
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
MsAcT in siliceous monolithic microreactors enables quantitative ester synthesis in water†
Katarzyna
Szymańska
*a,
Klaudia
Odrozek
a,
Aurelia
Zniszczoł
a,
Guzman
Torrelo
b,
Verena
Resch
bc,
Ulf
Hanefeld
b and
Andrzej B.
Jarzębski
ad
aDepartment of Chemical Engineering and Process Design, Silesian University of Technology, Ks. M. Strzody 7, 44-100 Gliwice, Poland. E-mail: Katarzyna.Szymanska@polsl.pl; Fax: +48322371461
bGebouw voor Scheikunde, Biokatalyse, Afdeling Biotechnologie, Technische Universiteit Delft, Julianalaan 136, 2628BL Delft, The Netherlands. E-mail: u.hanefeld@tudelft.nl
cOrganische und Bioorganische Chemie, Institut für Chemie, Karl-Franzens-Universität Graz, Heinrichstrasse 28, 8010 Graz, Austria
dInstitute of Chemical Engineering, Polish Academy of Sciences, Bałtycka 5, 44-100 Gliwice, Poland
First published on 29th February 2016
Abstract
Acyltransferase from Mycobacterium smegmatis (MsAcT) immobilised in continuous-flow microchannel (30–50 μm dia.) reactors with hierarchical pore structure (4 cm3 g−1 total pore volume) enabled quantitative, full and rapid transesterification of neopentylglycol (NPG) with ethyl acetate in a biphasic 50/50% system in less than one minute. MsAcT was attached either covalently via amino groups or by a specific His-tag-mediated adsorption on Ni or Co sites. Both methods gave similar results for enzyme loading (ca. 3 mg g−1 carrier, 60–70% immobilisation yield) and specific activity. The experiments revealed that the rate of monoester formation in the microreactor was exceedingly fast compared to that of diester synthesis and also the native enzyme behaviour in a batch reactor. The studies show that the course of transesterification was fully controlled by the biocatalytic properties of MsAcT confined in the mesoporous environment. These findings may be of significant interest from both fundamental and practical perspectives.
Introduction
Acyltransferase from Mycobacterium smegmatis (MsAcT) catalyses ester synthesis in water (Scheme 1).1,2 Its unique active site is rather apolar, thus creating a local situation in which the course of a reaction is not dominated by the thermodynamics of the aqueous medium but by the hydrophobicity of the active site. This asset makes MsAcT an excellent candidate for application in ester syntheses and transesterifications (such as biodiesel production) that are typically performed in aqueous or biphasic media.3 To date, only a few hydrolytic enzymes are known for their ability to catalyse synthesis reactions in aqueous environments, namely the very specialised polyketide thioesterases4–6 and a very limited number of lipases.7–9 To enable reaction engineering with MsAcT, the aim was to immobilise the enzyme. The octameric MsAcT (72 × 72 × 60 Å, Fig. 1) needs to be tightly bound to ensure its long-term use,2 and hydrophobic carriers (e.g. CNT) should be avoided since they may result in unfavourable partition effects and non-specific interactions of the enzyme or the products, which are detrimental for its catalytic properties and overall performance.10,11 | ||
| Scheme 1 Esterification of a primary diol, NPG, with ethyl acetate, catalysed by MsAcT in aqueous buffer. | ||
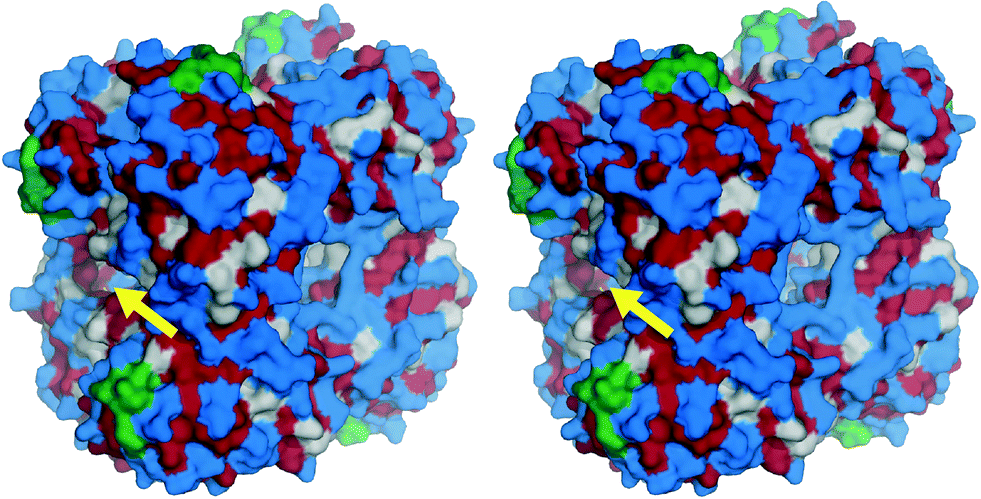 | ||
| Fig. 1 The octameric MsAcT (stereoview) displays an overall mixed hydrophilic surface: hydrophilic residues are highlighted in blue; grey residues are neither hydrophobic nor hydrophilic; hydrophobic residues are highlighted in red. The His-tag is located at the C-terminal (highlighted in green; please note that the His-tag is not included in the structure); the arrow indicates the entrance of the active site. Structural data from PDB: 2Q0S. The picture was generated using the PyMOL Molecular Graphics System. | ||
The most recent study of MsAcT's synthetic properties shed light on its application prospects to catalyse reactions in aqueous media.1 However, the reported experiments made use of cell-free extracts with low enzyme concentrations and were carried out in a batch system. Thus, while indicating the potential of MsAcT, the yields were modest and useful application was limited to primary alcohols. From an application perspective, continuous-flow systems with very high performance are desirable, in particular since they offer a new handle on reaction engineering.12
For this reason we deemed it important to undertake studies of the MsAcT behaviour in a continuous-flow transesterification of neopentylglycol (NPG) in a microchannel (micro)reactor. NPG is the substrate employed for the activity assay of MsAcT. It is particularly suitable for this study as it is a symmetric diol with two primary hydroxyl groups. This allows us to follow two conversion steps, mono- and diesterification. Additionally, it is a good model for other primary diols and even for glycerol that also has two primary hydroxyl groups.13 Thus, under these conditions the enzyme and the reactor should demonstrate their full capabilities, while at the same time offering an effective processing system. To preclude non-specific hydrophobic interactions between the enzyme and its support the microreactor was made of (hydrophilic) silica monolith with a hierarchical pore structure in micro- and nanometric size scales and meandering flow-through channels. This enables a facile flow of reactants and results in intensive mixing and mass transport to the enzyme. The latter was immobilised in very large mesopores using two distinctly different immobilisation concepts not only to improve operational stability but also to make use of the protein confinement effect by increasing the surface-to-volume ratio in microreactor technology.12
Experimental
Materials and methods
2,2-Dimethyl-1,3-propanediol (NPG), cethyltrimethylammonium bromide (CTAB), and tetraethoxysilane (TEOS) were from Acros Organics. Polyethylene glycol 35000 (PEG), ethyl acetate, cobalt(II) chloride, nickel(II) chloride, 1,4,8,11-tetraazacyclotetradecane (cyclam), 3-iodopropyl trimethoxysilane, and 3-aminopropyltrimethoxysilane were from Sigma-Aldrich. The other chemicals used in support synthesis, immobilisation and activity assay were from Avantor Poland.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm and 4 °C. The resulting cell-free extract (CFE) was lyophilised overnight, and the crude enzyme powder was stored at 4 °C. Activity assays were performed according to ref. 2 using neopentylglycol (NPG) as substrate and equal to 230 μmol min−1 (mg CFE)−1.
000 rpm and 4 °C. The resulting cell-free extract (CFE) was lyophilised overnight, and the crude enzyme powder was stored at 4 °C. Activity assays were performed according to ref. 2 using neopentylglycol (NPG) as substrate and equal to 230 μmol min−1 (mg CFE)−1.
 | ||
| Scheme 2 Functionalisation of silica carriers and immobilisation of MsAcT by covalent bonding (A) and specific His-tag-mediated adsorption on Ni/Co sites (B). | ||
Results and discussion
Characterisation of enzyme carriers
To date, MsAcT was immobilised only on carbon nanotubes (CNTs) and used for its perhydrolase activity in latex paint.10,23,24 Here we propose the application of hydrophilic macro/mesoporous monoliths with meandering/twisting flow-through channels, stimulating a chaotic movement (perturbations) of fluids and additionally enhancing mass transport.25 Employing a nonhydrophobic carrier would help in avoiding nonspecific interactions between the surface of the supports and the MsAcT active site.10Microscopic (SEM) and low-temperature nitrogen adsorption studies of silica monoliths revealed the prevalent presence of through-pores of 30–50 μm sizes and also mesopores of about 20 nm, and a total pore volume of over 4 cm3 g−1, determined by Hg porosimetry measurements as reported earlier15 (see the ESI†). This very open structure can also be seen from the images displayed in Fig. 2. Equally important, the modification of the monoliths appeared to have no effect on porosity in the macroscale. The specific surface area (SBET)26 of silica monolith measured by nitrogen adsorption was ca. 300 m2 g−1; after surface modification a slight decrease in this parameter was observed. Thus, not surprisingly, the measurements of backpressure vs. flow rate of water through the monoliths in the flow rate range of 0.03–3 mL min−1 gave a backpressure drop of only 0.028–2.8 kPa, which is significantly less than that in capillary reactors or packed bed columns filled with beads of micrometric sizes.27,28 These experiments also corroborated the very good stability of the monolith structure during at least two weeks of continuous flow operation, as observed in our earlier studies.18
 | ||
| Fig. 2 Silica monoliths with Ni (A) and Co (B). SEM image of monolith structure (C). | ||
Metal ions can be introduced onto a solid support by chelators such as iminodiacetic acid (IDA), nitriloacetic acid (NTA) or cyclam.19,20,29–33 As cyclam is known to be a stable ligand for several metals, MH-cyclam-Ni and MH-cyclam-Co were prepared as described previously20 (after minor modification). ICP mass spectrometry measurements gave the metal loading values of 8.96 mg Co (g silica)−1 ± 16% and 7.81 mg Ni (g silica)−1 ± 10%. After metal incorporation, the originally white silica monolith turned to yellow or blue for Ni and Co, respectively (Fig. 2). The same colour intensity, despite the considerable size (6 mm × 4 mm) of the monolith, indicated a homogenous distribution of the metal over its volume. To the best of our knowledge it is the first time that metal ions were homogenously incorporated into a monolithic column of that size, as to date only capillary or packed bed columns with metals were used.29–33
For non-specific MsAcT immobilisation the same silica monolith but functionalised with amino groups was used, and the presence of the latter was confirmed by FT-IR, as shown earlier,15 and elemental analysis (LECO CHN analyser). The amino group loading was 1.32 mmol (g silica)−1 (1.85 wt% of nitrogen). Before enzyme immobilisation the amino groups were activated with glutaraldehyde, after which the column changed colour uniformly to brown over the whole length of the monolith.
Performance of the monolithic microreactor
The recent extensive report on MsAcT-catalysed transesterification of NPG in buffer/ethyl acetate aqueous solutions in a batch reactor using the native enzyme1 provides an excellent platform to compare its behaviour with that of continuous-flow monolithic microreactors with the immobilised enzyme (Scheme 1). While effective immobilisation of the enzyme in the silica monolith's mesopores remained a critical factor, our studies primarily centered on the synthetic potential of the immobilised enzyme/microchannel reaction system. Two immobilisation methods were applied: (i) conventional covalent bonding tends to give more stable catalysts, but could reduce enzyme activity,11,18 and (ii) specific adsorption onto the Ni/Co-modified support via the His-tag,19,20,29–33 since the His-tag is remote from the active site (Fig. 1). At first, the activity, stability and enzyme loadings were studied using porous silica monoliths as enzyme support.Monolithic silica supports were functionalised with amino groups activated with glutaraldehyde (GLA) to produce a Schiff's base for covalent attachment and with Ni or Co for specific adsorption. However, we started with the evaluation of enzyme loadings obtained from different immobilisation methods, which appeared to be fairly similar: 2.96 mg g−1 for covalent, 3.15 and 3.07 mg g−1 carrier for adsorption on Ni and Co, respectively. That gives 60–70% of immobilisation yield. As it is not possible to predict whether the octameric MsAcT will be immobilised via one metal or one lysine amino group or rather by a multipoint attachment it was considered important to ensure that an excess of both metal and aldehyde groups was available for binding. This clearly was the case (Table 1). The performance of the biocatalysts in 50/50% v/v biphasic conditions of 0.05 M phosphate buffer (Na2HPO4/KH2PO4, pH 7.5) and ethyl acetate also appeared to be much the same (Fig. 3). We believe that this similarity in the enzyme loading and activity indicates a similar orientation of the enzyme attached by means of different immobilisation procedures and a free access to its active site. Bearing in mind that silica is hydrophilic by nature, it is likely that in a nonspecific covalent immobilisation, hydrophilic regions of the protein were involved, and hence its active center located in a very hydrophobic region (Fig. 1) was free and not distorted by immobilisation. As shown in Fig. 1, the His-tag residues are surrounded by hydrophilic amino acids, implying that the same region of protein was involved in both immobilisation procedures.
| Carrier | Functional group loading | Protein loading |
|---|---|---|
| MH-amino | 0.343 mmol | 0.77 mg |
| MH-Ni | 2.031 mg ± 10% | 0.82 mg |
| MH-Co | 2.329 mg ± 16% | 0.80 mg |
 | ||
| Fig. 3 Transesterification of neopentylglycol in a biphasic (50/50% v/v) system of buffer/ethyl acetate by MsAcT covalently immobilised on amino-modified monolith (A, B), adsorbed by His-tag on Ni-modified monolith (C, D) and on Co-modified monolith (E, F). Conditions: 0.05 M phosphate buffer pH 7.5; 96 mM neopentylglycol in ethyl acetate; room temperature. Red, substrate; black, diester; blue, monoester. | ||
A closer inspection of the data given in Fig. 3 indicates that the NPG transesterification in the microchannel reactor qualitatively portrays all trends observed earlier in the batch reactor studies.1 However, it also reveals a huge quantitative discrepancy between the operating characteristics of the batch reactor and the performance of the continuous-flow microchannel reactor with immobilised enzymes. The results displayed in Fig. 3B, D, and F demonstrate that the reaction of monoester formation in a biphasic system was exceedingly fast, in strong contrast to the following reaction of diester formation, the concentration of which grew steadily but slowly with time. Already after about 30 s of mean residence time the concentration of monoester reached 90 mM, whereas those of the substrate and diester were very low. Thus, very shortly after the reaction start-up (reactants' entry into the microreactor) substrate conversion exceeded 90%. After a further 15 s (longer residence time owing to the application of a lower flow rate) the substrate was fully converted and the concentration of the monoester reached a maximum of over 90 mM. For longer reaction/residence times (lower flow rates) the concentration of the diester at exit continued to grow as before, at the expense of the monoester. For the longest residence time of ca. 12 min, the diester yield was 50–60% for a single-pass flow. Thus, the rate of the enzyme-catalysed reaction appeared to be significantly enhanced in the proposed microchannel reactor compared to that in the batch reactor with the native enzyme,1 in which full substrate conversion was not achieved even during 7 hours irrespective of the enzyme loading (cf. Fig. 1 and 2 in ref. 1), and the mono-to-diester molar ratio was also much lower in the latter system. These results show not only the exceptional performance of the proposed monolithic microchannel reactor in boosting the enzyme-catalyzed reaction, which may be of major practical interest, but also that the course of transesterification was fully controlled by the catalytic properties of MsAcT confined in a well-defined, functionalised mesoporous environment. That again may be a major asset of the proposed solution, seen from both a fundamental and the most practical point of view.
We believe that this performance can be explained by two factors: (i) the flow-induced reaction effects typically observed in microfluidic/microchannel reactors, which in the case under study boosted monoester formation, additionally enhanced by (ii) the large concentration of the enzyme embedded in the mesoporous environment most favourable for expressing its activity. Similar effects were recently reported for the consecutive reactions carried out in microchannel reactors but without heterogeneous catalysts.12 Asano et al. showed that the application of microreactor technology could increase the yield of the monosubstitution product from 58.6% to 98.6% in the case of bromination, from 77% to 86.3% (nitration) and from 25.2% to 38.1% (ester reduction) in comparison with the batch method.12 A very high activity of enzymes embedded in large mesopores of silica monoliths and in particular mesoporous materials was also observed by other groups and us before.17,18,28,34
Remarkably, at least 50% conversion to the diester is achieved under these aqueous conditions, independent of the carrier. Equally remarkably, up to a flow rate of 1 mL min−1 no diol was observed. A residence time of 1 min was thus sufficient to achieve complete esterification of at least one alcohol group of NPG. This implies that this catalytic system has a great potential for rapid ester synthesis (including bio-diesel) under aqueous conditions.
For practical applications the stability of immobilised biocatalysts is a factor of major importance. In this respect, both the enzymes coupled by His-tag adsorption and by covalent binding showed a very good performance during almost 50 h of continuous operation (Fig. 4), and only a small decrease in activity was observed after 100 h (cf. ESI†). Also, intermediate washing of the reactor and storage of the reactor in buffer did not change the activity.
 | ||
| Fig. 4 Catalytic stability of MsAcT covalently immobilised on amino-modified monolith (A), adsorbed by His-tag on Ni-modified monolith (B) and on Co-modified monolith (C). For covalent immobilisation and His-tag adsorption on Co the cumulative flow rate was 0.1 mL min−1 (buffer/ethyl acetate 50/50% v/v); for His-tag adsorption on Ni the cumulative flow rate was 0.06 mL min−1 (buffer/96 mM neopentylglycol in ethyl acetate 50/50% v/v). Vertical lines show the interruptions in measurements; the immobilised enzyme was washed and stored in buffer: black, diester; blue, monoester. | ||
Another factor of significance for this reaction system is the water phase present. Typically this contains buffers leading to a significant salt load that has to be separated after the reaction and that increases corrosion. Therefore, the stability and the catalyst performance at different flow rates were studied for MsAcT immobilised on Ni-modified silica in the absence of a buffer (Fig. 5).
 | ||
| Fig. 5 Transesterification of neopentylglycol in the absence of buffer by MsAcT adsorbed by His-tag on Ni-modified monolith (A) and process stability (flow rate 0.1 mL min−1) (B). Conditions: 96 mM neopentylglycol in ethyl acetate; room temperature. Red, substrate; black, diester; blue, monoester. Vertical lines show the interruptions in measurements; the immobilised enzyme was washed and stored in buffer. | ||
Also in this case the increase in monoester and decrease in diester formation were observed when the flow rate increased (Fig. 5A). However, while in a biphasic system the largest concentration of monoester, at full substrate conversion, was observed for the flow rates in the range of 0.7–1.0 mL min−1 (Fig. 3), in the non-aqueous situation (Fig. 5A) it was only about 0.35 mL min−1. This is partly due not only to the higher NPG loading (no dilution by buffer) but also to a significant activity drop. Moreover, the non-aqueous system also detrimentally affected enzyme stability, as seen from a rapid decline in the enzyme activity after 4 h of continuous steady operation. MsAcT catalyses not only the formation of the esters but also some hydrolysis of ethyl acetate, leading to the formation of acetic acid and consequently a pH drop. After washing the enzyme with the buffer initial activity could be restored (Fig. 5B), indicating that the deactivation was reversible.
A destructive effect of the absence of a water phase was also observed by Mathews et al.,2 who found that below 2.5% water content MsAcT activity significantly decreased. The relatively high activity of the immobilised enzyme in the organic solvent system (Fig. 5) might be ascribed to water adsorbed to the silica carrier. Acetic acid formation and loss of water slowly led to a deactivation that could be reversed by treatment with a buffer (Fig. 5B). This demonstrates the great gain in enzyme stability achieved by immobilisation.
Conclusions
Our studies demonstrated that the metal ions and amino groups can be uniformly introduced onto the silica monolith surface and that post synthesis treatment has no detrimental effect on their macroporous morphology. Functionalised silica monoliths were successfully used to immobilise MsAcT and demonstrate its exceptional properties in a continuous-flow reaction. Complete ester synthesis could be achieved in aqueous media within very short reaction times. No differences in activity were observed for the enzyme attached using nonspecific covalent immobilisation and a very specific chelation procedure. This highlights the importance of a judicious selection of the carrier. The microreactor demonstrated very high activity and stability of catalytic and structural properties in the continuous esterification of NPG with ethyl acetate at room temperature and in biphasic conditions.Acknowledgements
The authors gratefully acknowledge the National Science Center (NCN) of Poland for the financial support for this work under Grant No. DEC-2013/09/D/ST8/04002 and Nicolas de Leeuw for help with the enzyme assay. V. Resch thanks the Austrian Science Fund (FWF) for an “Erwin-Schroedinger” Fellowship (J3292) and G. Torrelo thanks the Fundación Alfonso Martín Escudero (Spain) for a postdoctoral fellowship.Notes and references
- L. Wiermans, S. Hofzumahaus, C. Schotten, L. Weigand, M. Schallmey, A. Schallmey and P. Dominguez de Maria, ChemCatChem, 2013, 5, 3719 CrossRef CAS.
- I. Mathews, M. Soltis, M. Saldajeno, G. Ganshaw, R. Sala, W. Weyler, M. A. Cervin, G. Whited and R. Bott, Biochemistry, 2007, 46, 8969 CrossRef CAS PubMed.
- J. K. Poppe, R. Fernandez-Lafuente, R. C. Rodrigues and M. A. Z. Ayub, Biotechnol. Adv., 2015, 33, 511 CrossRef CAS PubMed.
- S.-C. Tsai, L. J. W. Miercke, J. Krucinski, R. Gokhale, J. C.-H. Chen, P. G. Foster, D. E. Cane, C. Khosla and R. M. Stroud, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14808 CrossRef CAS PubMed.
- K. J. Weissman, C. J. Smith, U. Hanefeld, R. Aggarwal, M. Bycroft, J. Staunton and P. F. Leadlay, Angew. Chem., Int. Ed., 1998, 37, 1437 CrossRef CAS.
- K. J. Weissman, Nat. Prod. Rep., 2015, 32, 436 RSC.
- M. Subileau, A.-H. Jan, H. Nozac'h, M. Pérez-Gordo, V. Perrier and E. Dubreucq, Biochim. Biophys. Acta, 2015, 1854, 1400 CrossRef CAS PubMed.
- S. Schmidt, C. Scherkus, J. Muschiol, U. Menyes, T. Winkler, W. Hummel, H. Gröger, A. Liese, H.-G. Herz and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2015, 54, 2784 CrossRef CAS PubMed.
- J. Mìller, M. A. Sowa, B. Fredrich, H. Brundiek and U. T. Bornscheuer, ChemBioChem, 2015, 16, 1791 CrossRef PubMed.
- C. Z. Dinu, G. Zhu, S. S. Bale, G. Anand, P. J. Reeder, K. Sanford, G. Whited, R. S. Kane and J. S. Dordick, Adv. Funct. Mater., 2010, 20, 392 CrossRef CAS.
- U. Hanefeld, L. Gardossi and E. Magner, Chem. Soc. Rev., 2009, 38, 453 RSC.
- Y. Asano, S. Togashi, H. Tsudome and S. Murakami, Pharm. Eng., 2010, 30, 32 Search PubMed.
- M. Paravidino, P. Böhm, H. Gröger and U. Hanefeld, Enzyme Catalysis in Organic Synthesis, ed. K. Drauz, H. Gröger and O. May, 3rd edn, Wiley-VCH, Weinheim, 2012, p. 249 Search PubMed.
- A. Koreniuk, K. Maresz, K. Odrozek, A. Jarzębski and J. Mrowiec-Białoń, Appl. Catal., A, 2015, 489, 203 CrossRef CAS.
- K. Szymańska, M. Pietrowska, J. Kocurek, K. Maresz, A. Koreniuk, J. Mrowiec-Białoń, P. Widłak, E. Magner and A. Jarzębski, Chem. Eng. J., 2016, 287, 148 CrossRef.
- K. Szymańska, J. Bryjak and A. B. Jarzębski, Top. Catal., 2009, 52, 1030 CrossRef.
- A. Jarzębski, K. Szymańska, J. Bryjak and J. Mrowiec-Białoń, Catal. Today, 2007, 124, 2 CrossRef.
- K. Szymańska, W. Pudło, J. Mrowiec-Białoń, A. Czardybon, J. Kocurek and A. B. Jarzębski, Microporous Mesoporous Mater., 2013, 170, 75 CrossRef.
- D. Gaffney, J. Cooney and E. Magner, Top. Catal., 2012, 55, 1101 CrossRef CAS.
- D. Gaffney, N. H. Abdallah, J. C. Cooney, F. R. Laffir, K. Engelmark Cassimjee, P. Berglund, U. Hanefeld and E. Magner, J. Mol. Catal. B: Enzym., 2014, 109, 154 CrossRef CAS.
- O. H. Lowry, N. J. Rosebrough, N. J. Farr and R. J. Randall, J. Biol. Chem., 1951, 193, 265 CAS.
- G. L. Peterson, Anal. Biochem., 1977, 83, 346 CrossRef CAS PubMed.
- C. Z. Dinu, I. V. Borkar, S. S. Bale, A. S. Campbell, R. S. Kane and J. S. Dordick, J. Mol. Catal. B: Enzym., 2012, 75, 20 CrossRef CAS.
- N. Grover, M. P. Douaisi, I. D. Borkar, L. Lee, C. Z. Dinu, R. S. Kane and J. S. Dordick, Appl. Microbiol. Biotechnol., 2013, 97, 8813 CrossRef CAS PubMed.
- A. D. Stroock, S. W. Dertinger, A. Ajdari, I. Mezic, H. A. Stone and G. M. Whitesides, Science, 2002, 295, 647 CrossRef CAS PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309 CrossRef CAS.
- E. Calleri, C. Temporini, F. Gasparrini, P. Simone, C. Villani, A. Ciogli and G. Massolini, J. Chromatogr. A, 2011, 1218, 8937 CrossRef CAS PubMed.
- J. Ma, Z. Liang, X. Qiao, Q. Deng, D. Tao, L. Zhang and Y. Zhang, Anal. Chem., 2008, 80, 2949 CrossRef CAS PubMed.
- A. A. Halim, N. Szita and F. Baganz, J. Biotechnol., 2013, 168, 567 CrossRef PubMed.
- M. Miyazaki, J. Kaneno, S. Yamaori, T. Honda, M. P. P. Briones, M. Uehara, K. Arima, K. Kanno, K. Yamashita, Y. Yamaguchi, H. Nakamura, H. Yonezawa, M. Fujii and H. Maeda, Protein Pept. Lett., 2005, 12, 207 CrossRef CAS PubMed.
- Z. Guo, S. Xu, Z. Lei, H. Zou and B. Guo, Electrophoresis, 2003, 24, 3633 CrossRef CAS PubMed.
- S. Matosevic, G. J. Lye and F. Baganz, J. Biotechnol., 2011, 155, 320 CrossRef CAS PubMed.
- S. Matosevic, G. J. Lye and F. Baganz, Biotechnol. Prog., 2010, 26, 118 CAS.
- L. Qiao, Y. Liu, S. P. Hudson, P. Yang, E. Magner and B. Liu, Chem. – Eur. J., 2008, 14, 151 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cy02067k |
| This journal is © The Royal Society of Chemistry 2016 |