
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hierarchical mesoporous Pd/ZSM-5 for the selective catalytic hydrodeoxygenation of m-cresol to methylcyclohexane†
James A.
Hunns
a,
Marta
Arroyo
b,
Adam F.
Lee
 a,
José M.
Escola
b,
David
Serrano
*bc and
Karen
Wilson
*a
a,
José M.
Escola
b,
David
Serrano
*bc and
Karen
Wilson
*a
aEuropean Bioenergy Research Institute, Aston University, Birmingham B4 7ET, UK. E-mail: k.wilson@aston.ac.uk; Tel: +44 (0)121 2044036
bChemical and Environmental Engineering Group, Rey Juan Carlos University, 28933, Móstoles, Madrid, Spain
cIMDEA Energy Institute, 28935 Móstoles, Madrid, Spain. E-mail: david.serrano@imdea.org
First published on 29th December 2015
Abstract
Mesopore incorporation into ZSM-5 enhances the dispersion of Pd nanoparticles throughout the hierarchical framework, significantly accelerating m-cresol conversion relative to a conventional microporous ZSM-5, and dramatically increasing selectivity towards the desired methylcyclohexane deoxygenated product. Increasing the acid site density further promotes m-cresol conversion and methylcyclohexane selectivity through efficient dehydration of the intermediate methylcyclohexanol.
Advances in the thermochemical processing of lignocellulosic biomass offer scalable routes to the production of sustainable liquid transportation biofuels from diverse waste-derived feedstocks.1 Pyrolytic thermal decomposition of biomass delivers a broad product distribution, which varies with feedstock, reaction temperature and residence time.2 Intermediate pyrolysis typically utilises low temperatures between 300–500 °C and long residence times which favour charcoal formation, whereas fast pyrolysis uses moderate temperatures of 450–550 °C and short vapour residence times <6 s to optimise liquid production. Higher temperatures and longer residence times favour gasification for direct power generation and upgrading of (purified) syngas via Fischer Tropsch and methanol synthesis. Of these routes, fast pyrolysis is the most promising for the direct conversion of agricultural waste and short rotation crops to bio-oils which retain up to 70% of the energy content of the raw biomass. Unfortunately, such fast pyrolysis bio-oils possess undesirable physicochemical properties including high oxygen contents of around 40 wt%, low pH, high viscosity and poor thermal stability, and relatively poor heating values of 16–19 MJ kg−1, which hinders their use as drop-in petroleum replacement fuels in conventional combustion engines.
A number of promising catalytic solutions to the upgrading of pyrolytic bio-oils are under development. Of these, solid acid (or base) catalysed esterification3 (or ketonisation) to ameliorate their low pH (arising principally from high acetic acid contents of 1–10%) through alkyl esters (or acetone) production, and subsequent hydrotreating to lower the oxygen content of ligneous and hemicellulosic depolymerisation products via hydrodeoxygenation (HDO),4,5 have proven most popular. HDO has been explored in batch and continuous upgrading mode, with early work utilising supported CoMo or NiMo sulfide catalysts6–8 analogous to hydrodesulfurization processes traditionally employed in petroleum refineries. Although such approaches permit oxygen removal, they are undesirable due to the requirement for H2S in order to continuously regenerate the active sulfide phase. Transition metal catalysts have also been widely screened for HDO, affording significant deoxygenation, but under forcing conditions of high hydrogen pressures and process temperatures to deliver alkane products under batch and continuous processing.9–12
Bifunctional transition metal-solid acid HDO catalysts are advantageous in combining a metallic component (usually Pt, Pd, Ni, Rh or Ru) to promote C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO hydrogenation, and an acid component to dehydrate aliphatic oxygenates (e.g. Al-SBA-15, ZSM-5 and beta zeolites, and sulfated metal oxides such as SO4/ZrO2). Consequently they permit lower hydrogen pressures than those required for direct hydrogenolysis over metal nanoparticles alone. However, the respective role of each active phase, and associated reaction mechanism and pathways, remains under debate.13–15 Hierarchical zeolites are a relatively new class of solid acids in which the incorporation of a second macroporous or mesoporous network may confer novel or improved catalytic performance. Mesoporous zeolites have previously shown activity for alkylation of benzene,16 conversion of methanol to hydrocarbons17 and other petrochemical cracking and isomerisation processes.18 Mesopores in particular can improve accessibility to acid sites predominantly localised within the micropores of zeolites, thereby (at least partially) overcoming mass-transport limitations, which are especially important in the transformation of larger bio-derived reactants,19 and enhancing catalytic activity. Shorter diffusion lengths, and consequent in-pore residence times, may also influence product selectivity through the suppression of secondary reaction pathways. Hierarchical mesoporous zeolites may also be advantageous for improving the dispersion of promoters (e.g. metal nanoparticles) throughout the porous architecture, rather than their localisation on the external surface of conventional microporous counterparts.20 Indeed, a Ni promoted, hierarchical nanocrystallite HBEA wide pore zeolite prepared via controllable base leaching was recently reported for stearic acid and palm oil hydrodeoxygenation which showed small improvements in stearic acid HDO selectivity to n-C17/C18 and iso-C17/C18 alkanes over microporous HBEA.21 Anisole HDO has also been performed over a hierarchical Ni/ZSM-5 catalyst,22 which exhibited high conversion and selectivity to cyclohexane.
C and CO hydrogenation, and an acid component to dehydrate aliphatic oxygenates (e.g. Al-SBA-15, ZSM-5 and beta zeolites, and sulfated metal oxides such as SO4/ZrO2). Consequently they permit lower hydrogen pressures than those required for direct hydrogenolysis over metal nanoparticles alone. However, the respective role of each active phase, and associated reaction mechanism and pathways, remains under debate.13–15 Hierarchical zeolites are a relatively new class of solid acids in which the incorporation of a second macroporous or mesoporous network may confer novel or improved catalytic performance. Mesoporous zeolites have previously shown activity for alkylation of benzene,16 conversion of methanol to hydrocarbons17 and other petrochemical cracking and isomerisation processes.18 Mesopores in particular can improve accessibility to acid sites predominantly localised within the micropores of zeolites, thereby (at least partially) overcoming mass-transport limitations, which are especially important in the transformation of larger bio-derived reactants,19 and enhancing catalytic activity. Shorter diffusion lengths, and consequent in-pore residence times, may also influence product selectivity through the suppression of secondary reaction pathways. Hierarchical mesoporous zeolites may also be advantageous for improving the dispersion of promoters (e.g. metal nanoparticles) throughout the porous architecture, rather than their localisation on the external surface of conventional microporous counterparts.20 Indeed, a Ni promoted, hierarchical nanocrystallite HBEA wide pore zeolite prepared via controllable base leaching was recently reported for stearic acid and palm oil hydrodeoxygenation which showed small improvements in stearic acid HDO selectivity to n-C17/C18 and iso-C17/C18 alkanes over microporous HBEA.21 Anisole HDO has also been performed over a hierarchical Ni/ZSM-5 catalyst,22 which exhibited high conversion and selectivity to cyclohexane.
Here we explore the impact of mesoporosity and acid strength upon m-cresol HDO over bifunctional, hierarchical Pd/ZSM-5 catalysts, which exhibit exceptional activity and selectivity towards the methylcyclohexane aliphatic product.
Hierarchical ZSM-5 zeolites were synthesised using the ‘protozeolitic units silanization’ method reported previously (ESI†).23 Briefly, a solution of tetraethylorthosilicate, tetrapropylammonium hydroxide, aluminum isopropoxide (AIP) and deionized water was prepared to obtain Si![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al atomic ratios of 30 and 100, and pre-crystallization was induced under reflux prior to the addition of a seed silanization agent and extended hydrothermal aging. Samples are denoted as h-ZSM-5(X), where X is the theoretical Si:Al atomic ratio in the precursor gel. A commercial nanocrystalline ZSM-5 (Südchemie) was employed for comparison and denoted as c-ZSM-5(30). Zeolites were subsequently impregnated via incipient-wetness with 1 wt% palladium from PdCl2, dried, calcined and reduced to yield Pd/h-ZSM-5(X) and Pd/c-ZSM-5(30). Bulk and surface physicochemical properties were characterized by elemental analysis, Ar porosimetry, powder XRD, and CO and NH3 chemisorption to titrate metal and acid active sites, respectively. Hydrodeoxygenation was undertaken in a stirred batch autoclave employing 50 mg of catalyst and 6 mmol m-cresol in n-dodecane at 200 °C and 20 bar H2, with kinetic profiling via off-line GC analysis. Conversion and selectivity are reported ±2%, with the latter calculated for the three major liquid phase products 3-methylcyclohexanone (MCHO), 3-methylcyclohexanol (MCOL), methylcyclohexane (MCX). Mass balances were between 90–99%.
:Al atomic ratios of 30 and 100, and pre-crystallization was induced under reflux prior to the addition of a seed silanization agent and extended hydrothermal aging. Samples are denoted as h-ZSM-5(X), where X is the theoretical Si:Al atomic ratio in the precursor gel. A commercial nanocrystalline ZSM-5 (Südchemie) was employed for comparison and denoted as c-ZSM-5(30). Zeolites were subsequently impregnated via incipient-wetness with 1 wt% palladium from PdCl2, dried, calcined and reduced to yield Pd/h-ZSM-5(X) and Pd/c-ZSM-5(30). Bulk and surface physicochemical properties were characterized by elemental analysis, Ar porosimetry, powder XRD, and CO and NH3 chemisorption to titrate metal and acid active sites, respectively. Hydrodeoxygenation was undertaken in a stirred batch autoclave employing 50 mg of catalyst and 6 mmol m-cresol in n-dodecane at 200 °C and 20 bar H2, with kinetic profiling via off-line GC analysis. Conversion and selectivity are reported ±2%, with the latter calculated for the three major liquid phase products 3-methylcyclohexanone (MCHO), 3-methylcyclohexanol (MCOL), methylcyclohexane (MCX). Mass balances were between 90–99%.
Elemental analysis confirmed that the experimental Si:Al ratios were in good agreement with their nominal values (Table 1). BET surface areas of both hierarchical zeolites were higher than their conventional counterpart, increasing by 16% between c-ZSM-5(30) and h-ZSM-5(30), and further increased for the high silicon sample, mirroring the total pore volumes and secondary mesopore volumes (Table S1 and Fig. S1†). Wide angle XRD confirmed the presence of ZSM-5 crystallites in the commercial and hierarchical materials (Fig. 1), and showed that the introduction of mesoporosity had little impact on overall crystalline domain size. In contrast, mass normalised acid site densities from NH3 chemisorption revealed that c-ZSM-5(30) possessed a slightly higher acid loading than h-ZSM-5(30) of 0.35 versus 0.3 mmol g−1 respectively, whereas h-ZSM-5(100) possessed only one third of the acid sites. There was little difference in the maximum ammonia desorption peak temperatures between the three materials, indicating that varying the Si:Al ratio only influenced the number of acid sites, but not their strength (Table 1).
| Sample | Surface areaa/m2 g−1 | Pore volumea/cm3 g−1 | Si:Al atomic ratio |
Zeolite crystal sizeb/nm | Pd particle sizec/nm | Acid site densityd/mmol g−1 | T Max NH3d/°C |
|---|---|---|---|---|---|---|---|
| a BET area from Ar physisorption. b Volume averaged crystallite domain size from XRD. c Mean diameter from CO chemisorption. d NH3 TPD. | |||||||
| c-ZSM-5(30) | 404 | 0.471 | 32 | 28 | — | 0.350 | 349 |
| Pd/c-ZSM-5(30) | 377 | 0.434 | — | — | 15.2 | 0.345 | — |
| h-ZSM-5(30) | 479 | 0.503 | 33 | 28 | — | 0.301 | 332 |
| Pd/h-ZSM-5(30) | 477 | 0.497 | — | — | 4.5 | 0.305 | — |
| h-ZSM-5(100) | 511 | 0.522 | 122 | 20 | — | 0.128 | 307 |
| Pd/h-ZSM-5(100) | 486 | 0.557 | — | — | 12.5 | 0.122 | — |
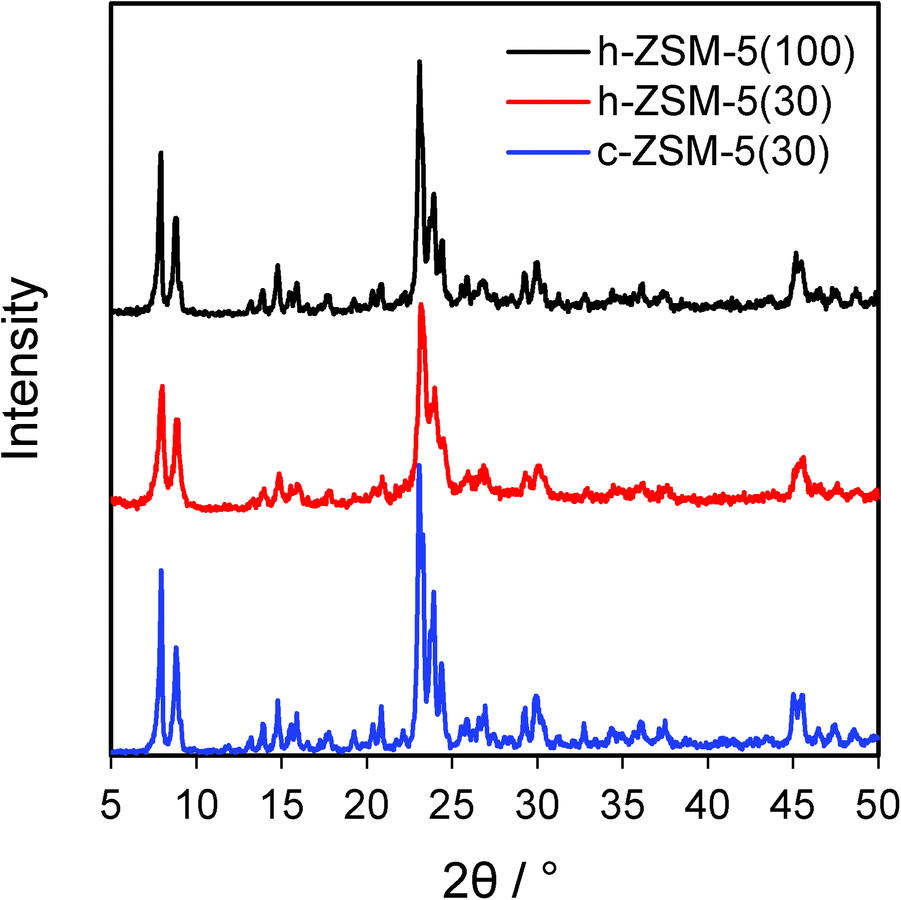 | ||
| Fig. 1 Powder XRD patterns of conventional and hierarchical ZSM-5. | ||
Pd impregnation resulted in similar metal loadings of ∼1 wt% for all three zeolites, and was accompanied by a small decrease in surface area consistent with a small degree of pore blockage, but had negligible impact on the intrinsic zeolite acidity. The introduction of mesoporosity lowered the mean Pd nanoparticle size, an effect more significant for lower silicon (higher aluminium) content h-ZSM-5(30) material, a phenomenon previously observed for palladium impregnated silica and alumina.24
Catalytic HDO of m-cresol proceeded efficiently over all three zeolites, with conversions varying between 78–100% achieved within 6 h reaction (Fig. 2). However, the acid site density and textural properties exerted a significant impact on the initial reaction rate. First considering the c-ZSM-5(30) and h-ZSM-5(30) catalysts, which exhibited very similar solid acid properties, mesopore incorporation doubled the rate of m-cresol HDO, although both catalysts attained full conversion within 6 h. We note that this rate enhancement did not directly correlate with the concomitant three-fold rise in palladium surface area (Table S1†), and hence is primarily attributed to improved mass transport and accessibility of in-pore acid sites. In contrast, increasing the silicon content (lowering the acid site density from 0.31 to 0.12 mmol g−1) resulted in a six-fold loss in activity for the h-ZSM-5(100) catalyst relative to h-ZSM-5(30); a high density of surface acid sites thus emerges as critical to efficient HDO under our conditions. The parent h-ZSM-5(100) material was inactive without palladium, confirming the requirement for metal centres to dissociatively adsorb hydrogen. However, there was no quantitative correlation between m-cresol HDO activity and the total palladium surface area. The metal surface area increased only three-fold between the Pd/h-ZSM-5(30) and Pd/h-ZSM-5(100) materials, far less than the corresponding rise in activity, evidencing a strong synergy between small Pd nanoparticles and strong acid sites.
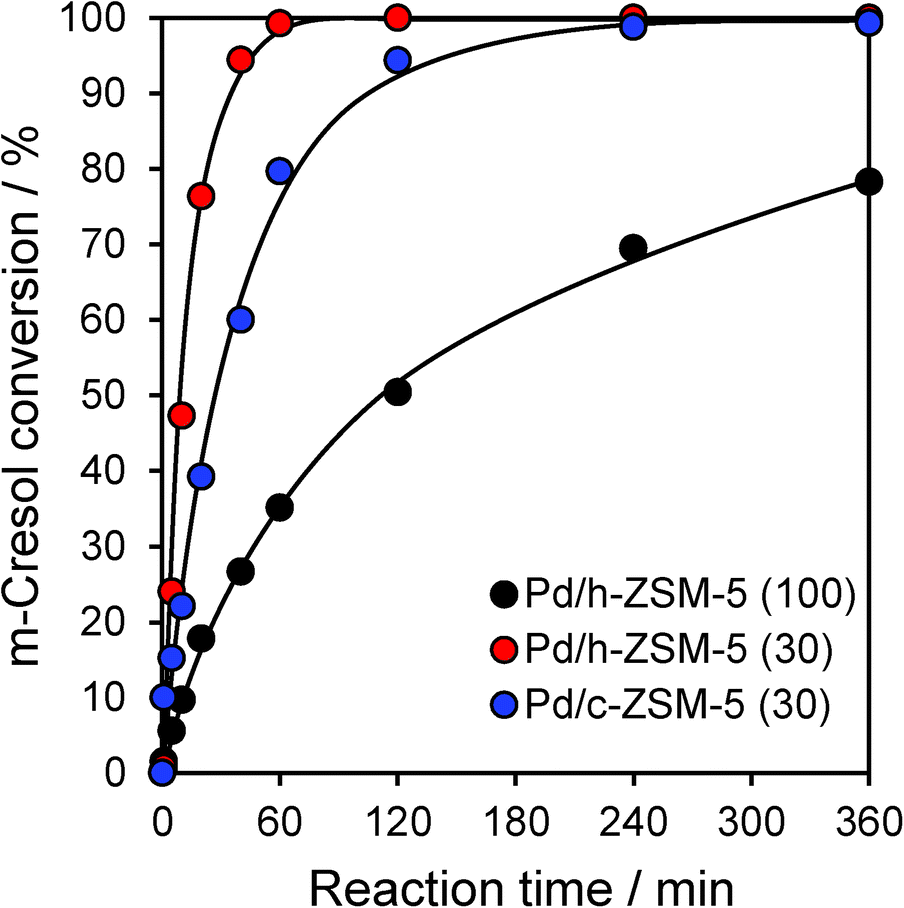 | ||
| Fig. 2 m-Cresol conversion over conventional and hierarchical Pd/ZSM-5 catalysts. | ||
All Pd/ZSM-5 catalysts yielded the same three liquid phase products MCHO, MCOL and MCX (Table 2). It has been previously proposed that m-cresol HDO over supported Pd and Pt (ref. 11–13) and Fe–Ni (ref. 25) proceeds via competitive alcohol deoxygenation versus ring hydrogenation (Scheme 1). However, in the present work, neither 3-methylcyclohexadienol nor toluene were observed, indicating that m-cresol HDO occurred exclusively via its tautomerisation to a cyclic dienone and subsequent ring hydrogenation to MCHO, which in turn underwent CO hydrogenation and dehydration to MCX. The latter was observed as the dominant product at extended reaction times over the more acidic Pd/c-ZSM-5(30) and Pd/h-ZSM-5(30) catalysts, consistent with rapid dehydration of the MCOL intermediate, with the hierarchical variant achieving 99% MCX selectivity. It is interesting to note that kinetic profiling revealed ring hydrogenation to MCHO as the dominant pathway during the first hour of reaction (Fig. S2†), at which point the selectivity of the more acidic catalysts transitioned from favouring CC hydrogenation to CO hydrogenation. This switchover coincided with high levels of m-cresol conversion, and suggests slower CO (of MCHO to MCOL) versus CC hydrogenation, as commonly observed for unsaturated carbonyls over transition metals.26 Interfacial interactions between metals and Lewis acid sites are reported to enhance CO hydrogenation,27 which may underpin the superior performance of the Pd/h-ZSM-5(30) versus Pd/h-ZSM-5(100) catalysts. We anticipate that the selectivity of Pd/c-ZSM-5 towards MCX would increase to mirror that of its hierarchical analogue at extended reaction times, reflecting the longer timescale for the former to achieve full m-cresol conversion. The less acidic Pd/h-ZSM-5(100) catalyst exhibited markedly different selectivity, favouring predominantly MCHO, accompanied by significant MCOL, as a consequence of its poorer activity for alcohol dehydration. The absence of the methylcyclohexene intermediate following MCOL dehydration demonstrates that alkene hydrogenation is rapid under these reaction conditions, as proposed for the stepwise transformation of related phenolics over Pd/zeolites. Further kinetic studies are currently underway to elucidate the rate-determining step in m-cresol HDO and the palladium structure sensitivity, in addition to the impact of palladium loading, H2 pressure and the selection of phenolic substrate (e.g. p-cresol), in order to optimise the yield of cyclic alkanes and establish the generic applicability of hierarchical ZSM-5 in bio-oil HDO. In summary, mesopore introduction into a microporous zeolite significantly promotes the selective HDO of m-cresol to the desirable methylcyclohexane fully deoxygenated product.
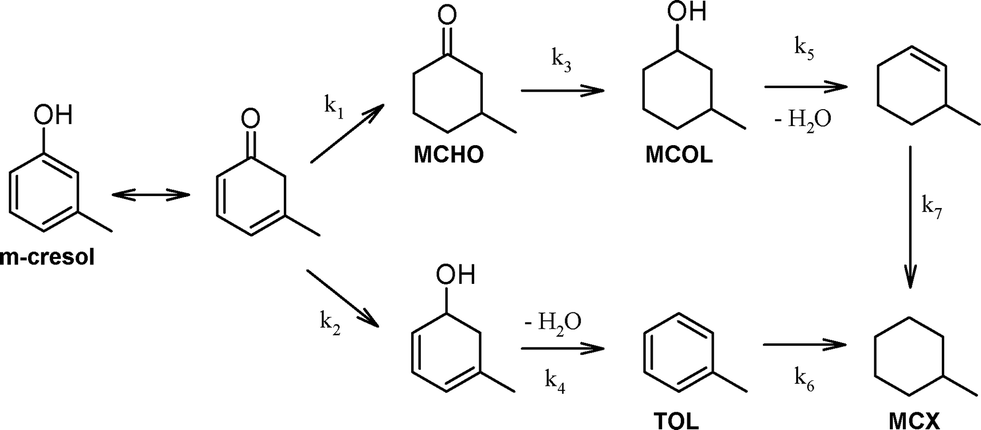 | ||
| Scheme 1 Proposed reaction pathways for m-cresol HDO over Pd/ZSM-5 catalysts. | ||
Conclusions
Hierarchical ZSM-5 zeolites synthesised with intrinsic mesoporosity via crystallization of silanized protozeolitic units exhibit superior surface area and porosity over their purely microporous counterparts. These enhanced textural properties are attained without any change in the Si:Al composition, and with only a small loss in total acid site density. Mesoporosity increases the dispersion of palladium nanoparticles throughout a strongly acidic hierarchical zeolite, and in combination with associated improved mass transport, confers exceptional activity and selectivity for m-cresol HDO to methylcyclohexane via stepwise hydrogenation and dehydration. m-Cresol HDO occurs exclusively via its tautomerisation to a cyclic dienone and subsequent ring hydrogenation to MCHO over conventional and hierarchical Pd/ZSM-5 catalysts. A high density of acid sites is essential to ensure complete dehydration of 3-methylcyclohexanone and 3-methylcyclohexanol reactively formed hydrogenated intermediates. Tailoring the pore architecture of solid acids offers a facile means to improve the catalytic upgrading of bio-oils.
Acknowledgements
We thank the EPSRC (EP/K036548/1 and EP/K014706/1) for financial support. JAH thanks Johnson Matthey for an Industrial CASE studentship. KW thanks the Royal Society for an Industry Fellowship. Support from the European Union Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 604307 is also acknowledged.Notes and references
- A. V. Bridgwater, Biomass Bioenergy, 2012, 38, 68–94 CrossRef CAS.
- W. N. R. W. Isahak, M. W. M. Hisham, M. A. Yarmo and T.-Y. Y. Hin, Renewable Sustainable Energy Rev., 2012, 16, 5910–5923 CrossRef CAS.
- L. Ciddor, J. A. Bennett, J. A. Hunns, K. Wilson and A. F. Lee, J. Chem. Technol. Biotechnol., 2015, 90, 780–795 CrossRef CAS.
- A. H. Zacher, M. V. Olarte, D. M. Santosa, D. C. Elliott and S. B. Jones, Green Chem., 2014, 16, 491–515 RSC.
- Y. Wang, T. He, K. Liu, J. Wu and Y. Fang, Bioresour. Technol., 2012, 108, 280–284 CrossRef CAS.
- A. L. Jongerius, R. Jastrzebski, P. C. A. Bruijnincx and B. M. Weckhuysen, J. Catal., 2012, 285, 315–323 CrossRef CAS.
- I. D. Mora, E. Mendez, L. J. Duarte and S. A. Giraldo, Appl. Catal., A, 2014, 474, 59–68 CrossRef CAS.
- Y. Wang, H. Lin and Y. Zheng, Catal. Sci. Technol., 2014, 4, 109–119 CAS.
- H. Ohta, H. Kobayashi, K. Hara and A. Fukuoka, Chem. Commun., 2011, 47, 12209–12211 RSC.
- C. Newman, X. Zhou, B. Goundie, I. T. Ghampson, R. A. Pollock, Z. Ross, M. C. Wheeler, R. W. Meulenberg, R. N. Austin and B. G. Frederick, Appl. Catal., A, 2014, 477, 64–74 CrossRef CAS.
- L. Nie and D. E. Resasco, J. Catal., 2014, 317, 22–29 CrossRef CAS.
- P. M. de Souza, L. Nie, L. E. P. Borges, F. B. Noronha and D. E. Resasco, Catal. Lett., 2014, 144, 2005–2011 CrossRef CAS.
- X. Zhu, L. Nie, L. L. Lobban, R. G. Mallinson and D. E. Resasco, Energy Fuels, 2014, 28, 4104–4111 CrossRef CAS.
- X. Zhu, L. L. Lobban, R. G. Mallinson and D. E. Resasco, J. Catal., 2011, 281, 21–29 CrossRef CAS.
- J. Horacek, G. St'avova, V. Kelbichova and D. Kubicka, Catal. Today, 2013, 204, 38–45 CrossRef CAS.
- H. Liu, S. Liu, S. Xie, C. Song, W. Xin and L. Xu, Catal. Lett., 2015, 145, 1972–1983 CrossRef CAS.
- Z. Liu, X. Dong, Y. Zhu, A.-H. Emwas, D. Zhang, Q. Tian and Y. Han, ACS Catal., 2015, 5, 5837–5845 CrossRef CAS.
- I. I. Ivanova, E. E. Knyazeva, A. A. Maerle and I. A. Kasyanov, Kinet. Catal., 2015, 56, 549–561 CrossRef CAS.
- H. W. Lee, B. R. Jun, H. Kim, D. H. Kim, J.-K. Jeon, S. H. Park, C. H. Ko, T.-W. Kim and Y.-K. Park, Energy, 2015, 81, 33–40 CrossRef CAS.
- C. H. Christensen, I. Schmidt, A. Carlsson, K. Johannsen and K. Herbst, J. Am. Chem. Soc., 2005, 127, 8098–8102 CrossRef CAS.
- B. Ma and C. Zhao, Green Chem., 2015, 17, 1692–1701 RSC.
- T. M. Sankaranarayanan, A. Berenguer, C. Ochoa-Hernández, I. Moreno, P. Jana, J. M. Coronado, D. P. Serrano and P. Pizarro, Catal. Today, 2015, 243, 163–172 CrossRef CAS.
- D. P. Serrano, J. Aguado, J. M. Escola, A. Peral, G. Morales and E. Abella, Catal. Today, 2011, 168, 86–95 CrossRef CAS.
- C. M. A. Parlett, L. J. Durndell, A. Machado, G. Cibin, D. W. Bruce, N. S. Hondow, K. Wilson and A. F. Lee, Catal. Today, 2014, 229, 46–55 CrossRef CAS.
- L. Nie, P. M. de Souza, F. B. Noronha, W. An, T. Sooknoi and D. E. Resasco, J. Mol. Catal. A: Chem., 2014, 388–389, 47–55 CrossRef CAS.
- L. J. Durndell, C. M. A. Parlett, N. S. Hondow, M. A. Isaacs, K. Wilson and A. F. Lee, Sci. Rep., 2015, 5, 9425 CrossRef CAS.
- A. Y. Stakheev and L. M. Kustov, Appl. Catal., A, 1999, 188, 3–35 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full synthetic, characterisation and reaction details. See DOI: 10.1039/c5cy02072g |
| This journal is © The Royal Society of Chemistry 2016 |