
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePd/zeolite-based catalysts for the preferential CO oxidation reaction: ion-exchange, Si/Al and structure effect†
Miriam
Navlani-García
a,
Izaskun
Miguel-García
a,
Ángel
Berenguer-Murcia
a,
Dolores
Lozano-Castelló
*a,
Diego
Cazorla-Amorós
a and
Hiromi
Yamashita
*b
aAlicante Materials Institute and Inorganic Chemistry Department, University of Alicante, 03080 Alicante, Spain. E-mail: d.lozano@ua.es; Fax: +34 96590 3454; Tel: +34 96590 3946
bDivision of Materials and Manufacturing Science, Graduate School of Engineering, Osaka University, 2-1 Yamada-oka, Suita, Osaka 565-0871, Japan. E-mail: yamashita@mat.eng.osaka-u.ac.ps
First published on 8th January 2016
Abstract
A screening of Pd/zeolite-based catalysts in the PrOx-CO reaction in H2-rich streams was performed using zeolites with different cations (H+, Na+ and Cs+) prepared by ion exchange and framework type (MFI and FAU). The assessment of the catalytic performance displayed by these zeolite-based samples revealed that both parameters play an important role in the catalytic behaviour. The optimisation of both parameters led to the preparation of an optimum catalyst, which showed high CO conversion and CO selectivity during long-term stability tests.
Introduction
In the last few years, energy consumption has risen to very high levels due to both worldwide population increase and technological development. The demand for energy is steadily growing and fossil fuel reserves are diminishing. Oil, coal, and natural gas supplies are not replenished as they are consumed, so an alternative energy source must be found. In this sense, the development of a hydrogen-based economy is gaining much attention, as H2 is an energetic vector with many advantages with respect to fossil fuels. The most important aspect is the possibility of obtaining clean energy directly from H2 and O2 that would produce water as the only product. To do that, one of the most viable perspectives is the implementation of hydrogen fuel cells. These are devices which would produce energy directly from the electrochemical reaction between H2 and O2. However, there are several drawbacks to overcome for the implementation of this technology. One of them is that each of the different H2 sources contains pollutants in different concentrations, and tolerance of the electrodes of fuel cells is very low to many of the pollutants contained in H2 streams. Therefore, it is mandatory to diminish the amount of the unwanted gases to levels that the fuel cells can tolerate. One of the gases which must be completely removed from the H2 streams is CO. This gas is obtained from the steam reforming reaction in which H2 is produced. To remove the CO generated, the first step is the water–gas shift (WGS) reaction, which usually takes place in two consecutive steps at different temperatures. However, the CO concentration exiting the WGS reactor is still too high for the entrance of the fuel cells. Therefore, an additional step is necessary to further diminish the CO concentration. Nowadays, several options are available, such as gas separation with membranes, pressure-swing adsorption, the methanation process and the preferential CO oxidation reaction (PrOx-CO). The objective of these processes is to diminish the CO levels to below 10 ppm, which is the tolerance level of the Pt electrodes used in the fuel cells.1The preferential CO oxidation reaction is one of the most promising and economic approaches to achieve acceptable CO concentrations in the H2 stream after the water–gas shift reactor, under temperatures between those of the WGS reactor and the fuel cell operation temperatures (80–200 °C).2,3 In order to perform the PrOx-CO reaction, several kinds of active phase catalysts have been studied, with those based on supported gold catalysts, supported Pt-based catalysts and other noble metals (Pd, Rh, Ru, Ir) being the most representative.4 In this regard, the low activity and selectivity of Pd-supported catalysts for PrOx-CO was confirmed by many authors.2,5,6 The low activity at low temperature was explained by the formation of a hydride suppressing the possibility of CO oxidation, whereas at higher temperature metallic Pd preferentially adsorbs hydrogen rather than CO, and the adsorbed H species are more easily oxidised by the surface Pd oxide species than CO. However, a study carried out by our research group indicated that the use of polymer-protected palladium-based nanoparticles supported on Al2O3 could be useful in the PrOx-CO reaction.7,8 In addition, recent studies demonstrated that the use of Pd in the PrOx reaction is very promising as it afforded the lowest activation energies among the platinum-group metals investigated.9
Concerning the catalyst support, several reports dealing with the support effect in the final catalytic performance can be found in the literature. For instance, a set of Au/CexZr1−xO2 catalysts was studied by Morfin and coworkers and they found that the catalytic performance was strongly dependent on the nature of the oxide.10 Kubacka investigated the role of the interface in Cu, Ni–CeO2 catalysts towards hydrogen production by means of several reactions (PrOx-CO, WGS and the reforming of bio-alcohols).11 The effect of the support was also investigated by Yamamoto in a study where bimetallic PtCu catalysts were supported on Fe2O3 and CeO2,12 and in this case the authors concluded that the supports have an important influence on both the structure of Pt–Cu and the reduction behaviour of the catalyst. It is well-known that alumina is the most common support for noble metal-based catalysts;13–15 however, it was found that the selectivity towards CO oxidation can be improved by using a different support and several studies have pointed out that the use of zeolites as support for noble metal catalysts could be promising to improve their performance.2,16,17 In addition, the zeolite matrix can facilitate the formation of active sites for different catalytic reactions, e.g. oxidation of aliphatic chlorinated volatile organic compounds,18 combustion of toluene,19 methylation of benzene by methanol20 and so forth. In fact, zeolites constitute the most important group of heterogeneous catalysts with large-scale application in refining and petrochemistry and are also widely used in environmental catalysis.21 The ability of zeolites to be used in many fields is related to their tunable properties, which can be controlled by selecting the charge-balancing cation, the framework structure and the zeolite composition. In particular, the use of zeolites as catalyst support in the PrOx reaction has been attributed to their high thermal stability and improved selectivity, which is related to molecular sieve effects.16,22 Most of the catalytic systems in which zeolites are studied as catalysts in the PrOx-CO reaction use Pt or Au as the active phase and the metal is incorporated to the zeolite structure by an ion exchange procedure22,23 or an impregnation method.22,24,25 However, the use of different active phases or different preparation methodologies is scarcely found in the literature and still constitutes a challenge for the research community. Thus, bearing these considerations in mind and the experience that our group has in the synthesis of palladium nanoparticles, the aim of the present work was to prepare catalysts based on palladium nanoparticles supported on zeolites with different ion-exchanged cations and framework type and assess the effect of these zeolite characteristics on the catalytic performance in the PrOx-CO reaction. To this end, Pd nanoparticles were firstly prepared in colloidal phase by using the polyol method and they were subsequently deposited on the zeolitic support by following a standard impregnation methodology. It should be highlighted that among the synthesis protocols encompassed in the chemical reduction of metal salt, the case of the reduction by solvent method has been reported as one of the most convenient experimental methodologies to prepare metal nanoparticles due to lack of the required templating materials, low cost and simplicity of the process. Apart from this, this synthetic protocol constitutes a very useful tool towards the preparation of size-, shape- and composition-controlled nanoparticles, which would be less favoured in the case of using the standard impregnation of the metal precursor and subsequent reduction.26 The selected zeolites were ZSM-5 (MFI framework type with 3-dimensional channels and a pore system consisting of 10-membered-rings with an opening channel diameter of 0.51 × 0.55 and 0.53 × 0.56 nm) and Y (FAU framework type with 3-dimensional channels and a pore system consisting of 12-membered rings with an opening channel diameter of 0.74 × 0.74 nm). Both zeolites were ion-exchanged with three different cations, H+, Na+ and Cs+. Once the catalysts were prepared and characterised, their catalytic performance in the PrOx-CO reaction was evaluated.
Experimental
Synthesis and purification of palladium nanoparticles
Palladium nanoparticles were synthesized following the reduction by solvent method as previously reported in a work published by our research group.7 The palladium precursor was Pd(OAc)2 (98%, Sigma-Aldrich) and the capping agent used was polyvinylpyrrolidone (PVP, 40 K, Sigma-Aldrich). The experiments were carried out in an inert argon atmosphere by means of a Schlenk system.For solution 1, 1.114 g (10 mmol) of PVP (capping agent) were added to 120 ml of ethylene glycol in a three-necked round-bottomed flask and the solution was stirred at 80 °C for 2 h using a magnetic stirrer and an oil bath.
For solution 2, 0.2245 g (1 mmol) of Pd(OAc)2 were dissolved in 50 ml of dioxane by stirring for 2 h at room temperature using a magnetic stirrer. This solution was light orange in colour.
Solution 1 was cooled to 0 °C with an ice bath and 3 ml of 1 M NaOH solution were added under stirring in order to adjust the pH of the resulting mixture to 9–10. Then, solution 2 was poured into solution 1 under vigorous stirring to ensure homogenisation and the final mixture was heated at 100 °C. The solution changed its colour from light orange to dark brown, which indicated that the metallic colloid had been formed.27 After that, the heating was maintained for 2 h and then the bath was removed and the colloidal suspension was cooled to room temperature.
The resulting suspension presented high stability against sintering and it could be stored under ambient conditions without formation of aggregates for periods of time over one year.
Once the colloids were prepared as described previously, the palladium nanoparticles were purified and redispersed in methanol. The purification was based on pouring an aliquot of nanoparticles colloid into a glass bottle, adding an excess of acetone and shaking the solution. As we previously reported,28 this treatment with acetone produced the extraction of the protecting polymer to the acetone phase and flocculation of the metallic nanoparticles. After discarding the supernatant organic phase, the purified nanoparticles were redispersed by gentle stirring in a known amount of methanol, so that the final suspension concentration was perfectly known.
Catalyst support preparation
In this work, six catalyst supports were used. In order to prepare the ion-exchanged ZSM-5 and Y zeolites (in H+, Na+ and Cs+ forms) a conventional ion-exchange method was used. The initial zeolites were NH4-ZSM-5 (provided by Zeolyst International) and Na-Y (provided by Sigma-Aldrich).In the case of ZSM-5 zeolite, H-ZSM-5 was obtained directly by conversion of the commercial form (NH4-ZSM-5) by heat treatment in air at 450 °C for 6 h with a heating rate of 5 °C min−1. Na-ZSM-5 zeolite was prepared by ion exchange of the H-zeolite with NaNO3 (0.2 M) as the Na+ precursor, and Cs-ZSM-5 zeolite was subsequently obtained by ion exchange of Na-ZSM-5 with CsNO3 (0.1 M) as the Cs+ precursor and keeping under stirring in a thermostatic bath at 60 °C for 1 h.
For Y zeolite, the H-Y form was obtained by ion exchange of the commercial form (Na-Y) by using NH4NO3 (1 M) and keeping under stirring in a thermostatic bath at 30 °C for 24 h and subsequent heat treatment in air at 450 °C for 6 h with a heating rate of 5 °C min−1. The Cs-Y zeolite was directly obtained by ion exchange of Na-Y with CsNO3 (0.1 M) as the Cs+ precursor and keeping under stirring in a thermostatic bath at 60 °C for 1 h.
After each ion exchange step, the samples were filtered and washed with distilled water and then the zeolites were dried at 100 °C and calcined at 550 °C for 4 h (heating rate of 1 °C min−1). The obtained catalyst supports were denoted as H-ZSM-5, Na-ZSM-5, Cs-ZSM-5, H-Y, Na-Y and Cs-Y.
Catalyst preparation
The different catalysts studied in this work were prepared by impregnation of the zeolitic supports with the palladium nanoparticles colloid. For this purpose, 1.5 g of each support was mixed with an adequate amount of the purified nanoparticles colloid suspension in order to obtain a final metallic loading of 1 wt%. The mixtures were vigorously stirred for two days using a magnetic stirrer to guarantee similar metal loading and dispersion in all the catalysts and then the samples were heat treated at 60 °C to eliminate the solvent (methanol). Finally, the catalysts were washed several times with a cold mixture of H2O/EtOH (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50, v/v) and dried at 60 °C for 12 h. The resulting catalysts were denoted as Pd/H-ZSM-5, Pd/Na-ZSM-5, Pd/Cs-ZSM-5, Pd/H-Y, Pd/Na-Y and Pd/Cs-Y.
:50, v/v) and dried at 60 °C for 12 h. The resulting catalysts were denoted as Pd/H-ZSM-5, Pd/Na-ZSM-5, Pd/Cs-ZSM-5, Pd/H-Y, Pd/Na-Y and Pd/Cs-Y.
Catalyst characterisation
Catalyst supports based on zeolites Y and ZSM-5 were characterised by XRD using a 2002 Seifert powder diffractometer to ensure that the ion exchange did not modify the zeolites' structure. The scanning rate was 2 °C min−1 and Cu-Kα radiation was used.The textural characterisation of the zeolitic supports was carried out by means of adsorption of N2 at −196 °C and CO2 at 0 °C (Autosorb 6, Quantachrome). Prior to the adsorption measurements, the supports were outgassed in situ under vacuum at 250 °C for 4 h in order to remove any adsorbed impurities. Apparent surface area values were calculated from nitrogen adsorption isotherms using the BET equation (SBET). Total micropore volume (VDR(N2)) and narrow micropore volume (VDR(CO2)) were calculated applying the Dubinin–Radushkevich (DR) equation to the N2 adsorption data at −196 °C and the CO2 adsorption data at 0 °C, respectively.
Transmission electron microscopy (TEM) images were obtained by using a JEOL (JEM-2010) transmission electron microscope equipped with an EDS analyser (OXFORD, model INCA Energy TEM 100) operating at 200 kV with a space resolution of 0.24 nm. In order to prepare the samples for TEM analysis, a small amount of the catalyst was suspended in ethanol and sonicated in an ultrasonic bath for a few minutes. After that, a drop of this suspension was deposited onto a 300 mesh Lacey copper grid and dried at room temperature. TEM images were also captured for the palladium colloid in order to compare the NP diameter and particle size distribution before and after the impregnation step.
The dispersion of the palladium nanoparticles on the catalysts (D), which is an estimation of the amount of metal exposed in the nanoparticles, was calculated from the following equation and assuming a spherical shape of the particles (d, particle diameter):29
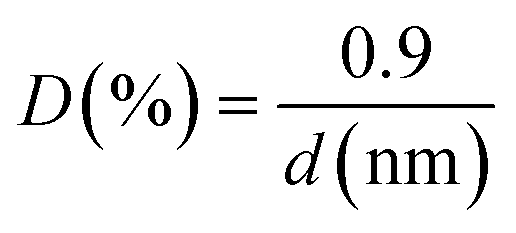
ICP-OES (inductively coupled plasma-optical emission spectroscopy) was the selected technique to determine the palladium loading. For this purpose, a Perkin-Elmer Optima 4300 system was used. The palladium extraction was carried out by oxidative treatment with aqua regia for 48 h. Then, the samples were filtered and diluted.
XPS (X-ray photoelectron spectroscopy) data were recorded using a VG-Microtech Multilab 3000 spectrometer equipped with a semispherical electron analyzer and a Mg Kα (hν = 1253.6 eV) 300 W X-ray source. Before recording the spectra, the samples were maintained in the analysis chamber until a residual pressure of 5 × 10−9 mbar was reached. The spectra were collected at a pass energy of 50 eV. Binding energies were referenced to the C 1 s line at 284.6 eV, which provide values with an accuracy of 0.2 eV. The Pd(0)/Pd(II) ratios were calculated from the integrated intensities of the spectra.
All the characterisation techniques were performed on the as-synthesised (non-reduced) catalysts.
Catalytic tests
All the catalysts were tested in the preferential CO oxidation reaction. The reactor used was a U-shaped reactor (16 mm inner diameter) where 150 mg of sample were placed. The samples were reduced with H2, using a flow of 50 ml min−1 of 10% H2/He in order to obtain reduced palladium nanoparticles. After that, the samples were cooled to room temperature and the reactant gas mixture (composed of 2% CO, 2% O2, 30% H2, balance He) was passed through the catalysts. The oxygen excess, defined by the following equation, was λ = 2 (being λ = 1 for stoichiometric conditions).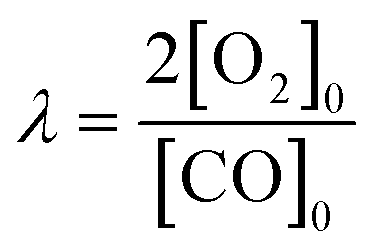
The gas composition is a hydrogen-rich feed in order to closely mimic the conditions used in the water–gas shift reactor in the hydrogen purification process for fuel cell applications. The catalytic tests were performed using a heating rate of 2 °C min−1 and a total gas flow rate of 100 ml min−1 that was set by means of mass flow controllers (MFCs, Bronkhorst). Catalytic activity was also expressed as TOF (in s−1), defined as moles of CO consumed per moles of surface metal per second. The exhaust gases were analysed by gas chromatography (Agilent Technologies 6890 N equipped with a CTRI column operating at 80 °C and a TCD detector). Moreover, the samples with the best catalytic behaviour were tested under a more complex gas mixture (composed of 2% CO, 2% O2, 30% H2, 20% CO2, 5% H2O and balance He) and by carrying out long-term stability experiments at 200 °C and with a total flow rate of 50 ml min−1 in order to check the effect of CO2 and H2O on the catalytic performance in the PrOx-CO reaction. The measured inlet and outlet concentrations were used to calculate the CO conversion and selectivity according to the following equations:
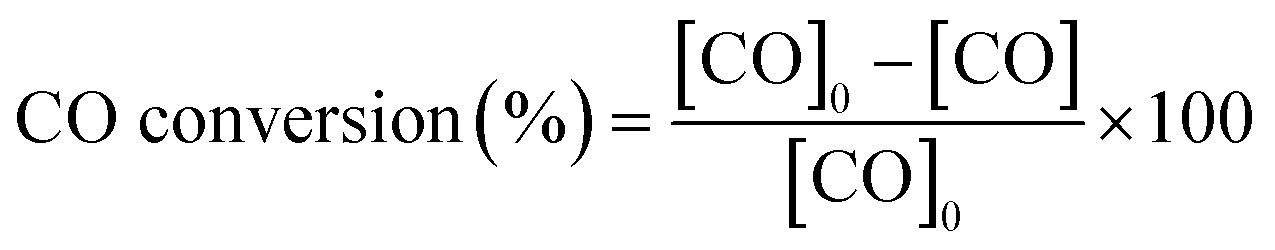
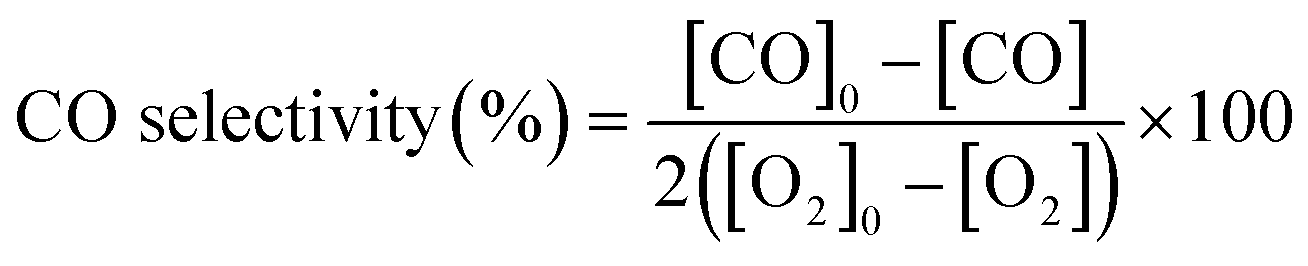
All experiments were performed in duplicate to ensure the reproducibility of the results.
Results
Colloid characterisation
A palladium colloid sample was prepared by the reduction by solvent method. This is a synthetic protocol that allows preparing metallic nanoparticles with a very small particle size and a narrow size distribution. Fig. 1 shows a TEM image of the Pd nanoparticles colloid prepared and purified with acetone and the histogram corresponding to the particle size distribution. As can be observed from the image, the nanoparticles show a small average particle size (d = 1.8 ± 0.3 nm) and a very narrow size distribution, as expected according to other authors' reports27 and observed by us in previous works.7,30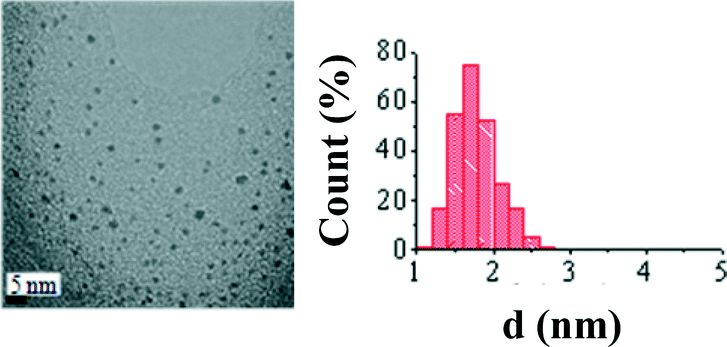 | ||
| Fig. 1 TEM image of the as-prepared Pd nanoparticle colloid prepared and purified with acetone, and the histogram corresponding to the particle size distribution. | ||
Nanoparticles prepared by the reduction by solvent method are highly stable against sintering and no agglomeration of the particles has been detected for periods of time over one year.
Support characterisation
In order to observe the effect of ion exchange in the crystallinity of the ZSM-5 and Y zeolites, XRD analyses were performed on the different supports. As was expected, the ion exchange steps (and the heat treatment performed to convert the ammonium form of the ZSM-5 zeolite in the acid counterpart) did not modify the zeolite crystallinity, as could be extracted from the comparison between the XRD patterns found in the literature (see Fig. S1 in the ESI†). Nevertheless when both zeolites were exchanged with cesium (supports denoted Cs-ZSM-5 and Cs-Y, respectively) a marked decrease in the number and intensity of the XRD peaks is observed compared to the parent commercial zeolites. It should be pointed out that when the ion exchange with cesium is carried out, the used salt contains trace amounts of Ba2+, which has been reported to significantly alter the zeolitic structure.31As reported in the literature, the change in the relative intensities of the peaks when ion-exchange is performed could be due to the fact that the inclusion of a larger cation in the zeolite network can partially distort the structure because it must be accommodated into the zeolite structure.32 Moreover, the XRD peak intensity decrease has been also attributed to the strong absorption of X-ray by Cs.33
Concerning the textural properties, Table 1 summarizes the BET surface area (SBET), the total micropore (VDR(N2)) and the narrow micropore volume (VDR(CO2)) of the supports and Fig. 2 shows the N2 adsorption–desorption isotherms at −196 °C.
| Sample | S BET (N2) (m2 g−1) | V DR (N2) (cm3 g−1) | V DR (CO2) (cm3 g−1) |
|---|---|---|---|
| H-ZSM-5 | 398 | 0.17 | 0.15 |
| Na-ZSM-5 | 373 | 0.17 | 0.14 |
| Cs-ZSM-5 | 344 | 0.16 | 0.12 |
| H-Y | 724 | 0.35 | 0.41 |
| Na-Y | 721 | 0.35 | 0.31 |
| Cs-Y | 499 | 0.24 | 0.20 |
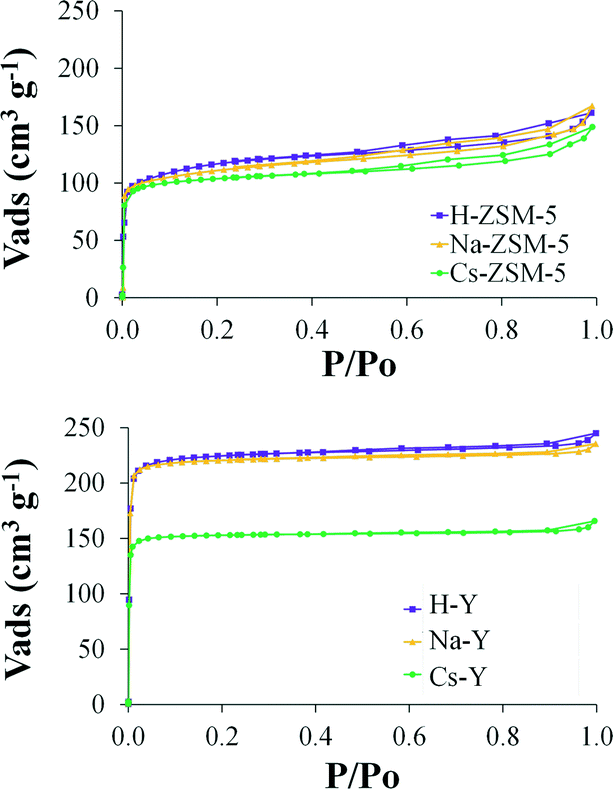 | ||
| Fig. 2 N2 adsorption–desorption isotherms at −196 °C corresponding to the catalyst supports. | ||
Comparing zeolites ZSM-5 and Y with the same cation exchanged, in all cases the surface area and pore volume of ZSM-5 samples are much lower than those of Y zeolite, as expected from the different structure.34 However, the MFI-type zeolites display a small amount of mesoporosity (as evidenced from the hysteresis loop found for relative pressure values between 0.5 and 0.95) which is not present in its FAU-type counterpart. On the other hand, in the subsequent ion-exchange steps (from H-zeolites to Na-zeolites and from Na-zeolites to Cs-zeolites) a decrease in surface area is observed. These results can be explained by the difference of cation sizes since the cation diameter of H+ (0.012 Å) is much smaller than that of Na+ (1.9 Å) and Cs+ (3.3 Å)35 and when these bigger cations occupy the sites inside the zeolite channel a decrease in surface area and pore volume is provoked.36
The decrease in surface area and micropore volume in sample Cs-Y is much more significant than in sample Cs-ZSM-5. It is important to take into account that in the ZSM-5 zeolite the cations are preferentially located in the intersection of the sinusoidal and straight channels at the edge of the four-membered ring, while in the Y zeolite, the cation can be located in three different sites: on the hexagonal prism faces between the sodalite units, in the open hexagonal faces and on the wall of the supercage.37 Thus, the presence of Cs+ in some of these ion-exchange sites in Y zeolite together with the higher ion-exchange capacity of this zeolite compared to ZSM-5 could lead to the partial blockage of the porosity.37
Catalyst characterisation
Concerning the catalyst characterisation, Fig. 3 shows the TEM micrographs of the fresh ion-exchanged ZSM-5 and Y catalysts and the pertinent histograms with the particle size distributions.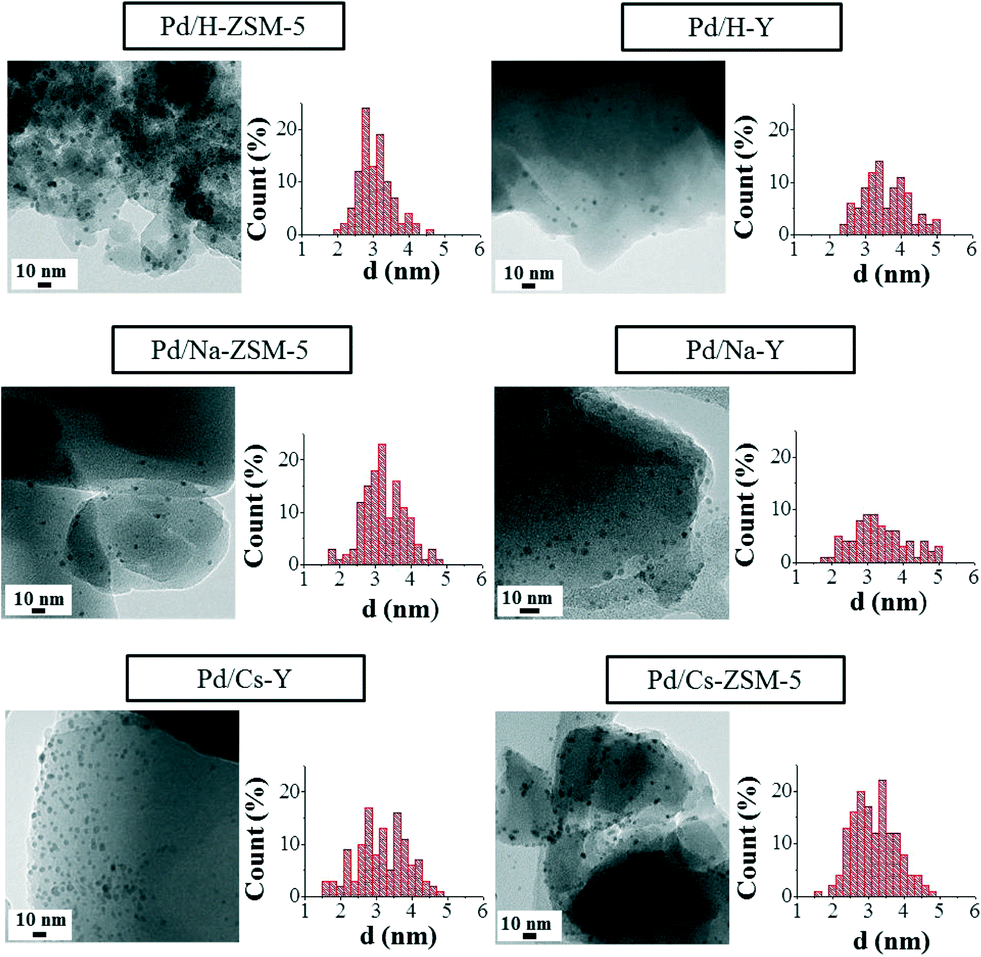 | ||
| Fig. 3 TEM images and Pd nanoparticle size distribution in the fresh ion-exchanged ZSM-5 and Y zeolites. | ||
As was previously reported by our research group for similar Pd/zeolite-based catalytic systems,30 Pd nanoparticles supported on zeolites show a very narrow size distribution and aggregates of palladium nanoparticles are not formed as a consequence of the impregnation or drying steps. However, a slight loss of their spherical shape when they are compared to the Pd nanoparticles in the metallic colloid can be appreciated, which might be favoured by the interaction between the metal and the support surface.7
In addition, in order to assess the effect of the experimental conditions used in the catalytic tests on the Pd nanoparticle morphology, TEM analysis of the used Pd/H-Y and Pd/Na-Y samples was also performed and the results are shown in Fig. 4.
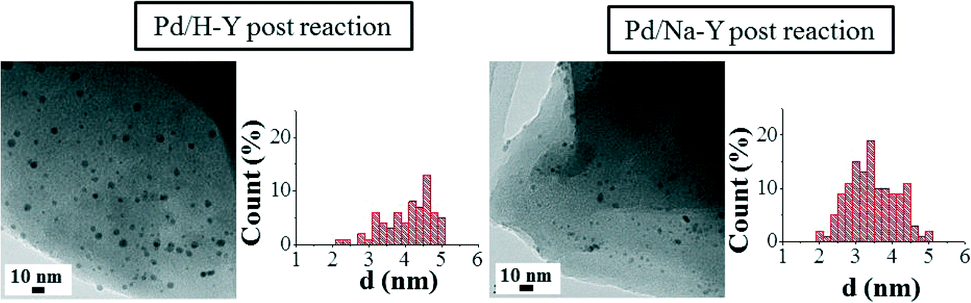 | ||
| Fig. 4 TEM images of samples Pd/H-Y and Pd/Na-Y after being used in the catalytic test and the pertinent histograms with the particle size distributions. | ||
The recorded TEM images of the used Pd/H-Y and Pd/Na-Y samples revealed that in both catalysts, the same palladium nanoparticle morphology as in the case of fresh samples was observed, but a significant particle size increase for the used Pd/H-Y sample was shown. Moreover, in order to check whether the nanoparticle size increase observed in sample Pd/H-Y after being tested was due to the reaction conditions used or to the previous catalyst pretreatment, TEM analysis of sample Pd/H-Y after being pretreated with 10% H2/He at 200 °C was also carried out (figure not shown here; see Table 2 for more details).
| Sample | d TEM (nm) | D TEM (%) | Metal loading ICP (wt%) |
|---|---|---|---|
| Pd/H-ZSM-5 (fresh) | 3.1 ± 0.5 | 29.0 | 0.68 |
| Pd/Na-ZSM-5 (fresh) | 3.2 ± 0.6 | 28.1 | 0.45 |
| Pd/Cs-ZSM-5 (fresh) | 3.2 ± 0.6 | 28.1 | 0.42 |
| Pd/H-Y (fresh) | 3.8 ± 0.9 | 23.7 | 0.46 |
| Pd/H-Y (pretreated) | 4.2 ± 1.0 | 21.4 | — |
| Pd/H-Y (post reaction) | 4.9 ± 1.1 | 18.4 | — |
| Pd/Na-Y (fresh) | 3.5 ± 1.0 | 25.7 | 0.57 |
| Pd/Na-Y (post reaction) | 3.5 ± 0.7 | 25.7 | — |
| Pd/Cs-Y (fresh) | 3.3 ± 0.8 | 27.3 | 0.71 |
Table 2 summarizes the nanoparticle sizes for all the studied catalysts (fresh and used in the case of Pd/H-Y and Pd/Na-Y samples), the dispersion calculated from the TEM images and the metal loading determined by means of ICP.
As can be seen from the data included in Table 2, the nanoparticle size and dispersion are very similar in all the fresh catalysts. Comparing the nanoparticles size in the catalysts and in the metal colloid, a significant particle size increase is produced when the palladium nanoparticles are supported onto the different zeolites (colloid, d = 1.8 ± 0.3 nm). This size increase could be a consequence of the impregnation process, where some nanoparticle deformation can take place, leading to the observation of apparent larger metal nanoparticles. Furthermore, considering the nanoparticle size observed in all the catalysts and the aforementioned zeolite opening channel diameters, it is expected that in all the catalysts the palladium nanoparticles are located on the zeolite external surface.
Regarding the post-reaction TEM analysis, some differences are observed for the used samples. In this sense, changes in nanoparticle size for sample Pd/Na-Y after being used in the catalytic test reaction were not observed, showing an average nanoparticle size of 3.5 nm for both fresh and used samples. On the other hand, in the case of sample Pd/H-Y the palladium nanoparticle size is not constant for the sample in all the stages. Fresh Pd/H-Y sample showed a nanoparticle size of 3.8 nm, while a slight increase was observed after the pretreatment with 10% H2/He at 200 °C (average particle size 4.2 nm). Moreover, TEM characterisation of the sample after being used in the catalytic tests indicated that the palladium nanoparticle size increases even more (average particle size 4.9 nm) after being tested under the experimental conditions used (gas mixture composed of 2% CO, 2% O2, 30% H2, and balance He and heated up to 200 °C). The results indicate that the palladium nanoparticles' stability in terms of anchorage on the support strongly depends on the cation present in the zeolite Y. In this regard, it seems that palladium nanoparticles are more stabilised on the Na-Y than on the H-Y support, which may be related to the stronger Pd/PVP-system interaction with Na/Y than with H/Y, which seems to have a metal nanoparticle sintering inhibition ability. In this sense, the dependence of the Pd/PVP nanoparticle system’s stability and particle size on the support characteristics for different metal oxide supports has already been reported.38
Concerning the Pd loading, the ICP spectroscopy results indicated that it ranged from 0.42 to 0.71 wt% even though all the catalysts were prepared to contain 1 wt% Pd.
In order to analyse the nature of the Pd contained in the different catalysts, XPS analyses of the fresh samples were performed. A typical XPS spectrum of a Pd-based catalyst shows two electronic transitions corresponding to the bands Pd 3d3/2 (at higher binding energies) and Pd 3d5/2 (at lower binding energies). Each of those transitions can be deconvoluted into two different peaks that are associated to electronically different Pd species, in agreement with previous results.7,8 The peak appearing at lower binding energies relates to Pd(0) and the peak at higher binding energies corresponds to Pd(II). In all the catalysts analysed in this study both Pd species (Pd(0) and Pd(II)) have been found by XPS analyses. Table 3 contains the results corresponding to the atomic percentage of the different Pd species present on the catalyst surface. Previous studies performed by our research group pointed out that after the reduction pre-treatment carried out before testing the catalysts, the Pd(II) (or Pd with some positive charge, δ+) contained in the samples is still present, so total metal reduction does not take place under the conditions used.7 This is due to the presence of PVP surrounding the nanoparticles. The excess of PVP that is used as surfactant during the synthesis of nanoparticles is removed in the purification step. However, some of the PVP molecules linked to the metal surface remain, and therefore when the nanoparticles are deposited onto the supports, some surfactant molecules are also present. The interaction between the PVP polymer and Pt and Rh metal nanoparticles has been widely studied in the literature.39,40 This interaction consists in the PVP withdrawing electronic density from the metal surface through the CO groups contained in the monomers. We have previously demonstrated that this is also the case with Pd nanoparticles.7,8 This interaction leaves the metal surface with a deficiency of electronic density, and in an XPS spectrum these metal species appear as partially oxidized Pd (Pd(II)). Moreover, the contribution of Pd(II) seems to depend on the catalyst support. In this sense, it could be said that in general, the Pd(II) contribution is slightly higher in the case of samples based on ZSM-5 zeolite than in the other catalysts.
| Sample | Pd(0) (atomic %) | Pd(II) (atomic %) |
|---|---|---|
| Pd/H-ZSM-5 | 76 | 24 |
| Pd/Na-ZSM-5 | 74 | 26 |
| Pd/Cs-ZSM-5 | 84 | 16 |
| Pd/H-Y | 80 | 20 |
| Pd/Na-Y | 91 | 9 |
| Pd/Cs-Y | 81 | 19 |
Catalytic tests
Catalytic tests were performed for all the catalysts prepared for this work. In addition, control experiments with the bare zeolitic supports were carried out to confirm the inactivity of the supports and, as expected, no catalytic activity was observed for any of them. Fig. 5 shows the catalytic performance (expressed as CO conversion and selectivity) of the palladium nanoparticles deposited on the differently ion-exchanged zeolites (ZSM-5 and Y, respectively).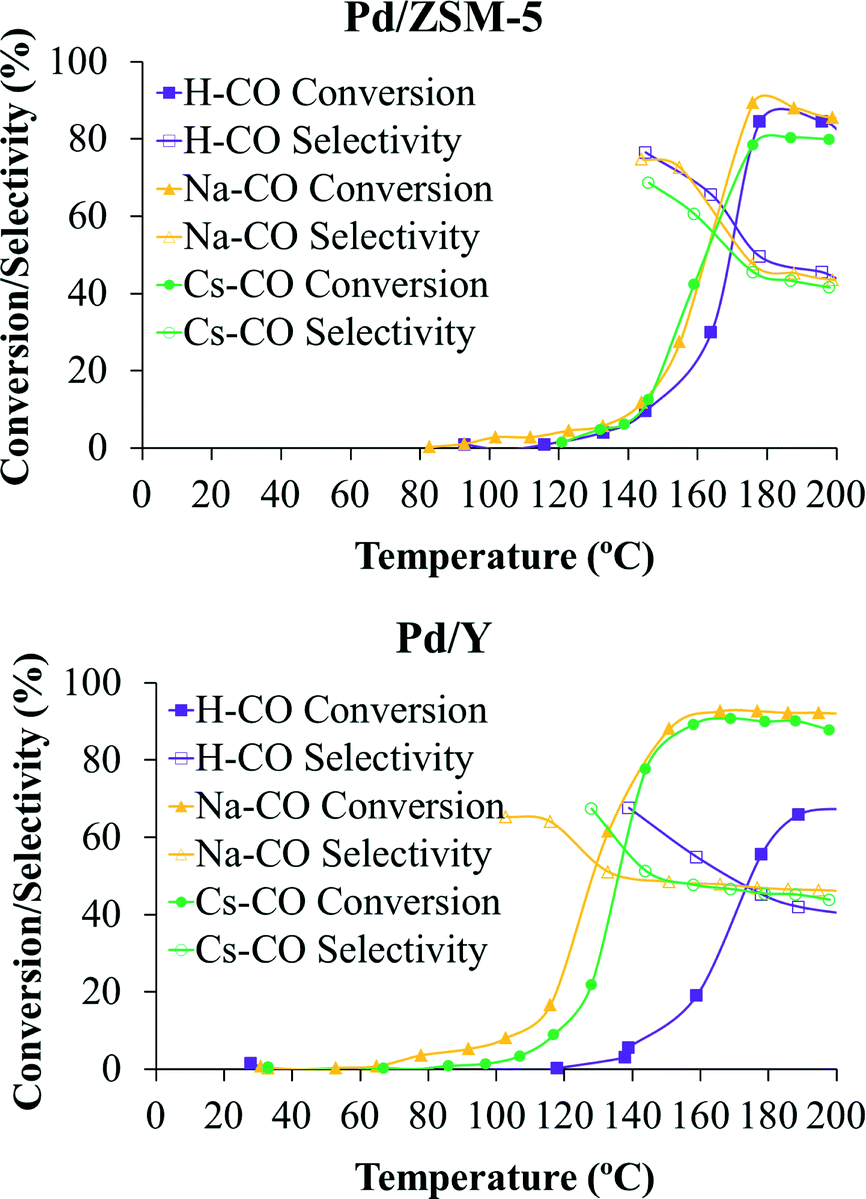 | ||
| Fig. 5 CO conversion (%, solid symbols) and selectivity (%, open symbols) of the different ion-exchanged ZSM-5 and Y catalysts. | ||
Fig. 5 presents the CO conversion and selectivity of the different ion-exchanged ZSM-5 and Y catalysts as a function of temperature. Both profiles are very important to determine the suitability of the catalysts for the PrOx-CO reaction. In this sense, the selectivity towards CO oxidation as a function of the temperature is an important parameter in the PrOx reaction, since the O2 in the gas stream can be used not only for the CO oxidation to CO2 but also for the concurrent H2 oxidation, which leads to important drawbacks from both a technological and an economical point of view.7
It can be observed in Fig. 5 that both CO conversion and selectivity towards CO oxidation strongly depend on the temperature. In this sense, the CO conversion reaches a maximum at a certain temperature (which is different for all the samples) and then decreases slowly. The selectivity also decreases at high temperatures for all the catalysts, which suggests that the H2 oxidation becomes more favoured at high temperatures due to the higher activation energy of the H2 oxidation with respect to the CO oxidation reaction.7,24,41
In order to assess the catalytic performance from a more comparative point of view, TOF (s−1) values calculated at 125 °C are included in Table 4. In addition, maximum CO conversion (%), maximum CO selectivity (%) and selectivity (%) at 25% of CO conversion are also included in the same table.
| Sample | Maximum CO conversion (%) | Maximum CO selectivity (%) | Selectivity (%) at 25% of CO conversion | TOF (s−1) 125 °C |
|---|---|---|---|---|
| Pd/H-ZSM-5 | 85 (178 °C) | 77 (145 °C) | 67 (161 °C) | 0.01 |
| Pd/Na-ZSM-5 | 89 (176 °C) | 75 (144 °C) | 73 (154 °C) | 0.04 |
| Pd/Cs-ZSM-5 | 80 (187 °C) | 69 (146 °C) | 65 (152 °C) | 0.03 |
| Pd/H-Y | 67 (200 °C) | 67 (139 °C) | 53 (163 °C) | 0.02 |
| Pd/Na-Y | 93 (166 °C) | 65 (103 °C) | 61 (120 °C) | 0.16 |
| Pd/Cs-Y | 91 (169 °C) | 67 (117 °C) | 66 (129 °C) | 0.10 |
The maximum CO conversions obtained are very high for all the catalysts studied, except for sample Pd/H-Y, reaching values very close to 90% and even higher for samples with Y zeolite. However, the temperature at which this maximum conversion is reached is different for all the catalysts. In this sense, if samples with the same zeolite structure and different ion-exchange cations are considered, it seems that zeolites ion-exchanged with sodium reach higher CO conversion and that these maximum conversion values are achieved at lower temperatures than in the other samples. Regarding selectivity, relatively high values of selectivity are also reached for all the catalysts, being higher for catalysts prepared with ZSM-5 zeolite. To better evaluate these values it is important to point out that under the experimental conditions used, an adequate catalyst for this reaction should have selectivity values towards CO oxidation over 50%, which means that CO oxidation is preferential over H2 oxidation and that, theoretically, all CO could be removed from the H2 flow.42
In the studied samples, maximum selectivity values towards CO oxidation between 65% and 77% were achieved at low CO conversion (low temperatures) and selectivity values very close to 50% remain at higher temperatures (see Fig. 5). Thus, bearing these results in mind and comparing with those reported elsewhere, where different catalytic systems were studied,7,14,43 it could be said that the prepared catalysts are very suitable materials for their use in the PrOx-CO reaction, especially in the case of samples ion-exchanged with sodium. Taking into account the results shown in Table 4, it seems that the addition of cations with lower charge density in the zeolite structure plays a positive role in the catalytic behaviour of the sample. This improvement when cations with lower charge density are added has been observed by other authors for samples based on zeolites44 and other systems.45–47
As can be seen, this improvement mainly reflects on the selectivity at 25% CO conversion and TOF parameters. As can be extracted from Table 4, the selectivity values achieved for 25% CO conversion are between 53% and 73% for the six studied samples and, in general, the achieved values are higher in samples with ZSM-5 zeolite. In samples based on ZSM-5 zeolite, this value of CO conversion is achieved at very similar temperatures for the three studied samples, but the selectivity is higher in sample Pd/Na-ZSM-5. Regarding the samples based on Y zeolite, the 25% CO conversion is achieved at temperatures much lower in the case of the samples ion-exchanged with Na+ and Cs+ cations (120 and 129 °C for samples Pd/Na-Y and Pd/Cs-Y, respectively) than for the protonated counterpart (163 °C). In the case of sample Pd/H-Y an important shift towards higher temperatures is observed for CO conversion and selectivity curves. This poor behaviour of sample Pd/H-Y under the experimental conditions used may be related to sintering of the palladium nanoparticles observed for this sample (see Table 2) when it is pretreated in 10% H2/He at 200 °C and under the subsequent catalytic test experimental conditions (gas mixture composed of 2% CO, 2% O2, 30% H2, and balance He and heated up to 200 °C), probably as a consequence of the weaker interaction between the palladium nanoparticles and the H-Y support compared to the Na-Y support. This fact indicates that the catalytic performance of the studied samples in the preferential CO oxidation reaction strongly depends on the average palladium nanoparticle size, as was previously reported by our group.7
Concerning the TOF parameter, it can be observed that when the catalytic activity is normalised, taking into account the metallic loading of the samples and the size of the particles, all the ZSM-5 catalysts show a much poorer behaviour in terms of activity than the Y catalysts.
Bearing in mind the support pore size and the Pd nanoparticle size, it is clear that, as was previously said, Pd nanoparticles are located on the outer zeolite surface. Thus, the location of the nanoparticles pointed out that the inner surface area of the zeolites does not play a significant role in the oxidation of CO. However, the outstanding catalytic performance of samples based on Y zeolite, (mainly in terms of TOF) could be related to the larger surface area of this zeolite. According to the porous texture characterisation results (see Table 1), samples based on Y zeolite may adsorb larger amounts of CO molecules in their porous structure. This larger presence of CO accumulated within the porous structure in samples with Y zeolites could tentatively explain the improved catalytic activity of these samples.
Once the results obtained in this work have been presented and discussed, a comparison with results reported in the literature should be carried out in order to highlight the strength of the studied Pd/zeolite-based catalytic systems in the PrOx-CO reaction. In this regard, the use of zeolitic materials as support for catalysts used in the PrOx-CO reaction is not very widely reported, with Al2O3 combined with a different noble metal7,14,48 and CuO/CeO2 (ref. 49–51) being the most common catalytic systems used for this application. Concerning the zeolitic system, the metallic active phase is normally incorporated by an ion exchange procedure; thus the metal might be located inside the zeolite porosity, leading to a lower catalytic performance, as claimed by Luengnaruemitchai et al.24 These authors prepared A-type zeolite-supported Pt catalysts by the impregnation method, and the metal was placed outside the pore of LTA as in the case of the palladium nanoparticles used in the present study. In that study, the catalytic activity was evaluated using a gas mixture composed of 1% CO, 1% O2, 40% H2 and He (balance) at a total flow rate of 50 ml min−1. Under these experimental conditions, almost 100% CO conversion and ∼50% selectivity were achieved at 200 °C approximately. Moreover, Rosso et al.22 prepared Pt/3A catalysts by different experimental procedures, cation exchange, wet impregnation and incipient wetness impregnation. The obtained results also indicated that the location of the metal and the catalytic performance depend on the experimental method used to load the metallic phase. The result reported in that work claimed that the location of Pt particles outside the zeolite pores, resulting from the wet impregnation and incipient wetness impregnation methods, has a positive effect on the preferential CO oxidation. Similar results were also obtained by Rivallan et al.52 for Pt nanoparticles incorporated on ZSM-5. Kotobuki and coworkers53 studied a catalytic system based on Pt–Fe alloy nanoparticles supported on mordenite, with a very high metal loading in comparison with the present work (4 and 0.5 wt% for Pt and Fe, respectively) and they confirmed that the CO conversion depends on the oxygen excess (λ). When λ = 2, as in the present study, they achieved about 100% CO conversion and ∼50% CO selectivity at about 100 °C, but these values decreased drastically to ∼67% CO conversion and ∼25% CO selectivity at 200 °C.
The suitable catalytic performance of the samples studied in this work is also evidenced by the comparison with other catalytic systems studied in the literature, where different supports apart from zeolites are studied. In this sense, the comparison with the results previously obtained in our group using palladium nanoparticles supported on alumina catalysts,7 where 55% selectivity was achieved for a 25% CO conversion, reveals that the present catalytic systems are very suitable materials for their use in the PrOx-CO reaction.
After presenting the screening of the catalytic behaviour in the PrOx-CO reaction of the samples studied in this work and comparing with those previously reported in the literature, it can be said that the Pd/zeolite-based samples are very promising catalysts in the preferential CO oxidation reaction, with sample Pd/Na-Y being the most interesting catalyst. At this point, once the Pd/Na-Y sample was identified as the most effective catalyst, stability test of this sample was also performed and shown in Fig. 6.
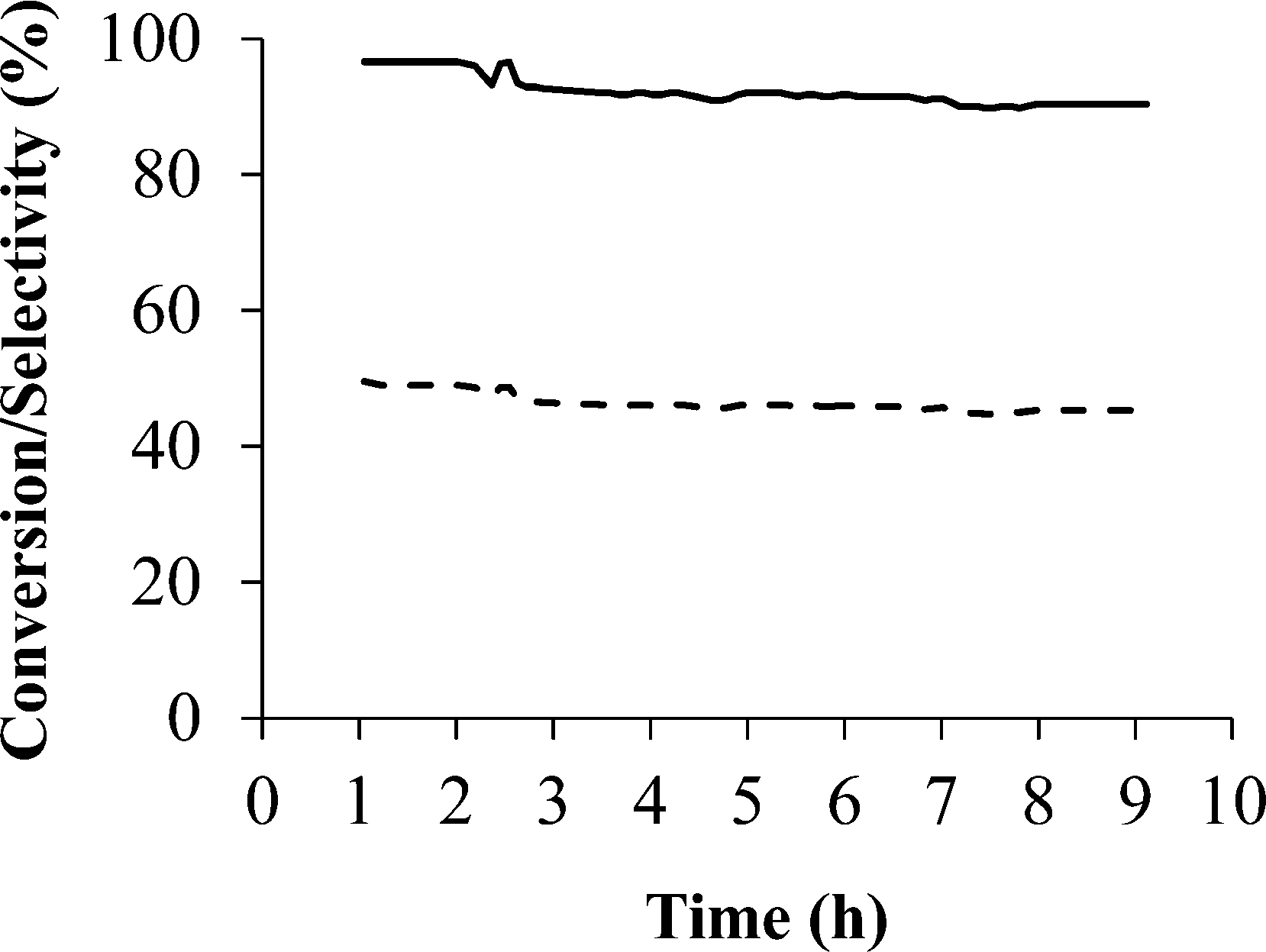 | ||
| Fig. 6 Stability test of sample Pd/Na-Y carried out at 200 °C and with a gas mixture composed of 2% CO, 2% O2, 30% H2, 20% CO2, 5% H2O and balance He. CO conversion (%) is plotted as a continuous line and selectivity (%) as a dotted line. | ||
The stability test results obtained for sample Pd/Na-Y indicated that the decrease in both CO conversion and selectivity is not very pronounced (CO conversion from 96% to 90%; CO selectivity from 50% to 45%) when the sample is subjected to the working conditions used. Thus, no deactivation was observed for sample Pd/Na-Y during the 9 h of experiment, even when CO2 and H2O are added in the gas mixture. The high stability observed for sample Pd/Na-Y suggests that there is no sintering or coke formation on the catalyst during the reaction. Therefore, it can be said that this is a promising material as a catalyst in the PrOx-CO reaction since high CO conversion and selectivity values are achieved, which remain over the long-term stability test.
Conclusions
Samples based on palladium nanoparticles supported on ion-exchanged ZSM-5 and Y zeolites have been prepared and tested in the PrOx-CO reaction. The obtained results seem to indicate that the catalytic performance of this type of materials depends on the zeolite structure, the composition and on the cation present in the zeolite.All the studied samples showed a very promising catalytic performance, with the exception of sample Pd/H-Y, which displayed a poor catalytic behaviour due to the palladium nanoparticles sintering, caused probably by the weaker interaction with the zeolitic support.
The screening of the Pd/zeolite-based catalysts in the PrOx-CO reaction in H2-rich streams showed the suitability of zeolites as supports for the PVP-capped Pd nanoparticles, as very interesting catalytic performances were observed for all the studied catalysts. In particular, the outstanding catalytic behaviour displayed by the sample Pd/Na-Y in the long-term stability tests indicated that this is a promising catalyst in the PrOx-CO reaction, as high CO conversion and CO selectivity values were achieved during a long period of time.
Acknowledgements
The authors thank the Spanish MINECO, GV and FEDER (PROMETEOII/2014/010, Projects CTQ2012-31762), for financial support.Notes and references
- A. Arango-Díaz, E. Moretti, A. Talon, L. Storaro, M. Lenarda, P. Núñez, J. Marrero-Jerez, J. Jiménez-Jiménez, A. Jiménez-López and E. Rodríguez-Castellón, Appl. Catal., A, 2014, 477, 54 CrossRef
.
- I. Rosso, C. Galletti, G. Saracco, E. Garrone and V. Specchia, Appl. Catal., B, 2004, 48, 195 CrossRef CAS
- K. Liu, A. Wang and T. Zhang, ACS Catal., 2012, 2, 1165 CrossRef CAS
- E. D. Park, D. Lee and H. C. Lee, Catal. Today, 2009, 139, 280 CrossRef CAS
- F. Mariño, C. Descorme and D. Duprez, Appl. Catal., B, 2004, 54, 59 CrossRef
- O. Pozdnyakova, D. Teschner, A. Wootsch, J. Kröhnert, B. Steinhauer, H. Sauer, L. Toth, F. C. Jentoft, A. Knop-Gericke, Z. Paál and R. Schlögl, J. Catal., 2006, 237, 17 CrossRef CAS
- I. Miguel-García, Á. Berenguer-Murcia and D. Cazorla-Amorós, Appl. Catal., B, 2010, 98, 161 CrossRef
- I. Miguel-García, Á. Berenguer-Murcia, T. García and D. Cazorla-Amorós, Catal. Today, 2012, 187, 2–9 CrossRef
- T. S. Nguyen, F. Morfin, M. Aouine, F. Bosselet, J. L. Rousset and L. Piccolo, Catal. Today, 2015, 253, 106 CrossRef CAS
- F. Morfin, A. Ait-Chaou, M. Lomello and J. L. Rousse, J. Catal., 2015, 331, 210 CrossRef CAS
- A. Kubacka, A. Martínez-Arias and M. Fernández-García, ChemCatChem, 2015, 7, 3614 CrossRef CAS
- J. Kugai, T. Moriya, S. Seino, T. Nakagawa, Y. Ohkubo, H. Nitani, Y. Mizukoshi and T. A. Yamamoto, Appl. Catal., B, 2012, 126, 306 CrossRef CAS
- M. Kipnis and E. Volnina, Appl. Catal., B, 2010, 98, 193 CrossRef CAS
- A. Manasilp and E. Gulari, Appl. Catal., B, 2002, 37, 17 CrossRef CAS
- Y. H. Kim and E. D. Park, Appl. Catal., B, 2010, 96, 41 CrossRef CAS
- H. Igarashi, H. Uchida, M. Suzuki, Y. Sasaki and M. Watanabe, Appl. Catal., A, 1997, 159, 159 CrossRef CAS
- M. Watanabe, H. Uchida, K. Ohkubo and H. Igarashi, Appl. Catal., B, 2003, 46, 595 CrossRef CAS
- R. López-Fonseca, J. I. Gutiérrez-Ortiz, M. A. Gutiérrez-Ortiz and J. R. González-Velasco, Catal. Today, 2005, 107–108, 200 CrossRef
- F. Ribeiro, J. M. Silva, E. Silva, M. F. Vaz and F. A. C. Oliveira, Catal. Today, 2011, 176, 93 CrossRef CAS
- Saepurahman, M. Visur, U. Olsbye, M. Bjørgen and S. Svelle, Top. Catal., 2011, 54, 1293 CrossRef CAS
- J. Čejka, G. Centi, J. Perez-Pariente and W. J. Roth, Catal. Today, 2012, 179, 2 CrossRef
- I. Rosso, C. Galletti, S. Fiorot, G. Saracco, E. Garrone and V. Specchia, J. Porous Mater., 2007, 14, 245 CrossRef CAS
- J. Xu, X.-C. Xu, L. Ouyang, X.-J. Yang, W. Mao, J. Su and Y.-F. Han, J. Catal., 2012, 287, 114 CrossRef CAS
- A. Luengnaruemitchai, M. Nimsuk, P. Naknam, S. Wongkasemjit and S. Osuwan, Int. J. Hydrogen Energy, 2008, 33, 206 CrossRef CAS
- P. Naknam, A. Luengnaruemitchai, S. Wongkasemjit and S. Osuwan, J. Power Sources, 2007, 165, 353 CrossRef CAS
- A. R. Tao, S. Habas and P. Yang, Small, 2008, 4, 310 CrossRef CAS
- P. Lu, T. Teranishi, K. Asakura, M. Miyake and N. Toshima, J. Phys. Chem. B, 1999, 103, 9673 CrossRef CAS
- S. Domínguez-Domínguez, Á. Berenguer-Murcia, D. Cazorla-Amorós and Á. Linares-Solano, J. Catal., 2006, 243, 74 CrossRef
-
M. Boudart, Kinetics of Heterogeneous Catalytic Reactions, Princeton University Press, Princeton, NJ, 1984 Search PubMed
- M. Navlani-Garcia, M. Martis, D. Lozano-Castello, D. Cazorla-Amoros, K. Mori and H. Yamashita, Catal. Sci. Technol., 2015, 5, 364 CAS
- F. J. Varela-Gandía, A. Berenguer-Murcia, D. Lozano-Castelló and D. Cazorla-Amorós, J. Membr. Sci., 2010, 351, 123 CrossRef
- R. Khaleghian-Moghadam and F. Seyedeyn-Azad, Microporous Mesoporous Mater., 2009, 120, 285 CrossRef CAS
- C. Bisio, G. Martra, S. Coluccia and P. Massiani, J. Phys. Chem. C, 2008, 112, 10520 CAS
- Y. T. Kim, K.-D. Jung and E. D. Park, Appl. Catal., A, 2011, 393, 275 CrossRef CAS
- B. Xu and L. Kevan, J. Phys. Chem., 1992, 96, 2642 CrossRef CAS
- R. M. Serra, E. E. Miró, P. Bolcatto and A. V. Boix, Microporous Mesoporous Mater., 2012, 147, 17–29 CrossRef
- J. Xu, B. L. Mojet and L. Lefferts, Microporous Mesoporous Mater., 2006, 9, 187 CrossRef
- B. Siyo, M. Schneider, J. Radnik, M.-M. Pohl, P. Langer and N. Steinfeldt, Appl. Catal., A, 2014, 478, 107 CrossRef CAS
- Y. Borodko, S. E. Habas, M. Koebel, P. Yang, H. Frei and G. A. Somorjai, J. Phys. Chem. B, 2006, 110, 23052 CrossRef CAS PubMed
- Y. Borodko, S. M. Humphrey, T. D. Tilley, H. Frei and G. A. Somorjai, J. Phys. Chem. C, 2007, 111, 6288 CAS
- O. H. Laguna, E. M. Ngassa, S. Oraá, A. Álvarez, M. I. Domínguez, F. Romero-Sarria, G. Arzamendi, L. M. Gandía, M. A. Centeno and J. A. Odriozola, Catal. Today, 2012, 180, 05 CrossRef
- M. J. Kahlich, H. A. Gasteiger and R. J. Behm, J. Catal., 1997, 171, 93 CrossRef CAS
- N. Iwasa, S. Arai and M. Arai, Appl. Catal., B, 2008, 79, 132 CrossRef CAS
- D. G. Lahr, J. Li and R. J. Davis, J. Am. Chem. Soc., 2007, 129, 3420 CrossRef CAS PubMed
- M. Kuriyama, H. Tanaka, S.-I. Ito, T. Kubota, T. Miyao, S. Naito, K. Tomishige and K. Kunimori, J. Catal., 2007, 252, 39 CrossRef CAS
- Y. Minemura, M. Kuriyama, S.-I. Ito, K. Tomishige and K. Kunimori, Catal. Commun., 2006, 7, 623 CrossRef CAS
- S. Derrouiche, P. Gravejat, B. Bassou and D. Bianchi, Appl. Surf. Sci., 2007, 253, 5894 CrossRef CAS
- J. Yin, J. Wang, T. Zhang and X. Wang, Catal. Lett., 2008, 125, 76 CrossRef CAS
- M. Monte, D. Gamarra, A. López Cámara, S. B. Rasmussen, N. Gyorffy, Z. Schay, A. Martínez-Arias and J. C. Conesa, Catal. Today, 2014, 229, 104 CrossRef CAS
- S. Zeng, Y. Wang, S. Ding, J. J. H. B. Sattler, E. Borodina, L. Zhang, B. M. Weckhuysen and H. Su, J. Power Sources, 2014, 256, 301 CrossRef CAS
- G. Landi, P. S. Barbato, A. Di Benedetto and L. Lisi, Appl. Catal., A, 2016, 181, 727–737 CrossRef CAS
- M. Rivallan, S. Thomas, M. Lepage, N. Takagi, H. Hirata and F. Thibault-Starzyk, ChemCatChem, 2010, 2, 1599 CrossRef CAS
- M. Kotobuki, A. Watanabe, H. Uchida, H. Yamashita and M. Watanabe, Appl. Catal., A, 2006, 307, 275 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Details of catalysts support characterisation. See DOI: 10.1039/c5cy02044a |
| This journal is © The Royal Society of Chemistry 2016 |