
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Methanol-to-olefins process over zeolite catalysts with DDR topology: effect of composition and structural defects on catalytic performance†
Irina
Yarulina
a,
Joris
Goetze
b,
Canan
Gücüyener
a,
Leonard
van Thiel
a,
Alla
Dikhtiarenko
a,
Javier
Ruiz-Martinez
b,
Bert M.
Weckhuysen
b,
Jorge
Gascon
*a and
Freek
Kapteijn
a
aCatalysis Engineering, Chemical Engineering Department, Delft University of Technology, Julianalaan 136, 2628 BL Delft, The Netherlands. E-mail: j.gascon@tudelft.nl
bInorganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands
First published on 25th January 2016
Abstract
A systematic study of the effect of physicochemical properties affecting catalyst deactivation, overall olefin selectivity and ethylene/propylene ratio during the methanol-to-olefins (MTO) reaction is presented for two zeolites with the DDR topology, namely Sigma-1 and ZSM-58. Both catalysts show high selectivity towards light olefins and completely suppress the formation of hydrocarbons bigger than C4, with selectivity to ethane not exceeding 1% and some traces of propane. By applying seeded growth approach, a series of Sigma-1 zeolites with tunable crystal size and acidity was synthesized. For this series the highest methanol throughput at 450 °C before deactivation was found for crystals 0.5 μm in size with an acidity corresponding to 0.5 Al atoms per zeolite cage, and a selectivity to ethylene and propylene reaching 90%. Comparison between ZSM-58 and Sigma-1 catalysts with similar morphologies and acidity under the same reaction conditions revealed a three times higher throughput of methanol in case of ZSM-58. The analysis of functional surface groups, assessed through FT-IR, revealed the presence of silanol defects in Sigma-1 responsible for faster catalyst deactivation. These silanol defects can be selectively removed (confirmed by FT-IR) from the zeolite framework by applying a mild treatment in presence of NaOH/CTAB, leading to an improved catalyst lifetime. Co-feeding experiments with short olefins and water show low reactivity of primary MTO products, which only react at the surface of the catalyst particles. These results demonstrate that migration of the reaction zone in case of DDR catalysts hardly affects catalyst stability, product composition and nature of deactivating species. The nature of these species depends mostly on reaction temperature: at low temperatures deactivation occurs mainly due to the formation of inert adamantane species, while at high temperatures poly-condensed aromatic hydrocarbons play the major role in deactivation.
1. Introduction
Due to the growing demand for propylene and ethylene, the search for efficient alternative routes to the synthesis of light olefins is now at the forefront of industrial and academic research.1 Among these routes, the methanol-to-hydrocarbons (MTH) conversion2 has gained a lot of attention during the last few decades.The reaction pathway includes conversion of methanol over acidic zeolites, with SAPO-34 (CHA) and ZSM-5 (MFI) being successfully implemented in industry.3 Several alternative processes are commercially available, and most of these rely on zeolite ZSM-5 as catalyst. These include the Mobil Oil MTG process, the Topsøe integrated gasoline synthesis (TIGAS) process, and the Lurgi methanol-to-propylene (MTP) process.3 In the latter case, the selectivity is optimized towards propylene rather than C5+ by tuning the reaction conditions (i.e., high temperature and low pressure) and by recycling long chain hydrocarbons.4 In the Norsk Hydro/UOP methanol-to-olefins (MTO) process, mostly ethylene and propylene are produced using SAPO-34 as the catalyst material.5
However, there are still several drawbacks associated with the relatively low stability of SAPO-34 towards multiple regenerations due to the presence of phosphorus in its framework, and to the broad product distribution when using ZSM-5. Furthermore, for both catalytic materials, the energy intensive separation of the olefin/paraffin product mixtures is a major disadvantage.
Facing the abovementioned issues, there is still a great interest in finding appropriate zeolite topologies that would deliver a narrower product distribution, while displaying an excellent stability. Preferably only ethylene and propylene should be produced, while control towards the formation of one of these products would be ideal. From this perspective, the search should continue taking small pore zeolites with large cavities into account. Such small pore zeolites are able to host inside their cavities the so-called “hydrocarbon pool”, the key intermediate,6–8 while allowing only small hydrocarbons to leave their cavities through smaller windows.
Recently, DDR was identified as a promising 8-ring zeolite structure for the MTO process.9 There are three zeolites belonging to the DDR family, namely DD3R,10 its pure silica version, and two Al-containing variants reported in literature, Sigma-1,11 and ZSM-58.12 Both DD3R and Sigma-1 are synthesized using adamantylamine as structure directing agent (SDA), while methyltropinium iodide is employed for ZSM-58 synthesis.
The key properties of DDR were discovered, while testing it in separation of olefin/paraffin mixtures.13,14 This material takes up only certain unsaturated molecules (ethylene, propylene, trans but-2-ene and butadiene),15,16 while saturated C3 and C4 molecules are excluded. This molecular sieving exclusion principle based on zeolite topology should be responsible for the high selectivity to ethylene and propylene during the MTO process. In turn, the ethylene-to-propylene ratio could be tuned by varying operating conditions and/or physicochemical properties of the zeolite.17–19 A great deal of work has been devoted to the investigation of the effect of crystal size and acid strength on the MTO performance of different zeolite frameworks focusing mainly on SAPO-34.20,21 However, in most cases the synthesis routes towards different morphologies and acidic composition involved a variation in SDAs, zeolite precursor composition or Si sources. Variation of these parameters might also lead to Al zoning,22,23 formation of structural defects in the zeolite framework,24 and other effects that would eventually affect reaction performance. Having said that, it is worth mentioning the recent findings for ZSM-5 (ref. 25 and 26) on the effect of coke formation governed by the presence of non-acidic silanol groups and internal defects. Though the correlation between amount of coke and internal defects has been established for ZSM-5, there are no studies confirming the same interdependence for other systems. As a consequence, there are no data available on healing/removing of these defects for other zeolites, especially for structures with 8-membered ring (8MR) pore openings.
In this work we systematically study the effect of physicochemical properties on catalyst deactivation, overall olefin selectivity and ethylene-to-propylene ratio using Sigma-1 and ZSM-58 zeolites with DDR topology as two case studies. All parameters were investigated independently, using always the same gel precursor composition, tuning zeolite morphology and acidity only by changing the amount of Al and by using as-synthesized material as seeds. By applying this approach, we could identify the effects of catalyst composition on reaction performance and the nature of species that lead to catalyst deactivation, with special emphasis on the effect of catalyst structural defects,24,27 something often overlooked in the open literature for 8MR zeolites.
2. Experimental
2.1. Synthesis of DDR zeolites
1-Adamantylamine (ADA, 97%), colloidal silica (Ludox HS-40, 40%), sodium aluminate (Al2O3 (50–56%), Na2O (40–45%)), ammonium nitrate (98%), hexadecyltrimethylammonium bromide (CTAB, ≥99%), sodium hydroxide (≥98%), methyl iodide (99.5%), tropine (≥97%), methanol (≥99.9%) were purchased from Sigma-Aldrich and used as received without any further purification.Firstly, Sigma-1 crystals used for the preparation of seeds were synthesized at 180 °C for 6 days using a molar composition of ADA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Na2O:SiO2:Al2O3:H2O = 20:3:60:1:2400 as described elsewhere, ADA being used as a template molecule.28 The obtained crystals were subjected to wet ball-milling by using the procedure reported by Charkhi et al.29 as basis.
:Na2O:SiO2:Al2O3:H2O = 20:3:60:1:2400 as described elsewhere, ADA being used as a template molecule.28 The obtained crystals were subjected to wet ball-milling by using the procedure reported by Charkhi et al.29 as basis.
A Retsch mixer mill MM2 equipped with zirconia grinding jar of 10 mL and zirconium oxide grinding balls of 2 mm diameter in size was used for wet-ball milling. In each step, 2 g of zeolite were wet ball milled using 9 g of zirconium oxide balls in 2 mL of deionized water medium for 4 h with a vibrational frequency of 50 Hz. After each wet-ball milling, zirconium oxide balls were washed with approximately 30 mL (±5 mL) of deionized water and separated from the solution. This solution was then centrifuged for 12 min to obtain a mother liquor with particles smaller than 200 nm. After removal of the mother liquor, precipitated powder was dried overnight at 80 °C and used in the next wet-ball milling step. Due to low yield in the first two wet-ball milling steps, the mother liquor obtained at these stages was added to the deionized water used in the next stages during cleaning of the zirconia balls to avoid unnecessary dilution. From the 3rd step on, approximately 8 mL of mother liquor was obtained from each step of the wet-ball milling. After 12 subsequent wet-ball milling steps, 80 mL of 0.5 wt% seeding solution with a 6–6.5 pH was obtained.
Subsequent syntheses were performed by addition of either ball-milled or as-synthesized crystals (0.1 wt%) with molar composition of ADA:Na2O:SiO2:Al2O3:H2O = 20:3:60:x:2400, reducing synthesis time from 6 days to 8–20 h depending on the Al content.
The as-synthesized materials were calcined at 650 °C for 10 h in a static oven. The calcined samples were ion-exchanged three times in aqueous NH4NO3 solution (1 M, 80 °C, 2 h, 100 mL per gram of zeolite) followed by calcination at 550 °C.
A modified sample was obtained by treating calcined at 650 °C Sigma-1 with 20 ml of solution per gram of zeolite of 0.075 M NaOH and 0.5 M CTAB solution.30 This modified sample was further subjected to calcination at 550 °C to remove CTAB followed by ion-exchange as outlined above.
ZSM-58 crystals were synthesized as reported elsewhere using methyltropinium iodide (MTI) as template,9 keeping the SiO2/Al2O3 ratio 110, and subjected to calcination and ion-exchange as described above.
MTI was prepared by adding drop-wise 25.1 g methyl iodide to a solution 25.0 g tropine in 100 g of ethanol at 0 °C. The resulted suspension was left for 3 days for stirring. After washing with 100 g of ethanol and filtering, the resulted powder was dried at 80 °C for 12 h.
2.2. Catalyst characterization
The XRD patterns of the powders are recorded in Bragg–Brentano geometry with a Bruker D8 Advance X-ray diffractometer equipped with a LynxEye position-sensitive detector. Measurements were performed at RT by using monochromatic CoKα (λ = 1.788970 Å) radiation between 2θ = 5° and 50°.The Pawley fitting of the experimental data was performed using Reflex Plus module of the Accelrys Material Studio software package. The refinement of unit cell parameters giving values of a = b = 13.8068(1) Å, c = 40.8500(1) Å, β = α = 90°, γ = 120° suggested a close agreement with the one expected for DDR zeolite as reported in the Atlas of Zeolite Framework Types.31 Zero offset, the scale factor, six background terms and profile parameters were refined during Pawley fitting. The profiles have been modeled as a pseudo-Voigt function. Corresponding refinement results are summarized in Table S1† and plotted in Fig. S1.†
Images where recorded using a JEOL JSM-6010LA with a standard beam potential of 10 kV and an Everhart–Thornley detector. X-ray microanalysis (SEM/EDX) confirmed the elemental composition in the sample by scanning microscopy (SEM) coupled with a dispersive X-ray microanalysis system (EDX) with a Silicon-drift detector.
Elemental analysis was performed with PerkinElmer Optima 4300 DV instrument. The samples were first digested in an aqueous mixture of 1% HF and 1.25% H2SO4. After dilution, analysis was done by inductively coupled plasma optical emission spectrometry (ICP-OES).
N2 adsorption was carried out using Tristar II 3020 Micromeritics sorptometer at 77 K. Prior to the experiment, samples were outgassed at 350 °C for 16 h.
Temperature-programmed NH3 desorption (NH3-TPD) was measured by AutoChem II chemisorption analyzer (Micromeritics). Approximately 0.2 g of the material was first degassed under He flow at 400 °C and then saturated with NH3 at 200 °C during 1 h using a flow of 1.65% NH3 in He. The gas mixture was then switched back to He and the sample was purged at 200 °C for about 1 h to remove weakly adsorbed NH3 molecules. TPD was subsequently recorded under He flow, from 200 °C to 800 °C. All flow rates were adjusted to 25 mL min−1, and the heating rates were 10 °C min−1 during different stages of experiment.
Transmission FT-IR spectroscopy was performed using a Nicolet Nexus spectrometer at 4 cm−1 resolution equipped with an extended KBr beam splitting and a mercury cadmium telluride (MCT) cryo-detector. The pellets were placed in an IR quartz cell equipped with CaF2 windows. A movable sample holder allows the sample to be placed in the infrared beam for the measurements or into the furnace for thermal treatments. The cell is connected to a vacuum line for pretreatment. The specimen is activated in vacuum at 400 °C for 16 h to remove adsorbed species. After this step, the samples were cooled down to −130 °C and CO was dosed up to 30 mbar.
Thermogravimetrical analysis (TGA) was performed by means of a Mettler Toledo TGA/SDTA851e on samples of 20 mg under flowing 100 mL min−1 of air at a heating rate of 5 °C min−1 up to 1000 °C. The amount of coke formed during the reaction was calculated from the weight loss between 150 °C and 800 °C for completely deactivated catalysts (after feeding at least 10 g methanol per g zeolite).
DRIFTS analysis was carried out in a Thermo Scientific Nicolet 8700 spectrometer, equipped with a high-temperature cell with CaF2 windows (HVC Praying Mantis Harrick) and a DGTS detector. The spectra were collected after 128 scans and a resolution of 4 cm−1, under 20 mL min−1 He flow. KBr spectra (at given temperatures) are used as background. Before collecting the spectra, samples deactivated after 5 h of MTO reaction were preheated in a He flow at 120 °C for 60 min to remove water.
Hydrocarbons that were retained in the catalyst after testing were investigated by dissolution experiments. Typically, 15 mg of the spent catalyst was dissolved in 1 mL 50% HF in a Teflon container. The organic compounds were extracted from the water phase by the addition of three times 1 mL CH2Cl2. Analysis of the extracted phase was performed on a GCMS-QP2010Ultra GC/MS system, equipped with a Zoex ZX1 thermal modulator and an Agilent VF-5 ms and VF-17 ms column in series. Hereinafter, the term ‘retained hydrocarbons’ or ‘coke’ is used to describe all compounds that are detectable by this method.
2.3. Catalyst testing
Catalytic experiments were carried out in a Microactivity Reference unit (PID Eng&Tech) at 380–475 °C and ambient pressure. The catalyst (1.75 g, 250–420 μm sieve fraction) was placed in a fixed-bed with internal diameter 9 mm. An HPLC pump (307 5-SC-type piston pump, Gilson) was used to feed methanol to the reactor system. The temperature was recorded by a thermocouple placed in the middle of the catalytic bed. A weight-hourly space velocity (WHSV) of 2 gMeOH gcat−1 h−1, a 1:1 molar feed composition of N2 and MeOH and atmospheric pressure are applied and referred to as standard operating conditions at the different temperatures.
The product mixture was analyzed online with an Interscience CompactGC equipped with a 15 m capillary RTX-1 (1% diphenyl-, 99% dimethylpolysiloxane) column and a flame ionization detector.
Conversion, selectivities and yields were calculated on a molar carbon basis. Thus, conversion was defined as the fraction of light oxygenates (methanol and dimethyl ether) consumed during the reaction:
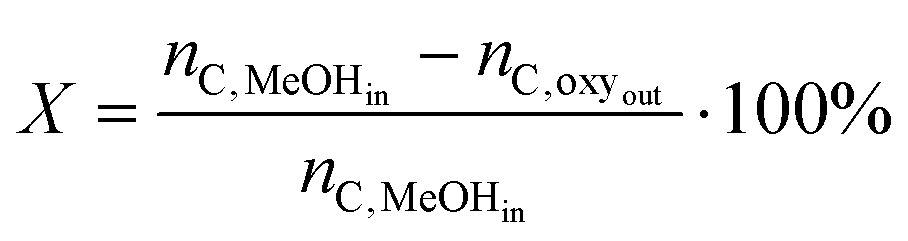 | (1) |
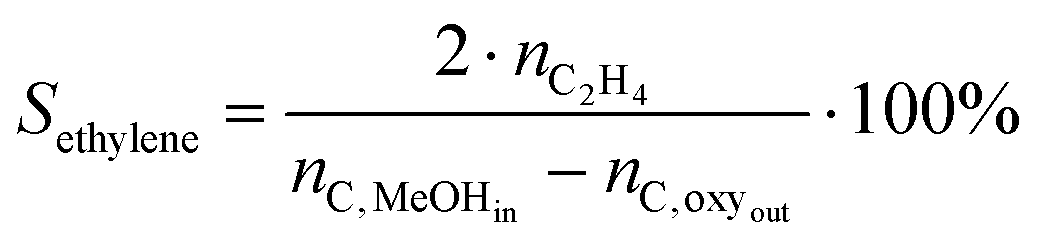 | (2) |
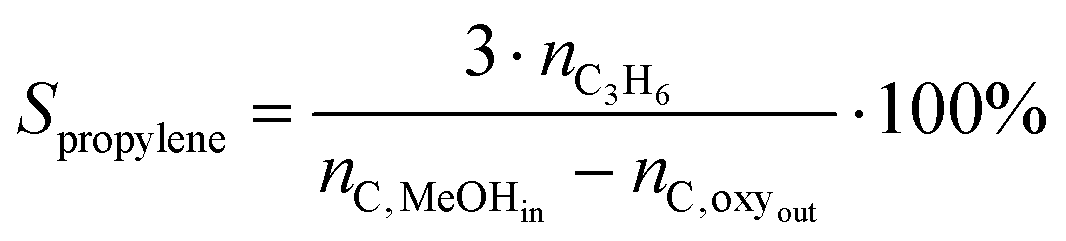 | (3) |
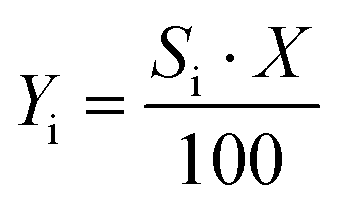 | (4) |
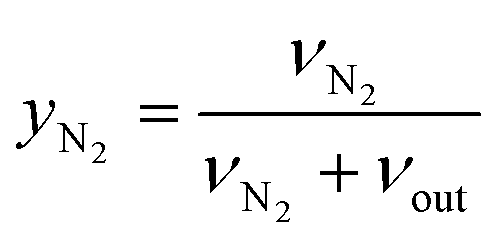 | (5) |
Hence, the molar flowrate of a component i (νi) could be calculated from the eqn (6):
| νi = yi·(νN2) + (νout) | (6) |
The performance results are presented in graphs as a function of the methanol throughput per amount of catalyst used (gMeOH gcat−1). The methanol conversion is an apparent one as it reflects the disappearance of this component, which can be due to chemical conversion or to retention on the catalyst. Parameters related to catalyst performance are summarized in Table 1.
| Sample | SiO2/Al2O3a (mol mol−1) | SiO2/Al2O3b (mol mol−1) | NH3 capacityb (mmol NH3 g−1) | Throughput before deactivation (gMeOH gcat−1) | TON (molMeOH molBAS−1) | C2=/C3=c (mol mol −1) | C2=/C2c (mol mol −1) | C3=/C3c (mol mol −1) | Coked (wt%) |
|---|---|---|---|---|---|---|---|---|---|
| a Determined from ICP. b Determined from NH3-TPD. c Calculated from Table S2 based on integral selectivities. d From TGA measurements of spent catalysts used at 450 °C. | |||||||||
| Sigma1-120-M | 120 | 117 | 0.281 | 1.3 | 144 | 0.7 | 21 | ≫103 | 8.1 |
| Sigma1-210-M | 200 | 207 | 0.160 | 2.0 | 391 | — | — | — | — |
| Sigma1-315-M | 399 | 314 | 0.105 | 2.5 | 740 | 1.0 | 39 | ≫103 | 10.5 |
| Sigma1-415-M | 724 | 417 | 0.080 | 2.5 | 982 | — | — | — | — |
| Sigma1-315-L | 406 | 317 | 0.105 | 1.6 | 478 | 0.8 | 34 | ≫103 | 9.6 |
| Sigma1-315-S | 420 | 317 | 0.105 | 3.6 | 1076 | 1.1 | 52 | ≫103 | 11.7 |
| Sigma1-315-S-m | 404 | 317 | 0.104 | 6.5 | 1944 | 1.0 | 62 | ≫103 | 12.1 |
| ZSM-58 | 110 | 111 | 0.296 | 4.5 | 476 | — | — | — | 11.8 |
Methanol throughput before deactivation (also referred to as MeOH uptake) is defined as overall MeOH throughput fed through the catalytic bed before the breakthrough of oxygenates (X ≈ 98%). Turnover numbers (TON) are defined as the total amount of MeOH converted per Brønsted acid site (BAS) before the conversion decreases below 98%. The amount of BAS was calculated from NH3-TPD. Ethylene/propylene (C2/C3) ratios (Table 1) are calculated for the experiments carried out at 450 °C taking into account integral selectivities.
Co-feeding experiments were performed keeping N2 flowrates equal to the ones used in MTO experiments and adding ethylene, propylene and/or water flows to obtain the desired mixtures.
Typically, after testing a catalyst for 5 h to ensure almost complete deactivation, the MeOH flow was stopped and the catalyst was cooled down under N2 flow. Later, the catalyst was transferred into a vial for further analysis of the trapped species.
3. Results
In this section the main properties and catalytic performance of the synthesized zeolites are described: (i) a series of Sigma-1 catalysts with different composition and particle size, (ii) a post-synthetically treated Sigma-1 catalyst and (iii) a series of ZSM-58 catalysts, produced with a different template.3.1. Synthesis and characterization of Sigma-1 zeolite
Powder XRD patterns shown in Fig. 1 together with reference peaks for DDR topology confirm that the synthesized catalysts consist only of pure DDR phase. SEM micrographs of zeolites synthesized with the same composition, but using different size of seeds are shown in Fig. 2. By using seeds with crystal sizes of 0.08, 0.5 and 2.5 μm, zeolites of 0.5, 2.5 and 4.6 μm with very narrow size distributions can be synthesized, respectively. The formed crystallites have a similar aspect ratio, with round slab shape. On the basis of their crystal size and NH3 TPD results (Tables 1 and 2), the zeolites are denoted as Sigma1-X–Y, were X refers to the actual SiO2/Al2O3 ratio (120, 210, 315, 415), while Y is related to crystal size (S – 0.5 μm; M – 2.5 μm; L – 4.6 μm). On the basis of the same nomenclature, the modified sample is denoted as Sigma1-315-S-m.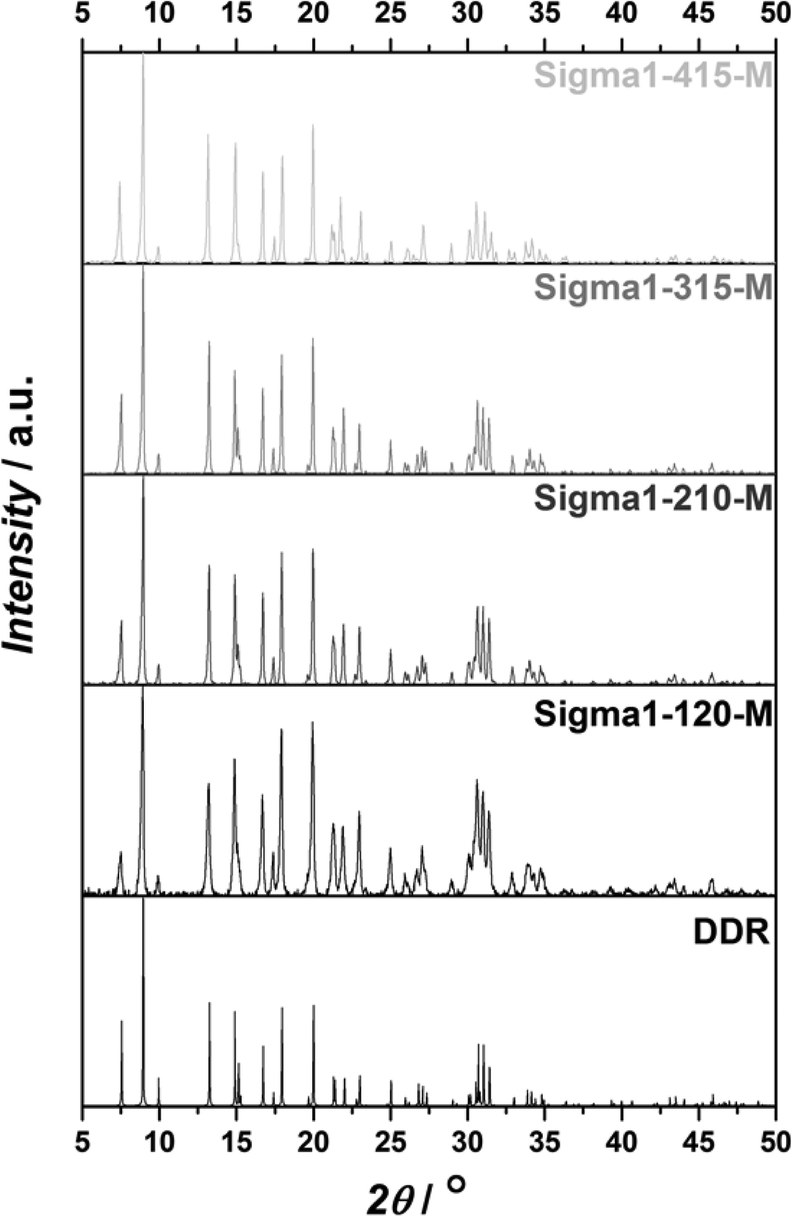 | ||
| Fig. 1 XRD patterns of calcined Sigma-1 zeolites compared with the DDR powder patterns. | ||
 | ||
| Fig. 2 SEM micrographs of (a) Sigma1-315-S (b) Sigma1-315-M (c) Sigma1-315-L with SiO2/Al2O3 = 315. | ||
| Sample | Particle sizea (μm) | S meso (m2 g−1) | S BET (m2 g−1) |
|---|---|---|---|
| a Determined from SEM. b t-Plot method. c BET plot applied to the N2 isotherm. | |||
| Sigma1-120-M | 2.7 | 20 | 313 |
| Sigma1-210-M | 2.7 | 14 | 349 |
| Sigma1-315-M | 2.4 | 7 | 364 |
| Sigma1-415-M | 2.5 | 5 | 371 |
| Sigma1-315-L | 4.6 | 12 | 340 |
| Sigma1-315-S | 0.5 | 15 | 357 |
| Sigma1-315-S-m | 0.5 | 51 | 403 |
| ZSM-58 | 0.6–1.8 | 22 | 372 |
When analyzing image of Sigma1-315-L (Fig. 2c), the observed intergrowth and aggregation infers an adhesive mechanism of growth, characteristic for the secondary growth mechanism taking place in the presence of seeds.33
The acidic properties of the synthesized materials were assessed by NH3 TPD (Fig. 3). For all samples, the desorption peak at high temperatures is assigned to the desorption of ammonia from Brønsted acid sites.9 Both the amount and the strength of acidity correlate well with the amount of Al in the zeolite, the more Al in the zeolite the more acid sites. Moreover, the maximum of the desorption peak shifts from 420 °C (Sigma1-120-M) to 375 °C (Sigma1-415-M), indicating the weakening of acid sites with decreasing Al content. Similar desorption temperatures for Sigma1-415-M, Sigma1-315-M and Sigma1-210-M indicate that their acid sites are isolated and possess a similar acid strength. The desorption peak of Sigma1-120-M is also characterized by a shoulder at 300 °C, indicating the presence of weaker Lewis acidity. Comparison of acidic properties of zeolites with similar composition, but different crystal size does not reveal any changes either in the amount of acid sites or in their acid strength. The observed mismatch in the amount of Al calculated from NH3 TPD and ICP for the high silica containing sample is associated to the small Al content, leading to a bigger error in the quantification.
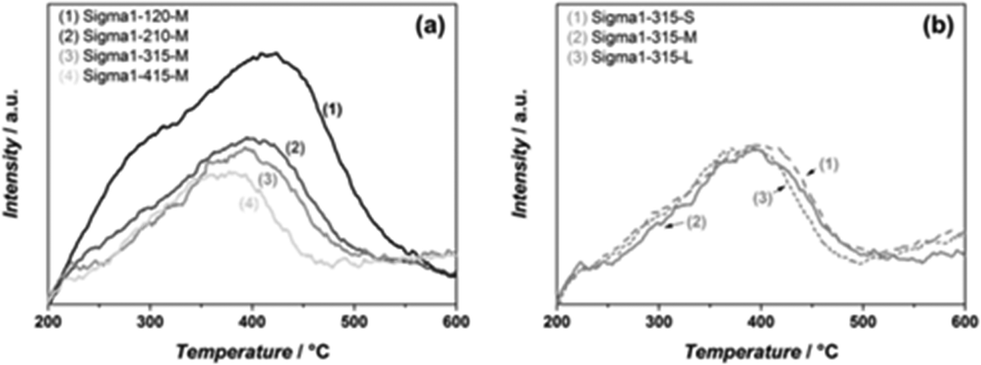 | ||
| Fig. 3 NH3 TPD profiles of Sigma-1 zeolites with different SiO2/Al2O3 ratio and medium crystal size (a), and of different crystal sizes with SiO2/Al2O3 ratio = 315 (b). | ||
Additionally, FT-IR spectroscopy of adsorbed CO (Fig. 4) was studied on Sigma1-120-M (a1, a2), ZSM-58 (b1, b2), Sigma1-315-S (c1, c2) and Sigma1-315-S-m (d1, d2) in order to assess surface groups and acid strength. The FT-IR spectra of pretreated samples show a complex absorption with a maximum at 3747–3745 cm−1 attributed to terminal Si–OH hydroxyl groups, located at the internal and external surface of the zeolite.34 Sigma1-315-S is also characterized by the presence of a triplet with maxima at 3747 cm−1 (HF), 3729 cm−1 (LF) and 3706 cm−1 (LF). The latter two represent vicinal silanol groups where only oxygen participates in H-bonding and the proton is relatively mobile.35 Further analysis reveals absorption bands around 3668 cm−1 for Sigma1-120-M and ZSM-58, pointing to the presence of extra-framework Al. An absorption band with a maximum at 3616 cm−1 is observed for all samples and represents Brønsted acidic OH groups. The broad band at about 3450 cm−1, which is especially prominent for Sigma1-120-M (Fig. 4a1) and Sigma1-315-S (Fig. 4c1), suggests the presence of silanol nests originating from structural defects.36 Upon interaction with CO, the bands associated with Brønsted acidity decrease at the same time as a band at ∼3300 cm−1 develops. The exact position of this band and shift in νOH is different for each sample: ΔνOH = 355 cm−1 (Sigma1-120-M), ΔνOH = 351 cm−1 (ZSM-58), ΔνOH = 313 cm−1 (Sigma1-315-S), ΔνOH = 317 cm−1 (Sigma1-315-S-m). The higher the SiO2/Al2O3 ratio, the lower is the ΔνOH, indicating lower acid strength, in agreement with NH3 TPD results.
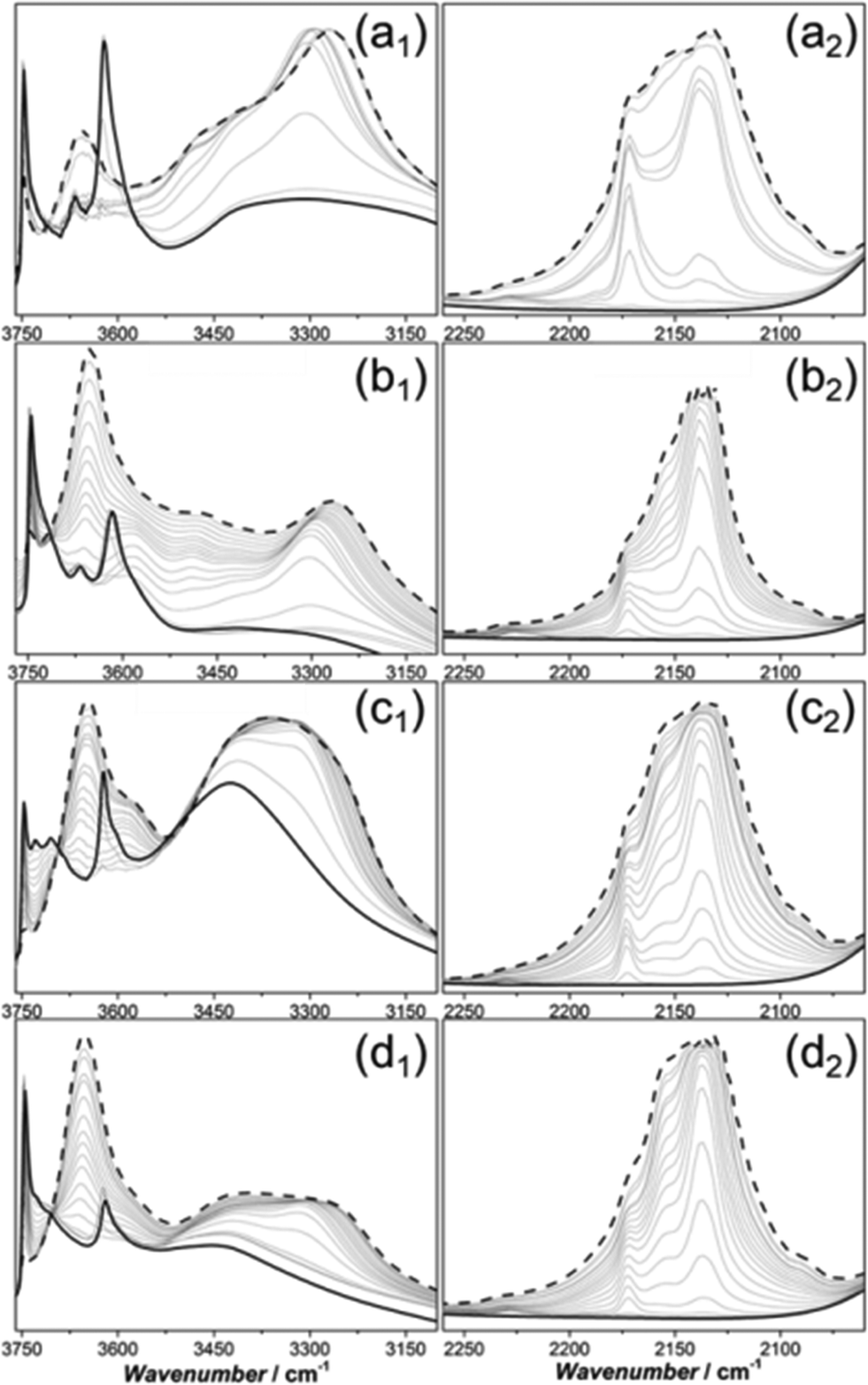 | ||
| Fig. 4 FT-IR spectra measured at −130 °C after adding increasing dosages of CO on (a) Sigma1-120-M; (b) ZSM-58; (c) Sigma1-315-S; (d) Sigma1-315-S-m. Panels a1, b1, c1 and d1 report the spectra in the hydroxyl group range (3760–3100 cm−1), while panels a2, b2, c2 and d2 report the CO vibrational mode region (2260–2060 cm−1). The bold solid line represents pretreated catalysts in vacuum, while the dashed black curves represent spectra collected at 30 mbar CO equilibrium. | ||
In the CO stretching region (Fig. 4, panels a2, b2, c2, d2), a band located at 2171 cm−1 appears at low CO coverage.17 This component arises from the interaction with strong Brønsted acid sites. At high CO coverage, the bands at about 2156 and 2138 cm−1 become more prominent, corresponding to CO interacting with silanol groups and liquid-like CO condensed in the zeolite pores, respectively. It should be also mentioned that the CO region is additionally characterized by the presence of components at 2227 cm−1, protuberant for Sigma1-120-M and ZSM-58 (Fig. 4, panels a2, b2). They are tentatively assigned to CO adsorbed on extraframework Al3+.37
3.2. Catalytic results
As also reported by Kumita et al.9 for DDR catalysts, no olefins bigger than C5 are observed when Sigma-1 is used as catalyst, independent of the applied reaction conditions. In addition, the formation of paraffins is very low, with ethane yields lower than 1% and some traces of propane (Table 1). Formation of DME is only observed once MeOH conversion drops below 100% becoming the main product at the catalyst deactivation stage. The overall product distribution of catalysts is reported in ESI† (Table S2).In the next paragraphs, the effect of reaction conditions on catalyst life-time and selectivity is extensively described.
 | ||
Fig. 5 Methanol conversion (a), yield of ethylene (b) and propylene at 380 °C ( , ,  , ,  ), 400 °C ( ), 400 °C ( , ,  , ,  ), 450 °C ( ), 450 °C ( , ,  , ,  ) and 475 °C (■, ▲, ♦) obtained in MTO reaction over Sigma1-315-M zeolite with SiO2/Al2O3 = 315 and crystal size of 2.5 μm at standard operation conditions. ) and 475 °C (■, ▲, ♦) obtained in MTO reaction over Sigma1-315-M zeolite with SiO2/Al2O3 = 315 and crystal size of 2.5 μm at standard operation conditions. | ||
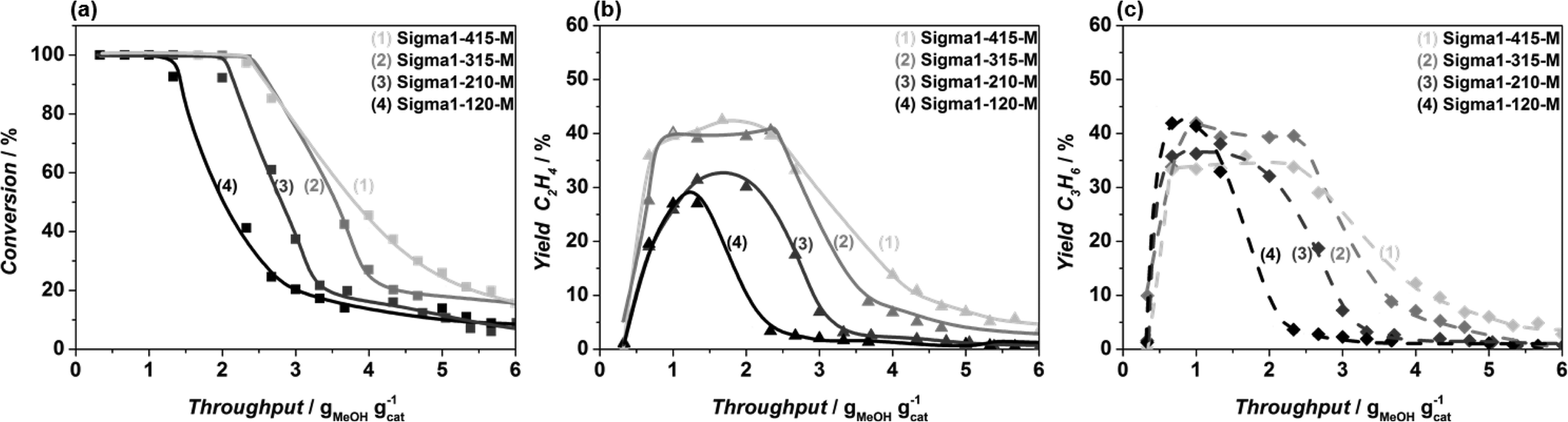 | ||
Fig. 6 Methanol conversion (a), yield of ethylene (b) and propylene at 450 °C, obtained in MTO reaction over Sigma1-415-M ( , ,  , ,  ), Sigma1-315-M ( ), Sigma1-315-M ( , ,  , ,  ), Sigma1-210-M ( ), Sigma1-210-M ( , ,  , ,  ), and Sigma1-120-M (■, ▲, ♦) zeolite at standard operation conditions. ), and Sigma1-120-M (■, ▲, ♦) zeolite at standard operation conditions. | ||
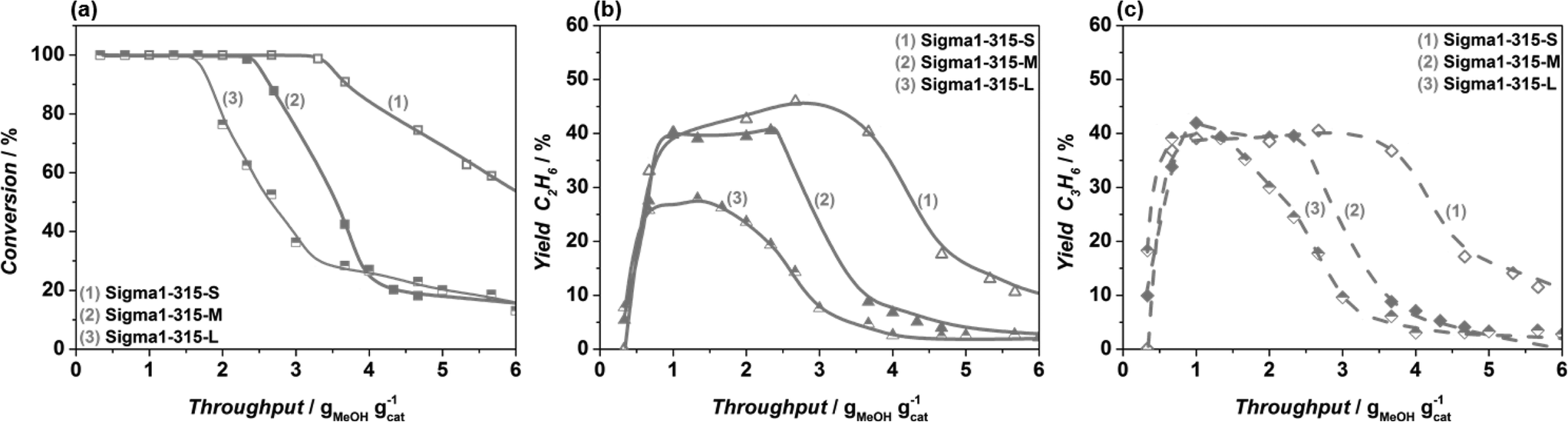 | ||
Fig. 7 Methanol conversion (a), yield of ethylene (b) and propylene at 450 °C, obtained in MTO reaction over Sigma1-315-S ( , ,  , ,  ), Sigma1-315-M ( ), Sigma1-315-M ( , ,  , ,  ), Sigma1-315-L ( ), Sigma1-315-L ( , ,  , ,  ) zeolite at standard operation conditions. ) zeolite at standard operation conditions. | ||
The catalytic performance of Sigma1-315-S-m, resulting from mild post-synthetic modification of Sigma1-315-S, shows a significantly increased overall MeOH throughput before deactivation from 3.6 to 6.6 g g−1 with a slight increase of propylene yield up to 45% (Fig. 8a).
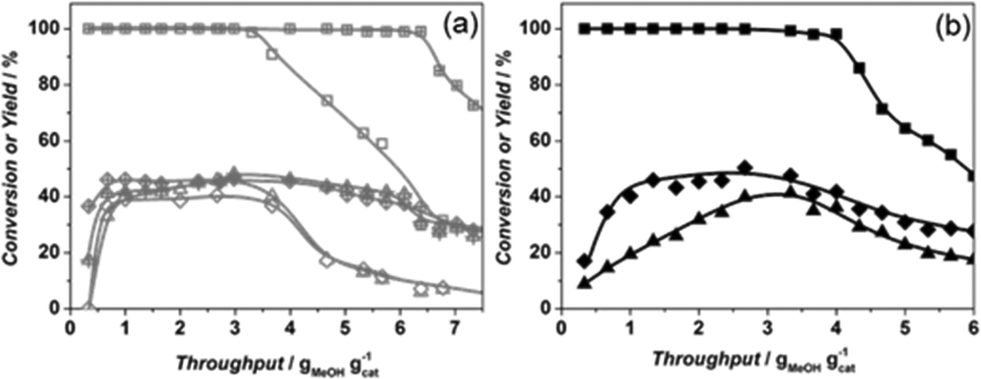 | ||
Fig. 8 Methanol conversion (■), ethylene (▲) and propylene (♦) yields of (a) Sigma1-315-S ( , ,  , ,  ) and Sigma1-315-S-m ( ) and Sigma1-315-S-m ( , ,  , ,  ); (b) ZSM-58 (■, ▲, ♦) (SiO2/Al2O3 = 110, 0.6–1.8 μm) at 450 °C and standard operation conditions. ); (b) ZSM-58 (■, ▲, ♦) (SiO2/Al2O3 = 110, 0.6–1.8 μm) at 450 °C and standard operation conditions. | ||
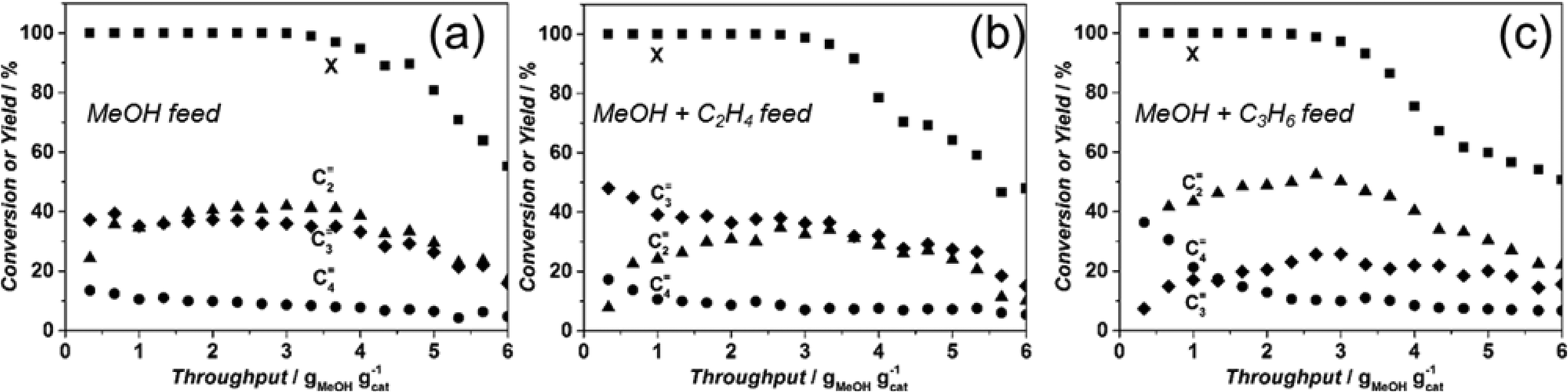 | ||
| Fig. 9 Methanol conversion (■), ethylene (▲), propylene (♦) and butenes (●) yields obtained over Sigma1-315-S tested at 450 °C and standard operating conditions (a) 50% MeOH + 50% N2; (b) 45% MeOH + 10% C2H4 + 45% N2; (c) 45% MeOH +10% C3H6 + 45% N2. | ||
This increase in propylene yield seems to be at the expense of ethylene formation. However, after having fed 2 g MeOH per gram of catalyst, the product distribution changes with decreasing propylene yield and increasing ethylene yield, becoming similar to the yields when only MeOH was fed (Fig. 9a and b). Co-feeding of propylene, however, affects mainly formation of butenes, especially during the first minutes of the reaction (40% vs. 20% without co-feeding). Nevertheless, the butenes yield rapidly drops to a constant level of 10%, while the ethylene yield reaches 53% at the expense of a lower formation of propylene (26%) suggesting, a higher probability of propylene to participate in consecutive reactions.
Furthermore, as was shown by Schulz,41 the reaction zone in a catalytic bed tends to migrate through the bed, i.e. first MeOH reacts in the first (upper) part of the catalytic bed, once the hydrocarbon pool is formed and this of the catalytic bed, i.e. feeding mixtures of ethylene and/or propylene with and without water. Layer has deactivated, the reaction front moves slowly downstream. Therefore it is essential to find out whether the formed olefins can undergo subsequent conversion in the absence of MeOH. For that reason, experiments were performed mimicking the bottom part.
Fig. 10(a and c) shows that feeding either ethylene or ethylene/water mixtures lead to a negligible conversion of around 2%, with the formation of propylene and butenes (80 and 20%, respectively). Addition of water only seems to maintain this ethylene conversion for longer periods (Fig. 10c). When propylene is fed in the absence of water, an initial conversion of 40% is observed, quickly dropping to 20% (Fig. 10b). The product stream in this case contains ethylene, butenes and C6 hydrocarbons. However, when water is co-fed together with propylene, simulating a situation much closer to that of the bottom bed during MTO operation, much lower initial conversions are observed (Fig. 10d). In this case, the initial conversion of 15% rapidly dropped to around 6% and stayed at the same level, with a product distribution being close to the one where only propylene was fed. Interestingly, apart from butenes and ethylene, the product composition also included some C5 and up to 45% C6 hydrocarbons, something not observed for the MTO experiments using catalysts with the DDR topology.
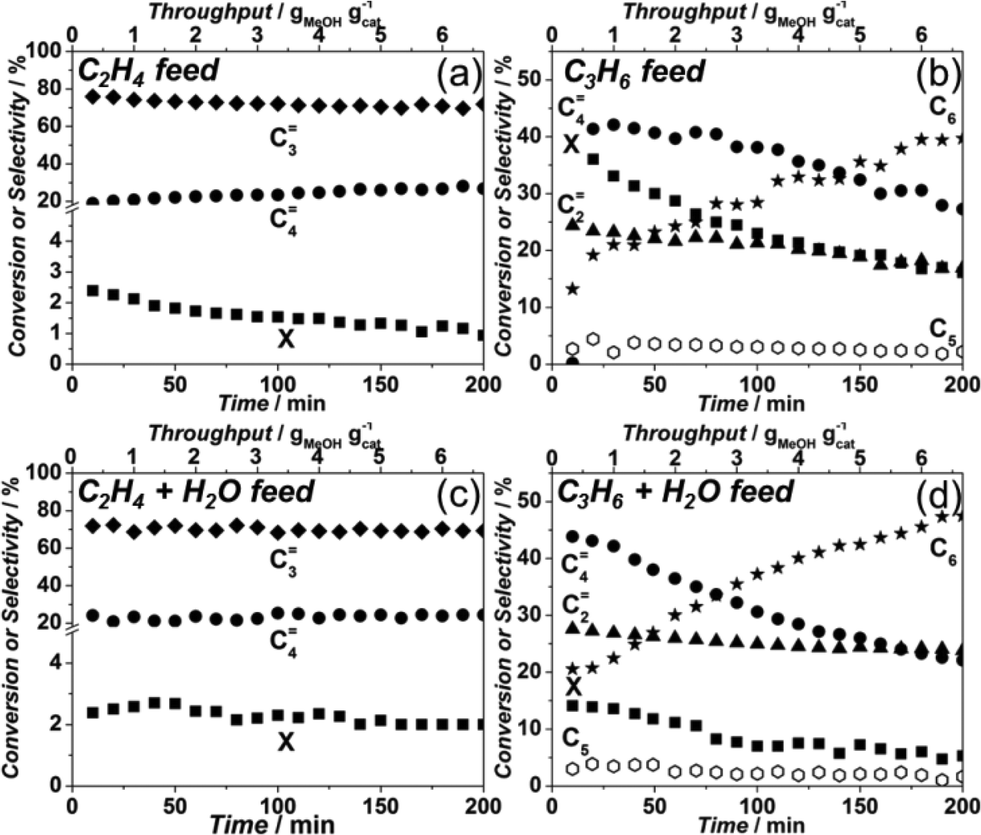 | ||
| Fig. 10 Product distribution in co-feeding experiments performed over Sigma1-315-S at 450 °C (a) 10% C2H4 + 90% N2; (b) 10% C3H6 + 90% N2; (c) 45% H2O + 10% C2H4 + 45% N2; (d) 45% H2O + 10% C3H6 + 45% N2. | ||
After feeding propylene, the catalyst was subjected to the MTO reaction at standard operating conditions to see whether the C5 and C6 hydrocarbon formation was indicative for catalyst deactivation. However, the observed catalyst lifetime and selectivities were identical to the ones shown on the Fig. 9a in an ordinary MTO experiment, indicating that these C5 and C6 species are formed on the external surface of the zeolite and do not affect coke formation related to MTO inside the zeolite cage.
Finally, as the product mixture obtained from Sigma1-315-S tested at 450 °C consisted mainly of ethylene and propylene, also an experiment co-feeding ethylene, propylene and water was performed to mimic a situation where olefins can react with each other in absence of MeOH. Fig. 11 shows that although initially the product mixture contained equal molar amounts of ethylene and propylene, both decreased upon formation of butenes and some very small amounts of C5 and C6 products. Initially, ethylene was consumed more than propylene in this experiment, and their conversion gradually declined until the composition returned to that of the feed mixture. The amount of butenes formed followed the ethylene and propylene conversion. Considering the converted amounts suggest only a minor participation of ethylene and propylene in consecutive reactions.
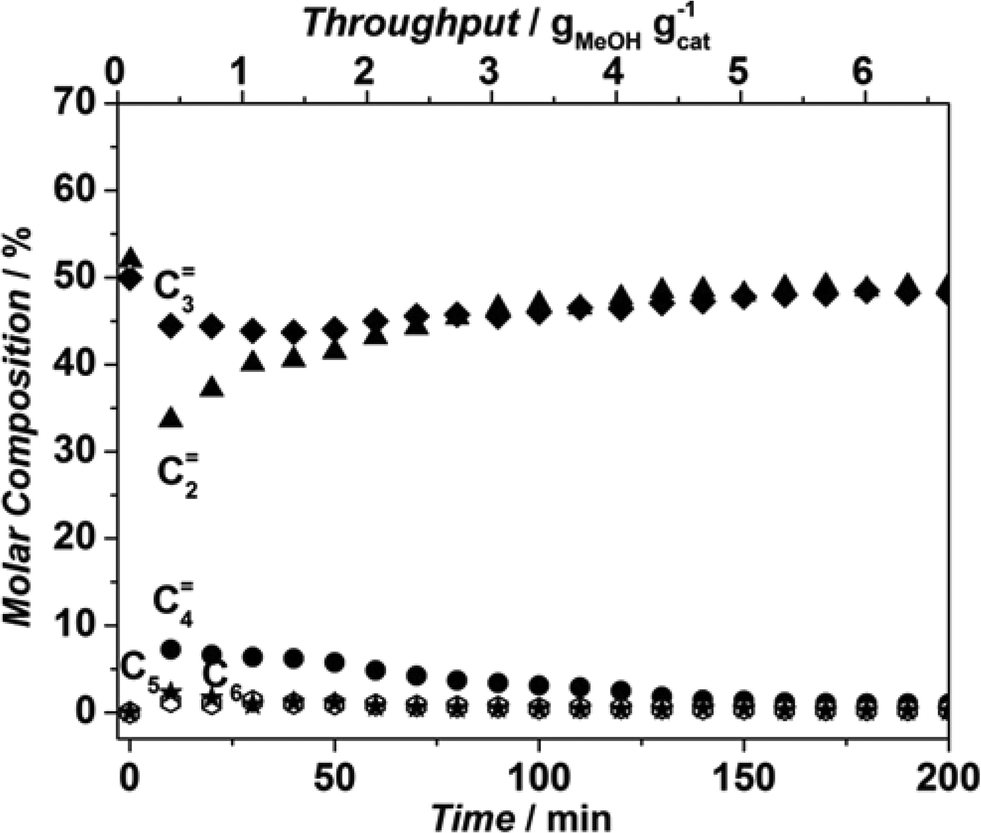 | ||
| Fig. 11 Product distribution in co-feeding C2H4 and C3H6 in the presence of water over Sigma1-315-S at 450 °C. | ||
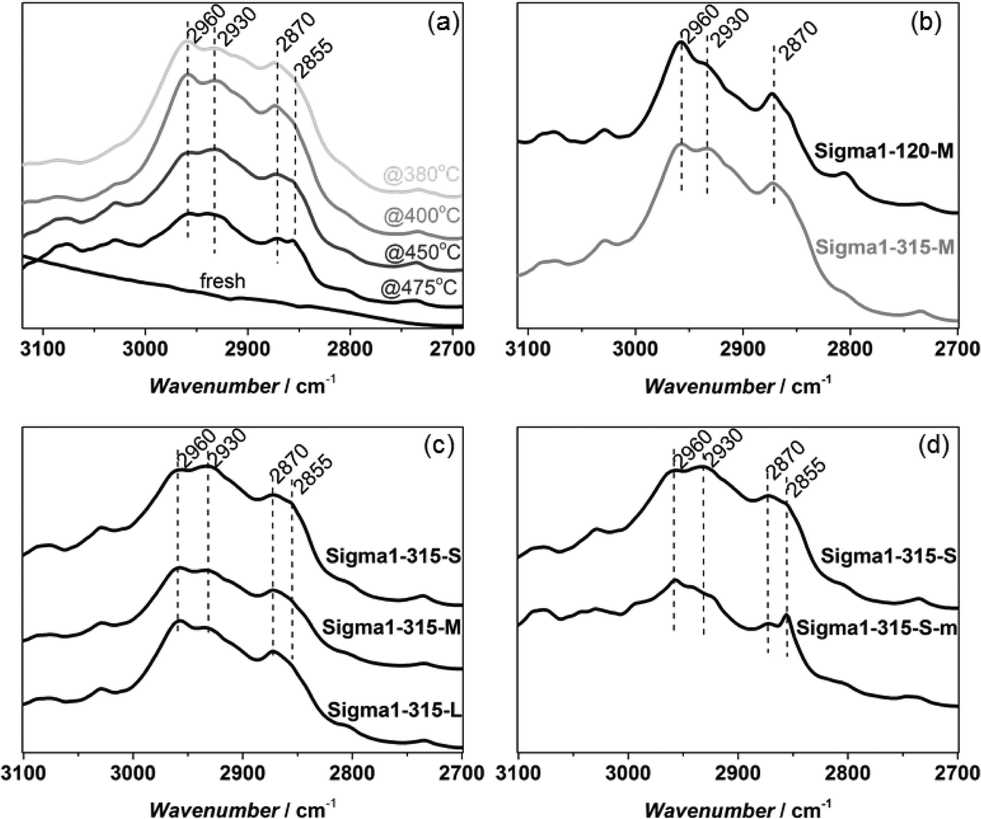 | ||
| Fig. 12 DRIFT spectra of spent Sigma1-315-S catalyst used at different temperatures (a); Sigma1-120-M and Sigma1-315-M at 450 °C (b); Sigma1-315-S, Sigma1-315-M and Sigma1-315-L at 450 °C (c); Sigma1-315-S and desilicated Sigma1-315-S-m at 450 °C (d). | ||
Bands at around 2960 cm−1, 2930 cm−1 and 2870 cm−1 are present in all deactivated samples. For spent catalyst used at 380 °C and 400 °C (Fig. 12a) the band at 2960 cm−1 is more intense than the one at 2930 cm−1. However, it steadily decreases with increasing reaction temperature and already at 450 °C the band at 2930 cm−1 is dominant. Fig. 9b and c show that the ratio between the intensities of bands at 2930 cm−1 and 2960 cm−1 depend on acidity and crystal size. The latter band is more pronounced in samples with higher acidity (Sigma1-120-M) and large crystal size (Sigma1-315-L). The spectra obtained for the spent desilicated Sigma1-315-S-m are different from the non-modified samples: an additional band appears at 2856 cm−1 and the band at 2960 cm−1 shifts to 2958 cm−1. Both bands at 2960 cm−1 and 2930 cm−1 are ascribed to the C–H stretching from methyl group in methylated benzenes and/or naphthalenes in the pores of the deactivated zeolites.42 The presence of bands in the region of 2960–2930 cm−1 shows the presence of aromatics with different substituents indicating different formed species depending on the acidity and morphology of the catalyst as well as the testing conditions. The broad band at 2930–2925 cm−1 is characteristic for highly alkylated benzenes, naphthalenes and phenanthrenes.42 The band at 2870 cm−1 represents asymmetric stretching of the C–H bond from methyl group CH(CH3),43 while at 2855 cm−1 C–H from methanol or DME.44
Additionally, a band at ∼3030 cm−1 could be identified in all catalysts, becoming more prominent in samples deactivated at temperatures higher than 400 °C. Bands in this region are attributed to aromatic C–H stretching vibrations, implying the presence of partially substituted aromatics.45
The region at 1250–1700 cm−1 (Fig. S2 in ESI†) is characterized by bands (1600 and 1567 cm−1) already described for deactivated DDR samples.9 In line with Kumita et al. observations, also a broad band at around 1600 cm−1 was observed, which is a combination of carbonaceous hydrogen deficient deposits46 and aromatic ν (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) modes.47 The intensity of the band at 1567 cm−1 varies from sample to sample, being the highest for the one with the highest acidity (Sigma1-120-M) indicating a higher concentration of alkylnaphthalenes and phenanthrene and pyrene structures48 trapped in the cages.
C) modes.47 The intensity of the band at 1567 cm−1 varies from sample to sample, being the highest for the one with the highest acidity (Sigma1-120-M) indicating a higher concentration of alkylnaphthalenes and phenanthrene and pyrene structures48 trapped in the cages.
The spent catalysts chosen for DRIFTS were further analysed by dissolving the samples in HF, followed by extraction with CH2Cl2 and analysis of the extract with GC–MS. Fig. 13 shows that the nature of retained species is dependent on both reaction conditions and physicochemical properties of the zeolites with naphthalenic species being dominant. The fraction of methylbenzenes was found to be negligibly small or even completely absent in some cases. Panels (a)–(d) show that the organic residue in the spent catalysts is characterized by mono-alkylated naphtalenes, being the main fraction for all samples except for Sigma1-315-S used at 380 °C, the lowest operation temperature. Interestingly, the main fraction of trapped species in this sample is represented by diadamantane, product of adamantane condensation. These species could also be found in all other deactivated samples, but their fraction significantly decreases with increasing reaction temperature. The formation of diamondoid hydrocarbons in cages of 8MR zeolites at lower temperatures was recently reported by Wei et al.49 Chen et al.50 also attributed the formation of adamantane hydrocarbons to spatial constraints imposed by the cage structure and attributed prompt deactivation at low temperatures to preferential formation of adamantanes over methylbenzenes. Further analysis of the organic residue in spent Sigma1-315-S reveals the formation of pyrene species, with an increased contribution at higher reaction temperatures.
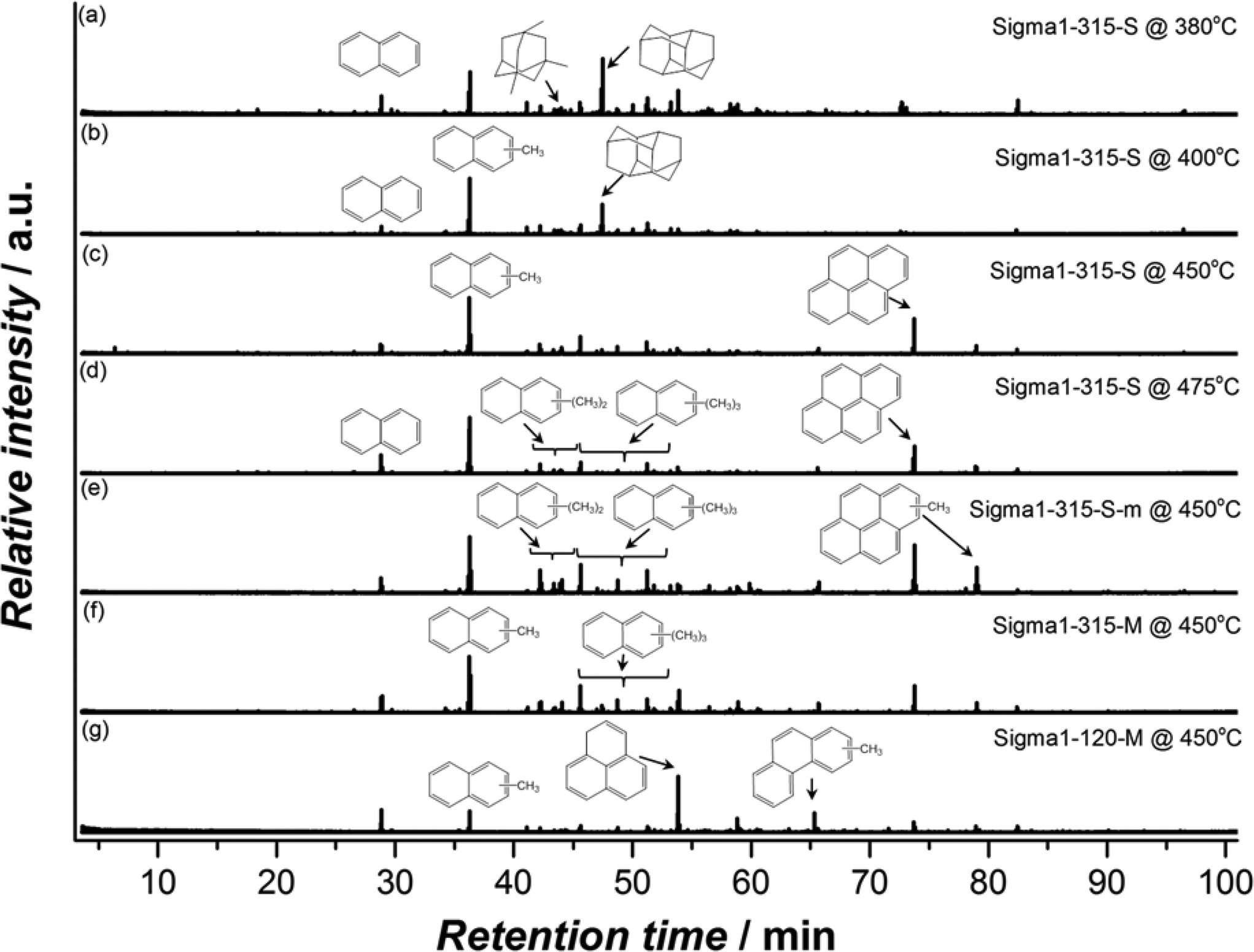 | ||
| Fig. 13 GC–MS chromatograms of the hydrocarbon compounds retained in the various spent (deactivated) Sigma-1 zeolites used at different temperatures. | ||
In contrast with Sigma1-315-M, species trapped in Sigma1-120-M deactivated at 450 °C are mostly represented by phenalene and phenanthrenic species and almost no alkylated naphthalene derivatives. Comparison of zeolites with the same acidity but different crystal size (Sigma1-315-S vs. Sigma1-315-M) does not reveal any difference in the species found in the spent catalysts.
The total amount of retained organic material analyzed by TGA is observed to be dependent on the amount of acid sites and crystal size. The amount of coke retained in zeolite cages increases from 8.1 to 10.5 wt% for samples with SiO2/Al2O3 = 120 and 400, respectively. Moreover, for samples with SiO2/Al2O3 = 400 the coke content measured after use at 450 °C varies from 9.6 to 11.7 wt% for samples with crystal size 4.6 and 0.5 μm, respectively. However, the amount of coke linearly increases for catalysts having a MeOH throughput before deactivation of less than 4 g g−1, reaching 12 wt% for materials that converted more than 4 g MeOH per gram catalyst, indicating that the catalytic improvement is due to the slowing down coke formation kinetics or due to the changing chemistry of the species itself (Fig. S3†).
4. Discussion
The MTO process is most probably among the most challenging catalytic processes from a fundamental perspective. In spite of the large number of investigations, still many questions regarding mechanism and reactivity are to be answered. One of the most striking observations is the difference in catalytic behavior among zeolite topologies. In case of 8MR zeolites, the carbon pool mechanism seems to dominate the formation of products.6 The nature of this mechanism, which proceeds via the formation of aromatics that obviously do not fit through small pore zeolites, makes catalyst testing rather challenging. Indeed, classical kinetic testing at low conversion levels, in contrast to recent examples with ZSM-5,38,51,52 is not possible when 8MR zeolites are used. Inspired by these recent examples, we have performed experiments at varying space velocities. In every case, 100% conversion was reached and similar throughputs before deactivation, in clear contrast to what was observed in case of ZSM-5. These experiments point at a much higher reactivity of 8MR zeolites and to the fact that kinetic control cannot be achieved during catalyst testing. As a consequence, comparison between different catalysts has traditionally been done at 100% conversion, see for example the review article of Stöcker2 about the comparison of different zeolites.A logical consequence of catalyst testing at 100% conversion is the appearance of the so-called migrating reaction zone, first proposed by Schulz.41 Along this line, one can envisage the existence of at least two different zones in the catalytic bed, namely: the part of the bed where methanol is still present and the part of the bed where primary MTO products (short-chain olefins and water) participate in secondary reactions in the absence of methanol. This phenomenon might imply the dependence of the catalyst lifetime on the ratios of these zones in the catalytic bed and makes mechanistic discussions based on such testing conditions rather doubtful, as demonstrated for ZSM-5 over the last few years.38,51,52
With this background, the assumption of a similar situation in case of 8MR zeolites is not straightforward. The migrating reaction zone considers that reaction products will diffuse into zeolite pores in the bottom part of the reactor and react further. This is indeed something feasible in case of 10MR or 12MR zeolites, where diffusivities of coefficients at reaction temperature for i.e. propylene have been estimated to be in the range of 10−8 m2 s−1. In contrast, for 8MR zeolites, such as CHA or DDR, diffusivities of at least two orders of magnitude smaller (10−10 m2 s−1) have been reported for propylene.53,54 When considering these numbers, one can anticipate that reaction of primary products in the bottom part of the reactor will be diffusion limited (i.e. to achieve half saturation capacity for propene of a 1 μm DDR crystal would take circa 20 minutes under reaction conditions, considering that there is no co-adsorption of other products like water). The co-feeding experiments confirm this hypothesis. On one hand, the presence of additional ethylene or propylene together with methanol does not alter the catalyst lifetime, pointing to a negligible interaction of formed olefins with coke precursors, which are of the same nature based on the results obtained from DRIFTS. Considering further the stage in the catalytic bed where the main products, ethylene, propylene and mixtures thereof, can undergo consecutive reactions in the presence of water, Fig. 10 indicates a negligible conversion of ethylene and a conversion of propylene with the production of butenes and C6 hydrocarbons. The appearance of longer hydrocarbons, which are not able to diffuse out of the DDR structure due to their big kinetic diameter,55 indicates that they are formed on the outer surface of the crystals and that propylene reacts mostly at the surface of the particles as it does not have enough time to diffuse into the pores of Sigma-1, while the less reactive ethylene passes through the reactor basically without being converted. Thus, on the outer surface of the crystals, propylene dimerization to C6 and cracking of C6 to C4 and C2 is taking place, both reactions being catalyzed by external acid sites.56
These results demonstrate, that a mechanistic analysis, especially when referring to coke formation in small pore systems (8MR), can, with a large degree of confidence, be made based on results obtained at 100% conversion. Clearly, in order to fully assess selectivity to different products, co-feeding experiments should be performed.
Coming back to the catalyst itself, the Sigma-1 zeolite with DDR topology is a noteworthy nominee among 8MR zeolites displaying outstanding selectivities to light olefins (i.e., propene) and hardly any formation of paraffins (Table 1), something highly relevant from the application perspective. Indeed, the selective formation of olefins means large savings as the separation of short olefin/paraffin mixtures is among the most energy intensive processes in chemical industry.57
Despite the fact that the synthesis of Sigma-1 zeolite was patented in 1987 by Stewart,11 the number of publications dealing with this material is rather low. This is probably due to the long synthesis and difficult reproducibility of materials with DDR topology, as extensively discussed by Gücüyener et al.14 However, the application of as-synthesized material as seeds does not only accelerate the synthesis13 but also leads to the pure phase with narrow crystal size distribution. The seed inductive effect described by Yu et al. for ZSM-5 (ref. 58) explains the role of seeds as a recognition effect, promoting nucleation of alike building units and acting as nuclei for the growth of zeolite crystals.
As already observed for ZSM-58,9 Sigma-1 displays up to 90% selectivity to ethylene and propylene almost without any propane being formed. The observed behavior lies in the unique structure of DDR cage and pore opening with dimensions of 4.4 × 3.6 Å, excluding molecules with kinetic diameter bigger than the window.15,55
The ethylene-to-propylene ratio of the product mixture could be tuned by both varying reaction conditions and catalyst composition and morphology. As a general observation, the parameters leading to the highest selectivity to ethylene also lead to the lowest selectivity to propylene, already pointing at competing reaction pathways to form these molecules.
At lower temperatures, the formation of propylene is favored over ethylene, while the MeOH throughput before deactivation is two times smaller. According to the generally accepted dual-cycle mechanism for the MTO process,59 the observed behavior is tentatively attributed to kinetic competition between the two cycles. First principle chemical kinetics calculations for the methylation of ethylene and propene show much higher activation energies for ethylene methylation pathway and hence methylation rates are one order magnitude higher when comparing ethylene and propylene.60 Quick catalyst deactivation observed at lower temperatures could be ascribed to the formation of diamondoid species. On the basis of literature evaluation49,50 and our own observations, one can suggest that at lower reaction temperatures 8MR zeolites with a cage-like structure promote formation of adamantane derivatives which are inert and lead to cage blocking. Their formation is probably favored due to the chemical and structural similarity to the SDAs (interaction of SDA with a cage is stable a priori) which points to the relatively low entropy contribution to the free energy barriers leading to high reaction rates.61 Higher reaction temperatures promote the cracking cycle together with elimination of ethylene from alkylated aromatics, but at this stage pyrene species (Fig. 13) start contributing to catalyst deactivation as these larger aromatic structures do not leave space for functionalization. The prevalence of dealkylation and/or cracking reactions at high temperatures was demonstrated by Qian et al.,62 who showed kinetic and mechanistic differences occurring in hydro-carbon pool at high and low temperatures by applying the combination of UV/Vis, confocal fluorescence, and synchrotron-based IR micro-spectroscopic techniques. It has to be mentioned as well that the presence of phenanthrene and pyrene species (Fig. 13) is surprising, as at most naphthalene derivatives fit in a DDR cage. This may indicate that polycyclic species should have been formed on the outer surfaces of the zeolite and/or in defects.17
The Si/Al ratio is another parameter that affects both MeOH throughput before deactivation of the catalyst and selectivity to light olefins. Application of zeolites with SiO2/Al2O3 = 120, which corresponds to one Al per cage of zeolite, leads to a fast deactivation, while the SiO2/Al2O3 = 315 with 0.5 Al atoms per cage gives the best results, in terms of MeOH uptake and ethylene and propylene yields. NH3 TPD results demonstrate that the Al content defines not only the amount but also the strength of acid sites. Application of zeolites with moderate acidity, characterized by an NH3 desorption peak in the range of 375–385 °C significantly increases MeOH throughput before deactivation. Interestingly, FT-IR spectroscopy and dissolution experiments demonstrate that, depending on the strength of acidity, the formed ‘coke’ species are different, the presence of methylbenzenes/methylnaphthalenes being more prominent present in spent Sigma-415-S, the zeolite with the lowest acidity. Moreover, the maximum concentration of methylbenzenes and methylnaphthalenes coincides with the maximum yield of ethylene, confirming the hypothesis that ethylene originates from the aromatic cycle. Furthermore, analysis of coke species formed over more acidic Sigma1-120-M shows the lowest amount of methyl-substituted aromatics corresponds to the lowest yield of ethylene than comparing Sigma1-120-M with other samples.
The observed findings regarding the effect of Si/Al ratio are in contradiction with the ZSM-58 results, where the best catalytic performance was found for zeolites with SiO2/Al2O3 = 110 and a further decrease in acidity (higher ratios) led to an abrupt loss of activity.9 However, in case of ZSM-58 the decreasing acidity was accompanied by an increased particle size, distorting the effect of acidity by additional diffusional limitations. In general, literature evaluation confirms that the MTO reaction requires application of zeolites with moderate to low acidity,48,63 but the catalytic activity at such high Si/Al ratios was, to the best of our knowledge, never reported before. According to Guisnet,64 a faster catalyst deactivation with higher Al content is related to a high density of acid sites, which provokes a higher interaction probability between intermediates, leading to faster formation of coke. TGA analysis of spent catalysts with different acidity shows that the most acidic zeolites contain the lowest amount of coke (8.1 wt% vs. 10.5 wt%), suggesting either a different nature of the coke species or a faster pore blockage in case of more acidic zeolites. Considering that the nature of occluded species for samples deactivated at the same temperature seems to be similar, vide supra, we speculate that more acidic Sigma-1 catalysts deactivate faster due to the preferential location of carbonaceous species in close proximity to the 8MR windows rather than inside the zeolite cavities, leading to a faster blockage of the 2D porosity.65
To confirm that differences observed for ZSM-58 are mainly due to a crystal size effect rather than to differences in the SiO2/Al2O3 ratio, the catalytic performance of zeolites with 3 different crystal sizes with the SiO2/Al2O3 = 315 ratio was investigated, while maintaining the same synthesis precursor gel composition. The comparison of these results with contradictory literature is not trivial. The absence of crystal size effect on overall selectivity to light olefins,66 an increased olefins selectivity with crystal size reduction,40,67 and an increasing propylene selectivity and decreasing ethylene selectivity when applying particles with a smaller crystal size39 have been reported by different authors in case of SAPO-34. In all cases the observed effects were attributed to an increasing catalyst effectiveness with decreasing particle size, i.e. the extent of particle utilization and/or shortening of diffusion pathway of products preventing their further conversion to undesired compounds in consecutive reactions.68
In contrast, Khare et al.51 have shown that for zeolites with MFI topology the increase of the light olefins selectivity was associated with an increase of the particle size due to propagation of aromatic-based catalytic cycle, i.e. an increased amount of methylbenzenes responsible for ethylene formation. In case of the DDR catalysts, based on the performed co-feeding experiments, we can conclude that the increase in ethylene selectivity with decreasing particle size is due to the higher external surface to volume ratio of the smaller particles. The co-feeding experiments undoubtedly demonstrate that propylene is prone to react in the external surface region of these particles, while the less reactive ethylene would pass the bottom part of the catalyst bed almost unaltered. In view of these results, we tentatively explain differences observed in the literature for SAPO-34 and ZSM-5 by different reactivity of surface sites and diffusion properties of these zeolites.
An increased MeOH uptake for smaller particles is attributed to the shortening of the diffusion pathway of reactants and products and related to the accessibility of the crystals during MTO. Qian et al.62,69 mapped the distribution of Brønsted acid sites and retained organic species in SAPO-34 and SSZ-13 50 μm in size crystals by applying synchrotron-based IR micro-spectroscopy. The 2D map shows that the concentration of alkylated aromatics is non-homogenous with the lowest concentration profile in the central part of the crystal, i.e. it is not utilized in the MTO reaction. The observed findings go in line with the proposed mechanism, namely firstly formed methoxy species are homogenously distributed in the crystals, while secondly formed aromatics alter the accessibility of the crystal, being mainly formed at the outer rim of the particle, blocking the core. The smaller the crystal the bigger its accessible area, which is confirmed by a quantitative coke analysis: smaller crystals hold around 11.7 wt% of coke, in contrast to bigger crystals, with a coke content of 9.6 wt%. Higher concentration of aromatics, in turn, leads to the higher amount of produced ethylene, which goes in line with our observations. This is further confirmed by the fact that, according to the GC–MS results (Fig 13(c) and (f)), the nature of organic species found in the deactivated catalysts is independent of crystal size.
Optimization of crystal size and acidity in Sigma-1 zeolites led to a significant improvement of MeOH throughput before catalyst deactivation. However the maximum MeOH uptake obtained for Sigma1-315-S still did not outperform the performance of ZSM-58 with an acidity comparable to Sigma1-120-M, confirmed by CO FTIR (Fig. 4a1 and b1) and NH3 TPD (Fig. S4 in ESI†). FT-IR spectra of pretreated samples show that Sigma-1 catalysts (Fig. 4a1 and c1) are characterized by the presence of a broad band assigned to silanol nests. In contrast, ZSM-58 samples do not contain these species (Fig. 4b1). According to Bordiga and co-workers,34 these nests could be eliminated by treating the sample at higher temperatures, but the fact that all as-synthesized zeolites were calcined at the same temperature, while during the CO FT-IR experiment they were pretreated under the same conditions points to their stability in case of Sigma-1. As the presence of silanol nests is the only difference observed for Sigma-1 and ZSM-58, we tentatively suggest that they are responsible for the increased coke formation rate and faster catalyst deactivation in case of Sigma-1. Moreover, FT-IR spectra of the desilicated Sigma1-315-S-m is characterized by significantly decreased intensity of the band at 3450 cm−1 in comparison with the parent Sigma1-315-S, implying that mild treatment in NaOH/CTAB solution led to the selective removal of Si from nests of hydrogen bonded silanol groups, leading to the improved catalytic performance. This modification only led to an increase in mesoporous surface area (15 m2 g−1vs. 52 m2 g−1, Fig. S5†), suggesting that internal defects were healed via Si rearrangement inside the zeolite.
Results reported by Barbera et al., confirm our observations.26 Recently, these authors showed that the deactivation of a series of ZSM-5 catalysts correlated with the relative intensities of silanols rather than with the amount of acid sites, suggesting possible “retention effects” of silanols on coke precursor that lead to a faster activity loss.
The presence of a larger number of defects in the Sigma-1 catalysts can be related to the fact that secondary growth was used for the synthesis of these solids, since faster crystal growth might lead to the formation of more defects either at the interphase seed-crystal or on the whole crystal lattice. Independently of the reason, the current results demonstrate the importance of lattice defects on the deactivation of zeolites during the MTO process. This fact, hardly considered in literature,70 has a much bigger importance than many other parameters normally studied and will certainly be key for the large scale implementation of MTO processes, since both industrial catalyst production and continuous regeneration may lead to the formation of additional lattice defects that will eventually affect the rate of coke formation.
In general, two-dimensional ZSM-58 and Sigma-1 zeolites with DDR topology exhibit catalyst lifetimes typical from the 8MR family,50,71,72 which are much shorter than for ZSM-5 belonging to 10MR.38,52 Catalyst lifetime expressed in grams of MeOH converted per gram of catalyst for these zeolites does not exceed 10 g g−1, CHA being an exception. It should be also mentioned, that other 2D 8MR like UFI71 or LEV50 show even shorter lifetimes in comparison with 2D DDR. A faster deactivation of 2D 8MR is related to the higher probability of blocking the pores in one direction in comparison with 3D 8MR zeolites. As was already stated above, the unique properties of 8MR zeolites originate from the cage and window dimensions, determining their catalytic/separation properties. Bhawe et al.72 have shown that ethylene selectivity increases with decreasing cage size, the highest ethylene yield being obtained for LEV having the smallest cage dimensions. In general, ethylene and propylene are the dominant products for 8MR zeolites, but formation of products up to C6 is also possible.50 From this perspective, DDR zeolites with very small pore openings have an advantage in comparison with other 8MR zeolites due to the negligible formation of paraffins and no products higher than C4.
Conclusions
The results here presented confirm the high selectivity of zeolites with the DDR topology for the formation of ethylene and propylene (up to a 90%) when used as catalysts in the MTO process. Such high selectivities are the result of an ideal combination between cage geometry and window size that allows only C2 and C3 olefins to leave the cages of this zeolite. From an application perspective, this size restriction is very important. Since pure C3 olefin is formed, the separation of products becomes much less energy intensive.One of the crucial parameters for catalyst stability is the overall zeolite acidity and strength. It was found that decreasing acidity of the zeolite from SiO2/Al2O3 = 120 to SiO2/Al2O3 = 315 leads to a two-fold increase in methanol throughput before full deactivation occurs. Furthermore, a decrease in the crystal size of Sigma-1 zeolite with optimized acidity led to almost two times improved throughput, ascribed to an improved utilization of the 2D porosity of this material.
Secondary reactions of the olefinic products occurred hardly, and only at the outer surface of the zeolite crystals. These processes did not affect the activity for the MTO reaction.
The influence of internal framework defects on MTO performance has been shown to be of the upmost importance. The presence of a larger number of silanol nests in Sigma-1 resulted in a faster deactivation than in case of ZSM-58, where such defects were hardly observed. According to our results, the presence of structural defects leads to a faster coke formation kinetics and therefore to a rapid deactivation. Upon removal of silanol nests from Sigma-1, the catalytic performance becomes comparable to the one of ZSM-58. Indeed, the presence of structural defects has a much bigger importance than many other parameters usually studied and will certainly be key for the large scale implementation of MTO processes based on these materials, since both industrial catalyst production and continuous regeneration may lead to the formation of additional crystal defects that will eventually influence coke formation rate.
Acknowledgements
This research received funding from The Netherlands Organisation for Scientific Research (NWO) in the framework of the TASC Technology Area “Syngas, a Switch to Flexible New Feedstock for the Chemical Industry (TA-Syngas)”.Notes and references
- J. S. Plotkin, Catal. Today, 2005, 106, 10–14 CrossRef CAS.
- M. Stöcker, Microporous Mesoporous Mater., 1999, 29, 3–48 CrossRef.
- F. J. Keil, Microporous Mesoporous Mater., 1999, 29, 49–66 CrossRef CAS.
- H. Ito, K. Honda, K. Oyama, N. Chikamatsu, K. Hiraoka and A. Okita, US 8530714 B2, Jgc Corporation, 2013 Search PubMed.
- J. Q. Chen, A. Bozzano, B. Glover, T. Fuglerud and S. Kvisle, Catal. Today, 2005, 106, 103–107 CrossRef CAS.
- I. M. Dahl and S. Kolboe, Catal. Lett., 1993, 20, 329–336 CrossRef CAS.
- I. M. Dahl and S. Kolboe, J. Catal., 1994, 149, 458–464 CrossRef CAS.
- C.-M. Wang, Y.-D. Wang, Y.-J. Du, G. Yang and Z.-K. Xie, Catal. Sci. Technol., 2016 10.1039/C5CY01419K.
- Y. Kumita, J. Gascon, E. Stavitski, J. A. Moulijn and F. Kapteijn, Appl. Catal., A, 2011, 391, 234–243 CrossRef CAS.
- H. Gies and B. Marler, Zeolites, 1992, 12, 42–49 CrossRef CAS.
- A. Stewart, GB2193202-A; GB2193202-B, Imperial Chem Ind Plc, 1988 Search PubMed.
- E. W. Valyocsik, US4709114 A, CA1286321-C; JP95068046-B2, Mobil Oil Corp, 1987 Search PubMed.
- J. Gascon, W. Blom, A. van Miltenburg, A. Ferreira, R. Berger and F. Kapteijn, Microporous Mesoporous Mater., 2008, 115, 585–593 CrossRef CAS.
- C. Gücüyener, J. van den Bergh, A. M. Joaristi, P. Magusin, E. J. M. Hensen, J. Gascon and F. Kapteijn, J. Mater. Chem., 2011, 21, 18386–18397 RSC.
- W. Zhu, F. Kapteijn, J. A. Moulijn, M. C. den Exter and J. C. Jansen, Langmuir, 2000, 16, 3322–3329 CrossRef CAS.
- W. Zhu, F. Kapteijn, J. A. Moulijn and J. C. Jansen, Phys. Chem. Chem. Phys., 2000, 2, 1773–1779 RSC.
- F. Bleken, M. Bjorgen, L. Palumbo, S. Bordiga, S. Svelle, K.-P. Lillerud and U. Olsbye, Top. Catal., 2009, 52, 218–228 CrossRef CAS.
- Q. Qian, J. Ruiz-Martinez, M. Mokhtar, A. M. Asiri, S. A. Al-Thabaiti, S. N. Basahel and B. M. Weckhuysen, Catal. Today, 2014, 226, 14–24 CrossRef CAS.
- T. Armaroli, L. J. Simon, M. Digne, T. Montanari, M. Bevilacqua, V. Valtchev, J. Patarin and G. Busca, Appl. Catal., A, 2006, 306, 78–84 CrossRef CAS.
- G. J. Yang, Y. X. Wei, S. T. Xu, J. R. Chen, J. Z. Li, Z. M. Li, J. H. Yu and R. R. Xu, J. Phys. Chem. C, 2013, 117, 8214–8222 CAS.
- K. Y. Lee, H. J. Chae, S. Y. Jeong and G. Seo, Appl. Catal., A, 2009, 369, 60–66 CrossRef CAS.
- R. von Ballmoos and W. M. Meier, Nature, 1981, 289, 782–783 CrossRef CAS.
- V. Gabova, J. Dedecek and J. Cejka, Chem. Commun., 2003, 1196–1197, 10.1039/b301634j.
- S. Svelle, L. Sommer, K. Barbera, P. N. R. Vennestrom, U. Olsbye, K. P. Lillerud, S. Bordiga, Y. H. Pan and P. Beato, Catal. Today, 2011, 168, 38–47 CrossRef CAS.
- P. Sazama, B. Wichterlova, J. Dedecek, Z. Tvaruzkova, Z. Musilova, L. Palumbo, S. Sklenak and O. Gonsiorova, Microporous Mesoporous Mater., 2011, 143, 87–96 CrossRef CAS.
- K. Barbera, F. Bonino, S. Bordiga, T. V. W. Janssens and P. Beato, J. Catal., 2011, 280, 196–205 CrossRef CAS.
- Z. Ristanovic, J. P. Hofmann, U. Deka, T. U. Schuelli, M. Rohnke, A. M. Beale and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2013, 52, 13382–13386 CrossRef CAS PubMed.
- M. F. De Lange, T. J. H. Vlugt, J. Gascon and F. Kapteijn, Microporous Mesoporous Mater., 2014, 200, 199–215 CrossRef CAS.
- A. Charkhi, H. Kazemian and M. Kazemeini, Powder Technol., 2010, 203, 389–396 CrossRef CAS.
- T. Biemelt, C. Selzer, F. Schmidt, G. Mondin, A. Seifert, K. Pinkert, S. Spange, T. Gemming and S. Kaskel, Microporous Mesoporous Mater., 2014, 187, 114–124 CrossRef CAS.
- C. Baerlocher, L. B. McCusker and D. H. Olson, Atlas of Zeolite Framework Types, Elsevier Science, 2007 Search PubMed.
- S. Sartipi, H. Jansma, D. Bosma, B. Boshuizen, M. Makkee, J. Gascon and F. Kapteijn, Rev. Sci. Instrum., 2013, 84, 124101 CrossRef PubMed.
- C. S. Cundy and P. A. Cox, Microporous Mesoporous Mater., 2005, 82, 1–78 CrossRef CAS.
- S. Bordiga, L. Regli, D. Cocina, C. Lamberti, M. Bjorgen and K. P. Lillerud, J. Phys. Chem. B, 2005, 109, 2779–2784 CrossRef CAS PubMed.
- S. Bordiga, P. Ugliengo, A. Damin, C. Lamberti, G. Spoto, A. Zecchina, G. Spano, R. Buzzoni, L. Dalloro and F. Rivetti, Top. Catal., 2001, 15, 43–52 CrossRef CAS.
- S. Bordiga, I. Roggero, P. Ugliengo, A. Zecchina, V. Bolis, G. Artioli, R. Buzzoni, G. Marra, F. Rivetti, G. Spano and C. Lamberti, J. Chem. Soc., Dalton Trans, 2000, 3921–3929, 10.1039/b004794p.
- A. Zecchina, G. Spoto and S. Bordiga, Phys. Chem. Chem. Phys., 2005, 7, 1627–1642 RSC.
- X. Sun, S. Mueller, Y. Liu, H. Shi, G. L. Haller, M. Sanchez-Sanchez, A. C. van Veen and J. A. Lercher, J. Catal., 2014, 317, 185–197 CrossRef CAS.
- B. P. C. Hereijgers, F. Bleken, M. H. Nilsen, S. Svelle, K.-P. Lillerud, M. Bjorgen, B. M. Weckhuysen and U. Olsbye, J. Catal., 2009, 264, 77–87 CrossRef CAS.
- D. Chen, K. Moljord, T. Fuglerud and A. Holmen, Microporous Mesoporous Mater., 1999, 29, 191–203 CrossRef CAS.
- H. Schulz and M. Wei, Top. Catal., 2014, 57, 683–692 CrossRef CAS.
- J. W. Park and G. Seo, Appl. Catal., A, 2009, 356, 180–188 CrossRef CAS.
- J. Pater, F. Cardona, C. Canaff, N. S. Gnep, G. Szabo and M. Guisnet, Ind. Eng. Chem. Res., 1999, 38, 3822–3829 CrossRef CAS.
- T. R. Forester and R. F. Howe, J. Am. Chem. Soc., 1987, 109, 5076–5082 CrossRef CAS.
- S. Chiaberge, G. Guglielmetti, L. Montanari, M. Salvalaggio, L. Santolini, S. Spera and P. Cesti, Energy Fuels, 2009, 23, 4486–4495 CrossRef CAS.
- M. Rozwadowski, M. Lezanska, J. Wloch, K. Erdmann, R. Golembiewski and J. Kornatowski, Chem. Mater., 2001, 13, 1609–1616 CrossRef CAS.
- H. S. Cerqueira, P. Ayrault, J. Datka, P. Magnoux and M. Guisnet, J. Catal., 2000, 196, 149–157 CrossRef CAS.
- J. W. Park, S. J. Kim, M. Seo, S. Y. Kim, Y. Sugi and G. Seo, Appl. Catal., A, 2008, 349, 76–85 CrossRef CAS.
- Y. X. Wei, J. Z. Li, C. Y. Yuan, S. T. Xu, Y. Zhou, J. R. Chen, Q. Y. Wang, Q. Zhang and Z. M. Liu, Chem. Commun., 2012, 48, 3082–3084 RSC.
- J. R. Chen, J. Z. Li, Y. X. Wei, C. Y. Yuan, B. Li, S. T. Xu, Y. Zhou, J. B. Wang, M. Z. Zhang and Z. M. Liu, Catal. Commun., 2014, 46, 36–40 CrossRef CAS.
- R. Khare, D. Millar and A. Bhan, J. Catal., 2015, 321, 23–31 CrossRef CAS.
- X. Sun, S. Mueller, H. Shi, G. L. Haller, M. Sanchez-Sanchez, A. C. van Veen and J. A. Lercher, J. Catal., 2014, 314, 21–31 CrossRef CAS.
- A. F. Combariza, G. Sastre and A. Corma, J. Phys. Chem. C, 2009, 113, 11246–11253 CAS.
- D. H. Olson, M. A. Camblor, L. A. Villaescusa and G. H. Kuehl, Microporous Mesoporous Mater., 2004, 67, 27–33 CrossRef CAS.
- W. Zhu, F. Kapteijn and J. A. Moulijn, Chem. Commun., 1999, 2453–2454 RSC.
- S. Sartipi, M. Makkee, F. Kapteijn and J. Gascon, Catal. Sci. Technol., 2014, 4, 893–907 CAS.
- J. van den Bergh, C. Gucuyener, E. A. Pidko, E. J. M. Hensen, J. Gascon and F. Kapteijn, Chem. – Eur. J., 2011, 17, 8832–8840 CrossRef CAS PubMed.
- Q. Yu, Q. Zhang, J. Liu, C. Li and Q. Cui, CrystEngComm, 2013, 15, 7680–7687 RSC.
- S. Svelle, F. Joensen, J. Nerlov, U. Olsbye, K. P. Lillerud, S. Kolboe and M. Bjorgen, J. Am. Chem. Soc., 2006, 128, 14770–14771 CrossRef CAS PubMed.
- J. Van der Mynsbrugge, J. De Ridder, K. Hemelsoet, M. Waroquier and V. Van Speybroeck, Chem. – Eur. J., 2013, 19, 11568–11576 CrossRef CAS PubMed.
- V. Van Speybroeck, K. De Wispelaere, J. Van der Mynsbrugge, M. Vandichel, K. Hemelsoet and M. Waroquier, Chem. Soc. Rev., 2014, 43, 7326–7357 RSC.
- Q. Qian, J. Ruiz-Martinez, M. Mokhtar, A. M. Asiri, S. A. Al-Thabaiti, S. N. Basahel, H. E. van der Bij, J. Kornatowski and B. M. Weckhuysen, Chem. – Eur. J., 2013, 19, 11204–11215 CrossRef CAS PubMed.
- Q. Zhu, J. N. Kondo, R. Ohnuma, Y. Kubota, M. Yamaguchi and T. Tatsumi, Microporous Mesoporous Mater., 2008, 112, 153–161 CrossRef CAS.
- M. Guisnet, L. Costa and F. R. Ribeiro, J. Mol. Catal. A: Chem., 2009, 305, 69–83 CrossRef CAS.
- M. Opanasenko, W. Roth and J. Cejka, Catal. Sci. Technol., 2015 10.1039/C5CY02079D.
- W. Dai, G. Wu, L. Li, N. Guan and M. Hunger, ACS Catal., 2013, 3, 588–596 CrossRef CAS.
- S. Wilson and P. Barger, Microporous Mesoporous Mater., 1999, 29, 117–126 CrossRef CAS.
- J. Gascon, J. R. van Ommen, J. A. Moulijn and F. Kapteijn, Catal. Sci. Technol., 2015, 5, 807–817 CAS.
- Q. Qian, J. Ruiz-Martinez, M. Mokhtar, A. M. Asiri, S. A. Al-Thabaiti, S. N. Basahel and B. M. Weckhuysen, ChemCatChem, 2014, 6, 772–783 CrossRef CAS.
- U. Olsbye, S. Svelle, M. Bjorgen, P. Beato, T. V. W. Janssens, F. Joensen, S. Bordiga and K. P. Lillerud, Angew. Chem., Int. Ed., 2012, 51, 5810–5831 CrossRef CAS PubMed.
- J. W. Park, J. Y. Lee, K. S. Kim, S. B. Hong and G. Seo, Appl. Catal., A, 2008, 339, 36–44 CrossRef CAS.
- Y. Bhawe, M. Moliner-Marin, J. D. Lunn, Y. Liu, A. Malek and M. Davis, ACS Catal., 2012, 2, 2490–2495 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cy02140e |
| This journal is © The Royal Society of Chemistry 2016 |