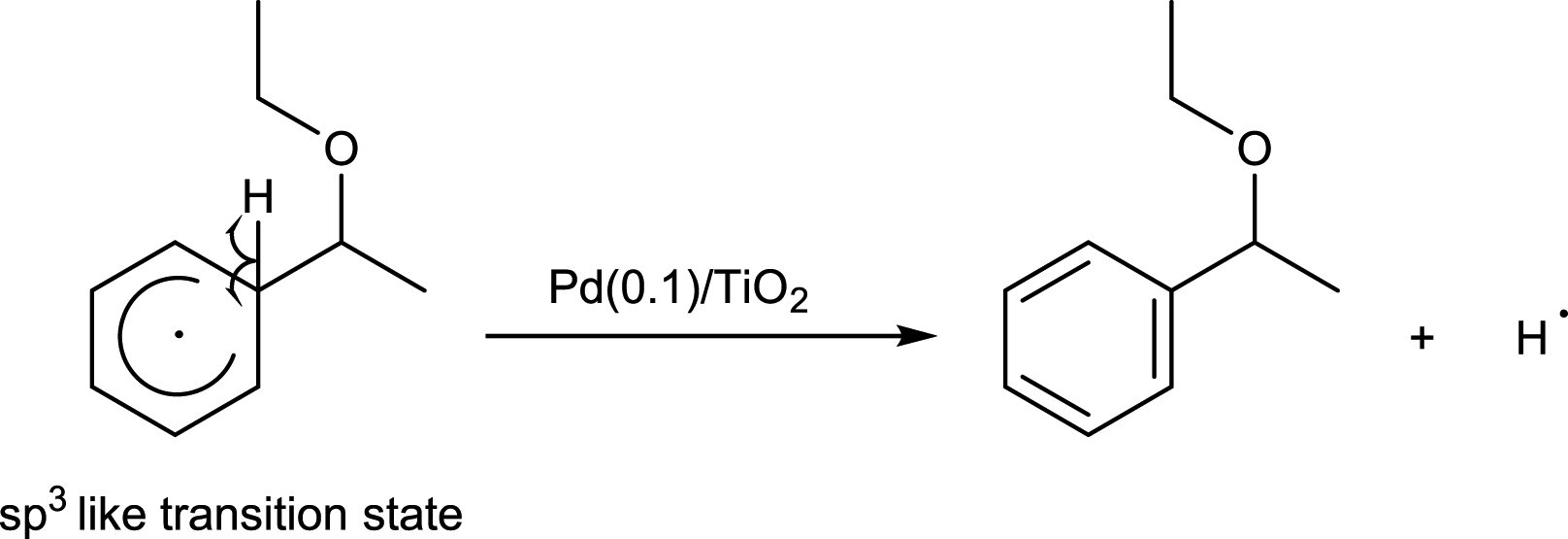 | (3) |
As a result, the amount of 1-EEB increased when the reaction was carried out at higher temperature under light (Table S2,† entries 1–4). However, no product was formed in the test at higher temperature in the dark (Table S2,† entry 5), eliminating the possibility that the thermal catalytic reaction with the Pd metal catalyst proceeds independently at higher temperatures. The apparent activation energy determined from the Arrhenius plot was 35.4 kJ mol−1 (Fig. 2a). It was further confirmed that the increase in the amount of 1-EEB at higher temperature was not due to its increased desorption from the catalyst surface in the adsorption–desorption equilibrium (Table S3†). The variation of the amount of desorbed 1-EEB with temperature gave a very gentle slope of 5.5 kJ mol−1 (Fig. 2b), which was much lower than the thermal activation energy during the reaction under light. Thus, the increase with temperature under light should be mainly due to thermocatalysis promoted further at higher temperatures. This result indicates that the rate-determining step would be this thermal reaction between benzene and the α-oxyalkyl radical originating from ether, which would be promoted by the Pd catalyst, in the form of metallic Pd nanoparticles, with the apparent thermal activation energy of 35.4 kJ mol−1 and hence would be accelerated at higher temperatures.
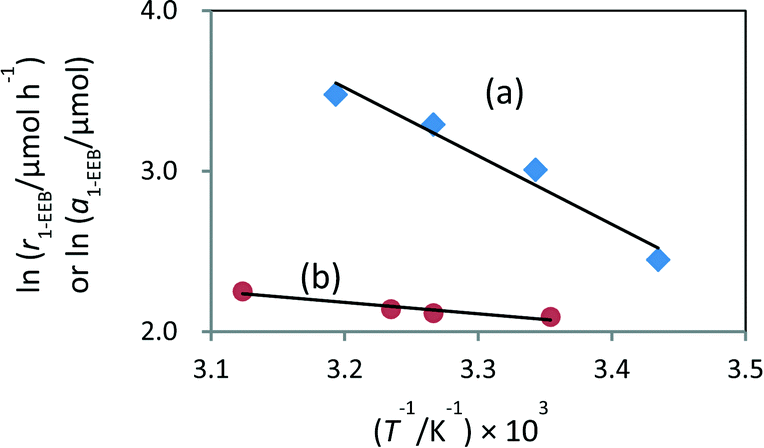 | ||
| Fig. 2 Pseudo-Arrhenius plots for (a) temperature-controlled reactions between benzene and DEE under light and (b) adsorption tests of 1-EEB in the dark, with the Pd(0.2)/TiO2 bifunctional photocatalyst. r1-EEB: formation rate of 1-EEB; a1-EEB: amount of desorbed 1-EEB in the solution. | ||
3.4. State of Pd nanoparticles over TiO2
The Pd K-edge XAFS spectra revealed the state of the Pd catalyst on TiO2. Fig. 3A shows the normalized XANES spectra of the Pd(0.2)/TiO2 catalyst sample and references, i.e., a metallic Pd foil and a PdO powder sample. The spectrum of the catalyst sample exhibited similar oscillation to metallic Pd at the higher energy region of more than 24.4 keV although the post-edge feature was not identical to that of the metallic Pd foil. Obviously, this was quite different from that of the PdO sample.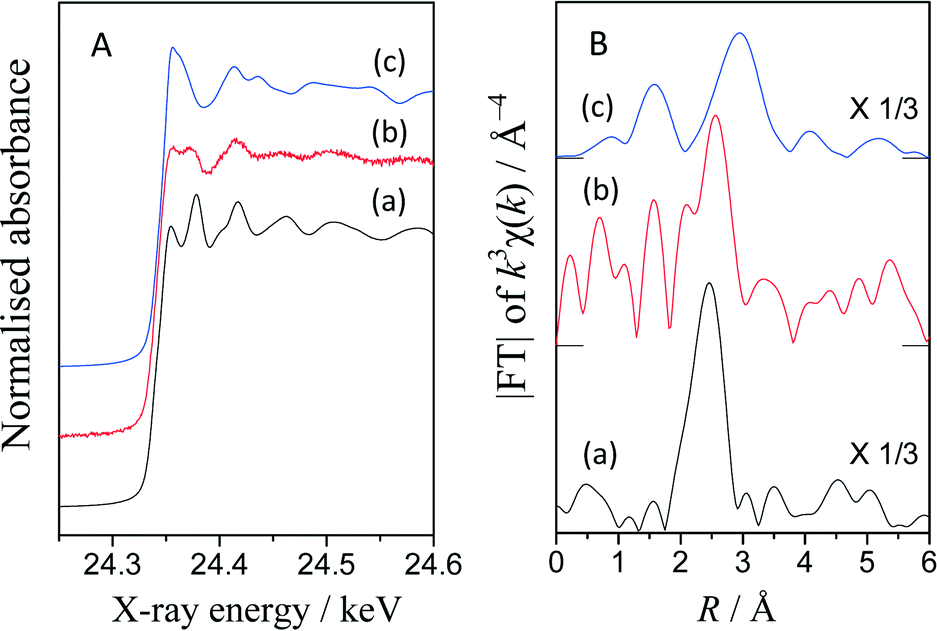 | ||
| Fig. 3 Normalized XANES spectra (A) and Fourier transforms of the k3-weighted EXAFS (B) of the metallic Pd foil (a), the Pd(0.2)/TiO2 sample after the reaction test between benzene and DEE (b), and PdO powder (c). The intensity of the FT-EXAFS of the reference samples was reduced to one third. | ||
Fig. S3† shows the differential spectra of these XANES. The differential spectrum of the Pd(0.2)/TiO2 catalyst sample exhibited a similar shape to that of the Pd metal and different from that of PdO. The absorption edge values were evaluated from these spectra. Interestingly, the Pd(0.2)/TiO2 catalyst sample showed a lower value (24.337 keV) than the Pd metal (24.346 keV) and PdO (24.349 keV), implying that the Pd species in the Pd(0.2)/TiO2 catalyst might be electron-rich. It is known that, when TiO2 and a precious metal nanoparticle form a semiconductor–metal junction, the electrons migrate to the metal particle, which would make the metal particle electron-rich. Thus, in the present case, the Pd nanoparticles loaded on the surface of TiO2 might be negatively charged to some extent. In addition, during the reaction under photoirradiation, the photoexcited electron stored in the metallic Pd nanoparticle might further enhance this property.
Fig. 3B shows the Fourier-transformed EXAFS spectra. The peaks around 1.6, 2.5 and 3.0 Å correspond to the Pd–O shell in PdO, the Pd–Pd shell in the Pd metal and the Pd–Pd shell in PdO. Although the catalyst sample showed a noisy spectrum, the Pd–Pd shell in the Pd metal was clearly visible, indicating that the Pd species would be metallic. However, an additional peak was observed at 1.6 Å. According to the curve fitting analysis (Table S4†), the coordination number was 3.9 for the Pd–Pd shell and 1.5 for the Pd–O shell. This suggests that the Pd species on the TiO2 surface would be metallic Pd nanoparticles connected to the TiO2 surface through oxygen atoms.
Thus, it can be proposed that the Pd species in the Pd(0.2)/TiO2 catalyst would be in the metallic state during the photocatalytic reaction between benzene and DEE. This further suggests that the Pd particles in the sample can function as an electron-rich metal catalyst during this reaction, making the catalyst a bifunctional photocatalyst. This electron-rich property might be advantageous to inducing electron donation to the anti-bonding π* molecular orbital of the adsorbed benzene to activate it.
3.5. Proposed mechanism
Based on these observations, it is clear that the Pd/TiO2 catalyst can be recognised as a bifunctional catalyst comprising both the TiO2 photocatalyst with the Pd co-catalyst and the Pd metal nanoparticle thermocatalyst. The reaction mechanism with the bifunctional catalyst can be proposed as follows.The TiO2 photocatalyst can be excited by photoirradiation to generate the excited electron (e−) and hole (h+) (Fig. 4, i). The hole would activate the α-C–H bond of ether to generate the α-oxyalkyl radical and a proton as shown in eqn (1) and (ii). Thereafter, the Pd nanoparticle would thermally catalyse the C–C bond formation between benzene and the α-oxyalkyl radical to form the reaction intermediate having the sp3-like carbon centre in the aromatic ring (eqn (2) and (iii)). This step would be the rate-determining step under these conditions. The intermediate would release a hydrogen radical so as to give the desired alpha-substituted product (eqn (3) and (iv)). A proton would be reduced to a hydrogen radical by the photoexcited electrons present on the Pd surface which would couple with the previously eliminated hydrogen radical to form hydrogen (eqn (4) and (v)).
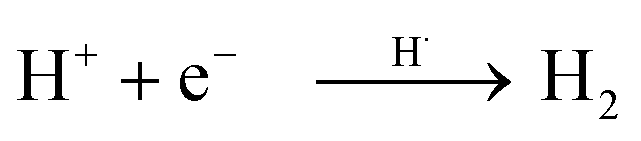 | (4) |
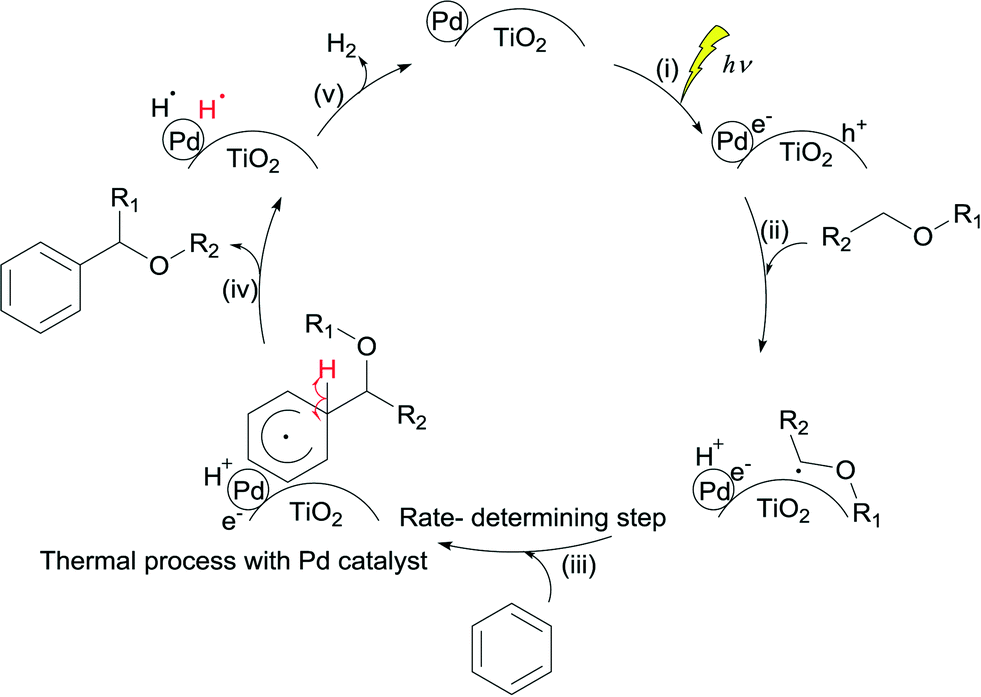 | ||
| Fig. 4 Proposed mechanism of the reaction between benzene and ether by a bifunctional Pd/TiO2 photocatalyst through the direct activation of the sp3 C–H bond of ether to form the C–C bond with benzene. | ||
These reaction steps are summarised in the scheme shown in Fig. 4. It is proposed that this reaction should be a one-photon process.
4. Conclusions
In conclusion, we successfully achieved the direct activation of the sp3 C–H bond in various ethers followed by their successive sp3 C–sp2 C bond formation with benzene by using a heterogeneous bifunctional catalyst that could function as both a photocatalyst and a palladium metal catalyst. The photocatalyst could activate the sp3 C–H bond at the α-carbon in various ethers to form the α-oxyalkyl radical species and the palladium catalyst would promote the reaction between the aromatic ring of benzene and the radical species with the formation of the sp3 C–sp2 C bond to produce the ether-substituted benzene, i.e., α-arylated ether.We hope that this new strategy for direct C–H activation and successive C–C bond formation will be widely applicable to other reaction systems.
Acknowledgements
The XAFS experiments were performed under the approval of the Photon Factory Program Advisory Committee (Proposal No. 2014G547). This work was supported by JSPS KAKENHI Grant Number 25105723 for Scientific Research on Innovative Areas “Molecular Activation Directed towards Straightforward Synthesis”.Notes and references
- R. G. Bergman, Nature, 2007, 446, 391 CrossRef CAS PubMed; K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef PubMed; Handbook of C–H transformations, ed. G. Diker, Wiley-VCH, Weinheim, Germany, 2005 Search PubMed.
- J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507 CrossRef CAS PubMed; D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2009, 110, 704 Search PubMed; M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704 CrossRef PubMed.
- M. Fagnoni, D. Dondi, D. Ravelli and A. Albini, Chem. Rev., 2007, 107, 2725 CrossRef CAS PubMed; G. Palmisano, V. Augugliaro, M. Pagliaro and L. Palmisano, Chem. Commun., 2007, 3425 RSC; Y. Shiraishi and T. Hirai, J. Photochem. Photobiol., C, 2008, 9, 157 CrossRef; H. Yoshida, in Environmentally benign photocatalysts: Applications of titanium oxide-based materials, ed. M. Anpo and P. V. Kamat, Springer, 2010, ch. 27, p. 647 CrossRef PubMed; H. Kisch, Angew. Chem., Int. Ed., 2013, 52, 812 CrossRef PubMed.
- H. Yoshida, H. Yuzawa, M. Aoki, K. Otake, H. Itoh and T. Hattori, Chem. Commun., 2008, 4634 RSC; H. Yuzawa, M. Aoki, K. Otake, T. Hattori, H. Itoh and H. Yoshida, J. Phys. Chem. C, 2012, 116, 25376 Search PubMed.
- H. Yuzawa and H. Yoshida, Chem. Commun., 2010, 46, 8854 RSC; H. Yuzawa, J. Kumagai and H. Yoshida, J. Phys. Chem. C, 2013, 117, 11047 Search PubMed.
- H. Yuzawa, S. Yoneyama, A. Yamamoto, M. Aoki, K. Otake, H. Itoh and H. Yoshida, Catal. Sci. Technol., 2013, 3, 1739 CAS.
- Y. R. Luo, Handbook of Bond Dissociation Energies in Organic Compounds, CRC Press, 2002 Search PubMed; D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press, London, 86th edn, 2006 Search PubMed.
- H. Yoshida, Y. Fujimura, H. Yuzawa, J. Kumagai and T. Yoshida, Chem. Commun., 2013, 49, 3793 RSC.
- P. G. M. Wuts and T. W. Greene, Protective Groups in Organic Synthesis, Wiley & Sons, New York, 4th edn, 2007, p. 102 CrossRef CAS PubMed; P. P. Pradhan, J. M. Bobbitt and W. F. Bailey, J. Org. Chem., 2009, 74, 9524 CrossRef CAS PubMed; Z. Song, J. Gong, M. Zhang and X. Wu, Asian J. Org. Chem., 2012, 1, 214 CrossRef; D. Lednicer, Strategies for Organic Drug Synthesis and Design, Wiley & Sons, Hoboken, 2008 Search PubMed.
- K. V. S. Rao, B. S. Srinivas, A. R. Prasad and M. Subrmahanyam, Chem. Commun., 2000, 1533 Search PubMed; S. Marinkovic and N. Hoffmann, Chem. Commun., 2001, 1576 RSC.
- T. Caronna, C. Gambarotti, L. Palmisano, C. Punta and F. Recupero, J. Photochem. Photobiol., A, 2005, 171, 237 CrossRef CAS.
- F. Agapito, B. J. C. Cabral and J. A. M. Simoes, J. Mol. Struct.: THEOCHEM, 2005, 719, 109 CrossRef CAS.
- H. Yuzawa and H. Yoshida, Chem. Lett., 2013, 42, 1336 CrossRef CAS; H. Yuzawa and H. Yoshida, Top. Catal., 2014, 57, 984 CrossRef.
- E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, ed. J. Murdzek, Edwards Brothers, 2005, ch. 8, p. 428 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Effect of TiO2 structure on the photocatalytic activity, catalytic reaction test in a flow reactor, ESR study, thermal effects on catalysis and adsorption and XAFS analysis of a Pd/TiO2 sample. See DOI: 10.1039/c5cy02290h |
| This journal is © The Royal Society of Chemistry 2016 |